

Abstract
Osteoporosis is a disorder in which loss of bone strength leads to fragility fractures. This review examines the fundamental pathogenetic mechanisms underlying this disorder, which include: (a) failure to achieve a skeleton of optimal strength during growth and development; (b) excessive bone resorption resulting in loss of bone mass and disruption of architecture; and (c) failure to replace lost bone due to defects in bone formation. Estrogen deficiency is known to play a critical role in the development of osteoporosis, while calcium and vitamin D deficiencies and secondary hyperparathyroidism also contribute. There are multiple mechanisms underlying the regulation of bone remodeling, and these involve not only the osteoblastic and osteoclastic cell lineages but also other marrow cells, in addition to the interaction of systemic hormones, local cytokines, growth factors, and transcription factors. Polymorphisms of a large number of genes have been associated with differences in bone mass and fragility. It is now possible to diagnose osteoporosis, assess fracture risk, and reduce that risk with antiresorptive or other available therapies. However, new and more effective approaches are likely to emerge from a better understanding of the regulators of bone cell function.
Osteoporosis, characterized by the loss of bone mass and strength that leads to fragility fractures, has probably existed throughout human history but only recently became a major clinical problem as human lifespan increased. In the early 19th century, Sir Astley Cooper, a distinguished English surgeon, noted “the lightness and softness that (bones) acquire in the more advanced stages of life” and that “this state of bone . . . favors much the production of fractures” (1). The term osteoporosis was coined by Johann Lobstein at about the same time, but the disorder he described was probably osteogenesis imperfecta (2). In 1940, the American physician and endocrinologist Fuller Albright described postmenopausal osteoporosis and proposed that it was the consequence of impaired bone formation due to estrogen deficiency (3). Subsequently, the concept that there are 2 forms of osteoporosis, one related to estrogen deficiency at the menopause and the other to calcium deficiency and aging of the skeleton, was proposed (4). This has been replaced by the current concept that osteoporosis represents a continuum, in which multiple pathogenetic mechanisms converge to cause loss of bone mass and microarchitectural deterioration of skeletal structure. These factors, coupled with an increased risk of falls, contribute to a high incidence of fragility fractures in osteoporotic patients.
There is a rapidly expanding amount of information, based on laboratory studies, that indicates that osteoporosis is likely to be caused by complex interactions among local and systemic regulators of bone cell function. The heterogeneity of osteoporosis may be due not only to differences in the production of systemic and local regulators, but also to changes in receptors, signal transduction mechanisms, nuclear transcription factors, and enzymes that produce or inactivate local regulators. Within the last decade, the identification of many of the regulatory mechanisms that have been linked to osteoporosis has been the result of genetic studies. Since the first human osteoporosis study indicated an association among bone mass, fragility, and polymorphisms in the vitamin D receptor (VDR) gene, more than 30 candidate genes have been reported that might influence skeletal mass and fragility (5, 6). However, these studies have presented conflicting data, due in part to small sample size and differences in the genetic background of control and disease subjects. Since osteoporosis is a complex, polygenic disorder, the contributions of specific gene polymorphisms are likely to be relatively small, but may still be clinically important. Large cohort studies using standardized genotyping methodology and genetically similar control and affected individuals are needed to better define the role of specific genes in osteoporosis pathogenesis, as exemplified by a recent study on estrogen receptor polymorphisms (7).
Basic pathogenetic mechanisms
Skeletal fragility can result from: (a) failure to produce a skeleton of optimal mass and strength during growth; (b) excessive bone resorption resulting in decreased bone mass and microarchitectural deterioration of the skeleton; and (c) an inadequate formation response to increased resorption during bone remodeling. In addition, the incidence of fragility fractures, particularly of the hip and wrist, is further determined by the frequency and direction of falls.
To understand how excessive bone resorption and inadequate formation result in skeletal fragility, it is necessary to understand the process of bone remodeling, which is the major activity of bone cells in the adult skeleton. The bone remodeling or bone multicellular units (BMUs) described many years ago by Frost and others (8) can occur either on the surface of trabecular bone as irregular Howship lacunae or in cortical bone as relatively uniform cylindrical haversian systems. As illustrated in Figure 1, the process begins with the activation of hematopoietic precursors to become osteoclasts, which normally requires an interaction with cells of the osteoblastic lineage. Because the resorption and reversal phases of bone remodeling are short and the period required for osteoblastic replacement of the bone is long, any increase in the rate of bone remodeling will result in a loss of bone mass. Moreover, the larger number of unfilled Howship lacunae and haversian canals will further weaken the bone. Excessive resorption can also result in complete loss of trabecular structures, so that there is no template for bone formation. Thus, there are multiple ways in which an increase in osteoclastic resorption can result in skeletal fragility. However, high rates of resorption are not always associated with bone loss; for example, during the pubertal growth spurt. Hence an inadequate formation response during remodeling is an essential component of the pathogenesis of osteoporosis.
Figure 1.

The BMU with possible sites of estrogen action. Bone remodeling on the surface of trabecular bone is illustrated here. The process is similar in the haversian systems in cortical bone. Osteoclast activation is ordinarily initiated by an interaction of hematopoietic precursors with cells of the osteoblast lineage but may also be initiated by inflammatory cells, particularly T cells (see Figure 2 for further details). Once osteoclasts are formed, there is a resorption phase of limited duration and a brief reversal phase, during which the bone surface is covered by mononuclear cells but bone formation has not yet begun. The formation phase is then initiated, possibly by factors produced by the osteoclast or reversal cells or released from the bone matrix. The formation phase, which is substantially longer than the first 3 phases, involves the production of matrix by progressive waves of osteoblasts. These then become flat lining cells, are embedded in the bone as osteocytes, or undergo apoptosis. Potential sites of action of estrogen include effects on T cell cytokine production (i); effects on stromal or osteoblastic cells to alter their production of RANKL or OPG (ii); direct inhibition of differentiated osteoclasts (iii); and effects on bone formation mediated by osteoblasts or osteocytes to enhance the response to mechanical forces initiated by these cells (iv). Note that the BMU is shown as being compartmentalized by an overlying layer of cells. It has been proposed that these are separated lining cells, but they may be of vascular origin.
Central role of estrogen
The concept that estrogen deficiency is critical to the pathogenesis of osteoporosis was based initially on the fact that postmenopausal women, whose estrogen levels naturally decline, are at the highest risk for developing the disease. Morphologic studies and measurements of certain biochemical markers have indicated that bone remodeling is accelerated at the menopause, as both markers of resorption and formation are increased (8, 9). Hence, contrary to Albright’s original hypothesis, an increase in bone resorption, and not impaired bone formation, appears to be the driving force for bone loss in the setting of estrogen deficiency. But the rapid and continuous bone loss that occurs for several years after the menopause must indicate an impaired bone formation response, since in younger individuals going through the pubertal growth spurt, even faster rates of bone resorption can be associated with an increase in bone mass. However, the increased bone formation that normally occurs in response to mechanical loading is diminished in estrogen deficiency, suggesting estrogen is both anti-catabolic and anabolic (10).
Estrogen deficiency continues to play a role in bone loss in women in their 70s and 80s, as evidenced by the fact that estrogen treatment rapidly reduces bone breakdown in these older women (11). Moreover, recent studies in humans have shown that the level of estrogen required to maintain relatively normal bone remodeling in older postmenopausal women is lower than that required to stimulate classic target tissues such as the breast and uterus (12). Fracture risk is inversely related to estrogen levels in postmenopausal women, and as little as one-quarter of the dose of estrogen that stimulates the breast and uterus is sufficient to decrease bone resorption and increase bone mass in older women (13). This greater sensitivity of the skeleton may be age related. In 3-month-old mice, the uterus appeared to be more responsive to estrogen than bone, whereas in 6-month-old mice, the reverse was found (14).
Estrogen is critical for epiphyseal closure in puberty in both sexes and regulates bone turnover in men as well as women. In fact, estrogen has a greater effect than androgen in inhibiting bone resorption in men, although androgen may still play a role (15). Estrogen may also be important in the acquisition of peak bone mass in men (16). Moreover, osteoporosis in older men is more closely associated with low estrogen than with low androgen levels (17).
Estrogen deficiency increases and estrogen treatment decreases the rate of bone remodeling, as well as the amount of bone lost with each remodeling cycle. Studies in animal models and in cell culture have suggested that this involves multiple sites of estrogen action, not only on the cells of the BMU, but on other marrow cells (Figure 1). Estrogen acts through 2 receptors: estrogen receptor α (ERα) and ERβ. ERα appears to be the primary mediator of estrogen’s actions on the skeleton (10). Osteoblasts do express ERβ, but the actions of ERβ agonists on bone are less clear. Some studies suggest that the effects of estrogen signaling through ERα and ERβ are in opposition, while other studies suggest that activation of these 2 receptors has similar effects on bone (18, 19).
Single nucleotide polymorphisms (SNPs) of ERα may affect bone fragility. In the largest study to date, 1 of the SNPs for this receptor was associated with a significant reduction in fracture risk, independent of bone mineral density (BMD) (7). Other studies have suggested that SNPs of ERα can affect BMD and rates of bone loss as well as fracture risk in both men and women (20, 21).
An orphan nuclear receptor, estrogen receptor–related receptor α (ERRα), with sequence homology to ERα and ERβ, is also present in bone cells (22). Despite its inability to bind estrogens, this receptor may interact with ERα and ERβ or act directly to alter bone cell function. A regulatory variant of the gene encoding ERRα was recently found to be associated with a significant difference between lumbar spine and femoral neck BMD in premenopausal women (23).
Sex hormone–binding globulin (SHBG), the major binding protein for sex steroids in plasma, may not only alter the bioavailability of estrogen to hormone-responsive tissues but also affect its entry into cells. Epidemiologic studies suggest that SHBG may have an effect on bone loss and fracture risk independent of the effect as a binding protein (24). Local formation of estradiol from aromatase could play an additional role (25).
While estrogen can act on cells of the osteoblastic lineage, its effects on bone may also be dependent on actions on cells of the hematopoietic lineage, including osteoclast precursors, mature osteoclasts, and lymphocytes. Local cytokines and growth factors may mediate these effects. Bone loss after ovariectomy in rodent models can be prevented by inhibiting IL-1 or TNF-α and does not occur in mice deficient in the IL-1 receptor or TNF-α (26, 27). The effects of estrogen on cytokine production may be mediated by T cells (28). A direct effect of estrogen in accelerating osteoclast apoptosis has been attributed to increased TGF-β production (29). Another possibility is that estrogen exerts its beneficial effects by suppressing ROS (30). In estrogen deficiency, thiol antioxidant defenses may be diminished, and the resultant increase in ROS may induce TNF-α (31). The relevance of these findings for human osteoporosis has yet to be determined.
Calcium, vitamin D, and parathyroid hormone
The concept that osteoporosis is due primarily to calcium deficiency, particularly in the elderly, was initially put forward as a counterproposal to Albright’s estrogen deficiency theory. Decreased calcium intake, impaired intestinal absorption of calcium due to aging or disease, as well as vitamin D deficiency can result in secondary hyperparathyroidism. The active hormonal form, 1,25 dihydroxy vitamin D (calcitriol), is not only necessary for optimal intestinal absorption of calcium and phosphorus, but also exerts a tonic inhibitory effect on parathyroid hormone (PTH) synthesis, so that there are dual pathways that can lead to secondary hyperparathyroidism (32). Vitamin D deficiency and secondary hyperparathyroidism can contribute not only to accelerated bone loss and increasing fragility, but also to neuromuscular impairment that can increase the risk of falls (33, 34). Clinical trials involving older individuals at high risk for calcium and vitamin D deficiency indicate that supplementation of both can reverse secondary hyperparathyroidism, decrease bone resorption, increase bone mass, decrease fracture rates, and even decrease the frequency of falling (32). However, in a large recent study, calcium and vitamin D supplementation did not reduce fracture incidence significantly, perhaps because this population was less deficient in vitamin D (35).
Polymorphisms of the VDR have been studied extensively, but the results have been variable. This may be in part because the effect of a given polymorphism in this receptor is dependent on an interaction with the environment, particularly with calcium (36). VDR polymorphisms are also associated with differences in the response to therapy with calcitriol (37). There is also evidence for an effect on fracture risk independent of bone density and bone turnover, which might be due to an alteration in the frequency of falls (38).
Secondary hyperparathyroidism presents when there is relative insufficiency of vitamin D, that is, where the levels of the circulating form — 25-hydroxy vitamin D — fall below 30 ng/ml, suggesting that the target for vitamin D supplementation should be at this level or higher (39). The seasonal decrease in vitamin D level and increase in PTH level during the winter months is associated with an increase in fractures, independent of the increase in rate of falls (40). In addition, increased PTH levels are associated with increased mortality in the frail elderly, independent of bone mass and vitamin D status. The precise mechanisms underlying this relationship have not yet been determined, but the risk of cardiovascular death was increased (41). Polymorphisms of the calcium-sensing receptor, which regulates calcium secretion by suppression of PTH translation and PTH secretion, have not yet been associated with any alteration in bone phenotype (42, 43).
Receptor activator of NF-κB, its ligand, and osteoprotegerin
The concept that stimulation of bone resorption requires an interaction between cells of the osteoblastic and osteoclastic lineages was put forward many years ago, but its molecular mechanism was only identified recently (43, 44). Three members of the TNF and TNF receptor superfamily are involved (Figure 2); osteoblasts produce RANKL, a ligand for the receptor activator of NF-κB (RANK) on hematopoietic cells, which activates the differentiation of osteoclasts and maintains their function. Osteoblasts also produce and secrete osteoprotegerin (OPG), a decoy receptor that can block RANKL/RANK interactions. Stimulators of bone resorption have been found to increase RANKL expression in osteoblasts, and some also decrease OPG expression (43). Bone cells appear to express the membrane-bound form of RANKL, and thus, osteoblasts must physically interact with osteoclasts precursors in order to activate RANK. Soluble RANKL can be produced by activated T lymphocytes and is as active as membrane-bound RANKL in binding to RANK (45). Studies in transgenic mice showed that overexpression of OPG produced osteopetrosis, while OPG-knockout mice had a phenotype of severe osteoporosis with a high incidence of fractures (46). Recently a monoclonal antibody against RANKL was shown to produce prolonged inhibition of bone resorption in postmenopausal women (47). It was also shown that RANKL levels were increased on the surface of bone marrow cells from early postmenopausal women who are estrogen deficient (48). However, it has been difficult to demonstrate a role for OPG deficiency in the pathogenesis of osteoporosis, since OPG levels are not consistently altered. OPG levels increase with age, and it is possible that OPG production rises as a homeostatic response to limit the bone loss that occurs with an increase in other bone-resorbing factors (21, 49). Polymorphisms in the OPG gene have been associated with osteoporotic fractures and differences in BMD (50). OPG polymorphisms have also been linked to coronary artery disease (51). This is consistent with recent information linking the RANKL/RANK/OPG system to vascular calcification (52).
Figure 2.

Regulation of osteoclast formation and activity. In physiologic remodeling, activation of bone resorption requires contact between cells of the osteoblast and osteoclast lineages. M-CSF, which may be either membrane bound or secreted, interacts with its receptor, c-fms, to stimulate differentiation and proliferation of hematopoietic progenitors, which then express RANK as preosteoclasts. Osteoclast differentiation and activity are stimulated by RANK/RANKL interaction, and this interaction can be blocked by soluble OPG. Bone-resorbing factors can also stimulate COX2 activity, which may amplify responses to RANKL and OPG by producing prostaglandins. In pathologic conditions, inflammatory and malignant cells can increase osteoclastogenesis by producing soluble or membrane-bound M-CSF and RANKL as well as PTH-related protein (PTHrP), cytokines, and prostaglandins.
The RANKL/RANK interaction is critical for both differentiation and maintenance of osteoclast activity and hence represents a final common pathway for any pathogenetic factor in osteoporosis that acts by increasing bone resorption. While it is assumed that cells of the stromal/osteoblastic lineage are the major source of RANKL in physiologic bone remodeling, other cells may act as a source of RANKL in pathologic states; for example, T cell production may play a role in osteoporosis as well as inflammatory bone loss (53).
Recently a second system that might affect the interaction between osteoblasts and osteoclasts has been identified (54). This involves the membrane adapter DNAX-activating protein 12 and the Fc receptor common γ chain. Deletion of these molecules results in severe osteopetrosis in mice. The molecules are involved in signaling through the immunoreceptor tyrosine-based activation motif (ITAM). Cooperation between RANKL and ITAM signaling may be essential for osteoclastogenesis, for which nuclear factor of activated T cells (NFAT) is the master transcription factor.
Genes determining osteoblast differentiation and function
The recent discoveries of signal transduction pathways and transcription factors critical for osteoblast differentiation and function have opened up new approaches to understanding the pathogenesis of osteoporosis. Gene deletion studies have shown that absence of runt-related transcription factor 2 (Runx2) or a downstream factor, osterix, are critical for osteoblast differentiation (55, 56). Interestingly, overexpression of Runx2 leads to a decrease in bone mass (57). A role for polymorphisms of these transcription factors in osteoporosis has not yet been identified.
The recent identification of the critical role for the Wnt signaling pathway in regulating osteoblast function is of particular interest, since it has been shown to play an important role in determining bone mass and strength (58–61) (Figure 3). LDL receptor–related protein 5 (LRP5) interacts with the frizzled receptor to transduce signaling by Wnt ligands. A mutation of LRP5 that leads to constitutive activation can result in an increase in bone density (58, 59). The phenotype of families with LRP5 activating mutations varies considerably, although all show a striking absence of fractures. Some have normal skeletal architecture, while others show abnormalities due to skeletal overgrowth (60). Deletion of LRP5 results in a severe osteoporotic syndrome associated with abnormal eye development (61). Polymorphisms of LRP5 have been associated with differences in bone mass and fractures (62, 63). Mutations in LRP5 have been identified in a few patients with idiopathic juvenile osteoporosis (64).
Figure 3.
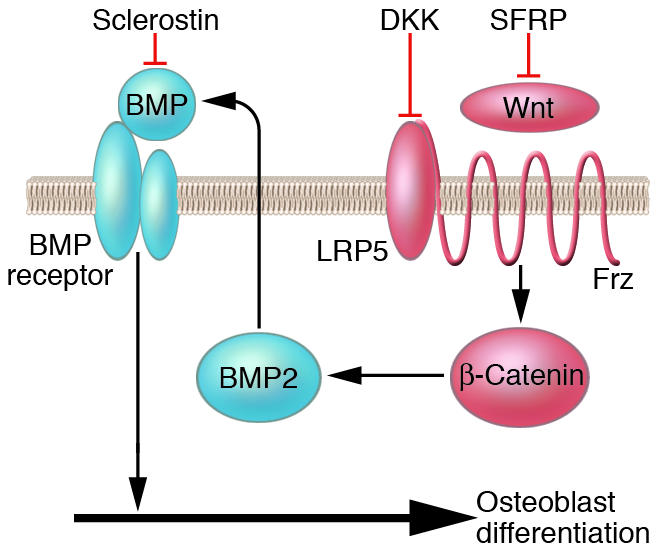
Interaction of the Wnt, BMP, and sclerostin pathways. Differentiation of osteoblasts during both development and remodeling is dependent on the activity of both the Wnt and BMP pathways. Wnt signaling requires the interaction of the LRP5 and frizzled receptors (Frz) and can be inhibited by Dickkopf (DKK; an inhibitor of LRP5) and secreted frizzled-related protein (SFRP). Antagonists such as sclerostin can block both BMP and Wnt signaling. The mediator of the canonical Wnt pathway, β-catenin, can synergize with BMP2 to enhance osteoblast differentiation and bone formation. Consistent with these interactions are the findings that high bone mass can result both from activating mutations of the Wnt pathway and deletion of SOST, the gene encoding sclerostin.
Animal and in vitro studies indicate that the Wnt signaling pathway is critical for osteoblast differentiation and function. Studies in mice suggest that the increase in bone mass in animals with activating mutations of LRP5 is due to an increased response to mechanical loading (65). The fact that fluid shear stress activates β-catenin signaling further supports the concept that Wnt signaling is critical in the response to mechanical loading (66). However, Wnt signaling is also critical in bone development and can affect peak bone mass (67). The inhibition of skeletal growth by glucocorticoids may be mediated by effects on Wnt signaling (68).
The precise mechanisms whereby Wnt signaling alters osteoblast function are not fully understood, but there is evidence that the canonical β-catenin pathway is involved and that there is an interaction with bone morphogenetic protein 2 (BMP2) (69) (Figure 3). There are a number of inhibitors that have been shown to interact with BMP2 and with the Wnt signaling pathway. One of these, sclerostin, the product of the SOST gene, has been shown to inhibit both BMP2 and Wnt signaling (70, 71). Inactivating mutations of this gene can produce the high-bone-mass disorder Van Buchem disease or sclerosteosis (72, 73). Another potential inhibitory factor is the production of secreted frizzled-related protein (SFRP) by osteoblasts (74).
Local and systemic growth factors
Remodeling imbalance, characterized by an impaired bone formation response to increased activation of bone remodeling, is an essential component of the pathogenesis of osteoporosis (8, 75). This may be due, in part, to an age-related decrease in the capacity of osteoblasts to replicate and differentiate. However, it seems likely that specific defects in the production or activity of local and systemic growth factors will also contribute to impaired bone formation. BMPs as well as other members of the TNF family have been implicated. IGF, which is both a systemic and local regulator, as well as TGF-β, can also alter bone formation. There is some association between BMD and the incidence of osteoporotic fractures and polymorphisms in the genes encoding IGF-1 and TGF-β (76–78), but the largest study to date, in Icelandic and Danish cohorts, suggests that polymorphisms of the BMP2 gene are linked to low BMD and fracture risk (79). Inhibition of local IGF-1 production may be an important component of glucocorticoid-induced osteoporosis as well as the inhibition of growth in childhood (80).
Cytokines, prostaglandins, NO, and leukotrienes
The concept that locally produced cytokines such as IL-1 and prostaglandins such as prostaglandin E2 (PGE2) can affect bone is more than 30 years old (81, 82). Subsequently, many cytokines were found to either stimulate or inhibit bone resorption and formation (83). Prostaglandins have both stimulatory and inhibitory actions; however, the predominant effect of PGE2, which is the major prostaglandin produced by bone cells, is to stimulate both resorption and formation (84). The possibility that these factors might also be involved in the pathogenesis of osteoporosis is based largely on animal studies of bone loss after ovariectomy (26, 27, 53, 85, 86); however, there is evidence that polymorphisms of IL-1, IL-6, TNF-α, and their receptors can influence bone mass in humans (87–89).
Prostaglandins, particularly PGE2, are produced by bone cells largely through the action of inducible cyclooxygenase 2 (COX2). COX2 is induced by most of the factors that stimulate bone resorption and thus may enhance the response to these agents (84). Treatment with COX inhibitors blunts the response to impact loading and fluid shear stress, indicating that prostaglandins play an important role in the response mechanical forces, and this may be enhanced by estrogen (90, 91). In epidemiologic studies, small increases in BMD and decreases in fracture risk have been reported in individuals using NSAIDS (92, 93).
NO is produced by bone cells and is a cofactor for the anabolic response to mechanical loading (91, 94). However, unlike prostaglandins, NO may inhibit bone resorption, perhaps by increasing OPG production (95). This effect may be responsible for the increase in BMD that has been demonstrated in patients treated with isosorbide mononitrate and other activators of the NO pathway (96).
Leukotrienes, the products of lipoxygenase, can affect bone by stimulating resorption and inhibiting formation (97). Recently, arachidonate 15-lipoxygenase (encoded by Alox15), was identified as a negative regulator of bone density in mice (98), and polymorphisms in the human gene, ALOX15, were found to be associated with differences in peak BMD in postmenopausal women (99).
Collagen abnormalities
A polymorphism of the first intron of the gene coding for the type I collagen α1 chain and increased levels of homocysteine can influence fracture risk independent of BMD (100–102). This may be due to differences in helix formation or cross-linking of collagen, challenging the concept that mineral and matrix composition are normal in osteoporosis and that only structural abnormalities account for skeletal fragility.
Leptin and neural pathways
Leptin deficiency or resistance is associated with high BMD in mice despite the fact that gonadal function is diminished (103). This has been attributed to a central effect on adrenergic signaling. Increased β-adrenergic activity can decrease bone mass, but other neural pathways may be involved (104, 105). Some, but not all, epidemiologic studies suggest that β-adrenergic blockers can decrease fracture risk and increase BMD (106, 107). Another neural pathway has recently been implicated by the finding that mice in which the cannabinoid type 1 receptor is inactivated, as well as mice treated with antagonist of this receptor, are protected from ovariectomy-induced bone loss (108).
Implication for the diagnosis and treatment of osteoporosis
Prior to the introduction of BMD measurements, the diagnosis of osteoporosis was only made when fragility fractures occurred in an appropriate clinical setting, largely in postmenopausal women and older men. Today we use BMD to diagnose osteoporosis and osteopenia before fractures occur, as well as to confirm the diagnosis in patients with fragility fractures (109). Our goal is to identify those individuals most likely to have fractures in the future and to treat them appropriately so that their fracture risk will be reduced.
While low BMD is an important risk factor, many other factors have been identified in epidemiologic studies, including age, low body weight, and smoking. Many drugs and diseases that can aggravate bone loss and fragility are also important in predicting future fracture risk, such as glucocorticoid or thyroid hormone excess and chronic inflammatory disorders (109). Another key predictor is the rate of bone remodeling. Increased rates of osteoclastic bone resorption, measured by the level of collagen breakdown products, as well as increased bone formation, measured by bone-specific alkaline phosphatase, osteocalcin, or procollagen peptide levels, are associated with an increase in risk for bone loss and fragility fractures. Techniques that can analyze the microarchitecture of bone are being developed that could further improve diagnosis (110). In the future, it may be possible to identify genetic profiles that predict greater or lesser risk of fractures.
At present the therapy of osteoporosis is directed at the major pathogenetic mechanisms outlined here, with a strong emphasis on prevention of bone loss and fractures (111). While the first major pathogenetic mechanism, failure to achieve optimal peak bone mass, is determined largely by our genes, nutrition and lifestyle can have an effect during growth and development. Moreover, adequate calcium and vitamin D intake and appropriate physical activity may not only increase peak bone mass but also slow bone loss and reduce fracture risk throughout life.
Pharmacotherapy for osteoporosis has been focused mainly on interventions that could reverse the second pathogenetic mechanism, excessive bone resorption (112). In the past, estrogen replacement was the most widely used therapy. Ironically, the first study that truly established the antifracture efficacy of estrogen has also resulted in its being largely abandoned as the primary therapy for osteoporosis (113). Data from the Women’s Health Initiative indicated that the estrogen, particularly when combined with progestins, increased the risk of breast cancer and cardiovascular disease, which outweighed the benefits to the skeleton. The fact that much lower doses of estrogen can prevent bone loss in postmenopausal women (13) suggests that the cost/benefit ratio might be reversed with such therapy, but appropriate long-term studies have not yet been carried out. One alternative is the use of selective estrogen receptor modulators. One of these, raloxifene, has been shown to decrease the risk of vertebral fracture and does not appear to increase the risk of breast cancer (114).
The discovery that calcitonin is a selective inhibitor of osteoclastic activity more than 40 years ago led to the concept that calcitonin deficiency might be the cause of osteoporosis and that its administration might be the cure. However, calcitonin deficiency was not found in osteoporotic patients, and calcitonin therapy has been less effective than other antiresorptive agents, possibly because osteoclasts can escape calcitonin inhibition (112, 114).
The clinical trials showing that bisphosphonates could provide effective and relatively safe antiresorptive therapy have had an enormous impact on the management of osteoporosis. The currently FDA-approved bisphosphonates — alendronate, risedronate, and ibandronate — have been shown to reduce the incidence of fractures, including both vertebral and nonvertebral fractures, particularly hip fractures (112, 114). These agents bind to the bone surface and then are taken up by osteoclasts, leading to their inactivation and programmed cell death. Potent bisphosphonates have now been in use for more than 10 years, and there is some concern that prolonged use may cause excessive inhibition of bone remodeling and slow the repair of fractures and of microdamage (115).
Effective treatment for the third pathogenetic mechanism, impaired bone formation, has only become available recently. Although it was shown more than 60 years ago that intermittent administration of low doses of PTH could increase bone mass in animals, an effective clinical approach was only developed and approved by the FDA in 2002. Daily subcutaneous injections of the synthetic N-terminal portion of PTH, teriparatide, can increase bone mass and strength by markedly increasing bone formation, while only modestly increasing bone resorption (116). This therapy is as effective as bisphosphonates in fracture reduction. Extensive studies on the mechanism of action of PTH are underway and should help us in identifying the critical pathways that regulate bone formation and are abnormal in osteoporotic patients. Another putative anabolic agent, strontium ranelate, has recently been approved for treatment of osteoporosis in several countries outside the US (117). Its mechanism of action is not well understood.
New antiresorptive approaches are being investigated, including selective inhibitors of osteoclastic hydrogen ion transport and cathepsin K as well as antagonists to the αvβ3 integrins that are necessary for osteoclast adherence and motility (118). New anabolic approaches are likely to emerge from further study of the transcriptional regulation of osteoblasts.
While improvements in diagnosis and therapy are important, it is equally true that we have sufficient tools today to assess fracture risk and prevent or treat osteoporosis to reduce that risk. Thus, there are 2 challenges: (a) to develop better diagnostic tools and treatment; and (b) to apply our current knowledge more broadly in the community. The latter is far from being met. Moreover, it is critical that we meet both these challenges in the coming decades if we are to deal effectively with the increase in osteoporosis and fractures that is projected as our population ages (111).
Footnotes
Nonstandard abbreviations used: BMD, bone mineral density; BMP2, bone morphogenetic protein 2; BMU, bone multicellular unit; COX2, cyclooxygenase 2; ERα, estrogen receptor α; LRP5, LDL receptor–related protein 5; OPG, osteoprotegerin; PGE2, prostaglandin E2; PTH, parathyroid hormone; RANK, receptor activator of NF-κB; RANKL, RANK ligand; SNP, single nucleotide polymorphism; VDR, vitamin D receptor.
Conflict of interest: Lawrence G. Raisz chairs Data Safety and Monitoring Boards for Novartis, which manufactures and distributes bisphosphonates for the treatment of bone metastases. He receives research support from Servier International, which manufactures and distributes strontium ranelate for the treatment of osteoporosis. He served as Scientific Editor for the US Surgeon General’s 2004 report “Bone Health and Osteoporosis” (111).
References
- 1.Cooper, A., and Cooper, B.B. 1822. A treatise on dislocations, and on fractures of the joints. Churchill. London, United Kingdom. 425 pp.
- 2.Schapira D, Schapira C. Osteoporosis: the evolution of a scientific term. Osteoporos. Int. 1992;2:164–167. doi: 10.1007/BF01623921. [DOI] [PubMed] [Google Scholar]
- 3.Albright F, Bloomberg E, Smith PH. Postmenopausal osteoporosis. Trans. Assoc. Am. Physicians. 1940;55:298–305. [Google Scholar]
- 4.Riggs BL, et al. Changes in bone mineral density of the proximal femur and spine with aging. Differences between the postmenopausal and senile osteoporosis syndromes. J. Clin. Invest. 1982;70:716–723. doi: 10.1172/JCI110667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu YZ, Liu YJ, Recker RR, Deng HW. Molecular studies of identification of genes for osteoporosis: the 2002 update. J. Endocrinol. 2003;177:147–196. doi: 10.1677/joe.0.1770147. [DOI] [PubMed] [Google Scholar]
- 6.Baldock PA, Eisman JA. Genetic determinants of bone mass. Curr. Opin. Rheumatol. 2004;16:450–456. doi: 10.1097/01.moo.0000127828.34643.b4. [DOI] [PubMed] [Google Scholar]
- 7.Ioannidis JP, et al. Differential genetic effects of ESR1 gene polymorphisms on osteoporosis outcomes. JAMA. 2004;292:2105–2114. doi: 10.1001/jama.292.17.2105. [DOI] [PubMed] [Google Scholar]
- 8.Parfitt AM, Villanueva AR, Foldes J, Rao DS. Relations between histologic indices of bone formation: implications for the pathogenesis of spinal osteoporosis. J. Bone Miner. Res. 1995;10:466–473. doi: 10.1002/jbmr.5650100319. [DOI] [PubMed] [Google Scholar]
- 9.Ebeling PR, et al. Bone turnover markers and bone density across the menopausal transition. J. Clin. Endocrinol. Metab. 1996;81:3366–3371. doi: 10.1210/jcem.81.9.8784098. [DOI] [PubMed] [Google Scholar]
- 10.Lee K, Jessop H, Suswillo R, Zaman G, Lanyon L. Endocrinology: bone adaptation requires oestrogen receptor-alpha. Nature. 2003;424:389. doi: 10.1038/424389a. [DOI] [PubMed] [Google Scholar]
- 11.Prestwood KM, et al. The short-term effects of conjugated estrogen on bone turnover in older women. J. Clin. Endocrinol. Metab. 1994;79:366–371. doi: 10.1210/jcem.79.2.8045949. [DOI] [PubMed] [Google Scholar]
- 12.Prestwood KM, Kenny AM, Unson C, Kulldorff M. The effect of low dose micronized 17ss-estradiol on bone turnover, sex hormone levels, and side effects in older women: a randomized, double blind, placebo-controlled study. J. Clin. Endocrinol. Metab. 2000;85:4462–4469. doi: 10.1210/jcem.85.12.7001. [DOI] [PubMed] [Google Scholar]
- 13.Prestwood KM, Kenny AM, Kleppinger A, Kulldorff M. Ultralow-dose micronized 17beta-estradiol and bone density and bone metabolism in older women: a randomized controlled trial. JAMA. 2003;290:1042–1048. doi: 10.1001/jama.290.8.1042. [DOI] [PubMed] [Google Scholar]
- 14.Modder UI, et al. Dose-response of estrogen on bone versus the uterus in ovariectomized mice. Eur. J. Endocrinol. 2004;151:503–510. doi: 10.1530/eje.0.1510503. [DOI] [PubMed] [Google Scholar]
- 15.Falahati-Nini A, et al. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J. Clin. Invest. 2000;106:1553–1560. doi: 10.1172/JCI10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khosla S, Melton LJ, 3rd, Atkinson EJ, O’Fallon WM. Relationship of serum sex steroid levels to longitudinal changes in bone density in young versus elderly men. J. Clin. Endocrinol. Metab. 2001;86:3555–3561. doi: 10.1210/jcem.86.8.7736. [DOI] [PubMed] [Google Scholar]
- 17.Van Pottelbergh I, Goemaere S, Zmierczak H, Kaufman JM. Perturbed sex steroid status in men with idiopathic osteoporosis and their sons. J. Clin. Endocrinol. Metab. 2004;89:4949–4953. doi: 10.1210/jc.2003-032081. [DOI] [PubMed] [Google Scholar]
- 18.Windahl SH, et al. Female estrogen receptor beta–/– mice are partially protected against age-related trabecular bone loss. J. Bone Miner. Res. 2001;16:1388–1398. doi: 10.1359/jbmr.2001.16.8.1388. [DOI] [PubMed] [Google Scholar]
- 19.Sims NA, et al. Deletion of estrogen receptors reveals a regulatory role for estrogen receptors-beta in bone remodeling in females but not in males. Bone. 2002;30:18–25. doi: 10.1016/s8756-3282(01)00643-3. [DOI] [PubMed] [Google Scholar]
- 20.Albagha OM, et al. Association of oestrogen receptor alpha gene polymorphisms with postmenopausal bone loss, bone mass, and quantitative ultrasound properties of bone. J. Med. Genet. 2005;42:240–246. doi: 10.1136/jmg.2004.023895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khosla S, et al. Relationship of estrogen receptor genotypes to bone mineral density and to rates of bone loss in men. J. Clin. Endocrinol. Metab. 2004;89:1808–1816. doi: 10.1210/jc.2003-031448. [DOI] [PubMed] [Google Scholar]
- 22.Bonnelye E, Aubin JE. Estrogen receptor-related receptor alpha: a mediator of estrogen response in bone. J. Clin. Endocrinol. Metab. 2005;90:3115–3121. doi: 10.1210/jc.2004-2168. [DOI] [PubMed] [Google Scholar]
- 23.Laflamme N, et al. A frequent regulatory variant of the estrogen-related receptor alpha gene associated with BMD in French-Canadian premenopausal women. J. Bone Miner. Res. 2005;20:938–944. doi: 10.1359/JBMR.050203. [DOI] [PubMed] [Google Scholar]
- 24.Goderie-Plomp HW, et al. Endogenous sex hormones, sex hormone-binding globulin, and the risk of incident vertebral fractures in elderly men and women: the Rotterdam Study. J. Clin. Endocrinol. Metab. 2004;89:3261–3269. doi: 10.1210/jc.2002-022041. [DOI] [PubMed] [Google Scholar]
- 25.Van Pottelbergh I, Goemaere S, Kaufman JM. Bioavailable estradiol and an aromatase gene polymorphism are determinants of bone mineral density changes in men over 70 years of age. J. Clin. Endocrinol. Metab. 2003;88:3075–3081. doi: 10.1210/jc.2002-021691. [DOI] [PubMed] [Google Scholar]
- 26.Kimble RB, et al. Simultaneous block of interleukin-1 and tumor necrosis factor is required to completely prevent bone loss in the early postovariectomy period. Endocrinology. 1995;136:3054–3061. doi: 10.1210/endo.136.7.7789332. [DOI] [PubMed] [Google Scholar]
- 27.Lorenzo JA, et al. Mice lacking the type I interleukin-1 receptor do not lose bone mass after ovariectomy. Endocrinology. 1998;139:3022–3025. doi: 10.1210/endo.139.6.6128. [DOI] [PubMed] [Google Scholar]
- 28.Gao Y, et al. Estrogen prevents bone loss through transforming growth factor beta signaling in T cells. Proc. Natl. Acad. Sci. U. S. A. 2004;101:16618–16623. doi: 10.1073/pnas.0404888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes DE, et al. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat. Med. 1996;2:1132–1136. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- 30.Lean JM, et al. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J. Clin. Invest. 2003;112:915–923. doi:10.1172/JCI200318859. doi: 10.1172/JCI18859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lean JM, Jagger CJ, Kirstein B, Fuller K, Chambers TJ. Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology. 2005;146:728–735. doi: 10.1210/en.2004-1021. [DOI] [PubMed] [Google Scholar]
- 32.Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: consequences for bone loss and fractures and therapeutic implications. Endocr. Rev. 2001;22:477–501. doi: 10.1210/edrv.22.4.0437. [DOI] [PubMed] [Google Scholar]
- 33.Bischoff-Ferrari HA, et al. Effect of vitamin D on falls: a meta-analysis. JAMA. 2004;291:1999–2006. doi: 10.1001/jama.291.16.1999. [DOI] [PubMed] [Google Scholar]
- 34.Sambrook PN, et al. Serum parathyroid hormone predicts time to fall independent of vitamin D status in a frail elderly population. J. Clin. Endocrinol. Metab. 2004;89:1572–1576. doi: 10.1210/jc.2003-031782. [DOI] [PubMed] [Google Scholar]
- 35.Grant AM, et al. Oral vitamin D3 and calcium for secondary prevention of low-trauma fractures in elderly people (Randomised Evaluation of Calcium Or vitamin D, RECORD): a randomised placebo-controlled trial. Lancet. 2005;365:1621–1628. doi: 10.1016/S0140-6736(05)63013-9. [DOI] [PubMed] [Google Scholar]
- 36.Ferrari SL, Rizzoli R, Slosman DO, Bonjour JP. Do dietary calcium and age explain the controversy surrounding the relationship between bone mineral density and vitamin D receptor gene polymorphisms? J. Bone Miner. Res. 1998;13:363–370. doi: 10.1359/jbmr.1998.13.3.363. [DOI] [PubMed] [Google Scholar]
- 37.Morrison NA, et al. Vitamin D receptor genotypes influence the success of calcitriol therapy for recurrent vertebral fracture in osteoporosis. Pharmacogenet. Genomics. 2005;15:127–135. doi: 10.1097/01213011-200502000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Garnero P, Munoz F, Borel O, Sornay-Rendu E, Delmas PD. Vitamin D receptor gene polymorphisms are associated with the risk of fractures in postmenopausal women, independently of bone mineral density. The OFELY study. J. Clin. Endocrinol. Metab. 2005;90:4829–4835. doi: 10.1210/jc.2005-0364. [DOI] [PubMed] [Google Scholar]
- 39.Lips P. Which circulating level of 25-hydroxyvitamin D is appropriate? J. Steroid Biochem. Mol. Biol. 2004;89–90:611–614. doi: 10.1016/j.jsbmb.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 40.Pasco JA, et al. Seasonal periodicity of serum vitamin D and parathyroid hormone, bone resorption, and fractures: the Geelong Osteoporosis Study. J. Bone Miner. Res. 2004;19:752–758. doi: 10.1359/JBMR.040125. [DOI] [PubMed] [Google Scholar]
- 41.Sambrook PN, et al. Serum parathyroid hormone is associated with increased mortality independent of 25-hydroxy vitamin d status, bone mass, and renal function in the frail and very old: a cohort study. J. Clin. Endocrinol. Metab. 2004;89:5477–5481. doi: 10.1210/jc.2004-0307. [DOI] [PubMed] [Google Scholar]
- 42.Bollerslev J, et al. Calcium-sensing receptor gene polymorphism A986S does not predict serum calcium level, bone mineral density, calcaneal ultrasound indices, or fracture rate in a large cohort of elderly women. Calcif. Tissue Int. 2004;74:12–17. doi: 10.1007/s00223-002-0066-1. [DOI] [PubMed] [Google Scholar]
- 43.Suda T, et al. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999;20:345–357. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- 44.Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption — a hypothesis [letter] Calcif. Tissue Int. 1982;34:311. doi: 10.1007/BF02411258. [DOI] [PubMed] [Google Scholar]
- 45.Kanamaru F, et al. Expression of membrane-bound and soluble receptor activator of NF-kappaB ligand (RANKL) in human T cells. Immunol. Lett. 2004;94:239–246. doi: 10.1016/j.imlet.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Bucay N, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bekker PJ, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J. Bone Miner. Res. 2004;19:1059–1066. doi: 10.1359/JBMR.040305. [DOI] [PubMed] [Google Scholar]
- 48.Eghbali-Fatourechi G, et al. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J. Clin. Invest. 2003;111:1221–1230. doi:10.1172/JCI200317215. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khosla S, et al. Correlates of osteoprotegerin levels in women and men. Osteoporos. Int. 2002;13:394–399. doi: 10.1007/s001980200045. [DOI] [PubMed] [Google Scholar]
- 50.Langdahl BL, Carstens M, Stenkjaer L, Eriksen EF. Polymorphisms in the osteoprotegerin gene are associated with osteoporotic fractures. J. Bone Miner. Res. 2002;17:1245–1255. doi: 10.1359/jbmr.2002.17.7.1245. [DOI] [PubMed] [Google Scholar]
- 51.Soufi M, et al. Osteoprotegerin gene polymorphisms in men with coronary artery disease. J. Clin. Endocrinol. Metab. 2004;89:3764–3768. doi: 10.1210/jc.2003-032054. [DOI] [PubMed] [Google Scholar]
- 52.Collin-Osdoby P. Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ. Res. 2004;95:1046–1057. doi: 10.1161/01.RES.0000149165.99974.12. [DOI] [PubMed] [Google Scholar]
- 53.Weitzmann MN, et al. T cell activation induces human osteoclast formation via receptor activator of nuclear factor kappaB ligand-dependent and -independent mechanisms. J. Bone Miner. Res. 2001;16:328–337. doi: 10.1359/jbmr.2001.16.2.328. [DOI] [PubMed] [Google Scholar]
- 54.Takayanagi H. Mechanistic insight into osteoclast differentiation in osteoimmunology. J. Mol. Med. 2005;83:170–179. doi: 10.1007/s00109-004-0612-6. [DOI] [PubMed] [Google Scholar]
- 55.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 56.Nakashima K, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 57.Geoffroy V, Kneissel M, Fournier B, Boyde A, Matthias P. High bone resorption in adult aging transgenic mice overexpressing cbfa1/runx2 in cells of the osteoblastic lineage. Mol. Cell. Biol. 2002;22:6222–6233. doi: 10.1128/MCB.22.17.6222-6233.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Little RD, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet. 2002;70:11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Boyden LM, et al. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 60.Van Wesenbeeck L, et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am. J. Hum. Genet. 2003;72:763–771. doi: 10.1086/368277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gong Y, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 62.Koay MA, et al. Influence of LRP5 polymorphisms on normal variation in BMD. J. Bone Miner. Res. 2004;19:1619–1627. doi: 10.1359/JBMR.040704. [DOI] [PubMed] [Google Scholar]
- 63.Bollerslev J, et al. LRP5 gene polymorphisms predict bone mass and incident fractures in elderly Australian women. Bone. 2005;36:599–606. doi: 10.1016/j.bone.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 64.Hartikka H, et al. Heterozygous mutations in the LDL receptor-related protein 5 (LRP5) gene are associated with primary osteoporosis in children. J. Bone Miner. Res. 2005;20:783–789. doi: 10.1359/JBMR.050101. [DOI] [PubMed] [Google Scholar]
- 65.Akhter MP, et al. Bone biomechanical properties in LRP5 mutant mice. Bone. 2004;35:162–169. doi: 10.1016/j.bone.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 66.Norvell SM, Alvarez M, Bidwell JP, Pavalko FM. Fluid shear stress induces beta-catenin signaling in osteoblasts. Calcif. Tissue Int. 2004;75:396–404. doi: 10.1007/s00223-004-0213-y. [DOI] [PubMed] [Google Scholar]
- 67.Kronenberg H, Kobayashi T. Transcriptional regulation in development of bone. Endocrinology. 2004;146:1012–1017. doi: 10.1210/en.2004-1343. [DOI] [PubMed] [Google Scholar]
- 68.Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem. Biophys. Res. Commun. 2005;329:177–181. doi: 10.1016/j.bbrc.2005.01.117. [DOI] [PubMed] [Google Scholar]
- 69.Mbalaviele G, et al. beta-Catenin and BMP-2 synergize to promote osteoblast differentiation and new bone formation. J. Cell. Biochem. 2005;94:403–418. doi: 10.1002/jcb.20253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li X, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005;280:19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 71.Winkler DG, et al. Sclerostin inhibition of Wnt-3a-induced C3H10T1/2 cell differentiation is indirect and mediated by BMP proteins. J. Biol. Chem. 2004;280:2498–2502. doi: 10.1074/jbc.M400524200. [DOI] [PubMed] [Google Scholar]
- 72.Loots GG, et al. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15:928–935. doi: 10.1101/gr.3437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Balemans W, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum. Mol. Genet. 2001;10:537–543. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 74.Bodine, P.V., et al. 2005. The Wnt antagonist secreted frizzled-related protein-1 controls osteoblast and osteocyte apoptosis. J. Cell. Biochem. doi:10.1002/jcb.20599. [DOI] [PubMed]
- 75.Eriksen EF, et al. Cancellous bone remodeling in type I (postmenopausal) osteoporosis: quantitative assessment of rates of formation, resorption, and bone loss at tissue and cellular levels. J. Bone Miner. Res. 1990;5:311–319. doi: 10.1002/jbmr.5650050402. [DOI] [PubMed] [Google Scholar]
- 76.Lau EM, et al. Osteoporosis and transforming growth factor-beta-1 gene polymorphism in Chinese men and women. J. Bone Miner. Metab. 2004;22:148–152. doi: 10.1007/s00774-003-0463-1. [DOI] [PubMed] [Google Scholar]
- 77.Langdahl BL, Carstens M, Stenkjaer L, Eriksen EF. Polymorphisms in the transforming growth factor beta 1 gene and osteoporosis. Bone. 2003;32:297–310. doi: 10.1016/s8756-3282(02)00971-7. [DOI] [PubMed] [Google Scholar]
- 78.Kim JG, Roh KR, Lee JY. The relationship among serum insulin-like growth factor-I, insulin-like growth factor-I gene polymorphism, and bone mineral density in postmenopausal women in Korea. Am. J. Obstet. Gynecol. 2002;186:345–350. doi: 10.1067/mob.2002.120483. [DOI] [PubMed] [Google Scholar]
- 79.Styrkarsdottir U, et al. Linkage of osteoporosis to chromosome 20p12 and association to BMP2. PLoS Biol. 2003;1:E69. doi: 10.1371/journal.pbio.0000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mehls O, Himmele R, Homme M, Kiepe D, Klaus G. The interaction of glucocorticoids with the growth hormone-insulin-like growth factor axis and its effects on growth plate chondrocytes and bone cells. J. Pediatr. Endocrinol. Metab. 2001;14(Suppl. 6):1475–1482. [PubMed] [Google Scholar]
- 81.Klein DC, Raisz LG. Prostaglandins: stimulation of bone resorption in tissue culture. Endocrinology. 1970;86:1436–1440. doi: 10.1210/endo-86-6-1436. [DOI] [PubMed] [Google Scholar]
- 82.Raisz LG, et al. Effect of osteoclast activating factor from human leukocytes on bone metabolism. J. Clin. Invest. 1975;56:408–413. doi: 10.1172/JCI108106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horwitz, M.C., and Lorenzo, J.A. 2002. Local regulators of bone: IL-1, TNF, lymphotoxin, interferon-γ, IL-8, IL-10, IL-4, the LIF/IL-6 family, and additional cytokines. In Principles of bone biology. J.P. Bilezikian, L.G. Raisz, and G.A. Rodan, editors. Academic Press. San Diego, California, USA. 961–977.
- 84.Pilbeam, C.C., Harrison, J.R., and Raisz, L.G. 2002. Prostaglandins and bone metabolism. In Principles of bone biology. J.P. Bilezikian, L.G. Raisz, and G.A. Rodan, editors. Academic Press. San Diego, California, USA. 979–994.
- 85.Kawaguchi H, et al. Ovariectomy enhances and estrogen replacement inhibits the activity of bone marrow factors that stimulate prostaglandin production in cultured mouse calvariae. J. Clin. Invest. 1995;96:539–548. doi: 10.1172/JCI118066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ammann P, et al. Transgenic mice expressing soluble tumor necrosis factor-receptor are protected against bone loss caused by estrogen deficiency. J. Clin. Invest. 1997;99:1699–1703. doi: 10.1172/JCI119333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tasker PN, Albagha OM, Masson CB, Reid DM, Ralston SH. Association between TNFRSF1B polymorphisms and bone mineral density, bone loss and fracture. Osteoporos. Int. 2004;15:903–908. doi: 10.1007/s00198-004-1617-2. [DOI] [PubMed] [Google Scholar]
- 88.Chung HW, et al. Association of interleukin-6 promoter variant with bone mineral density in pre-menopausal women. J. Hum. Genet. 2003;48:243–248. doi: 10.1007/s10038-003-0020-8. [DOI] [PubMed] [Google Scholar]
- 89.Ferrari SL, et al. Interactions of interleukin-6 promoter polymorphisms with dietary and lifestyle factors and their association with bone mass in men and women from the Framingham Osteoporosis Study. J. Bone Miner. Res. 2004;19:552–559. doi: 10.1359/JBMR.040103. [DOI] [PubMed] [Google Scholar]
- 90.Forwood MR. Inducible cyclo-oxygenase (COX-2) mediates the induction of bone formation by mechanical loading in vivo. J. Bone Miner. Res. 1996;11:1688–1693. doi: 10.1002/jbmr.5650111112. [DOI] [PubMed] [Google Scholar]
- 91.Bakker AD, et al. Additive effects of estrogen and mechanical stress on nitric oxide and prostaglandin E(2) production by bone cells from osteoporotic donors. Osteoporos. Int. 2004;16:983–989. doi: 10.1007/s00198-004-1785-0. [DOI] [PubMed] [Google Scholar]
- 92.Raisz LG. Potential impact of selective cyclooxygenase-2 inhibitors on bone metabolism in health and disease. Am. J. Med. 2001;110(Suppl. 3A):43S–45S. doi: 10.1016/s0002-9343(00)00684-7. [DOI] [PubMed] [Google Scholar]
- 93.Carbone LD, et al. Association between bone mineral density and the use of nonsteroidal anti-inflammatory drugs and aspirin: impact of cyclooxygenase selectivity. J. Bone Miner. Res. 2003;18:1795–1802. doi: 10.1359/jbmr.2003.18.10.1795. [DOI] [PubMed] [Google Scholar]
- 94.Chow JW, Fox SW, Lean JM, Chambers TJ. Role of nitric oxide and prostaglandins in mechanically induced bone formation. J. Bone Miner. Res. 1998;13:1039–1044. doi: 10.1359/jbmr.1998.13.6.1039. [DOI] [PubMed] [Google Scholar]
- 95.Wang FS, et al. Nitric oxide donor increases osteoprotegerin production and osteoclastogenesis inhibitory activity in bone marrow stromal cells from ovariectomized rats. Endocrinology. 2004;145:2148–2156. doi: 10.1210/en.2003-1074. [DOI] [PubMed] [Google Scholar]
- 96.Jamal SA, Cummings SR, Hawker GA. Isosorbide mononitrate increases bone formation and decreases bone resorption in postmenopausal women: a randomized trial. J. Bone Miner. Res. 2004;19:1512–1517. doi: 10.1359/JBMR.040716. [DOI] [PubMed] [Google Scholar]
- 97.Traianedes K, Dallas MR, Garrett IR, Mundy GR, Bonewald LF. 5-Lipoxygenase metabolites inhibit bone formation in vitro. Endocrinology. 1998;139:3178–3184. doi: 10.1210/endo.139.7.6115. [DOI] [PubMed] [Google Scholar]
- 98.Klein RF, et al. Regulation of bone mass in mice by the lipoxygenase gene Alox15. Science. 2004;303:229–232. doi: 10.1126/science.1090985. [DOI] [PubMed] [Google Scholar]
- 99.Urano T, et al. Association of a single nucleotide polymorphism in the lipoxygenase ALOX15 5′-flanking region (-5229G/A) with bone mineral density. J. Bone Miner. Metab. 2005;23:226–230. doi: 10.1007/s00774-004-0588-x. [DOI] [PubMed] [Google Scholar]
- 100.Mann V, Ralston SH. Meta-analysis of COL1A1 Sp1 polymorphism in relation to bone mineral density and osteoporotic fracture. Bone. 2003;32:711–717. doi: 10.1016/s8756-3282(03)00087-5. [DOI] [PubMed] [Google Scholar]
- 101.McLean RR, et al. Homocysteine as a predictive factor for hip fracture in older persons. N. Engl. J. Med. 2004;350:2042–2049. doi: 10.1056/NEJMoa032739. [DOI] [PubMed] [Google Scholar]
- 102.van Meurs JB, et al. Homocysteine levels and the risk of osteoporotic fracture. N. Engl. J. Med. 2004;350:2033–2041. doi: 10.1056/NEJMoa032546. [DOI] [PubMed] [Google Scholar]
- 103.Ducy P, et al. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100:197–207. doi: 10.1016/s0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- 104.Elefteriou F, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434:514–520. doi: 10.1038/nature03398. [DOI] [PubMed] [Google Scholar]
- 105.Takeda S. Central control of bone remodeling. Biochem. Biophys. Res. Commun. 2005;328:697–699. doi: 10.1016/j.bbrc.2004.11.071. [DOI] [PubMed] [Google Scholar]
- 106.Pasco JA, et al. Beta-adrenergic blockers reduce the risk of fracture partly by increasing bone mineral density: Geelong Osteoporosis Study. J. Bone Miner. Res. 2004;19:19–24. doi: 10.1359/JBMR.0301214. [DOI] [PubMed] [Google Scholar]
- 107.Reid IR, et al. beta-Blocker use, BMD, and fractures in the study of osteoporotic fractures. J. Bone Miner. Res. 2005;20:613–618. doi: 10.1359/JBMR.041202. [DOI] [PubMed] [Google Scholar]
- 108.Idris AI, et al. Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat. Med. 2005;11:774–799. doi: 10.1038/nm1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Raisz LG. Clinical practice. Screening for osteoporosis. N. Engl. J. Med. 2005;353:164–171. doi: 10.1056/NEJMcp042092. [DOI] [PubMed] [Google Scholar]
- 110.Benito M, et al. Deterioration of trabecular architecture in hypogonadal men. J. Clin. Endocrinol. Metab. 2003;88:1497–1502. doi: 10.1210/jc.2002-021429. [DOI] [PubMed] [Google Scholar]
- 111.Office of the Surgeon General. 2004. Bone health and osteoporosis: a report of the Surgeon General. US Department of Health and Human Services. Rockville, Maryland, USA. 404 pp. [PubMed]
- 112.Rosen CJ. Clinical practice. Postmenopausal osteoporosis. N. Engl. J. Med. 2005;353:595–603. doi: 10.1056/NEJMcp043801. [DOI] [PubMed] [Google Scholar]
- 113.Anderson GL, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701–1712. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 114.Cranney A, et al. Meta-analyses of therapies for postmenopausal osteoporosis. IX: summary of meta-analyses of therapies for postmenopausal osteoporosis. Endocr. Rev. 2002;23:570–578. doi: 10.1210/er.2001-9002. [DOI] [PubMed] [Google Scholar]
- 115.Ott SM. Long-term safety of bisphosphonates. J. Clin. Endocrinol. Metab. 2005;90:1897–1899. doi: 10.1210/jc.2005-0057. [DOI] [PubMed] [Google Scholar]
- 116.Neer RM, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N. Engl. J. Med. 2001;344:1434–1441. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 117.Meunier PJ, et al. The effects of strontium ranelate on the risk of vertebral fracture in women with postmenopausal osteoporosis. N. Engl. J. Med. 2004;350:459–468. doi: 10.1056/NEJMoa022436. [DOI] [PubMed] [Google Scholar]
- 118.Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289:1508–1514. doi: 10.1126/science.289.5484.1508. [DOI] [PubMed] [Google Scholar]