

Abstract
Several of the proinflammatory peptides involved in rheumatoid arthritis pathogenesis, including peptides induced downstream of tumor necrosis factor-α as well as the monocyte/T cell-attracting chemokines RANTES and stromal cell-derived factor (SDF)-1α and the neuropeptides vasoactive intestinal peptide (VIP) and substance P, have their biological half-lives controlled by dipeptidyl peptidase IV (DPPIV). Proteolysis by DPPIV regulates not only the half-life but also receptor preference and downstream signaling. In this article, we examine the role of DPPIV homologs, including CD26, the canonical DPPIV, and their substrates in the pathogenesis of rheumatoid arthritis. The differing specific activities of the DPPIV family members and their differential inhibitor response provide new insights into therapeutic design.
Introduction
A significant proportion of the biologically active peptides, including systemic and locally acting neuropeptides, lymphokines, cytokines and chemokines, contain an evolutionarily conserved amino-terminal penultimate proline residue as a proteolytic-processing regulatory element. This penultimate proline protects the peptide from general aminopeptidase activity, which has led to the view that the high specificity of the dipeptidyl aminopeptidases constitutes a critical regulatory 'check-point' [1]. Limited proteolysis of such peptides by dipeptidyl peptidase (DPP) IV and/or structural homolog (DASH)-related molecules may lead to both quantitative and, due to the diversification of their receptor preference, qualitative changes to their signaling potentials [2,3]. Molecules of the DASH family have been invoked in the pathogenesis of a range of autoimmune processes, including systemic lupus erythematosus (SLE) and multiple sclerosis, in particular [4]. As these proteases and their substrates play a fundamental role in the migration and activation of immune cells and their interactions with extracellular matrix, we examine here their likely role in the progression of rheumatoid arthritis (RA).
It is tempting, although unrealistic, to propose that the marked changes in DPPIV enzymatic activity in blood plasma, synovial fluid (SF) and immune cells observed during the course of RA might be causally related to the disease etiology. Nevertheless, it is not presumptuous to propose that once DPPIV levels are altered they would participate in a positive-feedback cycle that could rapidly accelerate to exacerbate damage and thus take part in RA pathogenesis. Although this leads to speculation of therapeutic modalities based upon inhibition of DPPIV enzymatic activity, gene knockout experiments suggest that DASH family members can to some extent compensate, but not fully substitute, for each other [2,5]. As a consequence, inhibition of DPPIV activity must be examined from the perspective of the enzymatic activities and interactions of all DASH family members rather than the functionality expressed by a single enzyme in an isolated biochemical framework.
The members of the DASH family
Initially, DPPIV activity was classified simply by the enzymatic reaction, cleavage of a dipeptide from the accessible amino terminus of proteins in which the second amino acid is a proline (EC 3.4.14.5). Its expression was high on endothelial cell membranes and also in tissues with strong secretory capacity, including ovary, pancreas, liver and particularly kidney where DPPIV constituted up to 14% of the total membrane protein. In these tissues, the DPPIV levels were constant and synthesis was believed to be constitutive. The field took a vast leap forward when it was established that a 105 to 110 kDa membrane-expressed human lymphocyte activation antigen, defined by the CD26 monoclonal antibody cluster, was identical to DPPIV and was subject to activation-induced regulation (for a review see [4]). This was rapidly followed by the discovery that CD26 is itself the high affinity lymphocyte adenosine deaminase (ADA)-binding protein. Because the anti-folate treatments for RA, exemplified by methotrexate, mediate their anti-inflammatory effects in part through locally increasing extracellular adenosine concentrations, the localization and functional activity of an adenosine-metabolizing protein on activated T lymphocytes would clearly be an undesirable state in RA [6-8].
Although the greatest part of systemic DPPIV activity resides in both membrane-bound and, to some degree, proteolytically cleaved soluble CD26, a significant amount of DPPIV activity can be attributed to a growing panel of other proteins, including fibroblast-activation protein α (FAP-α)/seprase, quiescent cell proline dipeptidase (QPP), DPP8, DPP9, attractin, N-acetylated α-linked acidic dipeptidases (NAALADases) and thymus-specific serine protease [9]. These proteins form the DASH group on the basis of having an associated DPPIV enzymatic activity with or without much structural homology, or being structurally similar but enzymatically inactive (DPP6, DPP10). In some instances, the DPPIV activity is clearly intrinsic whereas in others the nature of the DPPIV activity remains debatable. In these latter cases, DPPIV activity may represent association with minimal amounts of CD26, which results in enhanced substrate hydrolysis, or may represent a separate enzyme specificity that can also accept DPPIV substrates. The contamination issue has been extensively examined and, to date, in the case of attractin for example, there is no evidence of contamination of purified preparations by CD26 [10,11]. This, however, does not exclude association with other family members. The possibility of alternative specificity is exemplified by the NAALADases, where the primary function appears to be glutamate carboxypeptidase II activity rather than aminodipeptidase functionality. Alternatively, the FAP-α/seprase protein exhibits both DPPIV and gelatinase activity; this latter property has profound effects upon the invasive properties of the expressing cell [2].
Most DPPIV/CD26 activity is membrane-expressed. There is, however, a strong circulating activity, which may be of critical importance for systemic bioactive peptide activity. Although debate remains as to the relative contributions of cleaved membrane CD26, attractin and secreted QPP to the soluble circulating DPPIV-like enzymatic activity, the functional consequences will be identical. Accordingly, this may provide a mechanism for restricting activity of paracrine/autocrine bioactive peptides at the site of release, and may further ensure rapid down-regulation of physiologically activated peptides such as glucagon-like peptide (GLP)-1, which is released by the intestine but targets the pancreas. In inflammatory reactions, it may provide a mechanism for restricting chemokine-responding T cells to the inflamed region. For example, both RANTES (regulated upon activation normal T-cell expressed and secreted) and stromal cell-derived factor (SDF)-1α are already known to be regulated by DPPIV [12,13].
The DPPIV activity, gelatinase activity and ADA complex forming function are differentially represented within individual DASH proteins. In fact, some proteins such as DPP10 have clear structural homology but no DPPIV activity at all. They may retain some of the other activities, which include association with the hematopoietic-specific CD45RO tyrosine phosphatase, or interaction with collagen. In contrast to human DPPIV/CD26, the mouse enzyme has no ADA complex forming ability and is a marker of thymic differentiation but is not an activation antigen. Nevertheless, its membrane presence has profound influence over the signal transduction responses of the T cell. This cautions against extending too far CD26-related immune results in mice to conclusions concerning RA pathogenesis and therapy in humans.
DASH molecules in rheumatoid arthritis
Based upon the enzymatic activities of the DASH enzymes described above, it is immediately apparent that DPPIV-like and gelatinase activities, collagen binding and regulation of extracellular adenosine could all have substantial roles in every phase of an inflammatory response, from recognition and proliferation to cytokine/chemokine activity and chemotaxis. It is from this vantage point that we will now view RA as a chronic systemic inflammatory disease affecting mostly articular tissues. RA may be initiated by an unknown antigenic peptide derived either from an exogenous antigen or an autoantigen. Presentation of a putative arthritogenic peptide by RA-associated HLA-DRB1*0404 or HLA-DRB1*0401 results in release of IL-2 by CD4+ T cells and induction of clonal expansion of other CD4+ T cells [14]. Cytokine and growth factor release by the induced T cells stimulates B cells, synoviocytes, and monocytes/macrophages, which leads to enhanced leukocyte recruitment into the joint and synovitis. The synovial inflammation results in release of matrix metalloproteases and cysteine proteases, leading to proteolytic degradation of the connective tissue [15,16]. Each of these processes will be affected by the presence of DPPIV, not directly but, rather, indirectly through the enzymatic processing of the bioactive peptides that regulate each stage. Reflecting this, the use of new specific DPPIV inhibitors to block RA and SLE development has led in the past year to three issued patents and eight pending applications in the United States alone.
Dipeptidyl peptidase IV (CD26)
Although DPPIV activity is a generic term for the enzyme's catalytic functions, and may reside in several proteins, the use of DPPIV as a protein descriptor has become synonymous with CD26. As described above, the merging of the three separate research directions involving cell surface DPPIV activity, immune cell CD26 signal transduction, and ADA-complex forming activity into studies of the multifunctional CD26 have led to a more profound understanding of inflammation. Specifically, modulation of CD26 activity will affect chemotaxis, invasiveness, signal transduction, proliferation and recruitment of other immune cells; clearly, the interaction of these activities will be well orchestrated in vivo but unlikely to be recapitulated in assays in vitro.
CD26 is predominantly a type 2 integral plasma membrane molecule expressed as a homodimer or heterodimer with seprase, with or without bound ADA. A small proportion appears to circulate in the plasma, cleaved from membrane CD26 by an unknown mechanism, but at a site in the membrane proximal extracellular domain. This cleavage is intriguing because CD26 is relatively resistant to proteolytic cleavage. CD26 is well represented in kidney, liver, pancreas and ovary, on endothelial cells and is localized in secretory vesicles destined for plasma membrane fusion in synovial fibroblasts [17]. In cultured kidney cells, CD26 appears to cycle from the membrane through early and late endosomes back to the membrane and, through its DPPIV activity, is often used as a marker of secretory vesicle traffic [18]. The low secretory activity of resting T cells correlates well with the low CD26 surface expression and the rapid upregulation and sustained signal following activation supports a role for CD26 in maintaining and regulating stimulation. Although cross-linking of CD26 with CD3 leads to increased activation and proliferation, this cannot be a direct signal-transducing property of CD26 as its cytoplasmic tail consists of only six amino acids. This has led to the suggestion that CD26 directly associates with the memory T cell marker CD45RO to influence signaling. As intracellular CD26 moving in vesicles to the plasma membrane is localized within the cholesterol/sphingomyelin-rich lipid raft domains [19], it is likely that associating with CD26 will help recruit signaling costimulatory/regulatory molecules to the raft-localized T cell receptor. In support of a more general function during activation not restricted to T cells, CD26 is also found on activated B cells, activated natural killer (NK) cells [20,21] and some subpopulations of macrophages [22], where it plays a role in regulation of maturation and migration of NK and NKT cells, cytokine secretion, T cell-dependent antibody production and immunoglobulin isotype switching of B cells [5]. Reflecting this general upregulation and function in active immune processes, the number of circulating CD26 positive cells is higher in the active phases of autoimmune diseases but decreased in immunosuppressions of varying origin [23,24]. The ability of T cells to regulate their membrane levels of CD26 is in sharp contrast to expression on endothelial and renal cells, where CD26 is constitutively produced and membrane levels are relatively constant. This suggests that location may be critical and that locally situated peptides may be exposed and vulnerable to T cell-expressed DPPIV.
The source and regulation of soluble DPPIV is more difficult to ascertain. The possibility that secreted DPPIV may be important in regulating T cell reactivity is provided by the report that T cells from individuals with high serum DPPIV levels are refractory to costimulatory enhancement by exogenously added CD26 during response to tetanus toxoid [25]. This effect may occur through modulation of adhesion-or peptide-mediated costimulatory signaling since direct stimulation through the T cell receptor is unaffected. Soluble DPPIV stimulates proliferation of blood T cells induced by recall antigens indirectly via antigen presenting cells [26] and potentiates transendothelial migration of T cells [27], both effects being dependent on intrinsic hydrolytic activity of the enzyme. In general, lower DPPIV serum activity is associated with immunosuppression, pregnancy, several kinds of cancer, human immunodeficiency virus infection, and also with SLE and irradiation [9,28-30]. In contrast, an increase of serum DPPIV activity, together with an increased number of CD26 positive circulating cells, was observed during the rejection of allografts [31].
In RA, decreased DPPIV enzymatic activity was observed in blood plasma/sera compared with healthy controls [32]. Further studies demonstrated a significant inverse correlation of serum DPPIV activity with disease severity as determined by C-reactive protein concentration, the number of swollen joints and with the Disease Activity Score 28 [33-35]. Hypersialylation is associated with a decrease in the specific circulating DPPIV activity in RA patients, and the activity could be restored following neuraminidase treatment [36]. The same study demonstrated that serum DPPIV from SLE patients was also hypersialylated but activity was not restored following neuraminidase treatment, from which it may be inferred that differential post-translational glycosylation may affect enzyme activity and/or substrate preference. A single report notes no difference in DPPIV activity in the sera of RA patients compared to the normal controls, but the study groups in this report were not delineated by disease severity, type of therapy or active/inactive disease states [37].
The relationship of serum DPPIV activity to clinical severity is usually based on enzyme activity, which provides no information on relative contributions of individual DASH family members. Furthermore, the mechanism by which DPPIV activity is reduced in the blood of RA patients remains purely speculative at present. One possibility is that T cell activation down-regulates the as yet unidentified protease that cleaves and releases membrane CD26. Alterations in specific activity due to increases in other less active DASH forms may also account for apparent reduced serum activity. Indeed, we have observed patient-specific patterns of multiple molecular weight forms bearing DPPIV-like activity in human plasma [35].
DPPIV/CD26 is strongly upregulated on peripheral blood (PB) T cells of RA patients, where both the staining intensity and number of positive cells correlate with disease activity (Table 1). Despite reports that DPPIV/CD26 may be seen as a Th1 response marker [33,38,39] based on cytokine release following T cell receptor stimulation, this is arbitrary. First, antibody ligation of the T cell receptor is not physiological, and second, the delineation of human lymphocytes into Th1 and Th2 subsets is considerably less precise than that of the mouse. The consequences upon CD26 of activation by lymphokines and cytokines are, however, specific and depend upon the cell type responding. In T cells, CD26 is upregulated by IL-12 and IL-2 but not by tumor necrosis factor (TNF)-α, IFN-γ, IL-15 and IL-4, while TNF-α and IL-15 are efficient stimulators of CD26 expression on NK cells and fibroblasts [33,40-42]. Moreover, TNF-α neutralizing antibodies caused down-regulation of CD26 expression in T cells [39].
Table 1.
Dipeptidyl peptidase IV/CD26 in rheumatoid arthritis
| Peripheral blood CD3+CD26+ | Peripheral blood CD4+CD26+ | Synovial fluid CD3+CD26+ | Serum/plasma DPPIV activity (μmol/min/l) | Synovial DPPIV activity (μmol/min/l) | Notes | Reference | |
| Controls | 12.9 ± 4.7 | Ta1 antibody; all active with therapy | [47] | ||||
| RA (active) | 40.2 ± 10.6 | Ta1 antibody; all active with therapy | [47] | ||||
| Controls | 58.2 ± 10.6 | 1F7 antibody; all active with therapy | [47] | ||||
| RA (active) | 72.0 ± 8.8 | 1F7 antibody; all active with therapy | [47] | ||||
| Controls | 12.3 ± 4.2 | Ta1 antibody | [49] | ||||
| OA | 8.1 ± 2.0 | Ta1 antibody | [49] | ||||
| RA (inactive) | 41.6 ± 23.1 | Ta1 antibody | [49] | ||||
| RA (active) | 62.2 ± 10.1 | Ta1 antibody | [49] | ||||
| Controls | 58.8 ± 9.0 | 1F7 antibody | [123] | ||||
| OA | 49.0 ± 11.0 | 1F7 antibody | [123] | ||||
| RA (inactive) | 60.6 ± 4.0 | 1F7 antibody | [123] | ||||
| RA (active) | 75.9 ± 10.0 | 36.3 ± 10.0 | 1F7 antibody | [123] | |||
| OA | 36.7 ± 6.5 (CD26: 614 ± 157 ng/ml) | 14.6 ± 4.4 (CD26: 247 ± 25 ng/ml) | [55] | ||||
| RA | [55] | ||||||
| All | 26.7 ± 9.9 (CD26: 473 ± 176 ng/ml) | 16.1 ± 5.6 (CD26: 259 ± 80 ng/ml) | [55] | ||||
| Non-inflammatory | 31.8 ± 12.7 (CD26: 558 ± 195 ng/ml) | (n = 24; CRP < 20 mg/l) | [55] | ||||
| Inflammatory | 22.4 ± 4.7 (CD26: 404 ± 139 ng/ml) | (n = 17; CRP > 20 mg/l) | [55] | ||||
| Controls | 41.3 ± 4.7 | [32] | |||||
| RA (active) | 34.5 ± 3.2 | [32] | |||||
| SLE | 29.9 ± 6.5 | [32] | |||||
| OA | 702.6 ± 41.2 mU/mg | Synovial fluid | [60] | ||||
| RA (active) | 549.6 ± 28.3 mU/mg | Synovial fluid | [60] | ||||
| Controls | 634 μmol/min/mol | [36] | |||||
| Sjogren's syndrome | 969 μmol/min/mol | [36] | |||||
| SLE | 650 μmol/min/mol | [36] | |||||
| RA (active) | 224 μmol/min/mol | [36] | |||||
| OA | 1.0 ± 1.4 nmol/min/mg | Synovial membranes | [54] | ||||
| RA (active) | |||||||
| All grades | 1.1 ± 1.1 nmol/min/mg | Synovial membranes | [54] | ||||
| Grade 1 (0–10 ml fluid) | 1.8 ± 1.4 nmol/min/mg | Synovial membranes | [54] | ||||
| Grade 2 (10–20 ml fluid) | 0.9 ± 0.6 nmol/min/mg | Synovial membranes | [54] | ||||
| Grade 3 (> 20 ml fluid) | 0.6 ± 0.5 nmol/min/mg | Synovial membranes | [54] |
CRP, C-reactive protein; DPP, dipeptidyl peptidase; OA, osteoarthritis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus. Values for peripheral blood CD3+CD26+, peripheral blood CD4+CD26+ and synovial fluid CD3+CD26+ are shown as % gated lymphocytes.
The concentrations of IL-12 and IL-15 in sera of RA patients are increased independently of disease activity, while blood plasma DPPIV activity correlates inversely and T-cell DPPIV activity/CD26 expression correlates positively with disease severity. This serves only to confirm that regulation of surface CD26 expression and secreted DPPIV activity is dependent upon more than activation by a single stimulus, representing rather the threshold response to a number of inputs. Although it may seem counterintuitive to have an increase in circulating activated T cell surface DPPIV activity while free soluble enzyme is reduced in the plasma, this may be a simple partitioning effect reflecting reduced proteolytic release of the membrane form. Furthermore, such a situation would be advantageous to the development of RA, where local inflammation would be enhanced while the reduced plasma DPPIV activity would be less effective at restricting the bioactive peptides locally. Consequently, increased circulating chemotactic and lymphokine/cytokine activities will lead to inappropriate systemic activation. As increased CD26 expression is a marker strongly associated with extravasating activated T cells, a role for DPPIV is implied [43,44]. It is unlikely that the DPPIV activity per se is responsible for clearing a path through connective tissue. It has been demonstrated, however, that the DASH family member seprase forms a complex with CD26 that is a prerequisite for invasion and migration of fibroblasts through a collagenous matrix [45]. T cells do not express seprase, but CD26 can associate with other type II transmembrane prolyl serine peptidases that may function in the invadopodia of activated T cells [46]. The precise role of CD26 in invasion remains enigmatic; the high expression of CD26 on transendothelial migrating cells is a property of a previously determined CD4+ memory phenotype [43]. Extravasation is a continuous process, and by inactivating chemokines as the migration progesses, CD26 may keep the path at a surveillance level rather than a full-blown immune invasion.
T cells expressing high levels of CD26, which are abundant in the PB of RA patients, are believed to migrate into the rheumatoid synovium to initiate local inflammation and tissue destruction [47]. Within the SF, concentrations of IL-15, a known inducer of CD26 on both T and NK cells, are high [48]. Using the two monoclonal antibodies 1F7 and Ta1, both of which recognize epitopes in the 100 amino acids between residues 247 and 358 of DPPIV, Gerli et al. [49] observed strong Ta1 positivity, but 1F7 negativity, on SF T cells. The 1F7 antibody is able to cross-link CD26 and co-stimulate TcR-driven stimulation, which is not the case for Ta1. This difference in expression of the two epitopes probably does not represent alternative forms of CD26, as the epitopes are so close to each other. Other possibilities include supramolecular associations that affect antibody accessibility, or even cross-reactivity with unrelated epitopes. In fact, 1F7 has already been shown to react weakly with attractin [50].
Not only cross-reactivity but also differences in subcellular localization of DPPIV (i.e. secretory endosomes or plasma membrane) may have important consequences for T cell reactivity and pre-secretion modification of bioactive peptides. For example, total DPPIV activity within SF mononuclear cells from RA patients is no different from those from osteoarthritic (OA) patients, while the plasma membrane DPPIV activity is significantly higher on cells from the OA patients [51].
Significantly lower DPPIV was found in SF of RA patients compared with SF from OA patients or healthy donors [34,52]. Within the synovium, synovial fibroblasts strongly express DPPIV while the secreted activity is reduced [53], similar to the relationship of T cell DPPIV to plasma DPPIV. Nevertheless, as the joint fluid volume increases, the DPPIV activity of RA synovial membrane decreases [54]. The increased membrane DPPIV expression and reduced secreted activity in the plasma/serum is now an accepted and reproducible finding in RA, although not consistently observed in the synovial fluid (Table 1). Despite the inconsistency in reporting of differing DPPIV levels between non-inflammatory and inflammatory synovial fluid, there is a consistent, reproducible reduction in synovial fluid DPPIV levels of a third to a half in comparison with circulating levels (Table 1). Upon initiation of an inflammatory reaction within the synovium, the reduced representation of DPPIV will lead to a potentiation of the half-lives of immunoattractive and enhancing peptides, including RANTES, SDF-1, vasoactive intestinal peptide (VIP) and substance P (SP). The lower synovial DPPIV activity in the context of higher circulating levels in the periphery may contribute towards a gradient of functional chemokine activity that will maintain attraction of activated migrating immune cells into the synovium.
The plasma DPPIV levels appear to be strongly reduced in RA, and splitting patient samples into non-inflammatory and inflammatory by C-reactive protein levels reveals the reduction is predominantly observed in inflammatory RA [55]. Although we will describe later the therapeutic effects of DPPIV inhibitors on RA progression, this does not, however, necessarily imply a direct relationship between DPPIV activity and inflammation. The development of an inflamed environment more properly represents a cascade involving multiple heterogeneous cell types that may be maintained and exacerbated by the activity of DPPIV. Nevertheless, in two mouse models of inflammatory arthritis, the severity of joint damage correlated directly with levels of SDF-1α where the efficacy of this chemokine to attract T cells was directly and negatively controlled by serum levels of DPPIV, and the joint-infiltrating T cells had an increased expression of SDF-1α receptor (CXCR4). As observed in human samples, plasma levels of DPPIV were significantly reduced as the arthritis developed [55]. The severity of the induced arthritis was significantly increased in CD26-/- knockout mice, but in the complete absence of CD26/DPPIV, few conclusions can be drawn as to the relationship of systemic to synovial DPPIV enzyme activity. Nevertheless, the more severe pathology certainly supports a direct role for DPPIV in inactivating pro-inflammatory substrates such as SDF-1α, particularly as the mouse form lacks ADA binding activity. Due to the absence of an alternatively spliced terminal exon, mice do not produce a secreted form of attractin, but it has been suggested that sequestering and inactivation of circulating chemokine substrates is a function of the plasma secreted attractin isoform in humans [56].
The activities of CD26 are not limited to DPPIV enzymatic activity and its ADA binding capacity may be of importance. The relative decrease in CD26+ T cells in SF, coupled with a concomitant reduction in soluble DPPIV activity, relate inversely to the concentration of free ADA, which is high in RA patients [8,57]. As the methotrexate- and deoxyco-formycin-based therapies act as anti-inflammatory agents by increasing extracellular adenosine and blocking ADA, respectively, the presence of high soluble ADA suggests a pro-inflammatory environment independent of CD26. This contention is questionable, however, because extracellular ADA is not effective at overcoming high levels of adenosine inhibiting T cells unless it is in complex with CD26. Because the resident T cells appear to be CD26low, this suggests a crucial role for strongly CD26+ activated T cells infiltrating into the synovium from the periphery. This concept stresses the kinetic nature of synovial inflammation where a single time point assay may identify a synovial population of CD26low 'spent' T cells and miss a constant influx of CD26high activated T cells driven by their high levels in the PB. A mechanism driven in this manner would be consistent with the systemic nature of RA, and suggests that any process that results in long-term peripheral activation of T cells could at some point lead to RA. The complex genetics of RA with susceptibility loci on human chromosomes 1p13, 1p36, 5q31, 6p21.3 and 21q22.3 also point to multiple origins with a uniform end process. It must be stressed that T cell driven immune processes cannot be viewed in isolation as the fibroblast-like synoviocytes express high levels of both CD26 and associated ADA isoform 1, which may maintain a pro-inflammatory environment independent of the CD26low T cells. A further issue is whether the presence of high ADA will lead to maximal binding with CD26 because the affinity is strong, with a KD of 65 nM [58]. The formation of ADA-CD26 complexes leads to an increase in ADA specific activity that will directly reduce adenosine levels, reducing the efficacy of methotrexate- and deoxycoformycin-based therapies.
Dipeptidyl peptidase II/quiescent cell proline dipeptidase
Although sequence identities at nucleotide and tryptic peptide-mass spectroscopic levels suggest that DPPII, QPP and DPP7 are identical, there remain some differences in substrate specifity and pH optimum criteria that prevent a definitive conclusion of identity [9,59]. Nevertheless, until this is resolved we will retain the terminology used in the original reports. As both DPPII and QPP localize intracellularly, it is unlikely that they will participate in the processing of extracellular peptides, although it does not exclude the possibility that they may modify peptides prior to secretion. The presence of QPP in a post-Golgi vesicular compartment can be inferred by its release as a secreted protein following calcium mobilization. Initially identified in the human Jurkat T cell line, expression profiling suggests it is widespread, with strong representation in human blood, cervix, mammary gland, ovary, uterus, kidney, lung, pancreas and skin, suggesting an association with secretory processes related, in particular, to the female reproductive system.
Although the physiological substrates for QPP remain unknown, inhibition of QPP activity leads to an initiation of an atypical apoptotic pathway in quiescent lymphocytes. No significant sequence homology exists between CD26 and QPP; it is the DPPIV-like enzymatic activity of DPPII/QPP that classifies it as a DASH molecule. In contrast to the canonical DPPIV, DPPII/QPP does not cleave neuropeptide Y (NPY) [3].
The relationship of DPPII/QPP/DPP7 activity to CD26-related enzyme activity is complex in that DPPII activity increases in the plasma of RA patients, while DPPIV decreases [32]. This suggests that, overall, DASH activity may be less critical than individual substrate specificity, which may be influenced in large part by initial subcellular localization rather than plasma levels. Significantly higher DPPII activity was detected in SF [37,60] and in the synovial membrane from RA patients than in OA cases, in which the synovial membrane DPPII activity correlated positively with the SF volume [54]. Although DPPII activity is low in comparison to CD26, this suggests that DPPII is at least associated with an inflammatory-mediated secretory process.
Attractin
Attractin was initially identified as a serum protein with DPPIV activity secreted by activated T cells [10]. Expression profiling in several primary cells and cell lines revealed that, similar to DPPII/QPP, its expression was not limited to T cells but rather to all cells with strong secretory capacity [50]. A membrane form was subsequently identified in humans and its similarity with non-primate attractin, and the absence of secreted attractin in non-primates, suggest that the membrane-tethered form is the main functional entity [61]. Similar to CD26, attractin is a potent enhancer of recall antigen-driven T-cell proliferation. There is no sequence homology with CD26 and the classical serine protease catalytic site is not present, which has led to debate about its enzyme activity. Nevertheless, several studies have purified it to apparent homogeneity with no contamination by CD26 but with lower specific activity than that of native kidney-purified DPPIV [10,11]. This raises the possibility that the DPPIV activity of attractin may be secondary and that it has other more specific substrate specificities yet to be identified, similar to the NAALADases, which have both amino-terminal DPPIV activity as well as a more pronounced carboxy-terminal glutamate carboxylase activity. It has been suggested that the secreted circulating form in humans may play a role in systemic deactivation of chemotactic cytokines, ensuring that the peptides are only active at the local site where they are released. There are several instances where membrane DPPIV activity does not correlate well with detected CD26. In these cases, varying levels of other DASH molecules, including attractin, may be responsible, which may be indicated by alterations in DPPIV specific activity [28]. Alterations in the DPPIV levels in SF may represent an increase in the presence of attractin. Certainly, analysis of serum reveals an increase in attractin in RA patients with erosive disease, which is not observed in individuals with non-erosive RA or healthy individuals (B Guild, personal communication) [62].
Seprase/fibroblast activation protein-α
Seprase probably plays a critical role in exacerbating joint degeneration once it has been initiated. Expressed by fibroblasts activated in a wound healing environment, seprase has a strong gelatinase activity that aids breakdown of extracellular matrix. Either alone or in complex with DPPIV/CD26 as a heterodimer, it localizes on invadopodia at the forefront of extracellular matrix breakdown [45]. Once degeneration is initiated in the synovium, seprase will be upregulated in activated fibroblasts, where it will contribute to the degenerative process. Unlike wound healing in general that will be localized to a specific site and can thus be carefully regulated without disturbing the overall immune homeostasis, the presence of systemic activated T cells will lead to their continuous influx into the inflamed synovium, maintaining the inflammatory state. This will lead to a continuous activation of synovial fibroblast seprase expression, a process further maintained by the subsequent articular erosion.
Other DASH molecules
The appreciation that DPPIV-like enzymatic activity may reside in several molecules with differing specific activities and representation requires a reinterpretation of a considerable volume of data where DPPIV activity was measured independent of antigenic reactivity or, conversely, where CD26 antigenic activity but not DPPIV activity was measured. This is complicated further by a lack of understanding of the breadth of substrate and inhibitor specificities for the DASH family. This is illustrated well by the CD26-knockout mouse that still retains a circulating DPPIV activity almost 13% that of controls despite a complete absence of immunologically detectable CD26 [63]. Furthermore, the residual activity in the CD26 knockouts could not be inhibited by the DPPIV inhibitor valine-pyrrolidide. Accordingly, multifactorial analysis of inhibitor panels and antibody-binding specificities may prove to be a useful technique for weighting the contribution of individual DASH family proteins to DPPIV-mediated degradation of bioactive peptides.
The DASH family substrates: enzymatic regulation and their relationship with rheumatoid arthritis
The immune system receives input not only from the cells and messengers considered to be part of the classic immune network, but is clearly also influenced by neuroendocrine and reproductive signals. Consequently, the pathogenesis of the chronic disabling inflammatory diseases, of which RA is one, must take into account these extra-immune influences. By the same token, despite the understanding that multiple mediator abnormalities may contribute to RA development [64,65], it is difficult to assess whether effects in vivo related to a neuroendocrine peptide are direct upon the immune cell, or indirect through creating an environment that modulates immune activity. For example, malnutrition leads to an immunosuppression that was not well understood until the discovery that adipocyte-derived leptin levels fall in the fasting condition or during inflammation, and T cell responses independent of the nutritional state can be restored by administration of leptin, which binds directly to receptors on T cells [66]. In several instances, there is clear evidence for neuroendocrine receptors on immune cells, and many of the ligands for these receptors have been identified as substrates for DPPIV activity. It is already well established that DPPIV cleavage can have powerful effects upon cytokine and chemokine activity. In the sections below, we will examine how the increased DPPIV activity associated with activated T cells may exacerbate the systemic inflammatory reaction characteristic of RA by modifying neuropeptide, cytokine and chemokine functionalities.
Neuropeptides
To date, neuropeptides that have been shown to directly modulate immune function through expressed receptors include SP, the pancreatic polypeptide family (including NPY), calcitonin gene-related peptide (CGRP), VIP and gastrin-releasing peptide (GRP). Remarkably, not only do the levels of several of these neuroendocrine peptides undergo distinct alterations in active RA, but all have been identified as substrates for DPPIV activity where the highly specific clipping of the amino-terminal dipeptide with a penultimate proline may have profound effects upon receptor agonism and downstream functionality (Fig. 1). The presence of higher levels of surface DPPIV on systemic PB T cells will probably not have a significant effect upon the paracrine activities of these immune-activating neuropeptides, but the relative decrease of DPPIV/CD26 on the surface of SF-localized T cells may lead to a potentiation of the neuropeptide half-life, exacerbating the local inflammation.
Figure 1.
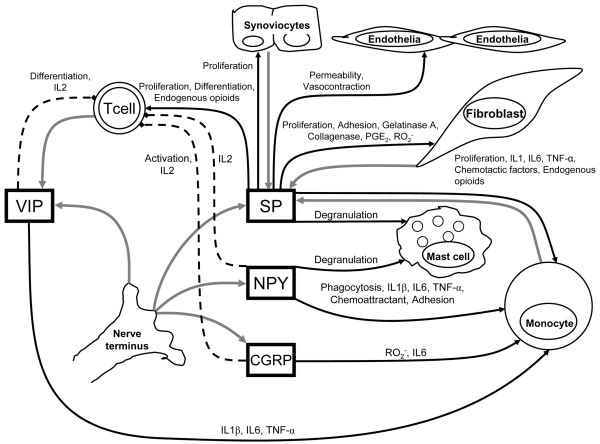
Contribution of dipeptidyl peptidase IV-sensitive neuropeptides to the control of inflammation in rheumatoid arthritis. Gray arrows indicate release of indicated mediator; black arrows indicate stimulation of indicated cell function; dashed lines indicate abrogation of cell function and/or release of indicated mediator. 'Endogenous opioids' include enkephalins, endorphins, dynorphin and endomorphin. CGRP, calcitonin gene-related peptide; IL, interleukin; NPY, neuropeptide Y; PGE2, prostaglandin E2; RO2-, reactive oxygen species; SP, substance P; TNF, tumor necrosis factor; VIP, vasoactive intestinal peptide.
Substance P
SP is an undecapeptide belonging, together with neurokinins A and B, to the tachykinin family. It is released by sensory neurons, fibroblasts, macrophages and fibroblast-like synovio-cytes. Although SP can bind to at least three membrane G protein-coupled receptors (the neurokinins NK1, NK2, NK3), its main target appears to be the NK1 receptor. The levels of SP in SF are high in RA [67], and downstream signaling leads to varying consequences depending upon the target cells. Mononuclear phagocytes respond with increased prostaglandin E2 and IL-1β, TNF-α and IL-6 secretion while mast cells respond by degranulation [68]. Rheumatoid synoviocyte proliferation is enhanced, and fibroblast-like synoviocytes release collagenases, IFN-γ, TNF-α, IL-1β, and oxygen radicals and upregulate surface adhesion proteins such as vascular cell adhesion molecule 1 in response to SP [69]. Intra-articular injection of IL-1 or TNF-α increases SP concentration in the SF and leads to cartilage degradation, while transforming growth factor (TGF)-β was shown to induce SP production in synovial fibroblasts [70]. In this respect, RA fibroblast SP release was more sensitive to TGF-β induction than were fibroblasts from OA subjects [69].
In responding to physiological levels of SP, leukocytes from patients with RA strongly upregulate release of IL-1β, TNF-α and IL-6 in contrast to a much lesser effect upon leukocytes from non-inflammatory OA [71]. This effect appears to be related to an increased representation of SP receptors and is supported by reports that pre-incubation of PB leukocytes from RA patients showed a stronger expression of T cell activation markers than did cells from controls [72], while SP stimulates T cell proliferation in RA patients more efficiently than in controls [73].
The biological effects of SP in vitro are mediated as effectively by the carboxy 4–11 fragment as by the full-length peptide, implying that DPPIV-mediated cleavage of an amino-terminal dipeptide will not affect receptor specificity or alter agonist-mediated downstream signaling. Nevertheless, the amino-terminal penultimate proline is resistant to cleavage by other aminopeptidases, lending protection against degradation. Consequently, removal of the amino-terminal dipeptide by DPPIV significantly shortens the in vivo biological half-life of SP [74].
The pancreatic polypeptide family
The pancreatic polypeptide family comprises NPY, peptide YY, and pancreatic polypeptide. NPY has pleiotropic activities across the endocrine, nervous and immune systems, signaling via at least five (Y1 to Y5) receptor subtypes. Hydrolytic processing of NPY by DPPIV changes its resulting receptor preference, converting it functionally from a Y1 to a Y2/Y5 agonist. Although NPY is very efficiently cleaved by DPPIV, it is highly resistant to hydrolytic attack by DPPII, another DASH member dysregulated in RA [3]. Both DPPIV and NPY are co-expressed not only in circulating immune cells but also in the vascular endothelium, supporting a role for DPPIV in the regulation of systemic NPY [75]. NPY acts, among other functions, as a chemoattractant and activator of mononuclear cells [76]. Together with SP, NPY induces phagocytosis and activation of macrophages as well as leukocyte production of TNF-α and other pro-inflammatory cytokines. Mononuclear blood cells from RA patients are more responsive to NPY than are similar cells from non-inflammatory OA patients, with strong increased secretion of IL-1β, TNF-α and IL-6, suggesting a change in receptor density as noted above for SP receptors.
Simultaneous application of TNF-α and IFN-γ to endothelial cells causes an increase in NPY, upregulation of the Y5 receptor, and complete loss of the Y2 receptor, together with an upregulation of DPPIV activity. Because the DPPIV-cleaved NPY binds effectively to the Y5 receptor, an autocrine loop is created [77] that might exacerbate activity of already activated RA T cells. These observations fit well with studies demonstrating NPY-associated chemoattractant and adhesion-inducing properties for leukocytes [78]. Certainly, increased levels of NPY are observed in SF of patients with RA [79]. Evidence for regulation of local inflammation is provided by the report that locally applied NPY potentiated, while NPY Y1 receptor antagonist abolished, concanavalin A-induced paw edema in rat [80]. In contrast, excessive stimulation of peritoneal macrophages by NPY suppresses TNF-α, and a similar effect has been noted upon IL-2 release by mouse leukocytes [71]. As mentioned above, however, results from mice must be treated cautiously because DPPIV/CD26 is not an activation antigen and does not bind ADA in them.
Calcitonin gene related protein
Compared to the other neuropeptides discussed, less is known about the immune effects of CGRP and the consequences of its proteolysis by DPPIV. This is in part due to CGRP functioning in the context of SP, with which it is usually colocalized and coreleased, providing an additive or synergistic effect. As for SP, increased concentrations of CGRP are observed in SF of RA patients [81], and it similarly induces high levels of IL-1β, TNF-α and IL-6 from RA PB leukocytes [71]. There is one report, however, of an anti-inflammatory CGRP effect leading to decreased IL-2 production [82].
Vasoactive intestinal peptide
VIP is predominantly viewed as an anti-inflammatory and anti-autoimmune mediator, executing its role mostly via down-regulation of pro-inflammatory mediators; in RA, this is observed for chemokines and TNF-α derived from isolated synovial cells [83]. Consistent with these observations, VIP administration decreased the severity of experimental arthritis in rodents [84]. Slightly elevated concentrations of VIP were observed in SF of RA patients compared to OA samples [81], suggesting an attempt by the immune system to down-regulate a cascading immune reaction. Nevertheless, at the systemic level, VIP probably can also induce strong production of some pro-inflammatory cytokines, including TNF-α, IL-6 and IL-1β, by RA PB cells at significantly higher levels than the release associated with noninflammatory cells from OA patients [71]. The balance of stimulatory and inhibitory effects of VIP and the effects of DPPIV activity is not a situation that can be duplicated in vitro, but rather represents the complexity of trying to model local environments where inflammation is being driven by an influx of activated cells from the systemic circulation.
Bombesin/gastrin-releasing peptide
Among its other functions, bombesin/GRP is believed to act as a tissue-specific paracrine growth factor that promotes proliferation of chondrocytes and stimulates antibody-dependent cellular cytotoxicity and NK cell activity [85,86]. In contrast with healthy controls, most RA patients, particularly those with an early arthritis, displayed measurable concentrations of GRP in the SF where the GRP concentration correlates with the number of SF leukocytes [67].
Cytokines
Cytokines are secreted by activated immune cells, mainly T cells and macrophages, as well as by other cell types such as fibroblasts. They have been found in synovial membrane and fluid in RA, psoriatic arthritis and OA, with quantitative differences observed dependent both upon disease type and severity [87-89].
Levels of TNF-α, IL-1 and IL-6 in RA SF and synovial tissues from RA patients are high and significantly higher than those in samples from controls and OA patients [90]. The ability of DPPIV to cleave these lymphokines is inversely correlated with the chain length [91]. It appears that DPPIV can cleave carboxy-shortened TNF-α, IL-1 and IL-6 but not the full-length mature peptides. The significance of this is difficult to interpret because the effects here of DPPIV would be anti-inflammatory. Further, the resistance to cleavage of full-length peptide has only been observed with CD26, and does not exclude cleavage by other DASH members, or their synergistic activity, or even an extracellular protein-assisted conformational change that would open the amino-terminal of the full-length protein to cleavage. In support of a mechanism more complex than simple CD26-mediated cleavage in vitro, full-length TNF-α undergoes a DPPIV-like cleavage in U937 human monocyte-like cells, and an identical activity was identified in both primary macrophages and monocytes [92]. Given the critical role ascribed to TNF-α in RA-associated inflammation [93], and that this lymphokine can itself induce fibroblasts and monocytes to produce downstream DPPIV substrates, including RANTES, macrophage inflammatory protein (MIP)-1 and IFN-γ inducible protein-10 (IP-10) [94-97], it is clear that DPPIV analysis should be global and not focused only on CD26 (Fig. 2).
Figure 2.
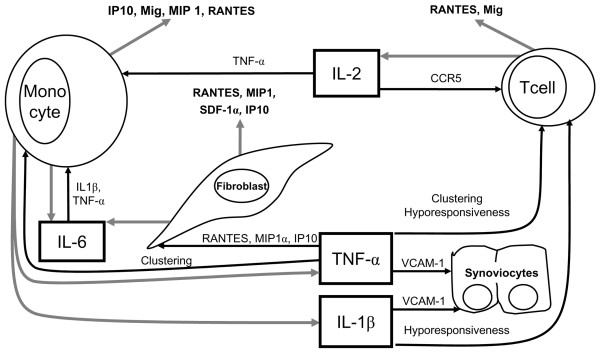
Contribution of dipeptidyl peptidase IV-sensitive cytokines to the control of inflammation in rheumatoid arthritis. Gray arrows indicate release of indicated mediator; black arrows indicate stimulation of indicated cell function; dashed lines indicate abrogation of cell function and/or release of indicated mediator. CCR, CC receptor; IL, interleukin; IP-10, IFN-γ inducible protein-10; Mig, monokine induced by interferon-γ; MIP, macrophage inflammatory protein; RANTES, regulated upon activation normal T-cell expressed and secreted; SDF, stromal cell-derived factor; TNF, tumor necrosis factor; VCAM-1, vascular adhesion molecule-1 (CD106).
TNF-α induces IL-1β secretion by macrophages and monocytes, leading to activation of synoviocytes, T and B cells and increased production of structural protein degrading enzymes in the RA joint environment. A positive correlation was found between IL-6 and SP levels as well as between IL-6 and the cell count in SF of patients with RA. In contrast with SP, VIP and CGRP, an elevated IL-6 concentration was detectable also in blood plasma of RA patients [81]. Because DPPIV activity should be inhibitory for most of these processes, the reduced DPPIV activity observed within the enflamed synovium would lead to longer biological half-lives for all these lymphokines, with the consequent maintenance of the activated state.
Chemokines
Chemokines constitute a large superfamily of paracrine/autocrine 'chemotactic cytokines' that bind to G protein-coupled seven-span transmembrane receptors and control leukocyte migration and homing as well as maturation and release of inflammatory mediators. They are classified into four subfamilies (CXC, CC, C, and CX3C) based on the number and spacing of the first two or four cysteine residues. The pro-inflammatory cytokines such as TNF-α and IL-1β, which are believed to play a critical role in the pathogenesis of RA, have been shown to upregulate a number of chemokines from several cell types within the synovium. Although the relative contribution made by individual chemokines is not yet clear, a growing number of reports indicate that RANTES (CC ligand (CCL)5), MIP-1β (CCL4), monokine induced by interferon-γ (Mig; CXC ligand (CXCL)9), IP-10 (CXCL10) and SDF-1α (CXCL12) actively participate in RA pathogenesis and have been shown to be substrates for DPPIV activity [94-96,98-101]. Amino-terminal processing of these molecules modifies, both quantitatively and qualitatively, their receptor preference and consequent functional properties.
In the RA synovial environment, Mig was found mainly in monocytic cells, whereas RANTES is expressed predominantly by CD3+ lymphocytes. Mig and IP-10, both ligands for the CXC receptor (CXCR)3, are attractants for lymphocytes mediating Th1-type responses in the RA enflamed joint. Their presence and expression create a gradient from the joint to the blood favoring Th1 cell migration into the tissue. In OA cases, the gradient appears to be generated in the opposite direction [102,103]. Memory CD4+ T cells, the major cell population present in the synovium infiltrate, strongly express the chemokine receptors CXCR3, CXCR4, CXCR6 and CCR5, which all bind pro-inflammatory chemokines. SDF-1 is important for retaining cells in the inflamed joint, and its receptor (CXCR4) is strongly upregulated in the synovium by IL-15 and TGF-β, both of which are highly expressed in SF [95]. Production of SDF-1 by rheumatoid synovial fibroblasts is probably critical in maintaining the recruitment of CXCR4 expressing T cells from the periphery to inflamed synovium [104,105]. Strong evidence that DPPIV-mediated inactivation of SDF-1 is an integral part of SDF-1 signaling and regulation of T cell attraction is provided by the report that CXCR4 and CD26 form a tight molecular complex and are internalized together following ligation and co-precipitate together in antibody-based pulldown assays [106]. Confirmation of these inferential findings has been demonstrated in mouse articular inflammatory models where DPPIV proteolysis of SDF-1α in vivo was shown to directly regulate T cell recruitment to the inflamed regions [55].
Robinson et al. [96] demonstrated elevated levels of RANTES, another DPPIV substrate, in PB, SF, and synovial tissues of RA patients. Expression of RANTES was undetectable in OA synovia, where inflammatory lymphocyte infiltration is not observed. Binding of RANTES to the chemokine receptors CC receptor (CCR)1, CCR3 and CCR5 leads to selective recruitment of T cells and monocytes into the synovium [94]. CCR3 and CCR5 receptors are upregulated on RA-derived cells, both in the periphery and in the inflamed synovium, rendering the cells more sensitive to local RANTES attractive gradients. Perhaps helping to exacerbate the initial inflammation, the receptor density increases as the SF leukocyte count increases. CCR5 expression is detected on the majority of cell types in the synovial environment, including macrophages, fibroblasts, vascular smooth muscle cells and perivascular lymphocytes [99]. Its expression on peripheral and synovial CD4+ cells in RA patients is further upregulated by IL-15, a pro-inflammatory cytokine [48]. RANTES is not the only DPPIV substrate elevated in RA; MIP-1α is also increased and also binds to CCR5 [97,98]. Examination of CCR5 antagonists as a therapeutic modality have been shown to inhibit collagen-induced arthritis in mice, an effect ascribed to interference with T-cell migration [107].
Following cleavage of RANTES by DPPIV, loss of the amino-terminal dipeptide did not have a profound effect upon T cell chemotaxis, but resulted in a loss of monocyte attraction [108]. This may represent a shift in receptor expression on monocytes as there is evidence of an affinity shift for RANTES from CCR1 to CCR5 after DPPIV cleavage. In addition, these results examined only the effect of CD26 upon RANTES without considering other DASH enzyme activities. Analysis using MALDI-TOF of full-length RANTES following incubation with either attractin or CD26 reveals that attractin cleaves only the amino-terminal dipeptide while CD26 may then release a further dipeptide from the amino terminus consisting of amino acids Tyr3 and Ser4, and will similarly release the same dipeptide from synthetic (3–68) RANTES, a further digestion that may also influence receptor preference [56].
DPPIV enzymatic activity inhibitors in rheumatoid arthritis
Therapeutic options for modifying DPPIV activity in RA would, at first glance, seem facilitated by the recent advances in designing a large panel of inhibitors to block degradation of glucagon-like peptide 1 (GLP1), another DPPIV substrate that plays a critical role in controlling glucose metabolism [109]. Nevertheless, there are several concerns. DPPIV levels are low to normal in the inflamed synovium, with the consequence that chemokines such as SDF-1 and RANTES will have enhanced longevity. Administration of systemic DPPIV inhibitors will serve only to enhance the biological half-life, potentiating influx of activated T cells from the periphery. Conversely, the high level of DPPIV/CD26 on activated systemic T cells is essential for efficient transendothelial migration [44], and blocking of DPPIV activity may be critical for blocking migration into the synovium. The blocking of migration and enhanced degradation of chemokines will need to be carefully balanced, and experimental animal models may be limited for this purpose for several reasons. First, it remains to be shown that experimentally induced mouse arthritis really represents the systemic infiltration process seen in the human disease as opposed to a local inflammation that just happens to have been induced in the joint. Second, the role and functions of CD26 in the rodent are quite different to those in humans. Third, it is clear that members of the DASH family are not uniformly sensitive to inhibitors, each member expressing a unique spectrum of inhibition responses to a panel of DPPIV-specific inhibitors. Finally, as alluded to above, there are several peptide modulators of metabolism that are substrates for DPPIV activity, and systemic administration will affect these processes as well as immune processes.
The importance of these complicating factors relative to the desired reaction to be controlled cannot be predicted, and needs to be experimentally determined. Certainly, T cell proliferation and TNF-α production in vitro can be abrogated by DPPIV inhibitors [110]. Inhibition of DPPIV suppressed both cellular CD26 expression, serum DPPIV activity and prolonged allograft survival [31,111]. Similar DPPIV inhibition in vivo led to an increase of immunosuppressive cytokine TGF-β1 in plasma, but did not cause a nonspecific general immunosuppression. Furthermore, DPPIV inhibitors have been shown to suppress T lymphocyte subpopulation migration into the inflamed tissue, as well as suppressing T cell DNA synthesis, and TNF-α, IL-1 and antibody production, all processes that may need to be controlled in RA [112]. Conversely, in some monocyte-derived cell populations, DPPIV enzymatic activity inhibitors may stimulate production of TNF-α [113].
Despite the compound and model-specific effects, there is increasing evidence that systemically distributed DPPIV inhibitors might have potent, dose-dependent anti-arthritic effects associated with down-regulation of a number of pro-inflammatory parameters both in vitro and in vivo in experimental animals [114-117]. Reinforcing the notion that systemic blocking of circulating T cell-associated DPPIV will be useful while synovial blocking might be counterproductive, DPPIV inhibitors were shown to increase the effect of SP on mitogen-induced proliferation of T cells, IL-2 production by T cells, immunoglobulin synthesis by B cells and TNF-α as well as other cytokine production by monocytes [68,118,119]. Similarly, the pro-inflammatory effects of NPY in concanavalin A-induced paw edema in rat was potentiated by co-application of a DPPIV inhibitor [80].
The potential to target systemic DPPIV and limit activity in the extracellular fluids would be desired pharmacologically, as would targeting of inhibitors to give broad spectrum inhibition of DASH family molecules. Ideally, preservation of DPPIV activity for peptides involved in metabolism and neurophysiology would be maintained. Such a trade-off may be accomplished not by complete broad inhibition of DPPIV activity, but simply by administration of inhibitor cocktails that would bring systemic cell-associated DPPIV activities within the low to normal range.
Conclusion
The chronic systemic inflammatory reaction characteristic of RA results in breakdown of cartilage and erosion of proximal bone. The initial insult that leads to development of autoreactive T cells remains enigmatic, but there can be no doubt that circulating activated T cells extravasate into the synovium where they release or express various factors that potentiate resorption of both cartilage and bone. The damaged synovial cells themselves release cellular components that mimic wound healing, leading to the further recruitment of immune cells. The predisposing factors, including genetic associations and possible initiating peptides, are not discussed here. This review instead addresses the involvement of DASH enzymatic activity in every step of the post-insult process, from T cell activation to extravasation, and from cartilage breakdown to the release of chemokines and cytokines that attract new T cells and monocytes, thereby increasing the inflammatory response and exacerbating degeneration. Some of the proposed interactions and mechanisms are outlined in Fig. 3.
Figure 3.
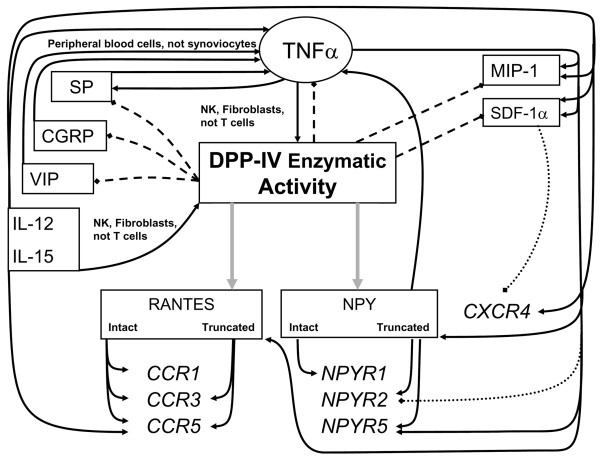
Overview of regulation of dipeptidyl peptidase (DPP)IV enzymatic activity and its substrates in rheumatoid arthritis-related inflammation. Gray arrows indicate modification of mediator receptor preference by proteolytic modification; black arrows indicate stimulation of indicated mediator or enzymatic activity; dashed lines indicate termination of indicated mediator function by proteolytic degradation; dotted lines indicate down-regulation of indicated receptor. Biologically active peptides are shown in regular font and receptors are shown in italics. See the text for references. CCR, CC receptor; CGRP, calcitonin gene-related peptide; CXCR, CXC receptor; IL, interleukin; MIP, macrophage inflammatory protein; NK, natural killer; NPY, neuropeptide Y; NPYR, neuropeptide Y receptor; SDF, stromal cell-derived factor; SP, substance P; TNF, tumor necrosis factor; VIP, vasoactive intestinal peptide.
The DASH family members, predominantly CD26 and attractin, are rapidly upregulated on T cell activation. Following increased expression on the T cell surface, DPPIV plays a critical role in allowing the activated T cells to extravasate into the extracellular space. Once in the vicinity of the synovium, the DPPIV activity would be able to initiate amino-terminal degradation of the increased levels of pro-inflammatory neuropeptides, cytokines and chemokines that are present and are substrates for such activity. This degradation might be effective in down-regulating inflammation were it not for the remarkable observation that DPPIV activity in the RA inflamed synovium is low to normal and does not reflect the higher levels in the peripheral circulation, which themselves are lower than normal. The lower level of secreted DPPIV in RA is matched by a reciprocal increase in T cell membrane expression (Table 1), which leads to an increase in the extravasation potential of a T cell while simultaneously extending the chemoattractive capacity of DPPIV-sensitive RANTES and SDF-1α. Why is secreted CD26 low in the synovium? Ligation and cross-linking of CD26 leads to its internalization, and because CD26 can bind collagen, there exists a strong possibility that synovio-cytes internalize CD26 cross-linked by fragments from the degenerating synovial lining. Reduction of the membrane levels in this way would also reduce the amount available for cleavage and release. Proteoglycans released into the synovial fluid by articular degeneration may contribute further to chemoattraction because their presence is necessary for efficient presentation of basic-charged chemokines such as RANTES and SDF-1α to their respective receptors [120,121]. The binding of chemokines by the proteoglycan fragments may also shield them from DPPIV proteolysis.
Therapeutic options include the systemic administration of DPPIV inhibitors together with TNF-α antagonists such as etanercept, adalimumab, and infliximab. The synergistic activity upon TNF-α and its downstream DPPIV-sensitive substrates would allow administration of lower doses of the TNF-α antagonists, thus reducing the incidence of adverse side effects [122]. Other potentially more innovative but complex approaches would include the systemic administration of DPPIV inhibitors to reduce circulating activated T cell activity with direct injection of recombinant DPPIV into the inflamed synovium to inactivate chemokines and reduce T cell and monocyte recruitment. Although collagen-induced arthritis in rodent models is of limited usefulness for modeling systemic RA processes, it would nevertheless be useful for testing of such novel approaches to the control of local inflammation in the synovium.
Abbreviations
ADA = adenosine deaminase; CCL = CC ligand; CCR = CC receptor; CGRP = calcitonin gene-related peptide; CXCL = CXC ligand; CXCR = CXC receptor; DASH = dipeptidyl peptidase-IV activity and/or structure homologues; DPP = dipeptidyl peptidase; FAP-α = fibroblast-activation protein α/seprase; GLP-1 = glucagon-like peptide-1; GRP = gastrin releasing peptide; HLA = human leukocyte antigens; IFN = interferon; IL = interleukin; IP-10 = IFN-γ inducible protein-10; Mig = monokine induced by interferon-γ; MIP = macrophage inflammatory protein; NAALADase = N-acetylated α-linked acidic dipeptidase; NK = natural killer; NPY = neuropeptide Y; OA = osteoarthritis; PB = peripheral blood; QPP = quiescent cell proline dipeptidase; RA = rheumatiod arthritis; RANTES = regulated upon activation normal T-cell expressed and secreted; SDF = stromal cell-derived factor; SF = synovial fluid; SLE = systemic lupus erythematosus; SP = substance P; TGF = transforming growth factor; TNF = tumor necrosis factor; VIP = vasoactive intestinal peptide.
Competing interests
JSD-C is inventor on United States patents 6,265,551 and 6,933,132 assigned to the Dana-Farber Cancer Institute concerning the use of attractin as an immunodiagnostic. The other authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
Work was supported by grant 7746-3 of the Grant Agency of the Ministry of Health of the Czech Republic (AS, EB) and by a Barr Award in Basic and Innovative Cancer Research (JSD-C). We thank Dr B Guild (Millennium Pharmaceuticals, Cambridge MA, USA) for permission to cite unpublished results.
Contributor Information
Aleksi Sedo, Email: Aleksi@mbox.cesnet.cz.
Jonathan S Duke-Cohan, Email: jonathan_Duke-Cohan@dfci.harvard.edu.
References
- Vanhoof G, Goossens F, De Meester I, Hendriks D, Scharpe S. Proline motifs in peptides and their biological processing. Faseb J. 1995;9:736–744. [PubMed] [Google Scholar]
- Busek P, Malik R, Sedo A. Dipeptidyl peptidase IV activity and/or structure homologues (DASH) and their substrates in cancer. Int J Biochem Cell Biol. 2004;36:408–421. doi: 10.1016/S1357-2725(03)00262-0. [DOI] [PubMed] [Google Scholar]
- Mentlein R. Dipeptidyl-peptidase IV (CD26)–role in the inactivation of regulatory peptides. Regul Pept. 1999;85:9–24. doi: 10.1016/S0167-0115(99)00089-0. [DOI] [PubMed] [Google Scholar]
- Boonacker E, Van Noorden CJ. The multifunctional or moonlighting protein CD26/DPPIV. Eur J Cell Biol. 2003;82:53–73. doi: 10.1078/0171-9335-00302. [DOI] [PubMed] [Google Scholar]
- Yan S, Marguet D, Dobers J, Reutter W, Fan H. Deficiency of CD26 results in a change of cytokine and immunoglobulin secretion after stimulation by pokeweed mitogen. Eur J Immunol. 2003;33:1519–1527. doi: 10.1002/eji.200323469. [DOI] [PubMed] [Google Scholar]
- Cronstein BN, Naime D, Ostad E. The antiinflammatory effects of methotrexate are mediated by adenosine. Adv Exp Med Biol. 1994;370:411–416. doi: 10.1007/978-1-4615-2584-4_89. [DOI] [PubMed] [Google Scholar]
- Montesinos MC, Desai A, Delano D, Chen JF, Fink JS, Jacobson MA, Cronstein BN. Adenosine A2A or A3 receptors are required for inhibition of inflammation by methotrexate and its analog MX-68. Arthritis Rheum. 2003;48:240–247. doi: 10.1002/art.10712. [DOI] [PubMed] [Google Scholar]
- Nakamachi Y, Koshiba M, Nakazawa T, Hatachi S, Saura R, Kurosaka M, Kusaka H, Kumagai S. Specific increase in enzymatic activity of adenosine deaminase 1 in rheumatoid synovial fibroblasts. Arthritis Rheum. 2003;48:668–674. doi: 10.1002/art.10956. [DOI] [PubMed] [Google Scholar]
- Sedo A, Malik R. Dipeptidyl peptidase IV-like molecules: homologous proteins or homologous activities? Biochim Biophys Acta. 2001;1550:107–116. doi: 10.1016/s0167-4838(01)00278-3. [DOI] [PubMed] [Google Scholar]
- Duke-Cohan JS, Morimoto C, Rocker JA, Schlossman SF. A novel form of dipeptidylpeptidase IV found in human serum. Isolation, characterization, and comparison with T lymphocyte membrane dipeptidylpeptidase IV (CD26) J Biol Chem. 1995;270:14107–14114. doi: 10.1074/jbc.270.23.14107. [DOI] [PubMed] [Google Scholar]
- Friedrich D, Kuhn-Wache K, Hoffmann T, Demuth HU. Isolation and characterization of attractin-2. Adv Exp Med Biol. 2003;524:109–113. doi: 10.1007/0-306-47920-6_14. [DOI] [PubMed] [Google Scholar]
- Proost P, De Meester I, Schols D, Struyf S, Lambeir AM, Wuyts A, Opdenakker G, De Clercq E, Scharpe S, Van Damme J. Amino-terminal truncation of chemokines by CD26/dipeptidyl-pepti-dase IV. Conversion of RANTES into a potent inhibitor of monocyte chemotaxis and HIV-1-infection. J Biol Chem. 1998;273:7222–7227. doi: 10.1074/jbc.273.13.7222. [DOI] [PubMed] [Google Scholar]
- Shioda T, Kato H, Ohnishi Y, Tashiro K, Ikegawa M, Nakayama EE, Hu H, Kato A, Sakai Y, Liu H, et al. Anti-HIV-1 and chemotactic activities of human stromal cell-derived factor 1alpha (SDF-1alpha) and SDF-1beta are abolished by CD26/dipeptidyl peptidase IV-mediated cleavage. Proc Natl Acad Sci USA. 1998;95:6331–6336. doi: 10.1073/pnas.95.11.6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy EHS, Kingsley GH, Panayi GS. Immunotherapies: T cell, B cell and complement. In: Hochberg MC, Silman AJ, Smolen J, Weinblatt ME, Weisman MH, editor. Rheumatology. 3. Edinburgh: Mosby; 2003. pp. 449–460. [Google Scholar]
- Giannelli G, Erriquez R, Iannone F, Marinosci F, Lapadula G, Antonaci S. MMP-2, MMP-9, TIMP-1 and TIMP-2 levels in patients with rheumatoid arthritis and psoriatic arthritis. Clin Exp Rheumatol. 2004;22:335–338. [PubMed] [Google Scholar]
- Yan S, Sloane BF. Molecular regulation of human cathepsin B: implication in pathologies. Biol Chem. 2003;384:845–854. doi: 10.1515/BC.2003.095. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gronow M, Gawdi G, Pizzo SV. Characterization of the plasminogen receptors of normal and rheumatoid arthritis human synovial fibroblasts. J Biol Chem. 1994;269:4360–4366. [PubMed] [Google Scholar]
- Casanova JE, Mishumi Y, Ikehara Y, Hubbard AL, Mostov KE. Direct apical sorting of rat liver dipeptidylpeptidase IV expressed in Madin-Darby canine kidney cells. J Biol Chem. 1991;266:24428–24432. [PubMed] [Google Scholar]
- Riemann D, Hansen GH, Niels-Christiansen L, Thorsen E, Immerdal L, Santos AN, Kehlen A, Langner J, Danielsen EM. Caveolae/lipid rafts in fibroblast-like synoviocytes: ectopepti-dase-rich membrane microdomains. Biochem J. 2001;354:47–55. doi: 10.1042/0264-6021:3540047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhling F, Junker U, Reinhold D, Neubert K, Jager L, Ansorge S. Functional role of CD26 on human B lymphocytes. Immunol Lett. 1995;45:47–51. doi: 10.1016/0165-2478(94)00230-O. [DOI] [PubMed] [Google Scholar]
- Buhling F, Kunz D, Reinhold D, Ulmer AJ, Ernst M, Flad HD, Ansorge S. Expression and functional role of dipeptidyl pepti-dase IV (CD26) on human natural killer cells. Nat Immun. 1994;13:270–279. [PubMed] [Google Scholar]
- Jackman HL, Tan F, Schraufnagel D, Dragovic T, Dezso B, Becker RP, Erdos EG. Plasma membrane-bound and lysosomal pepti-dases in human alveolar macrophages. Am J Respir Cell Mol Biol. 1995;13:196–204. doi: 10.1165/ajrcmb.13.2.7626287. [DOI] [PubMed] [Google Scholar]
- Bertotto A, Gerli R, Spinozzi F, Muscat C, Fabietti GM, Crupi S, Castellucci G, De Benedictis FM, De Giorgi G, Britta R, et al. CD26 surface antigen expression on peripheral blood T lymphocytes from children with Down's syndrome (trisomy 21) Scand J Immunol. 1994;39:633–636. doi: 10.1111/j.1365-3083.1994.tb03424.x. [DOI] [PubMed] [Google Scholar]
- De Meester I, Korom S, Van Damme J, Scharpe S. CD26, let it cut or cut it down. Immunol Today. 1999;20:367–375. doi: 10.1016/S0167-5699(99)01486-3. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Duke-Cohan JS, Kameoka J, Yaron A, Lee I, Schloss-man SF, Morimoto C. Enhancement of antigen-induced T-cell proliferation by soluble CD26/dipeptidyl peptidase IV. Proc Natl Acad Sci USA. 1994;91:3082–3086. doi: 10.1073/pnas.91.8.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuma K, Munakata Y, Ishii T, Iwata S, Kobayashi S, Hosono O, Kawasaki H, Dang NH, Morimoto C. Soluble CD26/dipeptidyl peptidase IV induces T cell proliferation through CD86 up-regulation on APCs. J Immunol. 2001;167:6745–6755. doi: 10.4049/jimmunol.167.12.6745. [DOI] [PubMed] [Google Scholar]
- Ikushima H, Munakata Y, Iwata S, Ohnuma K, Kobayashi S, Dang NH, Morimoto C. Soluble CD26/dipeptidyl peptidase IV enhances transendothelial migration via its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cell Immunol. 2002;215:106–110. doi: 10.1016/S0008-8749(02)00010-2. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Hosono O, Mimori T, Kawasaki H, Dang NH, Tanaka H, Morimoto C. Reduction of serum soluble CD26/dipeptidyl peptidase IV enzyme activity and its correlation with disease activity in systemic lupus erythematosus. J Rheumatol. 2002;29:1858–1866. [PubMed] [Google Scholar]
- Krepela E, Kasafirek E, Vicar J, Kraml J. An assay of dipeptidyl peptidase IV activity in human serum and serum of pregnant women with glycyl-L-proline-1-naphthylamide and other glycyl-L-proline-arylamides as substrates. Physiol Bohemoslov. 1983;32:334–345. [PubMed] [Google Scholar]
- Sedo A, Malik R, Votruba M, Hanic T, Hanic O. Serum dipeptidyl peptidase IV activities in Czernobyl zone inhabitants exposed to the power-station catastrophe. Eur J Clin Chem Clin Biochem. 1996;34:447–448. [PubMed] [Google Scholar]
- Korom S, De Meester I, Stadlbauer TH, Chandraker A, Schaub M, Sayegh MH, Belyaev A, Haemers A, Scharpe S, Kupiec-Weglinski JW. Inhibition of CD26/dipeptidyl peptidase IV activity in vivo prolongs cardiac allograft survival in rat recipients. Transplantation. 1997;63:1495–1500. doi: 10.1097/00007890-199705270-00021. [DOI] [PubMed] [Google Scholar]
- Hagihara M, Ohhashi M, Nagatsu T. Activities of dipeptidyl pep-tidase II and dipeptidyl peptidase IV in mice with lupus ery-thematosus-like syndrome and in patients with lupus erythematosus and rheumatoid arthritis. Clin Chem. 1987;33:1463–1465. [PubMed] [Google Scholar]
- Cordero OJ, Salgado FJ, Mera-Varela A, Nogueira M. Serum interleukin-12, interleukin-15, soluble CD26, and adenosine deaminase in patients with rheumatoid arthritis. Rheumatol Int. 2001;21:69–74. doi: 10.1007/s002960100134. [DOI] [PubMed] [Google Scholar]
- Kullertz G, Boigk J. Dipeptidyl peptidase IV activity in the serum and synovia of patients with rheumatoid arthritis. Z Rheumatol. 1986;45:52–56. [PubMed] [Google Scholar]
- Scholzova E, Sedova L, Mares V, Balaziova E, Vlasicova K, Nytrova P, Sevcik J, Sedo A. Dipeptidyl peptidase IV activity and/or structure homologues (DASH) – the novel players in pathogenesis of rheumatoid and psoriatic arthritides. Eur J Biochem. 2004;271:104–105. [Google Scholar]
- Cuchacovich M, Gatica H, Pizzo SV, Gonzalez-Gronow M. Characterization of human serum dipeptidyl peptidase IV (CD26) and analysis of its autoantibodies in patients with rheumatoid arthritis and other autoimmune diseases. Clin Exp Rheumatol. 2001;19:673–680. [PubMed] [Google Scholar]
- Mantle D, Falkous G, Walker D. Quantification of protease activities in synovial fluid from rheumatoid and osteoarthritis cases: comparison with antioxidant and free radical damage markers. Clin Chim Acta. 1999;284:45–58. doi: 10.1016/S0009-8981(99)00055-8. [DOI] [PubMed] [Google Scholar]
- Cordero OJ, Salgado FJ, Vinuela JE, Nogueira M. Interleukin-12 enhances CD26 expression and dipeptidyl peptidase IV function on human activated lymphocytes. Immunobiology. 1997;197:522–533. doi: 10.1016/s0171-2985(97)80084-8. [DOI] [PubMed] [Google Scholar]
- Salgado FJ, Vela E, Martin M, Franco R, Nogueira M, Cordero OJ. Mechanisms of CD26/dipeptidyl peptidase IV cytokine-dependent regulation on human activated lymphocytes. Cytokine. 2000;12:1136–1141. doi: 10.1006/cyto.1999.0643. [DOI] [PubMed] [Google Scholar]
- Cordero OJ, Salgado FJ, Fernandez-Alonso CM, Herrera C, Lluis C, Franco R, Nogueira M. Cytokines regulate membrane adenosine deaminase on human activated lymphocytes. J Leukoc Biol. 2001;70:920–930. [PubMed] [Google Scholar]
- Sorrell JM, Brinon L, Baber MA, Caplan AI. Cytokines and gluco-corticoids differentially regulate APN/CD13 and DPPIV/CD26 enzyme activities in cultured human dermal fibroblasts. Arch Dermatol Res. 2003;295:160–168. doi: 10.1007/s00403-003-0417-4. [DOI] [PubMed] [Google Scholar]
- Yamabe T, Takakura K, Sugie K, Kitaoka Y, Takeda S, Okubo Y, Teshigawara K, Yodoi J, Hori T. Induction of the 2B9 antigen/dipeptidyl peptidase IV/CD26 on human natural killer cells by IL-2, IL-12 or IL-15. Immunology. 1997;91:151–158. doi: 10.1046/j.1365-2567.1997.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brezinschek RI, Lipsky PE, Galea P, Vita R, Oppenheimer-Marks N. Phenotypic characterization of CD4+ T cells that exhibit a transendothelial migratory capacity. J Immunol. 1995;154:3062–3077. [PubMed] [Google Scholar]
- Masuyama J, Berman JS, Cruikshank WW, Morimoto C, Center DM. Evidence for recent as well as long term activation of T cells migrating through endothelial cell monolayers in vitro. J Immunol. 1992;148:1367–1374. [PubMed] [Google Scholar]
- Ghersi G, Dong H, Goldstein LA, Yeh Y, Hakkinen L, Larjava HS, Chen WT. Regulation of fibroblast migration on collagenous matrix by a cell surface peptidase complex. J Biol Chem. 2002;277:29231–29241. doi: 10.1074/jbc.M202770200. [DOI] [PubMed] [Google Scholar]
- Chen WT, Kelly T. Seprase complexes in cellular invasiveness. Cancer Metastasis Rev. 2003;22:259–269. doi: 10.1023/A:1023055600919. [DOI] [PubMed] [Google Scholar]
- Mizokami A, Eguchi K, Kawakami A, Ida H, Kawabe Y, Tsukada T, Aoyagi T, Maeda K, Morimoto C, Nagataki S. Increased population of high fluorescence 1F7 (CD26) antigen on T cells in synovial fluid of patients with rheumatoid arthritis. J Rheumatol. 1996;23:2022–2026. [PubMed] [Google Scholar]
- McInnes IB, Liew FY. Interleukin 15: a proinflammatory role in rheumatoid arthritis synovitis. Immunol Today. 1998;19:75–79. doi: 10.1016/S0167-5699(97)01205-X. [DOI] [PubMed] [Google Scholar]
- Gerli R, Muscat C, Bertotto A, Bistoni O, Agea E, Tognellini R, Fiorucci G, Cesarotti M, Bombardieri S. CD26 surface molecule involvement in T cell activation and lymphokine synthesis in rheumatoid and other inflammatory synovitis. Clin Immunol Immunopathol. 1996;80:31–37. doi: 10.1006/clin.1996.0091. [DOI] [PubMed] [Google Scholar]
- Duke-Cohan JS, Morimoto C, Rocker JA, Schlossman SF. Serum high molecular weight dipeptidyl peptidase IV (CD26) is similar to a novel antigen DPPT-L released from activated T cells. J Immunol. 1996;156:1714–1721. [PubMed] [Google Scholar]
- Sedova L, Scholzova E, Vlasicova K, Stolfa J, Krystufkova O, Ruz-ickova S, Sevcik J, Sedo A. Dipeptidyl peptidase IV activity and/or structure homologues (DASH) in blood and synovial fluid mononuclear cells of patients with rheumatoid and pso-riatic arthritis. Ann Rheum Dis. 2004;63(Suppl I):124. [Google Scholar]
- Fujita K, Hirano M, Ochiai J, Funabashi M, Nagatsu I, Nagatsu T, Sakakibara S. Serum glycylproline p-nitroanilidase activity in rheumatoid arthritis and systemic lupus erythematosus. Clin Chim Acta. 1978;88:15–20. doi: 10.1016/0009-8981(78)90142-0. [DOI] [PubMed] [Google Scholar]
- Bathon JM, Proud D, Mizutani S, Ward PE. Cultured human synovial fibroblasts rapidly metabolize kinins and neuropeptides. J Clin Invest. 1992;90:981–991. doi: 10.1172/JCI115975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamori M, Hagihara M, Nagatsu T, Iwata H, Miura T. Activities of dipeptidyl peptidase II, dipeptidyl peptidase IV, prolyl endopeptidase, and collagenase-like peptidase in synovial membrane from patients with rheumatoid arthritis and osteoarthritis. Biochem Med Metab Biol. 1991;45:154–160. doi: 10.1016/0885-4505(91)90016-E. [DOI] [PubMed] [Google Scholar]
- Busso N, Wagtmann N, Herling C, Chobaz-Peclat V, Bischof-Delaloye A, So A, Grouzmann E. Circulating CD26 is negatively associated with inflammation in human and experimental arthritis. Am J Pathol. 2005;166:433–442. doi: 10.1016/S0002-9440(10)62266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke-Cohan JS, Tang W, Schlossman SF. Attractin: a cub-family protease involved in T cell-monocyte/macrophage interactions. Adv Exp Med Biol. 2000;477:173–185. doi: 10.1007/0-306-46826-3_20. [DOI] [PubMed] [Google Scholar]
- Iwaki-Egawa S, Watanabe Y, Matsuno H. Correlations between matrix metalloproteinase-9 and adenosine deaminase isozymes in synovial fluid from patients with rheumatoid arthritis. J Rheumatol. 2001;28:485–489. [PubMed] [Google Scholar]
- Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science. 1993;261:466–469. doi: 10.1126/science.8101391. [DOI] [PubMed] [Google Scholar]
- Senten K, Van Der Veken P, De Meester I, Lambeir AM, Scharpe S, Haemers A, Augustyns K. Gamma-amino-substituted analogues of 1-[(S)-2,4-diaminobutanoyl]piperidine as highly potent and selective dipeptidyl peptidase II inhibitors. J Med Chem. 2004;47:2906–2916. doi: 10.1021/jm031122f. [DOI] [PubMed] [Google Scholar]
- Gotoh H, Hagihara M, Nagatsu T, Iwata H, Miura T. Activities of dipeptidyl peptidase II and dipeptidyl peptidase IV in synovial fluid from patients with rheumatoid arthritis and osteoarthritis. Clin Chem. 1989;35:1016–1018. [PubMed] [Google Scholar]
- Tang W, Gunn TM, McLaughlin DF, Barsh GS, Schlossman SF, Duke-Cohan JS. Secreted and membrane attractin result from alternative splicing of the human ATRN gene. Proc Natl Acad Sci USA. 2000;97:6025–6030. doi: 10.1073/pnas.110139897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H, Wu J, Kuhn E, Chin W, Chang B, Jones MD, O'Neil S, Clauser KR, Karl J, Hasler F, et al. Use of mass spectrometry to identify protein biomarkers of disease severity in the synovial fluid and serum of patients with rheumatoid arthritis. Arthritis Rheum. 2004;50:3792–3803. doi: 10.1002/art.20720. [DOI] [PubMed] [Google Scholar]
- Marguet D, Baggio L, Kobayashi T, Bernard AM, Pierres M, Nielsen PF, Ribel U, Watanabe T, Drucker DJ, Wagtmann N. Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. Proc Natl Acad Sci USA. 2000;97:6874–6879. doi: 10.1073/pnas.120069197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez MA, Garcia ME, Rodriguez JA, Mardonez G, Jacobelli S, Rivero S. Hypothalamic-pituitary-adrenal axis function in patients with active rheumatoid arthritis: a controlled study using insulin hypoglycemia stress test and prolactin stimulation. J Rheumatol. 1999;26:277–281. [PubMed] [Google Scholar]
- Masi AT, Bijlsma JW, Chikanza IC, Pitzalis C, Cutolo M. Neuroen-docrine, immunologic, and microvascular systems interactions in rheumatoid arthritis: physiopathogenetic and therapeutic perspectives. Semin Arthritis Rheum. 1999;29:65–81. doi: 10.1016/S0049-0172(99)80039-0. [DOI] [PubMed] [Google Scholar]
- Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- Westermark T, Rantapaa-Dahlqvist S, Wallberg-Jonsson S, Kjorell U, Forsgren S. Increased content of bombesin/GRP in human synovial fluid in early arthritis: different pattern compared with substance P. Clin Exp Rheumatol. 2001;19:715–720. [PubMed] [Google Scholar]
- Lotz M, Vaughan JH, Carson DA. Effect of neuropeptides on production of inflammatory cytokines by human monocytes. Science. 1988;241:1218–1221. doi: 10.1126/science.2457950. [DOI] [PubMed] [Google Scholar]
- Lambert N, Lescoulie PL, Yassine-Diab B, Enault G, Mazieres B, De Preval C, Cantagrel A. Substance P enhances cytokine-induced vascular cell adhesion molecule-1 (VCAM-1) expression on cultured rheumatoid fibroblast-like synoviocytes. Clin Exp Immunol. 1998;113:269–275. doi: 10.1046/j.1365-2249.1998.00621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Shimoyama Y, Hirabayashi K, Kajigaya H, Yamamoto S, Oda H, Koshihara Y. Production of neuropeptide substance P by synovial fibroblasts from patients with rheumatoid arthritis and osteoarthritis. Neurosci Lett. 2001;303:149–152. doi: 10.1016/S0304-3940(01)01713-X. [DOI] [PubMed] [Google Scholar]
- Hernanz A, Medina S, de Miguel E, Martin-Mola E. Effect of calci-tonin gene-related peptide, neuropeptide Y, substance P, and vasoactive intestinal peptide on interleukin-1beta, interleukin-6 and tumor necrosis factor-alpha production by peripheral whole blood cells from rheumatoid arthritis and osteoarthritis patients. Regul Pept. 2003;115:19–24. doi: 10.1016/S0167-0115(03)00127-7. [DOI] [PubMed] [Google Scholar]
- Yokoyama MM, Fujimoto K. Role of lymphocyte activation by substance P in rheumatoid arthritis. Int J Tissue React. 1990;12:1–9. [PubMed] [Google Scholar]
- Covas MJ, Pinto LA, Pereira Da Silva JA, Victorino RM. Effects of the neuropeptide, substance P, on lymphocyte proliferation in rheumatoid arthritis. J Int Med Res. 1995;23:431–438. doi: 10.1177/030006059502300604. [DOI] [PubMed] [Google Scholar]
- Covas MJ, Pinto LA, Victorino RM. Effects of substance P on human T cell function and the modulatory role of peptidase inhibitors. Int J Clin Lab Res. 1997;27:129–134. doi: 10.1007/BF02912447. [DOI] [PubMed] [Google Scholar]
- Zukowska Z, Pons J, Lee EW, Li L. Neuropeptide Y: a new mediator linking sympathetic nerves, blood vessels and immune system? Can J Physiol Pharmacol. 2003;81:89–94. doi: 10.1139/y03-006. [DOI] [PubMed] [Google Scholar]
- Schwarz H, Villiger PM, von Kempis J, Lotz M. Neuropeptide Y is an inducible gene in the human immune system. J Neuroimmunol. 1994;51:53–61. doi: 10.1016/0165-5728(94)90128-7. [DOI] [PubMed] [Google Scholar]
- Silva AP, Cavadas C, Baisse-Agushi B, Spertini O, Brunner HR, Grouzmann E. NPY, NPY receptors, and DPP IV activity are modulated by LPS, TNF-alpha and IFN-gamma in HUVEC. Regul Pept. 2003;116:71–79. doi: 10.1016/S0167-0115(03)00191-5. [DOI] [PubMed] [Google Scholar]
- Straub RH, Mayer M, Kreutz M, Leeb S, Scholmerich J, Falk W. Neurotransmitters of the sympathetic nerve terminal are powerful chemoattractants for monocytes. J Leukoc Biol. 2000;67:553–558. doi: 10.1002/jlb.67.4.553. [DOI] [PubMed] [Google Scholar]
- Appelgren A, Appelgren B, Eriksson S, Kopp S, Lundeberg T, Nylander M, Theodorsson E. Neuropeptides in temporo-mandibular joints with rheumatoid arthritis: a clinical study. Scand J Dent Res. 1991;99:519–521. doi: 10.1111/j.1600-0722.1991.tb01063.x. [DOI] [PubMed] [Google Scholar]
- Dimitrijevic M, Stanojevic S, Vujic V, Kovacevic-Jovanovic V, Beck-Sickinger A, Demuth H, von Horsten S. Effect of neuropeptide Y on inflammatory paw edema in the rat: involvement of peripheral NPY Y1 and Y5 receptors and interaction with dipeptidyl-peptidase IV (CD26) J Neuroimmunol. 2002;129:35–42. doi: 10.1016/S0165-5728(02)00173-X. [DOI] [PubMed] [Google Scholar]
- Arnalich F, de Miguel E, Perez-Ayala C, Martinez M, Vazquez JJ, Gijon-Banos J, Hernanz A. Neuropeptides and interleukin-6 in human joint inflammation relationship between intraarticular substance P and interleukin-6 concentrations. Neurosci Lett. 1994;170:251–254. doi: 10.1016/0304-3940(94)90331-X. [DOI] [PubMed] [Google Scholar]
- Wang F, Millet I, Bottomly K, Vignery A. Calcitonin gene-related peptide inhibits interleukin 2 production by murine T lymphocytes. J Biol Chem. 1992;267:21052–21057. [PubMed] [Google Scholar]
- Juarranz MG, Santiago B, Torroba M, Gutierrez-Canas I, Palao G, Galindo M, Abad C, Martinez C, Leceta J, Pablos JL, et al. Vasoactive intestinal peptide modulates proinflammatory mediator synthesis in osteoarthritic and rheumatoid synovial cells. Rheumatology (Oxford) 2004;43:416–422. doi: 10.1093/rheumatology/keh061. [DOI] [PubMed] [Google Scholar]
- Delgado M, Abad C, Martinez C, Leceta J, Gomariz RP. Vasoac-tive intestinal peptide prevents experimental arthritis by downregulating both autoimmune and inflammatory components of the disease. Nat Med. 2001;7:563–568. doi: 10.1038/87887. [DOI] [PubMed] [Google Scholar]
- Del Rio M, De la Fuente M. Stimulation of natural killer (NK) and antibody-dependent cellular cytotoxicity (ADCC) activities in murine leukocytes by bombesin-related peptides requires the presence of adherent cells. Regul Pept. 1995;60:159–166. doi: 10.1016/0167-0115(95)00125-5. [DOI] [PubMed] [Google Scholar]
- Hill DJ, McDonald TJ. Mitogenic action of gastrin-releasing polypeptide on isolated epiphyseal growth plate chondro-cytes from the ovine fetus. Endocrinology. 1992;130:2811–2819. doi: 10.1210/en.130.5.2811. [DOI] [PubMed] [Google Scholar]
- Bertazzolo N, Punzi L, Stefani MP, Cesaro G, Pianon M, Finco B, Todesco S. Interrelationships between interleukin (IL)-1, IL-6 and IL-8 in synovial fluid of various arthropathies. Agents Actions. 1994;41:90–92. doi: 10.1007/BF01986402. [DOI] [PubMed] [Google Scholar]
- Partsch G, Wagner E, Leeb BF, Broll H, Dunky A, Smolen JS. T cell derived cytokines in psoriatic arthritis synovial fluids. Ann Rheum Dis. 1998;57:691–693. doi: 10.1136/ard.57.11.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchlin C, Haas-Smith SA, Hicks D, Cappuccio J, Osterland CK, Looney RJ. Patterns of cytokine production in psoriatic syn-ovium. J Rheumatol. 1998;25:1544–1552. [PubMed] [Google Scholar]
- Steiner G, Tohidast-Akrad M, Witzmann G, Vesely M, Studnicka-Benke A, Gal A, Kunaver M, Zenz P, Smolen JS. Cytokine production by synovial T cells in rheumatoid arthritis. Rheumatology (Oxford) 1999;38:202–213. doi: 10.1093/rheumatology/38.3.202. [DOI] [PubMed] [Google Scholar]
- Hoffmann T, Faust J, Neubert K, Ansorge S. Dipeptidyl pepti-dase IV (CD 26) and aminopeptidase N (CD 13) catalyzed hydrolysis of cytokines and peptides with N-terminal cytokine sequences. FEBS Lett. 1993;336:61–64. doi: 10.1016/0014-5793(93)81609-4. [DOI] [PubMed] [Google Scholar]
- Bauvois B, Sanceau J, Wietzerbin J. Human U937 cell surface peptidase activities: characterization and degradative effect on tumor necrosis factor-alpha. Eur J Immunol. 1992;22:923–930. doi: 10.1002/eji.1830220407. [DOI] [PubMed] [Google Scholar]
- Firestein GS. The T cell cometh: interplay between adaptive immunity and cytokine networks in rheumatoid arthritis. J Clin Invest. 2004;114:471–474. doi: 10.1172/JCI200422651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig A, Krenn V, Toksoy A, Gerhard N, Gillitzer R. Mig, GRO alpha and RANTES messenger RNA expression in lining layer, infiltrates and different leucocyte populations of synovial tissue from patients with rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Virchows Arch. 2000;436:449–458. doi: 10.1007/s004280050472. [DOI] [PubMed] [Google Scholar]
- Loetscher P, Moser B. Homing chemokines in rheumatoid arthritis. Arthritis Res. 2002;4:233–236. doi: 10.1186/ar412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson E, Keystone EC, Schall TJ, Gillett N, Fish EN. Chemokine expression in rheumatoid arthritis (RA): evidence of RANTES and macrophage inflammatory protein (MIP)-1 beta production by synovial T cells. Clin Exp Immunol. 1995;101:398–407. doi: 10.1111/j.1365-2249.1995.tb03126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CR, Liu MF. Regulation of CCR5 expression and MIP-1alpha production in CD4+ T cells from patients with rheumatoid arthritis. Clin Exp Immunol. 2003;132:371–378. doi: 10.1046/j.1365-2249.2003.02126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan E, Wang J, Norcross MA. Amino-terminal processing of MIP-1beta/CCL4 by CD26/dipeptidyl-peptidase IV. J Cell Biochem. 2004;92:53–64. doi: 10.1002/jcb.20041. [DOI] [PubMed] [Google Scholar]
- Katschke KJ, Jr, Rottman JB, Ruth JH, Qin S, Wu L, LaRosa G, Ponath P, Park CC, Pope RM, Koch AE. Differential expression of chemokine receptors on peripheral blood, synovial fluid, and synovial tissue monocytes/macrophages in rheumatoid arthritis. Arthritis Rheum. 2001;44:1022–1032. doi: 10.1002/1529-0131(200105)44:5<1022::AID-ANR181>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Lambeir AM, Durinx C, Scharpe S, De Meester I. Dipeptidyl-pep-tidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit Rev Clin Lab Sci. 2003;40:209–294. doi: 10.1080/713609354. [DOI] [PubMed] [Google Scholar]
- Proost P, Schutyser E, Menten P, Struyf S, Wuyts A, Opdenakker G, Detheux M, Parmentier M, Durinx C, Lambeir AM, et al. Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood. 2001;98:3554–3561. doi: 10.1182/blood.V98.13.3554. [DOI] [PubMed] [Google Scholar]
- Hanaoka R, Kasama T, Muramatsu M, Yajima N, Shiozawa F, Miwa Y, Negishi M, Ide H, Miyaoka H, Uchida H, et al. A novel mechanism for the regulation of IFN-gamma inducible protein-10 expression in rheumatoid arthritis. Arthritis Res Ther. 2003;5:R74–81. doi: 10.1186/ar616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DD, Zachariah JP, Whichard LP. CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin Immunol. 2001;98:39–45. doi: 10.1006/clim.2000.4957. [DOI] [PubMed] [Google Scholar]
- Bradfield PF, Amft N, Vernon-Wilson E, Exley AE, Parsonage G, Rainger GE, Nash GB, Thomas AM, Simmons DL, Salmon M, et al. Rheumatoid fibroblast-like synoviocytes overexpress the chemokine stromal cell-derived factor 1 (CXCL12), which supports distinct patterns and rates of CD4+ and CD8+ T cell migration within synovial tissue. Arthritis Rheum. 2003;48:2472–2482. doi: 10.1002/art.11219. [DOI] [PubMed] [Google Scholar]
- Buckley CD, Amft N, Bradfield PF, Pilling D, Ross E, Arenzana-Seis-dedos F, Amara A, Curnow SJ, Lord JM, Scheel-Toellner D, et al. Persistent induction of the chemokine receptor CXCR4 by TGF-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol. 2000;165:3423–3429. doi: 10.4049/jimmunol.165.6.3423. [DOI] [PubMed] [Google Scholar]
- Herrera C, Morimoto C, Blanco J, Mallol J, Arenzana F, Lluis C, Franco R. Comodulation of CXCR4 and CD26 in human lymphocytes. J Biol Chem. 2001;276:19532–19539. doi: 10.1074/jbc.M004586200. [DOI] [PubMed] [Google Scholar]
- Yang YF, Mukai T, Gao P, Yamaguchi N, Ono S, Iwaki H, Obika S, Imanishi T, Tsujimura T, Hamaoka T, et al. A non-peptide CCR5 antagonist inhibits collagen-induced arthritis by modulating T cell migration without affecting anti-collagen T cell responses. Eur J Immunol. 2002;32:2124–2132. doi: 10.1002/1521-4141(200208)32:8<2124::AID-IMMU2124>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Iwata S, Yamaguchi N, Munakata Y, Ikushima H, Lee JF, Hosono O, Schlossman SF, Morimoto C. CD26/dipeptidyl peptidase IV differentially regulates the chemotaxis of T cells and monocytes toward RANTES: possible mechanism for the switch from innate to acquired immune response. Int Immunol. 1999;11:417–426. doi: 10.1093/intimm/11.3.417. [DOI] [PubMed] [Google Scholar]
- Wiedeman PE, Trevillyan JM. Dipeptidyl peptidase IV inhibitors for the treatment of impaired glucose tolerance and type 2 diabetes. Curr Opin Investig Drugs. 2003;4:412–420. [PubMed] [Google Scholar]
- Steinbrecher A, Reinhold D, Quigley L, Gado A, Tresser N, Izikson L, Born I, Faust J, Neubert K, Martin R, et al. Targeting dipeptidyl peptidase IV (CD26) suppresses autoimmune encephalo-myelitis and up-regulates TGF-beta 1 secretion in vivo. J Immunol. 2001;166:2041–2048. doi: 10.4049/jimmunol.166.3.2041. [DOI] [PubMed] [Google Scholar]
- Korom S, De Meester I, Schmidbauer G, Pratschke J, Brendel MD, Durinx C, Schwemmle K, Haemers A, Scharpe S, Kupiec-Weglinski JW. Specific inhibition of CD26/DPP IV enzymatic activity in allograft recipients: effects on humoral immunity. Transplant Proc. 1999;31:778. doi: 10.1016/S0041-1345(98)02069-7. [DOI] [PubMed] [Google Scholar]
- Reinhold D, Hemmer B, Gran B, Steinbrecher A, Brocke S, Kahne T, Wrenger S, Born I, Faust J, Neubert K, et al. Dipeptidyl pepti-dase IV (CD26): role in T cell activation and autoimmune disease. Adv Exp Med Biol. 2000;477:155–160. doi: 10.1007/0-306-46826-3_17. [DOI] [PubMed] [Google Scholar]
- Reinhold D, Bank U, Buhling F, Kahne T, Kunt D, Faust J, Neubert K, Ansorge S. Inhibitors of dipeptidyl peptidase IV (DP IV, CD26) specifically suppress proliferation and modulate cytokine production of strongly CD26 expressing U937 cells. Immunobiology. 1994;192:121–136. doi: 10.1016/S0171-2985(11)80412-2. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Murakami T, Horikawa H, Sugiura M, Kawashima K, Sugita T. Suppression of arthritis by the inhibitors of dipep-tidyl peptidase IV. Int J Immunopharmacol. 1997;19:15–24. doi: 10.1016/S0192-0561(97)00004-0. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Murakami T, Nonaka N, Ohnuki T, Yamada M, Sugita T. Anti-arthritic effects of the novel dipeptidyl peptidase IV inhibitors TMC-2A and TSL-225. Immunopharmacology. 1998;40:21–26. doi: 10.1016/S0162-3109(98)00014-9. [DOI] [PubMed] [Google Scholar]
- Williams YN, Baba H, Hayashi S, Ikai H, Sugita T, Tanaka S, Miyasaka N, Kubota T. Dipeptidyl peptidase IV on activated T cells as a target molecule for therapy of rheumatoid arthritis. Clin Exp Immunol. 2003;131:68–74. doi: 10.1046/j.1365-2249.2003.02020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams YN, Sugita T, Nosaka Y, Kubota T. Suppression of fibroblast activity by an inhibitor of dipeptidyl peptidase IV: a possible therapeutic strategy for rheumatoid arthritis. Clin Exp Rheumatol. 2003;21:685. [PubMed] [Google Scholar]
- Calvo CF, Chavanel G, Senik A. Substance P enhances IL-2 expression in activated human T cells. J Immunol. 1992;148:3498–3504. [PubMed] [Google Scholar]
- Laurenzi MA, Persson MA, Dalsgaard CJ, Ringden O. Stimulation of human B lymphocyte differentiation by the neuropeptides substance P and neurokinin A. Scand J Immunol. 1989;30:695–701. doi: 10.1111/j.1365-3083.1989.tb02478.x. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH. Glycosaminogly-can binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA. 2003;100:1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollheim FA. Predictors of joint damage in rheumatoid arthritis. Apmis. 1996;104:81–93. doi: 10.1111/j.1699-0463.1996.tb00691.x. [DOI] [PubMed] [Google Scholar]
- Nestorov I. Clinical pharmacokinetics of tumor necrosis factor antagonists. J Rheumatol Suppl. 2005;74:13–18. [PubMed] [Google Scholar]
- Muscat C, Bertotto A, Agea E, Bistoni O, Ercolani R, Tognellini R, Spinozzi F, Cesarotti M, Gerli R. Expression and functional role of 1F7 (CD26) antigen on peripheral blood and synovial fluid T cells in rheumatoid arthritis patients. Clin Exp Immunol. 1994;98:252–256. doi: 10.1111/j.1365-2249.1994.tb06134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]