

Abstract
Evidence is increasing that oxygen debt and its metabolic correlates are important quantifiers of the severity of hemorrhagic and post-traumatic shock and and may serve as useful guides in the treatment of these conditions. The aim of this review is to demonstrate the similarity between experimental oxygen debt in animals and human hemorrhage/post-traumatic conditions, and to examine metabolic oxygen debt correlates, namely base deficit and lactate, as indices of shock severity and adequacy of volume resuscitation. Relevant studies in the medical literature were identified using Medline and Cochrane Library searches. Findings in both experimental animals (dog/pig) and humans suggest that oxygen debt or its metabolic correlates may be more useful quantifiers of hemorrhagic shock than estimates of blood loss, volume replacement, blood pressure, or heart rate. This is evidenced by the oxygen debt/probability of death curves for the animals, and by the consistency of lethal dose (LD)25,50 points for base deficit across all three species. Quantifying human post-traumatic shock based on base deficit and adjusting for Glasgow Coma Scale score, prothrombin time, Injury Severity Score and age is demonstrated to be superior to anatomic injury severity alone or in combination with Trauma and Injury Severity Score. The data examined in this review indicate that estimates of oxygen debt and its metabolic correlates should be included in studies of experimental shock and in the management of patients suffering from hemorrhagic shock.
Introduction
In a noninjured, nonseptic, healthy state, oxygen consumption (VO2) is a closely regulated process because oxygen serves as the critical carbon acceptor in the generation of energy from a wide variety of metabolic fuels. Post-traumatic hemorrhage leads to a hypovolemia in which blood flow and consequently oxygen delivery to vital organs are decreased. When oxygen delivery is decreased to a degree sufficient to reduce VO2 to below a critical level, a state of shock occurs, producing ischemic metabolic insuffiency [1-3]. This degree of restriction in VO2 can also be produced by cardiogenic or vasodilatory shock, in which oxygen delivery is restricted by low flow. When this critical level of oxygen restriction is reached, an oxygen debt (O2D) occurs. In the literature, the terms 'oxygen debt' and 'oxygen deficit' are used interchangeably and are defined as the integral difference between the prehemorrhage/pretrauma resting normal VO2 and the VO2 during the hypovolemic, hemorrhage period [4-9]. For purposes of simplification, the term O2D ('oxygen debt') is used in this review. The presence and extent of an O2D is further highlighted by an increase in the unmetabolized metabolic acids generated by the anaerobic processes. It is the close congruence of O2D and related metabolic acidemia that permits precise quantification of the severity of the ischemic shock process in both animals and humans.
The aim of this review is to demonstrate the quantitative similarity between experimental O2D shock and that induced in humans by post-traumatic or severe hemorrhagic, hypovolemic conditions. It also examines the use of metabolic correlates of O2D as indices of the severity of the shock process in two mammalian species and in humans, and the value of these correlates as guides to the adequacy of volume-mediated resuscitation.
This review is based on a search of the Medline and Cochrane Library databases from 1964 to December 2004. The search terms 'oxygen debt or deficit', 'base excess or deficit', 'lactate', 'hemorrhagic shock' and 'multiple trauma' were used. These terms were mapped to Medline Subject Headings (MESH) terms, as well as being searched for as text items. The following combinations were studied: 'oxygen debt' or 'oxygen deficit' and 'hemorrhagic shock', 'lactate' and 'multiple trauma', as well as 'base excess' or 'base deficit' and 'multiple trauma'. No language restrictions were applied.
The clinical problem of quantification of hemorrhagic shock severity and the effectiveness of resuscitation
That post-traumatic shock is initiated by acute volume loss was first noted by Cannon [10] and later demonstrated by the experimental studies conducted by Blalock [11]. Subsequently, Wiggers [12] and Guyton [13] developed a variety of animal models based on controlled hemorrhage. Other models involving uncontrolled bleeding [14,15], fixed volume loss [16-20], or a defined level of hypotension [16,19-22] have been used. In previous studies, the severity of shock was defined by the degree and duration of the resulting hypovolemia. Thus, attempts were made to quantify the effectiveness of resuscitation by assessing the improvement in blood pressure or perfusion occurring in response to different volumes of electrolyte, colloid, or blood-containing fluids, which are administered to prevent death during the immediate postshock period.
In the clinical arena, this issue became acute during World War II, when fluid transfusion and use of blood and blood products as a means of effectively restoring blood volume became a realistic possibility. Consequently, volume infusion and blood or blood product transfusion were used extensively for the first time during the North African Campaign by US and UK forces [23], and was a primary modality for treatment of shock in the Korean War [24]. These clinical advances led to extensive efforts to elucidate human hypovolemic shock and to establish experimental models that emulate clinical shock. The most extensive series of clinical/physiologic studies were performed in postoperative [25,26] and posttrauma [27] shock patients, in whom the response to volume infusion was evaluated. These and other studies [28,29] of resuscitation after hypovolemic shock demonstrated the fall in VO2 associated with the decrease in cardiac output, and demonstrated the arterial vasoconstriction that occurred in an attempt to compensate for the fall in blood pressure. They also demonstrated the postresuscitation hyperdynamic state, in which cardiac output rises to permit an increase in VO2, apparently compensating for and even exceeding the initial fall in VO2 [1,2,26]. These data appeared to validate in humans the 'oxygen deficit' concept initially enunciated by Crowell and Smith [4] based on experimental findings. Nevertheless, in spite of these animal and clinical physiological studies, controversy remains with regard to the optimal nature and magnitude of postshock volume resuscitation. Options include massive isotonic fluid replacement [30,31], use of intravascular colloid containing fluids [32], and substitution with small volume hypertonic saline after hemorrhage [33].
Recently, however, a new resuscitation concept has emerged for application when the degree of autogenous vascular control is uncertain, namely permissive hypotension; this is achieved by administering small volumes of resuscitation fluid, permitting only minimal increase in perfusion until full vascular control of hemorrhage can be achieved by surgical intervention [34,35]. Although the statistical validity of the initial human studies [34] has been questioned [36], the concept appears to have some utility, provided that sufficient levels of tissue VO2 can be achieved to prevent the acute consequences of cellular ischemia [37]. These issues focus on the need for accurate and easily measured correlates of O2D that can quantify the severity of O2D and that can be monitored on a continuing basis during resuscitation.
Experimental models of hemorrhagic hypovolemic shock
A large number of animal models have been developed to simulate the critical end-points of hemorrhagic shock. Deitch [38] divided these models into three general categories: uncontrolled bleeding, controlled bleeding volume, and controlled decrements in blood pressure.
A more physiologically relevant animal model is needed because of the clinical requirement to progress beyond the traditional end-points of volume loss and subsequent blood pressure levels [39]. Furthermore, such a model is needed to determine why a state of hyperdynamic cardiovascular compensation develops after hypovolemic shock [25,40]. Also, numerous clinical studies have shown that hypovolemic trauma patients can remain in a state of shock, with evidence of inadequate tissue perfusion and metabolic acidosis [29,41,42], even if the traditional end-points have been normalized [1,2,25,40]. This is reflected in the present definition promulgated by the American College of Surgeons: 'Shock is an abnormality of the circulatory system that results in inadequate organ perfusion and tissue oxygenation' [3]. This understanding of the relationship between shock and inadequate perfusion has led to the development of a possibly more clinically relevant fourth general category of experimental hemorrhagic shock models, based on the concept of repayment of shock-induced O2D. Table 1 summarizes the historical development of hemorrhagic shock models with O2D as an end-point. It is based on a systematic Medline/Cochrane Library literature search using the terms 'oxygen debt or deficit' and 'hemorrhagic shock'. From 52 suggested articles, only 13 that strictly dealt with defined O2D in a hemorrhagic shock model are included.
Table 1.
Historical development of hemorrhagic shock models with oxygen debt as an end-point
| Author (year) [ref.] | Model | Method | Result |
| Crowell and Smith (1964) [4] | Dog | Hypotension of 30 mmHg; various oxygen deficits were allowed to accumulate | O2D as an indicator of survival |
| Rush et al. (1965) [5] | Dog | 30 min hemorrhage with varying hemorrhage volumes; achieved O2D varied | O2D as an indicator of cardiovascular change; the end-point 'survival' was not evaluated |
| Goodyer (1967) [90] | Dog | Hypotension of 30–50 mmHg; various oxygen deficits were allowed to accumulate | Irreversibility of shock is determined by peripheral mechanisms; the end-point'survival' was not evaluated |
| Jones et al. (1968) [7] | Dog | Hypotension of 30 mmHg; an oxygen deficit of120 cm3/kg was allowed to accumulate | O2D as an indicator of survival |
| Rothe (1968) [6] | Dog | Hypotension of 30 mmHg; various oxygen deficits were allowed to accumulate | No correlation betweeen O2D and survival |
| Neuhof et al. (1973) [8] | Rabbit | 30 min hemorrhage (1 ml/kg per min); achieved O2D varied | O2D as an indicator of survival |
| Schoenberg et al. (1985) [21] | Dog | Hypotension of 30 mmHg; various oxygen deficits were allowed to accumulate | No correlation betweeen O2D and survival |
| Reinhart et al. (1989) [91] | Dog | Hypotension of 40 mmHg; various oxygen deficits were allowed to accumulate | Excess oxygen uptake in recovery with hydroxyethylstarch; the end-point 'survival' was not evaluated |
| Dunham et al. (1991) [9] | Dog | Predetermined O2D after 60 min; independent of blood pressure or hemorrhage volume | O2D as an indicator of survival and O2D probability of death defined for dog |
| Sheffer et al. (1997) [92] | Computer | Computer simulation of myocardial oxygen deficit | For hemorrhage of 100 ml/min: time interval from injury to cardiac O2D inversely related to infusion rate; the end-point 'survival' was not evaluated |
| Siegel et al. (1997) [43] | Dog | Predetermined O2D after 60 min; independent of blood pressure or hemorrhage volume | Superiority of recombinant hemoglobin over colloid or whole blood in resuscitation |
| Rixen et al. (2001) [44] | Pig | Predetermined O2D after 60 min; independent of blood pressure or hemorrhage volume | O2D as an indicator of survival and O2D probability of death defined for pig. |
| Siegel et al. (2003) [37] | Dog | Predetermined O2D after 60 min; independent of blood pressure or hemorrhage volume | Determination of critical level of partial resuscitation as 30% of blood volume loss to return O2D to survival levels without vital organ cellular injury |
O2D, oxygen debt.
Thus, development of models of hemorrhagic shock must follow current knowledge and must consider indices of inadequate organ perfusion and tissue oxygenation, which are more meaningful end-points in the clinical setting [4]. Up to the 1990s O2D was used as a secondary end-point in pressure-controlled or volume-controlled models of hemorrhagic shock (Table 1); in contrast, Dunham and coworkers [9] described a canine model of hemorrhagic shock in which O2D was used as the independent predictor of the probability of death and organ failure. This canine model, which was validated in subsequent studies [37,43], follows the hypothesis that the total magnitude of O2D reached during hemorrhage is the critical determinant of survival, and that this variable and its metabolic consequences of lactic acidemia and base deficit better reflect the severity of the cellular insult than do traditional variables such as bleeding volume and blood pressure. This hypothesis was also verified in a pig model of O2D hemorrhagic shock [44].
General principles in the identification and quantification of oxygen debt
In healthy young men, the resting VO2 has been shown to average 140 ml/min per m2. If this VO2 is decreased by reduced blood flow with restriction in organ and tissue perfusion, a critical level of ischemia is induced, with a disparity between the oxidative requirement mandated by the level of metabolism and the level of oxygen delivery – an O2D occurs. Physiologically, if resuscitation is performed before a fatal metabolic debt is incurred then there is rapid repayment of the O2D, with VO2 overshoot as the unmetabolized acids are oxidatively metabolized during the reperfusion period. This is effected by an increase in oxygen delivery mediated by a rise in cardiac output – the 'hyperdynamic state' [1,26]. However, as the O2D accumulates the likelihood of cellular injury increases, with reduction in cellular membrane integrity and consequent cell swelling as intracellular water increases. Later in the process intracellular organelles become damaged, cellular synthetic mechanisms cease, and finally lysosomes are activated, which results in cell necrosis and death [45]. Even at less severe O2D levels, mechanisms that initiate later apoptosis are activated [46]. Depending on the extent and severity of the cellular injury, specific features of multiple organ failure (MOF) are initiated. Cells with the greatest oxidative requirements (e.g. brain, liver, kidney, myocardium and immunologic tissues) appear to be most vulnerable to O2D-induced injury or cell death.
Although evidence of cellular and organ failure often appears at various time points after recovery from O2D, it has long been known that the relationship between O2D and acute death can be quantified. Crowell and Smith [4] were the first to describe the effect of O2D in terms of a lethal dose (LD) effect. In their canine studies, O2Ds of 100 ml/kg or less were not lethal; O2Ds of 120 ml/kg led to an LD50 (i.e. a dose sufficient to kill 50% of the population studied); and O2Ds of 140 ml/kg or more were invariably fatal. A more precise quantification of the probability of death with increasing O2D in the same animal species was conducted by Dunham and coworkers [9], who established a complete probability of death function (Fig. 1a). Their studies noted an exponential relationship between probability of death and O2D, such that although the LD25 was at an O2D of 95.5 ml/kg, the LD50 lay at 113.5 ml/kg and the LD75 was at 126.5 ml/kg. This relationship has repeatedly been confirmed in dogs by more recent studies [37,43]. Studies in pigs [44] have found a nearly identical relationship, although the LD50 for the pig is at a slightly lower O2D/kg (95 ml/kg; Fig. 1b), corresponding with values calculated by Hannon and coworkers [19] in the same species. This difference between the two animals appears to reflect the greater percentage of adipose tissue in the pig as compared with the much leaner hound dog over the same range of body weight.
Figure 1.
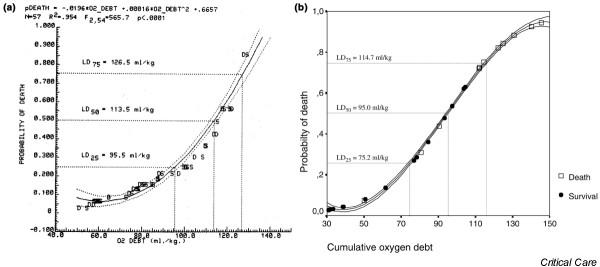
Probability of death as a function of oxygen debt. (a) Regression-derived relation of Kaplan–Meier probability of death as a function of increasing oxygen debt (O2D) in a canine O2D hemorrhagic shock model. Noted on the figure are the O2D values for lethal dose (LD)25 (i.e. a dose sufficient to kill 25% of the population studied), LD50, and LD75 probabilities. Points plotted along the regression line and its 95% confidence limits represent the actual Kaplan–Meier survival (S) values at 60 min of hemorrhage, or values at the time of death (D) for nonsurviving animals dying during the hemorrhage period or within 5 min of the 60 min hemorrhage sample. Note the good correlation of Kaplan–Meier points to the regression-estimated line. Reproduced with permission from Dunham and coworkers [9]. (b) Probability of death as a function of O2D in a pig O2D hemorrhagic shock model. Noted on the figure are the O2D values for LD25, LD50, and LD75 probabilities. Points plotted along the regression line and its 95% confidence limits represent the values of cumulative O2D (in ml/kg) at 60 min of hemorrhage for survivors (marked with circles) and nonsurvivors (marked with squares). Modified from Rixen and coworkers [44].
To understand better the concept of hemorrhage-induced O2D accumulation and its repayment by volume infusion, experimental animal responses were recently studied by Siegel and coworkers [37]. In that study 40 dogs were bled to achieve an O2D of 104 ± 7.6 ml/kg at 60 min after initiation of hemorrhage (estimated probability of death: 35.7% [9]; actual death rate: 40%; shed blood volume [SBV]: 71.0 ± 6.8% of the animals' estimated total blood volume [37]). Following hemorrhage, the animals were either given no initial resuscitation for 2 hours and then fully resuscitated with a volume of 5% colloid equivalent to 120% of their SBV. Alternatively, they were randomly assigned to initial resuscitation (R1) with a predetermined percentage of their SBV (again by infusing 5% colloid) equivalent to 8.4%, 15%, 30%, or 120% of their SBV. Then, after a 2 hour delay period in which no further volume resuscitation was given, the animals were given the remaining portion of the calculated 120% of the SBV lost during hemorrhage (delayed resuscitation: R2). This made the final quantity of volume replacement in each animal equal to 120% of the SBV. It is important to note that in those animals given no initial resuscitation, the O2D accumulation rate continued to rise either at the same (or slighly lower) rate as during the hemorrhage to or above the 90% mortality level, even though no further blood loss occurred. However, in all instances of R1, the O2D also continued to rise slightly until a critical quantity of R1 was given (at least 30% of the SBV), but the initial rate of recovery from the hemorrhage-induced O2D level was increased proportionately to the increase in R1. This relationship between the magnitude of initial resuscitation and the rate of O2D decrease was highly significant (P < 0.001) and predicted later evidence of cell death and organ failure in 7-day postshock survivors [37]. In contrast to the significant discrimination provided by O2D level, the simultaneously measured mean blood pressure responses were not found to be significant predictive of the adequacy of resuscitation [37].
These canine data and parallel data obtained in the O2D pig model [44] demonstrate that quantity of blood volume loss or replacement, blood pressure, and even cardiac output response do not reflect well the severity of shock or the effectiveness of volume resuscitation. However, these end-points are well defined in a quantitative manner by the magnitude of the O2D and by its rate of resolution during the resuscitation period, independent of species.
Metabolic correlates of oxygen debt
A considerable body of evidence has accumulated that strongly suggests, both in the animal setting [9,37,43,44] and in humans [29,41,47-49], that metabolic acids in blood or plasma are indices that reflect the degree of tissue hypoxia associated with hypovolemic ischemia. In this review the strict definition of base deficit (BD) – namely, a negative base excess – is used [29,49,50], with a decrease in base excess with increasing metabolic decompensation implying progressively negative values (e.g. -6 mmol/l to -10 mmol/l). However, because BD implies a negative base excess, only positive values of BD (without the minus sign) are used in the present review.
As the concept of O2D as the key process determining outcome evolved, one of the major goals of experimental studies was to examine the relationship between lactate or BD and hemorrhage-induced O2D [9,37,43,44]. This significant relationship was repeatedly demonstrated in progressive hemorrhage, with increases in BD or lactate being paralleled by increases in O2D [9,37,43,44]. Similar significant relationships were noted between decreases in these metabolic variables; the O2D fell during volume resuscitation, regardless of whether the fluid was crystalloid, hypertonic saline, carbonate/gelatine, colloid, or whole blood [9,37,43,44,51]. The rate of decline in O2D (and BD and lactate) was significantly more rapid when an oxygen-carrying solution of recombinant hemoglobin was employed for resuscitation [43]. The relationship between BD and O2D tended to reflect better the effectiveness of increases in initial volume resuscitation, whereas lactate reflected the overall trend in effectiveness of resuscitation but with less discrimination [37]. Very similar, albeit more variable, significant relationships (P < 0.0001) for the two metabolic correlates of O2D were also noted in the pig model [44]. The greater variability found in the pig may reflect a closer similarity to broad range of adipose tissue found in humans. Nevertheless, BD and lactate appear to correlate best with O2D in experimental hemorrhagic shock. This relationship is significant across species [9,44].
Finally, the relationship between O2D and BD can be used to address the problem of quantifying the effectiveness of small volume resuscitation during permissive hypotension. In other words, the paramedic, surgeon, or intensivist could resuscitate a hypovolemic patient to a level at which perfusion will yield a reduction in O2D that will allow critical organ oxidative metabolism to be maintained, at a blood pressure that will not encourage further hemorrhage until all open vessels are Although it is generally not practical to measure O2D in humans, a model for this approach using BD can be derived from animal data. In a canine O2D shock model, Siegel and coworkers [37] demonstrated that animals that were effectively volume resuscitated moved progressively down the O2D/BD regression line to lower values compatible with a reduced probability of death [37]. However, those animals that received inadequate volume resuscitation, particularly those that died during the 2 hour postshock period, moved to progressively higher points in the O2D/BD relationship. A similar but less quantifiable relationship was found for the O2D/lactate relationship.
In this respect attention must be paid to the recent development of hemorrhagic shock models with a target endpoint of metabolic acidosis [52-54]. Schultz [52] and Powell [53] and their groups studied bacterial translocation and restoration of central venous oxygen saturation after BD-guided hemorrhagic shock in rats. Also, DeAngeles and coworkers [54] studied resuscitation from BD/lactate guided hemorrhagic shock with diaspirin cross-linked hemoglobin, blood, and hetastarch in sheep. Thus, the use of BD and lactate as clinically useful surrogates for O2D is strongly supported by experimentation in numerous animal species.
Metabolic correlates of oxygen debt in determining the severity of shock and the effectiveness of resuscitation in humans Lactate
The search for identifiable and easily measured metabolic correlates of shock that could be used to quantify the severity of human circulatory failure began with the pioneering work of Huckabee [55], Weil and Afifi [56] and Harken [57]. These studies confirmed that the circulating level of lactate provided an indication of the anaerobic component induced by the shock process. Bakker and coworkers [58] reported evidence that the dependency on oxygen supply to body tissues was associated with increasing lactate levels.
Table 2 provides a summary of literature on lactate as an outcome predictor in adult multiple trauma patients based on a systematic Medline/Cochrane Library literature search, using the terms 'lactate' and 'multiple trauma'. Of 59 originally retrieved articles, 27 are specifically noted in the present review because they strictly deal with lactate as an outcome predictor in multiple trauma patients. In almost 3000 multiple trauma patients lactate was shown to predict outcome following postoperative complications, intracranial pressure, infection, sepsis, adult respiratory distress syndrome (ARDS), MOF, injury and hemorrhage severity, and survival.
Table 2.
Literature on lactate as an outcome predictor in adult multiple trauma patients
| Author (year) [ref.] | Trauma patients | Outcome prediction |
| Oestern et al. (1978/1979) [93,94] | 50 | Survival |
| Brandl et al. (1989) [95] | 51 | Survival |
| Siegel et al. (1990) [29] | 185 | Survival |
| Woltmann and Kress (1991) [96] | 35 | Survival |
| Nast-Kolb et al. (1992) [97] | 100 | Survival |
| Waydhas et al. (1992) [98] | 100 | MOF, sepsis |
| Roumen et al. (1993) [99] | 56 | MOF, ARDS |
| Abramson et al. (1993) [61] | 76 | Survival |
| Sauaia et al. (1994) [100] | 394 | MOF |
| Dunham et al. (1994) [101] | 17 | MOF, ARDS |
| Scalea et al. (1994) [102] | 30 | Intracranial pressure |
| Manikis et al. (1995) [103] | 129 | MOF, survival |
| Ivatury et al. (1995) [104] | 27 | Survival |
| Regel et al. (1996) [105] | 342 | MOF |
| Mikulaschek et al. (1996) [64] | 52 | Survival |
| Charpentier et al. (1997) [106] | 20 | Survival |
| Nast-Kolb et al. (1997) [107] | 66 | MOF |
| Cairns et al. (1997) [85] | 24 | MOF |
| Sauaia et al. (1998) [108] | 411 | MOF |
| Blow et al. (1999) [109] | 116 | MOF, survival |
| Claridge et al. (2000) [110] | 364 | Infection, survival |
| Crowl et al. (2000) [111] | 77 | 'Postoperative complications' |
| Rixen et al. (2000) [77] | 80 | ARDS |
| Ertel et al. (2001) [112] | 20 | Severity of hemorrhage, survival |
| Cerovic et al. (2003) [113] | 98 | Injury severity, survival |
| Egger et al. (2004) [114] | 26 | Injury severity |
ARDS, acute respiratory distress syndrome; MOF, multiple organ failure.
Clinically, however, it is important to note that not all cases of hyperlactatemia are accompanied by acidosis, and neither are all cases of hyperlactatemia caused by O2D. Other metabolic dysfunctions may also be associated with hyper-lactatemia [59] and can confuse assessment of the O2D effect, as can excessive alcohol intake and acute cocaine use. The most prominent group of patients with increased lactate levels in the absense of hypovolemia are patients with severe sepsis [28]. However, diabetic patients with keto-acidosis have increased lactate, and in patients with impaired hepatic function lactate uptake may be reduced and lactate levels may rise. Of specific importance in patients resuscitated from hemorrhagic shock is that administration of large quantities of exogenous lactate (e.g. via mass infusion of Ringer's lactate) has been shown to increase lactate to levels significantly greater than those expected to result from the shock process alone [60]. This clearly may distort interpretation of lactate levels as a clinical diagnostic tool. Furthermore, the reduction in oxygen delivery that induces O2D also causes other metabolic acids to accumulate in the extracellular/intravascular components, and so plasma lactate levels may not always quantitatively reflect the O2D process. Thus, the origin of a hyperlactatemia is clinically important and has direct implications for treatment choice.
Although the use of lactate-free resuscitation fluids may become routine in the future [60], the current widespread use of Ringer's lactate may be a further reason why it remains unclear whether the lactate level on hospital admission is prognostically significant in multiple trauma patients. Several studies have noted the predictive value of the initial lactate level [58,61,62], but others have shown other variables to be equivalent [63] or even better [29] in outcome prediction. In contrast, more than one study found no significant correlation between initial lactate level and posttrauma outcome [49,64-66]. Nevertheless, in a study of 375 trauma patients admitted directly from the scene of injury to a level I trauma center [62], simultaneously obtained arterial and peripheral venous lactate levels were shown to be highly correlated, and a lactate threshold level of > 2 mmol/l appeared to predict the likelihood of the Injury Severity Score (ISS) being 13 or greater with a high degree of accuracy. Thus, lactate appears to represent a good triage tool.
Base deficit
In the search for a more precise quantifier of severity of posttrauma hemorrhagic shock, Siegel [29], Rutherford [47], Davis [50], and Rixen [49] and their groups studied the value of BD as a single predictor of the severity of posttrauma hemorrhagic shock. The findings of those studies, which represent more than 8000 trauma patients with varying severities of injury, are shown in Fig. 2. All of the studies indicate that BD can be used to stratify trauma patients with respect to their likelihood of dying, and suggest that BD can also be used to provide an index of the effectiveness of resuscitation in humans as well as in experimental animals.
Figure 2.
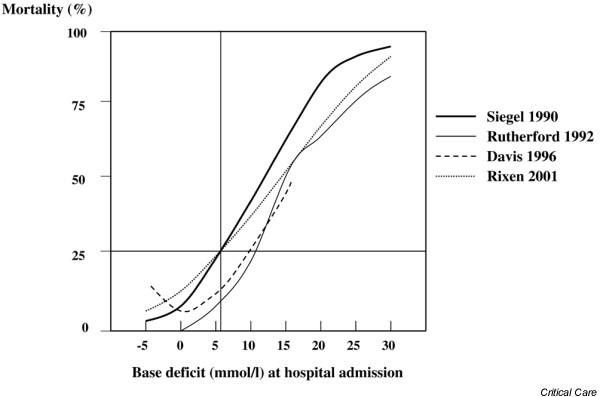
Mortality as a function of base deficit. Mortality curves presented as a function of the admission base deficit in more than 8000 multiple trauma patients derived from four independent studies. Modified from Zander [89].
With respect to studies in patients with greater injury severity, Table 3 provides a summary of literature on BD as an outcome predictor in adult multiple trauma patients based on a systematic Medline/Cochrane Library literature search using the terms 'base excess' or 'base deficit' and 'multiple trauma'. From 34 originally retrieved articles, 15 are noted because they strictly deal with base excess/BD as an outcome predictor in adult multiple trauma patients. Among the 6567 multiple trauma patients represented in Table 3, BD was found to predict outcomes in terms of hemodynamics, transfusion requirements, metabolism, coagulation, volume deficit, neutrophil chemiluminescence and CD11b expression, complement activation, acute lung injury, ARDS, hepatic dysfunction, MOF, and survival.
Table 3.
Literature on base excess/base deficit as an outcome predictor in adult multiple trauma patients
| Author (year) [ref.] | Trauma patients | Outcome prediction |
| Oestern et al. (1978/1979) [93,94] | 50 | Survival |
| Davis et al. (1988) [41] | 209 | Blood pressure, severity of volume deficit |
| Siegel et al. (1990) [29] | 508 | Survival |
| Sauaia et al. (1994) [100] | 394 | MOF |
| Regel et al. (1996) [105] | 342 | MOF |
| Botha et al. (1997) [48] | 17 | Neutrophil CD11b expression |
| Davis et al. (1998) [115] | 674 | Survival |
| Krishna et al. (1998) [116] | 40 | Survival |
| Fosse et al. (1998) [117] | 108 | Complement activation |
| Brown et al. (1999) [118] | 12 | PMN chemiluminescence |
| Eberhard et al. (2000) [119] | 102 | Acute lung injury |
| Rixen et al. (2000) [77] | 80 | ARDS |
| Rixen et al. (2001) [49] | 2069 | Hemodynamic, transfusion requirements, metabolism, coagulation, survival |
| Harbrecht et al. (2001) [120] | 1962 | Hepatic dysfunction |
ARDS, acute respiratory distress syndrome; MOF, multiple organ failure.
Although multiple trauma patients were not included exclusively, attention must be given to the studies conducted by Mackersie [67] and Davis [68] and their groups in more than 6000 trauma patients; those investigators showed that BD may also be considered an indicator of significant abdominal injury. Furthermore, the admission BD was also found to be an important prognostic indicator with respect to injury severity and death in pediatric [69-71] and elderly [72] trauma populations.
However, Siegel and coworkers [29] demonstrated that BD alone did not provide the best prediction of posttrauma mortality, and that it could be quantitatively coupled with an estimate of head injury, such as that provided by the Glasgow Coma Scale (GCS). The interaction between GCS score, BD, and mortality is illustrated in Fig. 3a, which was developed from findings in 185 patients whose major injury was blunt hepatic trauma. Also shown is the relationship of predicted to observed deaths based on the regression model (Fig. 3b). This relationship was verified in an independent group of 323 multiple trauma patients with pelvic fracture [29]. Indeed, the substantial differences in the proportion of trauma patients with severe head injury in the studies shown in Fig. 2 may account for the variation in the LD25 and LD50 points seen in these different clinical studies.
Figure 3.
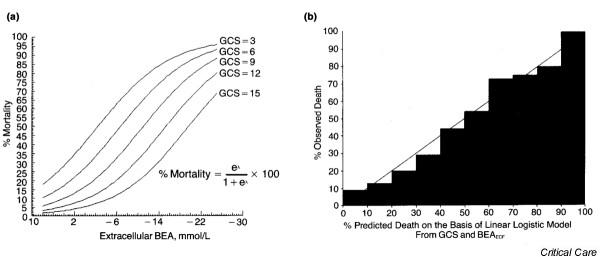
Interaction between base excess, Glasgow Coma Scale (GCS) and mortality. (a) Linear logistic model for predicting mortality from GCS and admission extracellular base excess (BEA) for 185 patients with blunt traumatic hepatic injury (λ = - 0.21 [GCS] - 0.147 [BEAECF] + 0.285; P < 0.0001 for model). (b) Predicted versus observed mortality in linear logistic model from GCS and BEA for patients with blunt traumatic hepatic injury. ECF, extracellular fluid. Reproduced with permission from Siegel and coworkers [29].
The validity of the use of BD in conjunction with other predictive variables was extended to a larger series of 2069 multiple trauma patients included in the German Trauma Society registry [73]. That study validated the probability of death relationship between BD and GCS, but it also showed that additional improvement in the sensitivity/specificity receiver operating characteristic (ROC) curve (ROC = 0.904, with greatest sensitivity and specificity of 82.3% and 83.0%, respectively) could be obtained by the addition of prothrombin time, age, and ISS to the equation. In this multifactorial analysis, the admission BD was one of the five best predictors for outcome (BD, GCS, age, prothrombin time, and ISS). Each of these five variables contributed significantly to the derived multifactorial regression model:
pDeath = 1/1 + e{-(intercept + β1[BD] + β2[GCS] + β3[prothrombin time] + β4[age] + β5[ISS])}
Where pDeath = probability of death, BD = hospital admission BD, intercept = -0.1551, β1 = 0.0840, β2 = -0.2067, β3 = -0.0359, β4 = 0.0438, and β5 = 0.0252.
However, when the three physiologic variables and age were added sequentially into the regression model, the ISS contributed only an additional 0.4% to the correctness of prediction. These data suggest that, because the full extent of the patient's injuries and their severities may not be readily evident on hospital admission, a reasonable immediate estimate of severity can be made on the basis of the patient's physiologic/metabolic response adjusted for age.
This prediction model was validated prospectively in an independent set of 1745 additional multiple trauma patients included in the German Trauma Society registry [73]. In both the development set (2069 patients) and in the independent validation set (1745 additional multiple trauma patients), the probability of death predicted by the model was compared with the observed mortality rate. The validation set yielded an area under the ROC curve for the model of 0.901, with greatest sensitivity and specificity of 82.2% and 83.3%, respectively (Fig. 4a). Using the goodness-of-fit test, there was no significant difference between the observed and predicted distributions of mortality. The model predicted the numbers of observed and expected events equally well across all strata in the development and validation sets (Fig. 4b), and therefore the model appeared to be well calibrated in both development and validation sets of multiple trauma patients.
Figure 4.
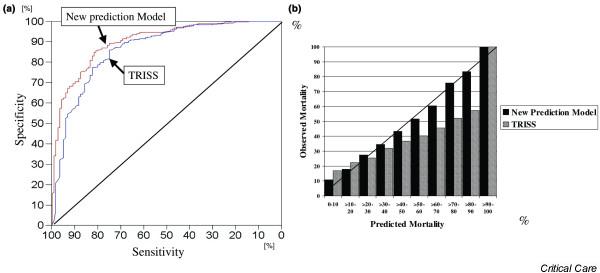
Discrimination and calibration of the multivariate outcome prediction model. (a) Discrimination. Receiver operating characteristic curve of the multivariate outcome prediction model based on base deficit, Glasgow Coma Scale score, prothrombin time, age and Injury Severity Score, compared with that derived from the Trauma and Injury Severity Score (TRISS) in the validation set of 1745 multiple trauma patients. The diagonal line corresponds to a test that is sensitive or specific just by chance. The area under the curve for the multivariate outcome prediction model is 0.901 and that for the TRISS score is 0.866. (b) Calibration. Predicted versus observed mortality for the multivariate outcome prediction model and the TRISS score in the validation set of 1745 multiple trauma patients.
The validation of this outcome prediction model for multiple trauma patients was completed by its comparison with the predictive ability of an international gold standard, namely the Trauma and Injury Severity Score (TRISS) score [74]. In the validation set of patients discussed above, TRISS discrimination yielded an area under the ROC curve of 0.866 (Fig. 4a). Although this difference in overall predictive ability may appear to be small, when the predicted versus observed death rates are examined in detail it is apparent that there is under-prediction by the TRISS score from the 30% to the 90% mortality range, which is the region of greatest clinical interest (Fig. 4b). Using the goodness-of-fit test [75] there was a significant difference between the observed and predicted mortality distributions in the TRISS score. Thus, the TRISS score did not predict well the number of observed events across all strata as compared with the prediction model based on BD, GCS, prothrombin time, age, and ISS. This weakness of TRISS and other scoring systems based on the Revised Trauma Score and ISS alone, without inclusion of specific patient metabolic data, has been extensively examined in comparison with other systems and is consistent with these observations [76].
The use of BD allows critical thresholds to be established by which the clinician can be alerted to the beginning of a deleterious trend in O2D or to progression of putative shock to a condition of life-threatening potential. In this regard, both the studies conducted by Davis [41] and Siegel [29] and their groups, as well as the more recent multicenter trial data [49], have shown that a critical threshold exists at or slightly above a BD of 6.0 mmol/l (Fig. 2). When the probability of death is analyzed as a function of BD [29], this is the point at which the exponential rise in probability of death begins, and it is also the point in the experimentally derived BD/O2D relationship [9] at which the O2D begins to rise exponentially. In contrast to the animal studies, in posttrauma humans, where there is frequently an associated brain injury, the observed mortality induced by a rise in BD to 6.0 mmol/l is also a function of the level of impairment in GCS, rising from a probability of death of 15% with a GCS score of 15 to 30% with a GCS score of 9 and 45% at a GCS score of 6 (Fig. 3). Thus, the overall probability of death both in the initial study conducted by Siegel and coworkers [29] and in the more recent report by Rixen and coworkers [49] exceeded 25% (LD25) when the admission BD was increased to 6.0 mmol/l or greater, independent of GCS score.
Furthermore, change in BD over time is an important variable in the prediction of outcome following hypovolemic post-traumatic shock. Rixen and coworkers [49] noted that the change in BD between hospital and intensive care unit (ICU) admission was a further significant predictor of outcome. Those investigators analyzed the development of BD over the period from hospital to ICU admission with respect to mortality rate. The trauma patients were subdivided into two groups at the time of hospital and subsequent ICU admission with respect to the LD25 threshold value of 6 mmol/l, which was previously noted to be the critical level [29,41,49]; patients with a BD below 6 mmol/l were considered to have 'good prognosis', and patients with a BD of 6 mmol/l or greater were considered to have 'bad prognosis'. Patients with a BD below 6 mmol/l on hospital admission and who subsequently had a BD below 6 mmol/l on ICU admission had the lowest mortality rate (13%). Patients with a BD above 6 mmol/l on hospital admission and who subsequently had a BD of 6 mmol/l or greater on ICU admission had the highest mortality rate (45%; P < 0.0001). Finally, the level of admission BD was shown to predict the probability of development of post-traumatic ARDS, with the incidence rising exponentially above a BD of 6.6 mmol/l [77].
Conclusion
The data reported above strongly indicate a need to add quantitative estimates of the effectiveness of perfusion and VO2 to hemorrhagic shock studies. Currently, indirect measurement techniques that reflect cellular oxygen utilization and perfusion either systemically (lactate and BD) or locally (gastric intramucosal pH and microdialysis [78]) predominate. It would be ideal to measure O2D at the cellular level as an end-point of experimental and clinical hemorrhagic shock. The muscle beds, subcutaneous tissue, and even skin have been advocated as sites at which perfusion may be more directly measured at the tissue level. Hartmann and coworkers [79] found good correlations between subcutaneous and transcutaneous partial oxygen tension (PO2), and with gastric tonometry in pigs. Subcutaneous PO2 tissue probes have been used in the experimental setting [80] as well as in severely injured patients [81,82]. McKinley and coworkers [83] studied skeletal muscle PO2, partial carbon dioxide tension, and pH using fiberoptic technology in hemorrhaged dogs, and Knudson and coworkers [84] examined the posthemorrhage and resuscitation oxygen tension response in muscle and liver in pigs. Another technique that holds promise for the future is that of near infrared spectroscopy [85]. All of these techniques, along with others currently being developed, may move the endpoints of hemorrhagic shock models to the organ, cellular, and subcellular levels. However, at present these newer technologies require the use of relatively complex and expensive or invasive methodologies, whereas relatively inexpensive handheld devices now exist for rapid field, emergency room, or ICU determinations of lactate and BD [86].
However, it is clear from experimental [87] and clinical studies [81,88] that some vascular beds may be more vasoconstricted than others; the skin, subcutaneous and muscle tissue, and intestinal perfusion are sacrificed to preserve cardiac, central nervous system, renal, and hepatic perfusion. Consequently, probes placed in the physiologically expendable tissues may not reflect the true total body situation, and especially vital organ O2Ds. This contention is supported by the findings reported by Siegel and coworkers [37], which showed that adequate resuscitation with a volume of 30% of SBV could preserve essential organ histology and physiologic function from an LD35–40 of O2D without increasing the cardiac index above control preshock levels. Only when the remaining volume of delayed full resuscitation was given did the cardiac index and oxygen consumption rise to hyperdynamic levels, suggesting that a large percentage of this hyperdynamic state is devoted to repayment of the O2D in less essential organs, which collectively represent the greater portion of body cell mass.
Nevertheless, both animal and clinical data strongly suggest that the overall O2D and/or its metabolic correlates (BD and lactate) better reflect the severity of shock than do currently available measures of local tissue or organ perfusion. This is shown by the probability of death curves for individual species, and by the relative consistency of LD25 and LD50 points for BD across species and especially in humans, when adjusted for GCS and other significant variables.
We require a more precise technique for assessing total body O2D, or at least that of critical organs, that can easily and repeatedly be applied in the clinical setting. Until such a technique becomes available the use of BD, either alone or in combination with GCS score and other significant variables of high predictive accuracy (e.g. prothrombin time and age), represents the best present system for clinical assessment of shock severity and success of resuscitation. These variables may be used to obtain information rapidly on a patient's level of compensation in response to posttrauma or hemorrhagic shock either by immediate reference to a predetermined graph (Fig. 3) or by entry of data into a handheld computer for computation of an estimate of probability of death using the regression equation shown above. This would facilitate clinical decision making at the bedside, in the emergency room, or in the ICU.
In conclusion, the data examined in this review strongly indicate that there is a need to add quantitative estimates of O2D and resulting metabolic acidosis to clinical studies, and that these variables should be considered in the management of patients sustaining severe hemorrhagic shock. The data also suggest that evaluation of metabolic correlates of the total body O2D (BD and, to a lesser extent, lactate) may be more useful in quantifying the responses of trauma or nontrauma patients to hemorrhage than are estimates of blood loss, quantitative measurements of volume replacement, or blood pressure and heart rate. Finally, we believe that further research based on the parameters of oxygen utilization and O2D will achieve even better, clinically suitable variables by which to assess the magnitude and severity of human stress physiology and to quantify the effectiveness of resuscitation therapies in the multiple trauma patient or the patient with life-threatening hemorrhage from a gastrointestinal lesion.
Abbreviations
ARDS = acute respiratory distress syndrome; BD = base deficit; GCS = Glasgow Coma Scale; ICU = intensive care unit; ISS = Injury Severity Score; LD = lethal dose; MOF = multiple organ failure; O2D = oxygen debt; PO2 = partial oxygen tension; ROC = receiver operating characteristic; SBV = shed blood volume; TRISS = Trauma and Injury Severity Score; VO2 = oxygen consumption.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
Both authors (DR and JHS) made substantial contributions to the conception and design of this review, and to the acquisition, analysis, and interpretation of data. Furthermore, both authors were involved in drafting the article and revising it critically for important content, and gave final approval of the version to be published.
Acknowledgments
Acknowledgements
Supported in part by the Deutsche Forschungsgemeinschaft in a grant to Dr Rixen and by the New Jersey Medical School: Wesley J. Howe Professorship in Trauma Surgery held by Dr Siegel.
Contributor Information
Dieter Rixen, Email: dieter.rixen@uni-wh.de.
John H Siegel, Email: siegeljh@umdnj.edu.
References
- Siegel JH, Linberg SE, Wiles CE. Therapy of low-flow shock states. In: Siegel JH, editor. Trauma: Emergency Surgery and Critical Care. Churchill Livingston: New York; 1987. pp. 201–284. [Google Scholar]
- Siegel JH. Through a glass darkly: the lung as a window to monitor oxygen consumption, energy metabolism, and severity of critical illness. Clin Chem. 1990;36:1585–1593. [PubMed] [Google Scholar]
- American College of Surgeons . Advanced Trauma Life Support Manual. American College of Surgeons; 1997. Shock; pp. 87–108. [Google Scholar]
- Crowell JW, Smith EE. Oxygen deficit and irreversible hemorrhagic shock. Am J Physiol. 1964;206:313–316. doi: 10.1152/ajplegacy.1964.206.2.313. [DOI] [PubMed] [Google Scholar]
- Rush BF, Rosenberg JC, Spencer FC. Changes in oxygen consumption in shock. J Surg Res. 1965;5:252–255. doi: 10.1016/s0022-4804(65)80013-0. [DOI] [PubMed] [Google Scholar]
- Rothe CF. Oxygen deficit in hemorrhagic shock in dogs. Am J Physiol. 1968;214:436–442. doi: 10.1152/ajplegacy.1968.214.3.436. [DOI] [PubMed] [Google Scholar]
- Jones CE, Crowell JW, Smith EE. A cause-effect relationship between oxygen deficit and irreversible hemorrhagic shock. Surg Gynecol Obstet. 1968;127:93–96. [PubMed] [Google Scholar]
- Neuhof H, Wolf H, Rohtermundt R, Glaser E, Lasch HG. Oxygen consumption of the organism in hemorrhagic shock. Experimental studies [in German] Z Kardiol. 1973;62:663–683. [PubMed] [Google Scholar]
- Dunham CM, Siegel JH, Weireter L, Fabian M, Goodarzi S, Guadalupi P, Gettings L, Linberg SE, Vary TC. Oxygen debt and metabolic acidemia as quantitative predictors of mortality and the severity of the ischemic insult in hemorrhagic shock. Crit Care Med. 1991;19:231–243. doi: 10.1097/00003246-199102000-00020. [DOI] [PubMed] [Google Scholar]
- Cannon WB. Traumatic Shock. New York: D Appleton Co; 1923. [Google Scholar]
- Blalock A. Acute circulatory failure as exemplified by shock and hemorrhage. Surg Gynec Obstet. 1934;58:551–566. [Google Scholar]
- Wiggers CJ. The present status of the shock problem. Physiol Rev. 1942;22:74–123. [Google Scholar]
- Guyton AC. Cardiac output in circulatory shock. In: Guyton AC, editor. Circulatory Physiology: Cardiac Output and its Regulation. Philadelphia, PA: WB Saunders; 1963. pp. 333–351. [Google Scholar]
- Fleischer GR, Templeton J, Delgado-Paredes C. An animal model for the study of hemorrhagic shock from abdominal trauma in children. Pediatr Emerg Care. 1987;3:18–21. doi: 10.1097/00006565-198703000-00005. [DOI] [PubMed] [Google Scholar]
- Bickell WH, Bruttig SP, Wade CE. Hemodynamic response to abdominal aortotomy in the anesthetized swine. Circ Shock. 1989;28:321–332. [PubMed] [Google Scholar]
- Vivaldi E, Macinelli S, Günther B. Experimental hemorrhagic shock in dogs: standardization. Res Exp Med (Berl) 1983;182:127–137. doi: 10.1007/BF01851118. [DOI] [PubMed] [Google Scholar]
- Traverso LW, Moore CC, Tillman FJ. A clinically applicable exsanguination shock model in swine. Circ Shock. 1984;12:1–7. [PubMed] [Google Scholar]
- Carroll RG, Iams SG, Pryor WH, Allison EJ. Single hemorrhage: a clinically relevant canine model of hemorrhagic shock. Resuscitation. 1988;16:119–126. doi: 10.1016/0300-9572(88)90076-7. [DOI] [PubMed] [Google Scholar]
- Hannon JP, Wade CE, Bossone CA, Hunt MM, Loveday JA. Oxygen delivery and demand in conscious pigs subjected to fixed-volume hemorrhage and resuscitated with 7.5% NaCl in 6% Dextran. Circ Shock. 1989;29:205–217. [PubMed] [Google Scholar]
- Mittmann U, Schmidt JD, Schmier J, Wirth RH. Hemorrhagic shock with fixed hypotension and with spontaneous recovery of blood pressure: a comparison of two shock models. Basic Res Cardiol. 1976;71:47–59. doi: 10.1007/BF01907782. [DOI] [PubMed] [Google Scholar]
- Schoenberg MH, Smedegard G, Gerdin B, Messmer K, Arfors KE. Hemorrhagic shock in the dog: I. Correlation between survival and severity of shock. Res Exp Med. 1985;185:21–33. doi: 10.1007/BF01851524. [DOI] [PubMed] [Google Scholar]
- Schlichting E, Lyberg T. Monitoring of tissue oxygenation in shock: an experimental study in pigs. Crit Care Med. 1995;23:1703–1710. doi: 10.1097/00003246-199510000-00015. [DOI] [PubMed] [Google Scholar]
- Beecher HK. Surgery in World War II The Physiologic Effects of Wounds. Washington, DC: Office of the Surgeon General, Department of the Army; 1952. [Google Scholar]
- Artz CP, Howard JM, Sako A, Bronwell AW, Prentice T. Clinical experiences in the early management of the most severely injuried battle casualties in the Korean war. Ann Surg. 1955;141:285–301. doi: 10.1097/00000658-195503000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker WC, Czer LSC. Evaluation of the biologic importance of various hemodynamics and oxygen transport variables: which variables should be monitored in postoperative shock? Crit Care Med. 1979;7:424–431. doi: 10.1097/00003246-197909000-00015. [DOI] [PubMed] [Google Scholar]
- Shoemaker WC, Appel PL, Kram HB, Waxman K, Lee TS. Prospective trial of supranormal values of survivors as therapeutic goals in high-risk surgical patients. Chest. 1988;94:1176–1186. doi: 10.1378/chest.94.6.1176. [DOI] [PubMed] [Google Scholar]
- Bishop MH, Shoemaker WC, Appel PL, Meade P, Ordog GJ, Wasserberger J, Wo CJ, Rimle DA, Kram HB, Umali R, et al. Prospective, randomized trial of survival values of cardiac index, oxygen delivery, and oxygen consumption as resuscitation end points in severe trauma. J Trauma. 1995;38:780–787. doi: 10.1097/00005373-199505000-00018. [DOI] [PubMed] [Google Scholar]
- Siegel JH, Cerra FB, Coleman B, Giovannini I, Shetye M, Border JR, McMenamy RH. Physiological and metabolic correlations in human sepsis. Surgery. 1979;86:163–193. [PubMed] [Google Scholar]
- Siegel JH, Rivkind AI, Dala S, Goodarzi S. Early physiologic predictors of injury severity and death in blunt multiple trauma. Arch Surg. 1990;125:498–508. doi: 10.1001/archsurg.1990.01410160084019. [DOI] [PubMed] [Google Scholar]
- Shires GT, Canizaro PC. Fluid resuscitation in the severely injured. Surg Clin North Am. 1964;55:1341–1389. doi: 10.1016/s0039-6109(16)40183-0. [DOI] [PubMed] [Google Scholar]
- Canizaro PC, Prager MD, Shires GT. The infusion of Ringer's lactate solution during shock. Am J Surg. 1971;122:494–502. doi: 10.1016/0002-9610(71)90474-0. [DOI] [PubMed] [Google Scholar]
- Roberts I, Alderson P, Bunn F, Chinnock P, Ker K, Schierhout G. Colloids versus crystalloids for fluid resuscitation in critically ill patients. Cochrane Database Syst Rev. 2004;4:CD000567. doi: 10.1002/14651858.CD000567.pub2. [DOI] [PubMed] [Google Scholar]
- Bunn F, Roberts I, Tasker R, Akpa E. Hypertonic versus near isotonic crystalloid fluid resuscitation in critically ill patients. Cochrane Database Syst Rev. 2004;3:CD002045. doi: 10.1002/14651858.CD002045.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickell WH, Wall MJ, Pepe PE, Martin RR, Ginger VF, Allen MK, Mattox KL. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. N Engl J Med. 1994;331:1105–1109. doi: 10.1056/NEJM199410273311701. [DOI] [PubMed] [Google Scholar]
- Silbergleit R, Satz W, McNamara RM, Lee DC, Schoffstall JM. Effect of permissive hypotension in continuous uncontrolled intra-abdominal hemorrhage. Acad Emerg Med. 1996;3:922–926. doi: 10.1111/j.1553-2712.1996.tb03320.x. [DOI] [PubMed] [Google Scholar]
- Siegel JH. Immediate versus delayed fluid resuscitation in patients with trauma [letter] N Engl J Med. 1995;332:681. doi: 10.1056/NEJM199503093321013. [DOI] [PubMed] [Google Scholar]
- Siegel JH, Fabian M, Smith JA, Kingston EP, Steele KA, Wells MR, Kaplan LJ. Oxygen debt criteria quantify the effectiveness of early partial resuscitation after hypovolemic hemorrhagic shock. J Trauma. 2003;54:862–880. doi: 10.1097/01.TA.0000066186.97206.39. [DOI] [PubMed] [Google Scholar]
- Deitch EA. Animal models of sepsis and shock: a review and lessons learned. Shock. 1998;9:1–11. doi: 10.1097/00024382-199801000-00001. [DOI] [PubMed] [Google Scholar]
- Porter JM, Ivatury RR. In search of the optimal end points of resuscitation in trauma patients: a review. J Trauma. 1998;44:908–914. doi: 10.1097/00005373-199805000-00028. [DOI] [PubMed] [Google Scholar]
- Siegel JH, Farrell EJ, Miller M, Goldwyn RM, Friedman HP. Cardiorespiratory interactions as determinants of survival and the need for respiratory support in human shock states. J Trauma. 1973;13:602–619. doi: 10.1097/00005373-197307000-00005. [DOI] [PubMed] [Google Scholar]
- Davis JW, Shackford SR, Mackersie RC, Hoyt DB. Base deficit as a guide to volume resuscitation. J Trauma. 1988;28:1464–1467. doi: 10.1097/00005373-198810000-00010. [DOI] [PubMed] [Google Scholar]
- Davis JW, Shackford SR, Holbrook TL. Base deficit as a sensitive indicator of compensated shock and tissue oxygen utilization. Surg Gynecol Obstet. 1991;173:473–476. [PubMed] [Google Scholar]
- Siegel JH, Fabian M, Smith JA, Costanino D. Use of recombinant hemoglobin solution in reversing lethal hemorrhagic hypovolemic oxygen debt shock. J Trauma. 1997;42:199–212. doi: 10.1097/00005373-199702000-00005. [DOI] [PubMed] [Google Scholar]
- Rixen D, Raum M, Holzgraefe B, Sauerland S, Nagelschmidt M, Neugebauer EA, Shock and Trauma Study Group A pig hemorrhagic shock model: oxygen debt and metabolic acidemia as indicators of severity. Shock. 2001;16:239–244. doi: 10.1097/00024382-200116030-00012. [DOI] [PubMed] [Google Scholar]
- Cowley RA, Mergner WJ, Fischer RS, Jones RT, Trump BF. The subcellular pathology of shock in trauma patients: studies using the immediate autopsy. Am Surg. 1979;45:255–269. [PubMed] [Google Scholar]
- Guan J, Jin DD, Jin LJ, Lu Q. Apoptosis in organs of rats in early stage after polytrauma combined with shock. J Trauma. 2002;52:104–111. doi: 10.1097/00005373-200201000-00018. [DOI] [PubMed] [Google Scholar]
- Rutherford EJ, Morris JA, Reed GW, Hall KS. Base deficit stratifies mortality and determines therapy. J Trauma. 1992;33:417–423. doi: 10.1097/00005373-199209000-00014. [DOI] [PubMed] [Google Scholar]
- Botha AJ, Moore FA, Moore EE, Peterson VM, Goode AW. Base deficit after major trauma directly relates to neutrophil CD11b expression: a proposed mechanism of shock-induced organ injury. Intensive Care Med. 1997;23:504–509. doi: 10.1007/s001340050365. [DOI] [PubMed] [Google Scholar]
- Rixen D, Raum M, Bouillon B, Lefering R, Neugebauer E, Arbeitsgemeinschaft 'Polytrauma' of the Deutschen Gesellschaft für Unfallchirurgie Base deficit development and its prognostic significance in posttrauma critical illness: an analysis by the trauma registry of the Deutsche Gesellschaft für Unfallchirurgie. Shock. 2001;15:83–89. doi: 10.1097/00024382-200115020-00001. [DOI] [PubMed] [Google Scholar]
- Davis JW, Parks SN, Kaups KL, Gladen HE, O'Donnell-Nicol S. Admission base deficit predicts transfusion requirements and risk of complications. J Trauma. 1996;41:769–774. doi: 10.1097/00005373-199611000-00001. [DOI] [PubMed] [Google Scholar]
- Raum M, Rixen D, Gregor S, Linker R, Holzgräfe B, Neugebauer E. Crystalloids vs. hypertonic saline vs. carbonate/gelatine: volume therapy after hemorrhagic shock – a controlled, randomised experimental study on pigs. Shock. 2002. p. 21.
- Schultz SC, Powell CC, Bernard E, Malcolm DS. Diaspirin crosslinked hemoglobin (DCLHb) attenuates bacterial translocation. Artif Cells Blood Substit Immobil Biotechnol. 1995;23:647–664. doi: 10.3109/10731199509117978. [DOI] [PubMed] [Google Scholar]
- Powell CC, Schultz SC, Malcolm DS. Diaspirin crosslinked hemoglobin (DCLHb): more effective than lactated Ringers solution in restoring central venous oxygen saturation after hemorrhagic shock in rats. Artif Cells Blood Substit Immobil Biotechnol. 1996;24:197–200. doi: 10.3109/10731199609117435. [DOI] [PubMed] [Google Scholar]
- DeAngeles DA, Scott AM, McGrath AM, Korent VA, Rodenkirch LA, Conhaim RL, Harms BA. Resuscitation from hemorrhagic shock with diaspirin cross-linked hemoglobin, blood, or starch. J Trauma. 1997;42:406–412. doi: 10.1097/00005373-199703000-00007. [DOI] [PubMed] [Google Scholar]
- Huckabee WE. Abnormal resting blood lactate. I. The significance of hyperlactatemia in hospitalized patients. Am J Med. 1961;30:833–839. doi: 10.1016/0002-9343(61)90171-1. [DOI] [PubMed] [Google Scholar]
- Weil MH, Afifi AA. Experimental and clinical studies on lactate and pyruvate as indicators of the severity of acute circulatory failure (shock) Circulation. 1970;16:989–1001. doi: 10.1161/01.cir.41.6.989. [DOI] [PubMed] [Google Scholar]
- Harken AH. Lactic acidosis. Surg Gynecol Obstet. 1976;142:593–606. [PubMed] [Google Scholar]
- Bakker J, Coffernils M, Leon M, Gris P, Vincent JL. Blood lactate levels are superior to oxygen-derived variables in predicting outcome in septic shock. Chest. 1991;99:956–962. doi: 10.1378/chest.99.4.956. [DOI] [PubMed] [Google Scholar]
- Vary TC, Siegel JH, Rivkind A. Clinical and therapeutic significance of metabolic patterns of lactic acidosis. Perspect Crit Care. 1988;1:85–132. [Google Scholar]
- Raum M, Rixen D, Linker R, Gregor S, Holzgraefe B, Neugebauer E. Influence of lactate infusion solutions on the plasma lactate concentration. Anasthesiol Intensivmed Notfallmed Schmerzther. 2002;37:356–358. doi: 10.1055/s-2002-32241. [DOI] [PubMed] [Google Scholar]
- Abramson D, Scalea TM, Hitchcock R, Trooskin SZ, Henry SM, Greenspan J. Lactate clearance and survival following injury. J Trauma. 1993;35:584–589. doi: 10.1097/00005373-199310000-00014. [DOI] [PubMed] [Google Scholar]
- Lavery RF, Livingston DH, Tortella BJ, Sambol JT, Slomovitz BM, Siegel JH. The utility of venous lactate to triage injured patients in the trauma center. J Am Coll Surg. 2000;190:656–994. doi: 10.1016/S1072-7515(00)00271-4. [DOI] [PubMed] [Google Scholar]
- Abou-Khalil B, Scalea TM, Trooskin SZ, Henry SM, Hitchcock R. Hemodynamic responses to shock in young trauma patients: need for invasive monitoring. Crit Care Med. 1994;22:633–639. doi: 10.1097/00003246-199404000-00020. [DOI] [PubMed] [Google Scholar]
- Mikulaschek A, Henry SM, Donovan R, Scalea TM. Serum lactate is not predicted by anion gap or base excess after trauma resuscitation. J Trauma. 1996;40:218–222. doi: 10.1097/00005373-199602000-00008. [DOI] [PubMed] [Google Scholar]
- Kollmorgen DR, Murray KA, Sullivan JJ, Mone MC, Barton RG. Predictors of mortality in pulmonary contusion. Am J Surg. 1994;168:659–664. doi: 10.1016/s0002-9610(05)80140-0. [DOI] [PubMed] [Google Scholar]
- James JH, Luchette FA, McCarter FD, Fischer JE. Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis. Lancet. 1999;354:505–508. doi: 10.1016/S0140-6736(98)91132-1. [DOI] [PubMed] [Google Scholar]
- Mackersie RC, Tiwary AD, Shackford SR, Hoyt DB. Intraabdominal injury following blunt trauma. Identifying the high-risk patient using objective risk factors. Arch Surg. 1989;124:809–813. doi: 10.1001/archsurg.1989.01410070063013. [DOI] [PubMed] [Google Scholar]
- Davis JW, Mackersie RC, Holbrook TL, Hoyt DB. Base deficit as an indicator of significant abdominal injury. Ann Emerg Med. 1991;20:842–844. doi: 10.1016/s0196-0644(05)81423-4. [DOI] [PubMed] [Google Scholar]
- Kincaid EH, Chang MC, Letton RW, Chen JG, Meredith JW. Admission base deficit in pediatric trauma: a study using the National Trauma Data Bank. J Trauma. 2001;51:332–335. doi: 10.1097/00005373-200108000-00018. [DOI] [PubMed] [Google Scholar]
- Randolph LC, Takacs M, Davis KA. Resuscitation in the pediatric trauma population: admission base deficit remains an important prognostic indicator. J Trauma. 2002;53:838–842. doi: 10.1097/00005373-200211000-00006. [DOI] [PubMed] [Google Scholar]
- Peterson DL, Schinco MA, Kerwin AJ, Griffen MM, Pieper P, Tepas JJ. Evaluation of initial base deficit as a prognosticator of outcome in the pediatric trauma population. Am Surg. 2004;70:326–328. [PubMed] [Google Scholar]
- Davis JW, Kaups KL. Base deficit in the elderly: a marker of severe injury and death. J Trauma. 1998;45:873–877. doi: 10.1097/00005373-199811000-00005. [DOI] [PubMed] [Google Scholar]
- Rixen D, Raum M, Bouillon B, Schlosser LE, Neugebauer E, Arbeitsgemeinschaft Polytrauma der Deutschen Gesellschaft für Unfallchirurgie Predicting the outcome in severe injuries: an analysis of 2069 patients from the trauma register of the German Society of Traumatology (DGU) [in German] Unfallchirurg. 2001;104:230–239. doi: 10.1007/s001130050719. [DOI] [PubMed] [Google Scholar]
- Boyd CR, Tolson MA, Copes WS. Evaluating trauma care: The TRISS method. J Trauma. 1987;27:370–378. [PubMed] [Google Scholar]
- Lemeshow S, Hosmer DW. A review of goodness of fit statistics for use in the development of logistic regression models. Am J Epid. 1982;115:92–106. doi: 10.1093/oxfordjournals.aje.a113284. [DOI] [PubMed] [Google Scholar]
- Siegel JH, Rixen D. Clinical and physiologic scoring systems for sepsis and organ dysfunction. In: Deitch EA, Vincent J-L, Windsor A, editor. Sepsis and Multiple Organ Dysfunction: A Multidiscipliary Approach. New York: WB Saunders; 2002. pp. 165–178. [Google Scholar]
- Rixen D, Siegel JH. Metabolic correlates of oxygen debt predict posttrauma early acute respiratory distress syndrome and the related cytokine response. J Trauma. 2000;49:392–403. doi: 10.1097/00005373-200009000-00003. [DOI] [PubMed] [Google Scholar]
- Rixen D, Raum M, Holzgraefe B, Schäfer U, Hess S, Tenhunen J, Tuomisto L, Neugebauer EA, Shock and Trauma Study Group Local lactate and histamine changes in small bowel circulation measured by microdialysis in pig hemorrhagic shock. Shock. 2002;18:355–359. doi: 10.1097/00024382-200210000-00011. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Montgomery A, Jonsson K, Haglund U. Tissue oxygenation in hemorrhagic shock measured as transcutaneous oxygen tension, subcutaneous oxygen tension, and gastrointestinal intramucosal pH in pigs. Crit Care Med. 1991;19:205–210. doi: 10.1097/00003246-199102000-00016. [DOI] [PubMed] [Google Scholar]
- Powell CC, Schultz SC, Burris DG, Drucker WR, Malcolm DS. Subcutaneous oxygen tension: a useful adjunct in assessment of perfusion status. Crit Care Med. 1995;23:867–873. doi: 10.1097/00003246-199505000-00015. [DOI] [PubMed] [Google Scholar]
- Beerthuizen GIJM, Goris RJA, Kreuzer FJA. Early detection of shock in critically ill patients by skeletal muscle PO2 assessment. Arch Surg. 1989;124:853–855. doi: 10.1001/archsurg.1989.01410070113022. [DOI] [PubMed] [Google Scholar]
- Hopf HW, Glass-Heidenreich L, Silva J, Pearce F, Ochsner MG, Rozycki G, Frankel H, Upton R, Champion H, Drucker W, et al. Subcutaneous tissue oxygen tension in 'well resuscitated' trauma patients. Crit Care Med. 1994;22:A60. [Google Scholar]
- McKinley BA, Parmley CL, Butler BD. Skeletal muscle PO2, PCO2, and pH in hemorrhage, shock, and resuscitation in dogs. J Trauma. 1997;44:119–127. doi: 10.1097/00005373-199801000-00015. [DOI] [PubMed] [Google Scholar]
- Knudson MM, Lee S, Erickson V, Morabito D, Derugin N, Manley GT. Tissue oxygen monitoring during hemorrhagic shock and resuscitation: a comparison of lactated Ringer's solution, hypertonic saline dextran, and HBOC-201. J Trauma. 2003;54:242–252. doi: 10.1097/01.TA.0000037776.28201.75. [DOI] [PubMed] [Google Scholar]
- Cairns CB, Moore FA, Haenel JB, Gallea BL, Ortner JP, Rose SJ, Moore EE. Evidence for early supply independent mitochondrial dysfunction in patients developing multiple organ failure after trauma. J Trauma. 1997;42:532–536. doi: 10.1097/00005373-199703000-00023. [DOI] [PubMed] [Google Scholar]
- Slomovitz BM, Lavery RF, Tortella BJ, Siegel JH, Bachl BL, Ciccone A. Validation of a hand-held device in determination of blood lactate in critically injured patients. Crit Care Med. 1998;26:1523–1528. doi: 10.1097/00003246-199809000-00019. [DOI] [PubMed] [Google Scholar]
- Gutierrez G, Brown SD. Response of the macrocirculation. In: Schlag G, Redl H, editor. Pathophysiology of Shock, Sepsis and Organ Failure. New York: Springer-Verlag; 1993. pp. 215–229. [Google Scholar]
- Gomersall CD, Joynt GM, Freebaim RC. Resuscitation of critically ill patients based on gastric tonometry: a prospective, randomized controlled trial. Crit Care Med. 2000;28:607–613. doi: 10.1097/00003246-200003000-00001. [DOI] [PubMed] [Google Scholar]
- Zander R. Relevance of base excess and lactate concentration on diagnosis and treatment [in German] Anasthesiol Intensivmed Notfallmed Schmerzther. 2002;37:343–346. doi: 10.1055/s-2002-32237. [DOI] [PubMed] [Google Scholar]
- Goodyer AVN. Left ventricular function and tissue hypoxia in irreversible hemorrhagic and endotoxic shock. Am J Physiol. 1967;212:444–450. doi: 10.1152/ajplegacy.1967.212.2.444. [DOI] [PubMed] [Google Scholar]
- Reinhart K, Rudolph T, Bredle DL, Cain SM. O2 uptake in bled dogs after resuscitation with hypertonic saline or hydroxyethylstarch. Am J Physiol. 1989;257:H238–H243. doi: 10.1152/ajpheart.1989.257.1.H238. [DOI] [PubMed] [Google Scholar]
- Sheffer N, Hirshberg A, Barnea O. Myocardial balance O2 during fluid resuscitation in uncontrolled hemorrhage: computer model. J Trauma. 1997;42:647–651. doi: 10.1097/00005373-199704000-00011. [DOI] [PubMed] [Google Scholar]
- Oestern HJ, Trentz O, Kolbow H, Hempelmann G, Trentz OA, Donay F. Predictive value of metabolic profiles in multiple trauma. Chir Forum Exp Klein Forsch. 1978;1:69–72. [PubMed] [Google Scholar]
- Oestern HJ, Trentz O, Hempelmann G, Trentz OA, Sturm J. Cardiorespiratory and metabolic patterns in multiple trauma patients. Resuscitation. 1979;7:169–184. doi: 10.1016/0300-9572(79)90024-8. [DOI] [PubMed] [Google Scholar]
- Brandl M, Pscheidl E, Amann W, Barjasic A, Pasch T. Biochemical and hormonal parameters with multiple trauma. Prog Clin Biol Res. 1989;308:743–749. [PubMed] [Google Scholar]
- Woltmann A, Kress HG. The prognostic value of the delayed cutaneous immune reaction following multiple trauma in comparison with other clinical parameters. Anaesthesist. 1991;40:276–281. [PubMed] [Google Scholar]
- Nast-Kolb D, Waydhas C, Jochum M, Duswald KH, Machleidt W, Spannagl M, Schramm W, Fritz H, Schweiberer L. Biochemical factors as objective parameters for assessing the prognosis in polytrauma [in German] Unfallchirurg. 1992;95:59–66. [PubMed] [Google Scholar]
- Waydhas C, Nast-Kolb D, Jochum M, Trupka A, Lenk S, Fritz H, Duswald KH, Schweiberer L. Inflammatory mediators, infection, sepsis, and multiple organ failure after severe trauma. Arch Surg. 1992;127:460–467. doi: 10.1001/archsurg.1992.01420040106019. [DOI] [PubMed] [Google Scholar]
- Roumen RM, Redl H, Schlag G, Sandtner W, Koller W, Goris RJ. Scoring systems and blood lactate concentrations in relation to the development of adult respiratory distress syndrome and multiple organ failure in severely traumatized patients. J Trauma. 1993;35:349–355. doi: 10.1097/00005373-199309000-00004. [DOI] [PubMed] [Google Scholar]
- Sauaia A, Moore FA, Moore EE, Haenel JB, Read RA, Lezotte DC. Early predictors of postinjury multiple organ failure. Arch Surg. 1994;129:39–45. doi: 10.1001/archsurg.1994.01420250051006. [DOI] [PubMed] [Google Scholar]
- Dunham CM, Frankenfield D, Belzberg H, Wiles CE, Cushing B, Grant Z. Inflammatory markers: superior predictors of adverse outcome in blunt trauma patients? Crit Care Med. 1994;22:667–672. [PubMed] [Google Scholar]
- Scalea TM, Maltz S, Yelon J, Trooskin SZ, Duncan AO, Sclafani SJ. Resuscitation of multiple trauma and head injury: role of crystalloid fluids and inotropes. Crit Care Med. 1994;20:1610–1615. [PubMed] [Google Scholar]
- Manikis P, Jankowski S, Zhang H, Kahn RJ, Vincent JL. Correlation of serial blood lactate levels to organ failure and mortality after trauma. Am J Emerg Med. 1995;13:619–622. doi: 10.1016/0735-6757(95)90043-8. [DOI] [PubMed] [Google Scholar]
- Ivatury RR, Simon RJ, Havriliak D, Garcia C, Greenbarg J, Stahl WM. Gastric mucosal pH and oxygen delivery and oxygen consumption indices in the assessment of adequacy of resuscitation after trauma: a prospective, randomized study. J Trauma. 1995;39:128–134. doi: 10.1097/00005373-199507000-00017. [DOI] [PubMed] [Google Scholar]
- Regel G, Grotz M, Weltner T, Sturm JA, Tscherne H. Pattern of organ failure following severe trauma. World J Surg. 1996;20:422–429. doi: 10.1007/s002689900067. [DOI] [PubMed] [Google Scholar]
- Charpentier C, Audibert G, Dousset B, Weber M, Garric J, Wel-fringer P, Laxenaire MC. Is endotoxin and cytokine release related to a decrease in gastric intramucosal pH after hemorrhagic shock. Intensive Care Med. 1997;23:1040–1048. doi: 10.1007/s001340050454. [DOI] [PubMed] [Google Scholar]
- Nast-Kolb D, Waydhas C, Gippner-Steppert C, Schneider I, Trupka A, Ruchholtz S, Zettl R, Schweiberer L, Jochum M. Indicators of the posttraumatic inflammatory response correlate with organ failure in patients with multiple injuries. J Trauma. 1997;42:446–454. doi: 10.1097/00005373-199703000-00012. [DOI] [PubMed] [Google Scholar]
- Sauaia A, Moore FA, Moore EE, Norris JM, Lezotte DC, Hamman RF. Multiple organ failure can be predicted as early as 12 hours after injury. J Trauma. 1998;45:291–301. doi: 10.1097/00005373-199808000-00014. [DOI] [PubMed] [Google Scholar]
- Blow O, Magliore L, Claridge JA, Butler K, Young JS. The golden hour and the silver day: detection and correction of occult hypoperfusion within 24 hours improves outcome from major trauma. J Trauma. 1999;47:964–969. doi: 10.1097/00005373-199911000-00028. [DOI] [PubMed] [Google Scholar]
- Claridge JA, Crabtree TD, Pelletier SJ, Butler K, Sawyer RG, Young JS. Persistent occult hypoperfusion is associated with a significant increase in infection rate and mortality in major trauma patients. J Trauma. 2000;48:8–14. doi: 10.1097/00005373-200001000-00003. [DOI] [PubMed] [Google Scholar]
- Crowl AC, Young JS, Kahler DM, Claridge JA, Chrzanowski DS, Pomphrey M. Occult hypoperfusion is associated with increased morbidity in patients undergoing early femur fracture fixation. J Trauma. 2000;48:260–267. doi: 10.1097/00005373-200002000-00011. [DOI] [PubMed] [Google Scholar]
- Ertel W, Keel M, Eid K, Platz A, Trentz O. Control of severe hemorrhage using C-clamp and pelvic packing in multiply injured patients with pelvic ring disruption. J Orthop Trauma. 2001;15:468–474. doi: 10.1097/00005131-200109000-00002. [DOI] [PubMed] [Google Scholar]
- Cerovic O, Golubovic V, Spec-Marn A, Kremzar B, Vidmar G. Relationship between injury severity and lactate levels in severely injured patients. Intensive Care Med. 2003;29:1300–1305. doi: 10.1007/s00134-003-1753-8. [DOI] [PubMed] [Google Scholar]
- Egger G, Aigner R, Glasner A, Hofer HP, Mitterhammer H, Zelzer S. Blood polymorphonuclear leukocyte migration as a predictive marker for infections in severe trauma: comparison with various inflammation parameters. Intensive Care Med. 2004;30:331–334. doi: 10.1007/s00134-003-2111-6. [DOI] [PubMed] [Google Scholar]
- Davis JW, Kaups KL, Parks SN. Base deficit is superior to pH in evaluating clearance of acidosis after traumatic shock. J Trauma. 1998;44:114–118. doi: 10.1097/00005373-199801000-00014. [DOI] [PubMed] [Google Scholar]
- Krishna G, Sleigh JW, Rahman H. Physiological predictors of death in exsanguinating trauma patients undergoing conventional trauma surgery. Aust N Z J Surg. 1998;68:826–829. doi: 10.1046/j.1440-1622.1998.01468.x. [DOI] [PubMed] [Google Scholar]
- Fosse E, Pillgram-Larsen J, Svennevig JL, Nordby C, Skulberg A, Mollnes TE, Abdelnoor M. Complement activation in injured patients occurs immediately and is dependent on the severity of the trauma. Injury. 1998;29:509–514. doi: 10.1016/S0020-1383(98)00113-2. [DOI] [PubMed] [Google Scholar]
- Brown GE, Silver GM, Reiff J, Allen RC, Fink MP. Polymorphonuclear neutrophil chemiluminescence in whole blood from blunt trauma patients with multiple injuries. J Trauma. 1999;46:297–305. doi: 10.1097/00005373-199902000-00017. [DOI] [PubMed] [Google Scholar]
- Eberhard LW, Morabito DJ, Matthay MA, Mackersie RC, Campbell AR, Marks JD, Alonso JA, Pittet JF. Initial severity of metabolic acidosis predicts the development of acute lung injury in severely traumatized patients. Crit Care Med. 2000;28:125–131. doi: 10.1097/00003246-200001000-00021. [DOI] [PubMed] [Google Scholar]
- Harbrecht BG, Doyle HR, Clancy KD, Townsend RN, Billiar TR, Peitzman AB. The impact of liver dysfunction on outcome in patients with multiple injuries. Am Surg. 2001;67:122–126. [PubMed] [Google Scholar]