

Abstract
The product of the von Hippel–Lindau gene, pVHL, targets the α subunits of the heterodimeric transcription factor hypoxia-inducible factor (HIF) for polyubiquitination in the presence of oxygen. The binding of pVHL to HIF is governed by the enzymatic hydroxylation of conserved prolyl residues within peptidic motifs present in the HIFα family members. By using a biochemical purification strategy, we have identified a human homolog of Caenorhabditis elegans Egl9 as a HIF prolyl hydroxylase. In addition, we studied the activity of a structurally diverse collection of low molecular weight inhibitors of procollagen prolyl 4-hydroxylase as potential inhibitors of the HIF hydroxylase. A model compound of this series stabilized HIF in a variety of cells, leading to the increased production of its downstream target, vascular endothelial growth factor.
Inactivation of the von Hippel–Lindau tumor suppressor gene is linked to the development of a variety of tumors, including clear cell carcinomas of the kidney and hemangioblastomas of the central nervous system (1). The product of this gene, pVHL, is part of a multiprotein complex that includes elongin B, elongin C, Rbx1, and Cul2 (reviewed in ref. 2). This complex functions as an E3 ubiquitin ligase which, in the presence of oxygen, targets the α subunits of the heterodimeric transcription factor HIF (hypoxia-inducible factor) for proteasome-dependent degradation (3–6). Consequently, tumors lacking pVHL overproduce HIF and its downstream targets. Among the latter are proteins implicated in angiogenesis, glucose uptake and metabolism, and mitogenesis (7–9).
Polyubiquitination of HIF by pVHL is inhibited by hypoxia as well as by hypoxia-mimetics, such as compounds that chelate or compete with iron. These observations can now be understood in light of the recent finding that the interaction of pVHL with HIF is linked to the enzymatic hydroxylation of conserved proline residues in HIF (10–13). The known prolyl hydroxylases require oxygen, iron, and the cofactor 2-oxoglutarate (14, 15). In addition, ascorbate is required to prevent the rapid inactivation of these enzymes by self-oxidation.
Several lines of evidence suggested that the HIF prolyl hydroxylase (HIF PH) was not identical to the well studied type I and type II mammalian prolyl hydroxylases, which modify collagen. First, the region of HIF that is hydroxylated does not resemble the (Pro-Pro-Gly)n motif that is recognized by the type I and type II prolyl hydroxylases (10–13). Second, type I and type II prolyl hydroxylases reside in the endoplasmic reticulum (ER), whereas HIF is an intracellular protein. Third, collagen prolyl hydroxylase activity is preserved under hypoxic conditions that are sufficient to inhibit HIF PH activity (16).
A variety of cell extracts contain HIF PH activity (10–13). By using a biochemical purification scheme we identified a human homolog of the Caenorhabditis elegans Egl9 gene as a mammalian HIF prolyl hydroxylase. While this work was in progress the same conclusion was reached by others using primarily bioinformatic and genetic approaches (17, 18). In parallel, a diverse set of low molecular weight compounds was identified that inhibit HIF PH activity in vitro, potentially opening the path for pharmacological manipulation of this oxygen-sensing pathway.
Materials and Methods
Preparation of Dialyzed Rabbit Reticulocyte Lysate (RRL).
One milliliter of RRL (Green Hectares, Oregon, WI) was dialyzed overnight against prolyl hydroxylation assay (PHA) buffer [40 mM Hepes (pH 7.4)/80 mM KCl/5% glycerol] by using a 3,000-Da cutoff dialyzer cassette (Pierce) and either used immediately, or snap frozen in dry ice/ethanol for later use.
In Vitro Transcription/Translation.
Coupled in vitro transcription/translation using wheat germ extract (TNT; Promega) or RRL (TNT; Promega) was carried out in the presence of [35S]methionine according to the manufacturer's instructions. Protein production was confirmed by SDS/PAGE, followed by autoradiography.
HIF Peptide Hydroxylation Assay.
One microgram of a synthetic, N-terminally biotinylated, peptide corresponding to HIF1α residues 556–575 (DLDLEMLAPYIPMDDDFQLR) was immobilized on 30 μl of streptavidin-agarose (Pierce) and incubated with either 50 μl of RRL, various amounts of RRL-derived column fractions, or recombinant EGLN1 for 2 hr, with tumbling, at room temperature. The total reaction volume was adjusted to 200 μl with PHA buffer and, where indicated, the following cofactors were added: FeCl2 (100 μM), ascorbate (2 mM), and 2-oxoglutarate (5 mM). The agarose was then washed four times with NETN [20 mM Tris⋅HCl (pH 8.0)/100 mM NaCl/1 mM EDTA/0.5% Nonidet P-40], and peptide hydroxylation was determined by pVHL-binding or by MS. For the former, 10 μl of 35S-labeled pVHL was added in a total volume of 500 μl of EBC [50 mM Tris⋅HCl (pH 8.0)/120 mM NaCl/0.5% Nonidet P-40] supplemented with complete protease inhibitor mixture (Roche Molecular Biochemicals, Indianapolis) and incubated for 1 hr at 4°C. After four washes with NETN, bound pVHL was eluted by boiling in SDS-containing sample buffer, resolved by PAGE, and detected by autoradiography. For the latter, a modified HIF1α peptide was used in which the two methionine residues were converted to alanine to avoid spurious oxidation events (10). After NETN washes, the peptide was washed once with PBS, eluted in 100 μl of 20 mM ammonium acetate (pH 7.0)/2 mM biotin, and analyzed by matrix-assisted laser desorption ionization–time-of-flight (MALDI-TOF) MS as described (10).
Purification of HIF PH.
RRL (0.3 liters) was diluted to 1.5 liters in 50 mM Tris⋅HCl (pH 7.4)/0.1 M KCl/5% (vol/vol) glycerol, and precipitated with 0.213 g/ml (NH4)2SO4. After centrifugation at 16,000 × g for 45 min, the supernatant was precipitated with an additional 0.153 g/ml (NH4)2SO4. The resulting pellet was resuspended in buffer A [40 mM Hepes (pH 7.4)/5% (vol/vol) glycerol], dialyzed against buffer A to conductivity equivalent to buffer A containing 0.1 M KCl, and applied at 0.4 l/hr to a 0.2 l phosphocellulose (Whatman, P11) column equilibrated in buffer A containing 0.1 M KCl. The phosphocellulose column was eluted stepwise at 0.4 liter/hr with buffer A containing 0.2 M and 0.5 M KCl, and 40-ml fractions were collected. Active fractions (assayed on biotinylated HIF peptide as above) were pooled and precipitated with 0.4 g/ml (NH4)2SO4. After centrifugation at 16,000 × g for 45 min, the pellet was resuspended in 4 ml of buffer A, centrifuged at 35,000 × g for 30 min, and applied at 2 ml/min to a TSK SW4000 HPLC column (TosoHaas, Montgomeryville, PA; 21.5 × 600 mm) equilibrated in buffer A containing 0.15 M KCl. The SW4000 column was eluted at 2 ml/min, and 4-ml fractions were collected. Pooled active fractions were diluted with buffer A to a conductivity equivalent to buffer A containing 0.1 M KCl, and applied at 1 ml/min to a TSK SP 5-PW HPLC column (TosoHaas; 7.5 × 75 mm). The SP 5-PW column was eluted at 1 ml/min with a 30-ml linear gradient from 0.1 to 0.5 M KCl in buffer A, and 1-ml fractions were collected. Active fractions were pooled, diluted 1:1 with buffer A containing 1.5 M (NH4)2SO4, and applied at 1 ml/min to a TSK phenyl 5-PW HPLC column (TosoHaas; 7.5 × 75 mm) equilibrated in buffer A containing 1.5 M (NH4)2SO4. The phenyl 5-PW column was eluted at 1 ml/min with a 30 ml linear gradient from 1.5 M to 0 M (NH4)2SO4 in buffer A, and 1-ml fractions were collected. Proteins from active fractions were precipitated with trichloroacetic acid, separated by SDS/PAGE, and stained with Coomassie blue. All of the visible bands were excised, and peptide sequences were obtained by MS.
Subcloning of EGLN1 cDNA.
A cDNA clone (FB2557_D04) from Origene Technologies (Rockville, MD) encoding full-length human EGLN1 was PCR amplified with the primers CGCGGATCCATGGCCAATGACAGCGGC (forward) and GCGCGAATTCCTAGAAGACGTCTTTACC (reverse), digested with BamHI and EcoRI, and ligated into pcDNA3-HA [introducing an N-terminal hemagglutinin (HA) epitope tag] (Invitrogen) and pGEX2TK (Amersham Pharmacia Biotech, Piscataway, NJ) vectors linearized with the same enzymes. Both plasmids were authenticated by DNA sequencing.
Production of GST-Fusion Proteins.
Logarithmic phase cultures of Escherichia coli (DH5α) transformed with pGEX2TK-EGLN1 were grown in LB supplemented with 0.1 mM isopropyl β-d-thiogalactoside (IPTG) for 3 hr at 37°C and then sonicated on ice in NETN buffer supplemented with complete protease inhibitor mixture (Roche). After centrifugation at 10,000 × g for 20 min at 4°C the clarified sonicate was incubated with glutathione-Sepharose (Amersham Pharmacia Biotech) for 1 hr with rocking at 4°C. After four washes with NETN, GST-EGLN1 was eluted in 100 mM Tris (pH 8.0)/120 mM NaCl/20 mM reduced glutathione. Production of GST-pVHL/elongin B/elongin C complexes was as described (10, 19). The integrity and yield of purified GST fusion proteins was assessed by SDS/PAGE followed by Coomassie blue staining.
Hydroxylation of Full-Length HIF1α.
Production and purification of His-tagged human HIF1α by using a baculoviral expression system was as described (4). The indicated amount of His-HIF1α was incubated with 50 μl of unprogrammed wheat germ extract or wheat germ extract programmed with pcDNA3 HA-EGLN1, for 2 hr at room temperature, in the presence of FeCl2 (100 μM), ascorbate (2 mM), and 2-oxoglutarate (5 mM). The reaction was then diluted to a total volume of 500 μl with EBC buffer supplemented with protease inhibitors and added to 40 μl of glutathione-Sepharose preloaded with GST-pVHL/elongin B/elongin C (GST-VBC). After 1 hr of incubation at 4°C, the Sepharose was washed four times with NETN. Bound HIF1α was eluted by boiling in SDS-containing sample buffer, resolved by SDS/PAGE, and detected by immunoblotting with an anti-HIF1α monoclonal antibody (clone H1α-67, 1:500 dilution, Novus Biologicals, Littleton, CO).
Prolyl Hydroxylase Inhibitors.
Prolyl hydroxylase inhibitors (FibroGen) were dissolved in DMSO at a stock concentration of 10 mM. For RRL activity inhibition, the RRL was preincubated with the indicated inhibitor for 15 min before use in the peptide hydroxylation assay. GST-EGLN1 was preincubated with the indicated inhibitor in 200 μl of PHA buffer for 15 min before addition of the biotinylated HIF peptide and cofactors.
Stabilization of HIF in Human Cell Lines.
Human foreskin fibroblasts (HFF), human lung fibroblasts (HLF), SCC-25 squamous carcinoma cells, and MCF7 mammary carcinoma cells were grown in DMEM with 10% FBS, 293 human embryonic kidney cells in DMEM with 5% FBS, human microvascular endothelial cells (HMEC-1) in RPMI medium 1640 with 10% FBS, human venous endothelial cells (AG10774B) and human umbilical vein endothelial cells (HUVEC) in endothelial growth media (EGM-2; BioWhittaker). Cells were grown in the presence of 20% O2 and 10% CO2 at 37°C. Confluent cells were washed twice with PBS and incubated overnight in OPTI-MEM medium (Invitrogen) containing 10 or 25 μM FG-0041 or 1% DMSO. The concentration of vascular endothelial growth factor (VEGF) in the conditioned medium from a comparable number of cells was measured by using a Quantikine immunoassay (R & D Systems) according to the manufacturer's instructions. The cells were washed twice with PBS and lysed in 10 mM Tris (pH 7.4)/150 mM NaCl/1 mM EDTA/0.5% IGEPAL CA-630 and protease inhibitor mix (Roche Molecular Biochemicals, Indianapolis) for 15 min on ice. After centrifugation for 5 min at 3,000 × g the nuclear pellet was lysed in 20 mM Hepes (pH 7.2)/400 mM NaCl/1 mM EDTA/1 mM DTT and protease inhibitor mix. The nuclear extracts were clarified by centrifugation for 5 min at 13,000 × g, resolved by SDS/PAGE, and immunoblotted with an anti-HIF1α monoclonal antibody (1:250 dilution; BD Biosciences-PharMingen, San Diego) and SuperSignal chemiluminescent detection system (Pierce). Comparable amounts of protein were loaded per well as determined by a BCA Protein Assay kit (Pierce) and confirmed by Coomassie blue staining of the membranes after transfer.
Results
RRL contains an enzymatic activity capable of hydroxylating an evolutionarily conserved proline residue present in HIF α subunits (10, 11, 13). Furthermore, this enzymatic activity does not appear to be attributable to the type I and type II prolyl hydroxylases, which hydroxylate collagen. The known prolyl hydroxylases require molecular oxygen and the cofactors iron, ascorbate, and 2-oxoglutarate. To determine the cofactor requirement of the HIF PH in anticipation of our attempts to purify this enzyme, we carried out HIF hydroxylation assays using RRL which was or was not dialyzed by using a membrane with a 3,000 Da molecular mass cutoff (Fig. 1). Hydroxylation was scored based on binding of an immobilized HIF1α peptide (corresponding to residues 556–575) to radiolabeled pVHL after incubation with RRL. Dialyzed RRL lost the ability to modify HIF (Fig. 1a). This activity could be restored, however, by the addition of ascorbate (Fig. 1b). The fact that addition of 2-oxoglutarate and iron was not required in these assays suggests either that these cofactors are not necessary for HIF PH activity or, more likely, that these cofactors are tightly bound by HIF PH and do not dissociate from the enzyme under the dialysis conditions used here.
Figure 1.

Cofactor requirement for HIF PH. (a) Binding of 35S-labeled pVHL in vitro translation products (IVT) to biotinylated HIF 556–575) peptide, which was preincubated with dialyzed (+) or undialyzed (−) RRL. (b and c) Binding of 35S-labeled pVHL to biotinylated HIF(556–575) peptide, which was preincubated with dialyzed RRL (b) or GST-EGLN1 (c), and supplemented with the indicated cofactors.
We next partially purified the HIF PH present in RRL by using the scheme depicted in Fig. 2a. HIF PH activity was monitored with the assay described above by using column fractions supplemented with iron, ascorbate, and 2-oxoglutarate. A representative analysis of TSK SP 5-PW column fractions is shown in Fig. 2b. After initial enrichment by ammonium sulfate precipitation and four rounds of column chromatography, the fractions exhibiting HIF PH activity contained approximately 15 proteins, as determined by Coomassie blue staining. MS sequencing of these proteins revealed a number of proteins, such as eukaryotic translation factors and microtubule-associated proteins, which are frequently identified as contaminants in biochemical purification schemes similar to the one used here and/or seemed unlikely to encode prolyl hydroxylases. On the other hand, several peptides were identified from EGLN1 (nomenclature according to Taylor, ref. 20) (Fig. 2c), which is a human homolog of C. elegans Egl9. The C. elegans Egl9 gene was initially identified in a genetic screen for genes that, when mutated, give rise to egg-laying defects (21). Because Egl9 was predicted earlier, by using in silico approaches, to encode a 2-oxoglutarate-dependent, iron-dependent, dioxygenase, we focused our studies on this protein (22).
Figure 2.

Purification of HIF PH. (a) Purification scheme. (b) Binding of 35S-labeled pVHL to biotinylated HIF (556–575) peptide, which was preincubated with the indicated column fractions after TSK SP 5-PW chromatography. FT, flowthrough. (c) Conceptual ORF of EGLN1. Peptides identified by MS analysis of partially purified HIF PH are shown with single or double underline.
EGLN1 produced in wheat germ extract (Fig. 3a) and in E. coli (Fig. 3b) hydroxylated the HIF1α-derived peptide. Hydroxylation was confirmed by MS (Fig. 3c). Importantly, recombinant EGLN1 was also capable of hydroxylating full-length HIF1α produced in insect cells (Fig. 3d). Recombinant EGLN1, like the HIF PH activity in RRL, was sensitive to the addition of ascorbate, but not to oxoglutarate, as single agents (compare Fig. 1 c and b). The former, unlike the latter, was also sensitive to the addition of FeCl2. This difference, among several possibilities, might reflect the abundance of free and hemoglobin-bound iron in RRL.
Figure 3.

Hydroxylation of HIF by recombinant EGLN1. (a and b) Binding of 35S-labeled pVHL to biotinylated HIF(556–575) peptide, which was preincubated with EGLN1, produced in wheat germ extract (a, lane 3), or with increasing amounts of GST-EGLN1 fusion protein produced in E. coli (b, lanes 2 and 3). In parallel, peptide was preincubated with unprogrammed wheat germ extract (a, lane 2) or glutathione elution buffer (b, lane 1). (c) Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) analysis of biotinylated HIF(556–575) peptide before (Left) and after (Right) preincubation with GST-EGLN1. (d) Binding of recombinant, full-length, HIF1α (200 ng or 1 μg as indicated by triangles) to immobilized GST-pVHL, elongin B, and elongin C complexes in the absence (lanes l and 2) or presence (lanes 3 and 4) of EGLN1 produced in wheat germ extract. Bound HIF was detected by anti-HIF1α immunoblot (IB) analysis.
A number of structurally diverse small molecule inhibitors of the collagen prolyl hydroxylases have been developed (23–27) (Table 1). Several of these compounds inhibited the HIF PH present in RRL (Fig. 4a), as well as recombinant EGLN1 (Fig. 4b and data not shown) in low micromolar concentrations. Of note, the sensitivity profile of the HIF PH present in RRL was qualitatively similar to that observed with recombinant EGLN1 (Fig. 4 and data not shown), strengthening the notion that EGLN1 is responsible for the HIF PH activity present in RRL. Treatment of a variety of mammalian cells with the model compound FG-0041 in low micromolar concentrations led to the dose-dependent accumulation of HIF1α (Fig. 5a), which appeared to be transcriptionally active, as evidenced by the increased production of VEGF in the cell lines tested (Fig. 5b). Collectively, these observations suggest that EGLN1 can be inhibited by pharmacological agents at concentrations achievable in vivo.
Table 1.
Prolyl hydroxylase inhibitors
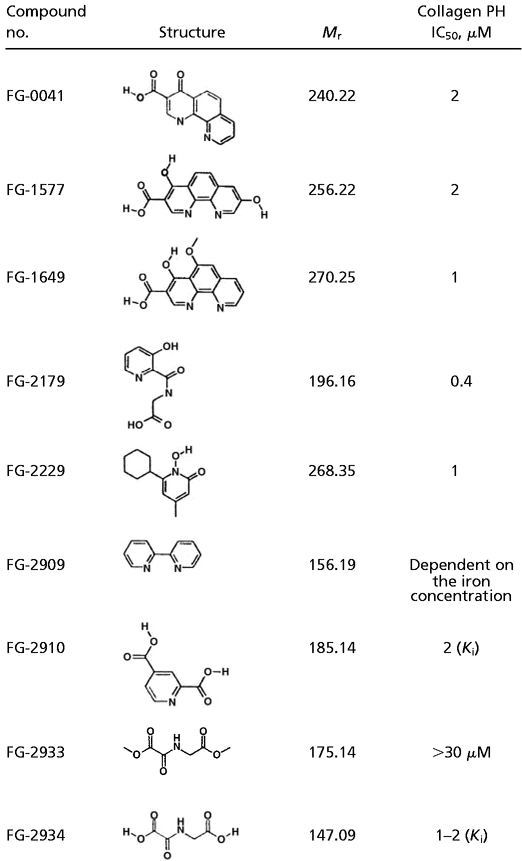 |
Figure 4.

Inhibition of EGLN1 activity by small molecules. (a) Binding of 35S-labeled pVHL to biotinylated HIF (556–575) peptide, which was preincubated with RRL in the presence of the indicated compounds (see Table 1). (b) Binding of 35S-labeled pVHL to biotinylated HIF(556–575) peptide, which was preincubated with GST-EGLN1 in the presence of the indicated compounds.
Figure 5.

Stabilization of transcriptionally active HIF in cells. (a) Anti-HIF1α immunoblot analysis of the indicated cells treated overnight with 0, 10, or 25 μM FG-0041, as indicated by the triangles. (b) VEGF levels of conditioned media from comparable numbers of cells treated as in a. Error bars indicate 1 standard deviation.
Discussion
The interaction of pVHL with HIF is governed by the enzymatic hydroxylation of conserved prolyl residues present in HIF paralogs and orthologs (10–13). We identified EGLN1 as a mammalian HIF PH by biochemical purification of the HIF PH activity present in RRL. While this work was in progress, two groups reported that EGLN1, EGLN2, and EGLN3, which are orthologs of C. elegans Egl9, represent HIF PHs (17, 18). Both of these groups used a candidate gene approach, based on the bioinformatic analysis of C. elegans and Drosophila melanogaster databases, coupled with genetic and biochemical validation studies. Thus, we arrived at the same conclusion as these earlier studies through the use of a complementary, unbiased, approach. Moreover, we show for the first time, to our knowledge, that a recombinant, human, Egl9 family member can hydroxylate full-length HIF, and we have begun to probe the universe of potential small molecule Egl9 inhibitors.
Inactivation of the lone Egl9 family member in C. elegans or D. melanogaster leads to HIF stabilization (17, 18). Neither our paper nor the earlier studies described above address the relative importance of the three mammalian Egl9 orthologs with respect to HIF hydroxylation in vivo. Thus, although we isolated EGLN1 from RRL we cannot exclude, with the reagents currently available, that EGLN2 and EGLN3 contribute to the total HIF hydroxylase activity present in such extracts. Furthermore we find that EGLN2, like EGLN1, can hydroxylate HIF1α in vitro, in keeping with earlier studies (refs. 17 and 18; data not shown). Conceivably, the relative contributions of the three Egl9 orthologs differ in various tissues and in diverse stages of development. Moreover, there are data suggesting that the three family members differentially hydroxylate two different proline residues in HIF (17). Finally, it will be important to determine whether any of the three mammalian Egl9 orthologs have targets other than HIF and, if so, whether such targets are unique to that family member or recognized by all three. In this regard, it is perhaps noteworthy that EGLN3 appears to be the ortholog of rat SM-20 (20). SM-20 resides in the mitochondria and has been implicated in the control of cell growth, differentiation, and apoptosis in cells such as muscle cells and neurons (28–32).
Recombinant EGLN1 is clearly sufficient to hydroxylate HIF1α. On the other hand, HIF PH activity present in RRL elutes with an apparent molecular mass of 320–440 kDa on size exclusion columns (M.I., R.C.C., and J.W.C., unpublished data). This raises the possibility that native HIF PH is composed of homomeric or heteromeric complexes that contain EGLN1. Further experiments are needed to address whether EGLN1 forms stable complexes with other proteins and, if so, whether such proteins modulate EGLN1 activity.
The requirement for oxoglutarate, iron, and ascorbic acid renders the prolyl hydroxylases susceptible to pharmacological attack. Ratcliffe and coworkers (11, 17) used the oxoglutarate analogs N-oxalyl-glycine (FG-2934) and dimethyloxalylglycine (FG-2933) to inhibit HIF PH in vitro and in worms We confirmed these earlier findings although clearly these agents are not potent enough to be suitable for clinical use. By studying a broader panel of procollagen prolyl hydroxylase inhibitors, we identified compounds that inhibit the HIF PH in low micromolar concentrations. Treatment of cells with one such compound, FG-0041, led to the accumulation of transcriptionally active HIF. Furthermore, we found that some inhibitors of collagen prolyl hydroxylase, such as Adriamycin (26) and 3,4-dihydroxybenzoic acid (33), did not inhibit HIF PH activity in vitro at the highest drug concentration tested (100 μM; data not shown). This finding suggests that it will be possible to design inhibitors that can discriminate between these two classes of hydroxylases.
HIF1α contains two transactivation domains called NTAD (N-terminal transactivation domain) and CTAD (C-terminal transactivation domain). A recent study showed that CTAD is inactive in the presence of oxygen because of the hydroxylation of a specific asparagine residue (34). The fact that FG-0041 led to the accumulation of transcriptionally active HIF suggests either that FG-0041 also inhibits asparagine hydroxylation, or that the NTAD is sufficient to activate transcription in this setting. Transcriptional activation by NTAD has also been invoked to account for the finding that disruption of the pVHL/HIF interaction, through inactivation of pVHL or the HIF pVHL-binding site, leads to robust activation of HIF target genes despite the presence of oxygen (35).
It has been suggested that stabilization of HIF through the inhibition of the HIF PH might be therapeutically beneficial in diseases characterized by acute or chronic ischemia (10, 36). Among these diseases are major causes of morbidity and mortality in the developed world, including myocardial infarction, stroke, peripheral vascular disease, and diabetes. Prolonged stabilization of HIF might lead to increased angiogenesis through the elaboration of HIF target genes such as VEGF and PDGF B. There have been concerns raised, however, as to whether HIF alone would be sufficient to induce mature, therapeutically useful, blood vessels (37). In this regard, a recent study found that transgenic dermal expression of a HIF mutant lacking the pVHL-binding domain led to accumulation of HIF and the induction of functional blood vessels (38). Short-term stabilization of HIF might lead to changes in cellular metabolism, such as changes in glucose uptake and metabolism, that would allow cells to survive acute hypoxic insults. Of note, FG-0041 was shown earlier to preserve left ventricular function in a rodent model of myocardial infarction (39). This effect was initially attributed to the inhibition of collagen prolyl hydroxylase (39). However, our finding that FG-0041 inhibits HIF PH activity, coupled with the fact that the beneficial effects observed in that study were already manifest within the first week after infarction (39), suggests that the salutary effect of FG-0041 was because of HIF stabilization rather than prevention of fibrosis.
Abbreviations
- HIF
hypoxia-inducible factor
- HIF PH
HIF prolyl hydroxylase
- RRL
rabbit reticulocyte lysate
- VEGF
vascular endothelial growth factor
References
- 1.Maher E, Kaelin W G., Jr Medicine (Baltimore) 1997;76:381–391. doi: 10.1097/00005792-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Kondo K, Kaelin W G., Jr Exp Cell Res. 2001;264:117–125. doi: 10.1006/excr.2000.5139. [DOI] [PubMed] [Google Scholar]
- 3.Cockman M, Masson N, Mole D, Jaakkola P, Chang G, Clifford S, Maher E, Pugh C, Ratcliffe P, Maxwell P. J Biol Chem. 2000;275:25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 4.Kamura T, Sato S, Iwain K, Czyzyk-Krzeska M, Conaway R C, Conaway J W. Proc Natl Acad Sci USA. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanimoto K, Makino Y, Pereira T, Poellinger L. EMBO J. 2000;19:4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohh M, Park C W, Ivan M, Hoffman M A, Kim T-Y, Huang L E, Chau V, Kaelin W G., Jr Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 7.Semenza G. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 8.Semenza G. Genes Dev. 2000;14:1983–1991. [PubMed] [Google Scholar]
- 9.Wenger R. J Exp Biol. 2000;203:1253–1263. doi: 10.1242/jeb.203.8.1253. [DOI] [PubMed] [Google Scholar]
- 10.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara J, Lane W, Kaelin W G., Jr Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 11.Jaakkola P, Mole D, Tian Y, Wilson M, Gielbert J, Gaskell S, Kriegsheim A, Hebestreit H, Mukherji M, Schofield C, et al. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 12.Yu F, White S, Zhao Q, Lee F. Proc Natl Acad Sci USA. 2001;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Masson N, Willam C, Maxwell P, Pugh C, Ratcliffe P. EMBO J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kivirikko K I, Myllyharju J. Matrix Biol. 1998;16:357–368. doi: 10.1016/s0945-053x(98)90009-9. [DOI] [PubMed] [Google Scholar]
- 15.Schofield C J, Zhang Z. Curr Opin Struct Biol. 1999;9:722–731. doi: 10.1016/s0959-440x(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi Y, Takahashi S, Shiga Y, Yoshimi T, Miura T. J Biol Chem. 2000;275:14139–14146. doi: 10.1074/jbc.275.19.14139. [DOI] [PubMed] [Google Scholar]
- 17.Epstein A, Gleadle J, McNeill L, Hewitson K, O'Rourke J, Mole D, Mukherji M, Metzen E, Wilson M, Dhanda A, et al. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 18.Bruick R, McKnight S. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 19.Stebbins C E, Kaelin W G, Jr, Pavletich N P. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- 20.Taylor M S. Gene. 2001;275:125–132. doi: 10.1016/s0378-1119(01)00633-3. [DOI] [PubMed] [Google Scholar]
- 21.Trent C, Tsuing N, Horvitz H R. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aravind, L. & Koonin, E. V. (2001) Genome Biol.2, research0007.1–0007.8. [DOI] [PMC free article] [PubMed]
- 23.Franklin T, Morris W, Edwards P, Large M, Stephenson R. Biochem J. 2001;353:333–338. doi: 10.1042/0264-6021:3530333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tschank G, Raghunath M, Gunzler V, Hanauske-Abel H M. Biochem J. 1987;248:625–633. doi: 10.1042/bj2480625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Majamaa K, Hanauske-Abel H M, Gunzler V, Kivirikko K I. Eur J Biochem. 1984;138:239–245. doi: 10.1111/j.1432-1033.1984.tb07907.x. [DOI] [PubMed] [Google Scholar]
- 26.Gunzler V, Hanauske-Abel H, Myllyla R, Kaska D, Hanauske A, Kivirikko K I. Biochem J. 1988;251:365–372. doi: 10.1042/bj2510365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baader E, Tschank G, Baringhaus K, Burghard H, Gunzler V. Biochem J. 1994;300:525–530. doi: 10.1042/bj3000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lipscomb E, Sarmiere P, Crowder R, Freeman R. J Neurochem. 1999;73:429–432. doi: 10.1046/j.1471-4159.1999.0730429.x. [DOI] [PubMed] [Google Scholar]
- 29.Lipscomb E, Sarmiere P, Freeman R. J Biol Chem. 2001;276:11775–11782. doi: 10.1074/jbc.M008407200. [DOI] [PubMed] [Google Scholar]
- 30.Moschella M, Menzies K, Tsao L, Lieb M, Kohtz J, Kohtz D, Taubman M. Gene Expression. 1999;8:59–66. [PMC free article] [PubMed] [Google Scholar]
- 31.Wax S, Tsao L, Lieb M, Fallon J, Taubman M. Lab Invest. 1996;74:797–808. [PubMed] [Google Scholar]
- 32.Wax S, Rosenfiled C, Taubman M. J Biol Chem. 1994;269:13041–13047. [PubMed] [Google Scholar]
- 33.Majamaa K, Gunzler V, Hanauske-Abel H M, Myllyla R, Kivirikko K I. J Biol Chem. 1986;261:7819–7823. [PubMed] [Google Scholar]
- 34.Lando D, Peet D, Whelan D, Gorman J, Whitelaw M. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 35.Kaelin W G., Jr Genes Dev. 2002;16:1441–1445. doi: 10.1101/gad.1003602. [DOI] [PubMed] [Google Scholar]
- 36.Bruick R, McKnight S. Genes Dev. 2001;15:2497–2502. doi: 10.1101/gad.931601. [DOI] [PubMed] [Google Scholar]
- 37.Carmeliet P. Nat Med. 2000;6:1102–1103. doi: 10.1038/80430. [DOI] [PubMed] [Google Scholar]
- 38.Elson D, Thurston G, Huang L, Ginzinger D, McDonald D, Johnson R, Arbeit J. Genes Dev. 2001;15:2520–2532. doi: 10.1101/gad.914801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nwogu J I, Geenen D, Bean M, Brenner M B, Huang X, Buttrick P M. Circulation. 2001;104:2216–2221. doi: 10.1161/hc4301.097193. [DOI] [PubMed] [Google Scholar]