

Abstract
Crosslinking of the T cell receptor has been proposed to be a prerequisite for T cell activation. Although the evidence supports this notion for CD4 T cells, the situation for CD8 T cells is less clear. Soluble class I monomers have been used to determine activation requirements in vitro with contradictory results. The possibility of transfer of peptide from soluble class I molecules onto class I molecules present on the surface of CD8 T cells, with ensuing presentation to other CD8 T cells, has been widely ignored. We show that monomeric and tetrameric class I molecules as well as free peptide can stimulate naïve CD8 T cells in vitro. We generate and characterize CD8 T cells that express the OT-I T cell receptor (for Kb/SIINFEKL) yet lack Kb and Db molecules, and show that their activation requirements differ from their class I positive counterparts when stimulated with soluble Kb molecules. By eliminating the confounding effect of peptide transfer, we unmask the true activation requirements for naïve CD8 T cells and show that multivalent engagement of T cell receptors, as well as costimulation, is required for optimal stimulation.
T cells are activated through their T cell receptor (TCR) when it combines with an MHC–peptide complex on an antigen-presenting cell (APC) (1). Interaction of MHC products with the coreceptors CD4 and CD8 provides essential additional signals. Crosslinking of TCRs using αCD3 monoclonal antibodies results in activation of T cells, an observation that led to the hypothesis that T cell activation requires dimerization of the TCR. Numerous studies have addressed the minimal requirements for T cell activation and the extent of crosslinking involved. Using MHC molecules that had been immobilized or multimerized in solution, kinetic and light-scattering experiments have yielded divergent outcomes. Kinetic data support a TCR dimerization model for both class I and class II restricted TCRs (2, 3), but data to the contrary have likewise been published (4). The results from cellular assays that explore TCR–MHC interactions support TCR oligomerization for CD4 T cells (5, 6), but experiments with CD8 T cells yield a less consistent picture. Monomeric class I molecules could induce calcium flux of primed CD8 T cells (7), suggesting that coligation of a single TCR and a CD8 molecule suffices for activation. However, data from Daniels and Jameson (8) obtained for primary OT-I CD8 T cells specific for Kb/SIINFEKL contradict this result and imply that activation by a monomeric MHC–peptide complex is not sufficient for CD8 T cell activation when using calcium flux as a readout. Oligomerization was also required for activation of two T cell clones expressing different TCRs (9, 10), although monomers were sufficient to trigger association of TCR with CD8/lck (10). Monomeric Kb molecules loaded with agonist peptide for the OT-I or the 2C TCR induce association of TCR, CD3ɛ, and CD8 (11), as assessed by fluorescence resonance energy transfer. In summary, published data suggest that TCR crosslinking is required for CD4 T cells. Data on the same issue are equivocal for CD8 T cells.
MHC-I tetramers have been used widely to stain antigen-specific T cells (12). Tetramers can also activate T cells in vitro (8), allowing differentiation of direct effects of the TCR–MHC interaction from those mediated by additional molecules present on APCs.
Peptide is bound to Kb noncovalently, albeit with a binding constant that suggests stable binding [the affinity of SIINFEKL to Kb is about 10−8 M (13)]. CD8 T cells can present peptide to each other and become target cells for adjoining CD8 T cells, referred to as “fratricide” (14, 15). There is no published evidence that documents this possibility for naïve CD8 T cells. On the contrary, reports on the stimulatory capacity of tetramers or transfected Kb molecules have found no evidence for direct activation by peptide (7, 8, 16, 17) or have largely ignored this issue (9, 11, 18). Also, the question of how much peptide is released from soluble class I molecules and whether that amount is sufficient to induce activation has not been answered. Here we use monomers and tetramers loaded with agonist peptides, null peptides, or a mixture of both to investigate the quantity and valency of peptide-loaded Kb molecules required for activation of CD8 T cells. We find that soluble Kb molecules in tetrameric and monomeric form, as well as free peptide, activate naïve CD8 T cells without need for costimulation. We then generate Kb-negative CD8 T cells to investigate the effects of soluble Kb molecules in the absence of confounding effects of free peptide by eliminating the possibility of peptide binding to class I molecules on CD8 T cells. We find that class I negative naïve CD8 T cells require multivalent engagement of the TCR as well as costimulation for optimal activation.
Materials and Methods
Animals and Cells.
Naïve CD8 T cells were isolated from spleens and lymph nodes of OT-I transgenic mice (19). OT-I is a transgenic TCR (Vα2/Vβ5) specific for an ovalbumin-derived peptide (SIINFEKL) restricted by H-2Kb. To obtain naïve OT-I CD8 T cells, cell suspensions from lymph nodes and spleens were depleted for B, NK, myeloid, erythroid, and CD4 T cells by magnetic separation using the CD8 isolation kit from Miltenyi Biotec (Auburn, CA). The purity of isolated CD8 T cells was determined by FACS after staining with αCD8-FITC antibody (PharMingen).
Class I negative CD8 T cells were generated by adoptive transfer of 1.3 × 107 bone marrow cells from OT-I KbDb−/− animals (20) into irradiated RAG−/− hosts (The Jackson Laboratory). Eight weeks after the transfer, lymph nodes and spleens were harvested, and CD8 T cells were enriched by depletion of Kb-positive cells derived from the RAG−/− host by using biotinylated Kb antibody (PharMingen) followed by streptavidin-coated magnetic beads (Miltenyi Biotec) and purification on Macs columns (Miltenyi Biotec). In a second step, CD8 T cells were enriched as described above. Purified cells were subjected to complement-mediated lysis after incubation with Kb antibody (PharMingen) to eliminate residual recipient-derived Kb-positive cells.
Cells were incubated at 105 cells/well in 96-well plates at 37°C for 20 h in the presence or absence of 10 μg/ml of αCD28 antibody (PharMingen) and mIL-2 (Roche Diagnostics) in the concentrations indicated. For control experiments, cells were activated by 5 μg/ml platebound αCD3 antibody (PharMingen) or 10 ng/ml phorbol myristate acetate and 700 ng/ml of ionomycin (Sigma).
DAP-3/Kb is a fibroblast cell line transfected with Kb. Cells were irradiated before incubation with peptide for 3 h. Cells were then washed three times before addition of CD8 T cells.
All experiments were performed in duplicates and are depicted as mean ± standard deviation. All figures represent data from at least two independent experiments.
Production of Kb/β2-Microglobulin/Peptide Multimers.
Kb monomers were prepared as described (21), by using the peptides SIINFEKL (agonist) and SSYSYSSL (polyS, null peptide) (19). Peptides were synthesized on an Advanced ChemTech peptide synthesizer and analyzed for purity by mass spectrometry. Peptides used directly in activation assays were further purified by rpHPLC. Refolded Kb/β2m–peptide complexes were purified by FPLC on a SuperDex 200 column (Amersham Pharmacia). To obtain Kb tetramers, monomers were biotinylated by using recombinant BirA enzyme and purified by FPLC. Kb molecules were mixed in a molar ratio of 4:1 with streptavidin or phycoerythrin (PE)-conjugated streptavidin (Molecular Probes). To obtain mixed tetramers, biotinylated monomers loaded with agonist peptide or null peptide were mixed in ratios of 3:1, 2:2, or 1:3 before addition of streptavidin. The tetramerized molecules were resolved as a single peak on a Superdex 200A column. Purity and absence of lower-order oligomers were confirmed by native gel electrophoresis. To prevent adsorption of monomers to the tissue culture plates that might allow polyvalent interaction with responsive T cells, all dilutions of monomers and tetramers were done in media (RPMI 1640) containing 10% FCS before addition to responsive T cells. To assess the degree of peptide release from purified monomers, freshly prepared monomers were diluted in full media to the maximal concentration used in activation assays and concentrated on a centricon concentrator (molecular mass cutoff 10,000 Da; Millipore). The flow-through fraction was used directly and in serial dilutions in activation assays.
Competition Binding Experiments.
Increasing concentrations of unlabeled monomers or tetramers loaded with either agonist or null peptides were mixed with a fixed suboptimal concentration (10−9 M) of agonist-loaded PE-labeled tetramers. CD8 T cells were plated in 96-well plates at 1 × 105 cells per well on ice before incubation with the mixture described above. After incubation on ice for 1 h, cells were washed twice and analyzed by FACS.
FACS Analysis.
Cells were collected and washed with FACS buffer (PBS/0.5% BSA/0.02% sodium azide). Cells were stained with αCD8-FITC (53-5.8), αCD69-PE (H1.2F3), and αCD44-Cy-Chrome (IM7) antibody (PharMingen) or with PE-labeled tetramers loaded with SIINFEKL or SSYSYSSL. Cells were washed twice in FACS buffer before FACS analysis.
Immunoprecipitation and Immunoblot.
CD8 T cells (8 × 106) were stimulated with 10−7 M of Kb molecules in tetrameric or monomeric form or with 10−7 M of SIINFEKL peptide, for the times indicated, at 37°C. Cells were harvested by a brief centrifugation at 800 × g and the pellet was lysed in buffer (1% BRIJ 96) containing protease inhibitors (1 mM 7-amino-1-chloro-3-tosylamido-2-heptanone/1 mM PMSF/0.02 mg/ml leupeptin/0.002 mg/ml aprotinin) and phosphatase inhibitors (1 mM orthovanadate/10 mM NaF/10 mM Na-pyrophosphate). Immunoprecipitation was carried out by using αCD3ζ antibody H146 (a kind gift from I. F. Luescher, Ludwig Institute for Cancer Research, Lausanne, Switzerland), in conjunction with fixed protein A-bearing Staphylococcus aureus. Immunoprecipitates were resolved on a 12.5% reducing SDS/PAGE before transfer to a polyvinylidene difluoride membrane (Millipore). Membranes were probed with α-phosphotyrosine (pTyr) antibody 4G10 (Upstate Biotechnology, Lake Placid, NY). Reaction products were visualized by probing with a horseradish peroxidase-conjugated anti-mouse IgG2b antiserum (Southern Biotechnology Associates) followed by enhanced chemiluminescence (NEN). After stripping, blots were reprobed with αCD3ζ antibody H146 followed by horseradish peroxidase-conjugated anti-Armenian hamster IgG (Jackson) to confirm equal loading of lanes.
Results
Monomers and Tetramers Activate Naïve OT-I CD8 T Cells.
Naïve CD8 T cells were isolated from OT-I TCR transgenic mice by depletion of B, NK, myeloid, erythroid, and CD4 T cells. The purity of CD8 T cells was >95% (Fig. 1A). More than 99% of CD8 T cells expressed the transgenic TCR, as shown by staining for Vα2 and Vβ5 (Fig. 1B), and displayed a naïve phenotype (data not shown).
Figure 1.

Monomers and tetramers activate naïve CD8 T cells with different potency. (A) CD8 T cells were purified from lymph nodes and spleens of OT-I mice. Preparations used are >95% CD8 positive. (B) CD8 T cells express the transgenic TCR OT-I as determined by staining with Vα2 and Vβ5. (C) Tetramers (Kb4) and monomers (Kb1) were resolved by FPLC. Monomers (Kb1[SIINFEKL]) and tetramers (Kb4[SIINFEKL]) loaded with agonist peptide stimulate naïve CD8 T cells with different potency. Tetramers loaded with an irrelevant peptide (Kb4[polyS]) fail to activate. Activation markers CD69 (D) and CD44 (E) were measured after stimulation for 20 h. (F) Purified naïve CD8 T cells were incubated with increasing amounts of PE-labeled tetramers loaded with agonist peptide (Kb4[SIINFEKL]) or null peptide (Kb4[polyS]) before FACS analysis. (G) Naïve CD8 T cells were incubated with 10−9 M PE-labeled agonist-loaded tetramers (4 × 10−9 M Kb molecules) and increasing amounts of unlabeled tetramers loaded with agonist peptide (Kb4[SIINFEKL]) or null peptide (Kb4[polyS]), or unlabeled monomers loaded with agonist peptide (Kb1[SIINFEKL]) before FACS analysis
CD8 T cells stimulated with tetrameric or monomeric Kb molecules (Fig. 1C) for 20 h without addition of costimulatory factors acquired activation markers (CD44 and CD69; Fig. 1 D and E) and initiated production of IFN-γ (data not shown). Stimulation by tetramers yielded slightly higher activation levels than stimulation by monomers. Also the kinetics of activation were more rapid for tetramers than for monomers (data not shown), presumably due to the lower avidity of the latter.
Monomers and Tetramers Bind to the OT-I TCR with Different Avidity.
We addressed binding of monomers and tetramers in a competition assay by using PE-coupled streptavidin to label tetramers. For competition experiments, we used PE-labeled tetramers at a concentration that yields maximum stimulation, whereas no unspecific staining with tetramers loaded with null peptide was seen at this concentration (Fig. 1F). Kb molecules loaded with agonist peptide but not those loaded with null peptide competed with K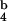 [SIINFEKL]-PE for TCR binding, either in monomeric or in tetrameric form (Fig. 1G). However, tetramers competed with K
[SIINFEKL]-PE for TCR binding, either in monomeric or in tetrameric form (Fig. 1G). However, tetramers competed with K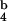 [SIINFEKL]-PE for TCR binding at a lower concentration than monomers. As reported by others (5), these data reflect the differences in avidity between monomers and tetramers for TCR binding. The binding kinetics of PE-labeled tetramers show some nonspecific binding at high concentrations and preclude quantitative analysis and calculation of IC50 values.
[SIINFEKL]-PE for TCR binding at a lower concentration than monomers. As reported by others (5), these data reflect the differences in avidity between monomers and tetramers for TCR binding. The binding kinetics of PE-labeled tetramers show some nonspecific binding at high concentrations and preclude quantitative analysis and calculation of IC50 values.
Peptide Is Released from Kb Molecules and Can Activate Naïve CD8 T Cells.
Because CD8 effector T cells can present peptide to other CD8 T cells and thereby target each other for lysis (“fratricide”) (14, 15), we investigated whether SIINFEKL peptide alone is sufficient to induce activation of a highly purified population of naïve CD8 T cells. CD8 T cells incubated with K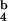 [SIINFEKL] or SIINFEKL-peptide showed very similar patterns of activation, whereas activation with K
[SIINFEKL] or SIINFEKL-peptide showed very similar patterns of activation, whereas activation with K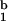 [SIINFEKL] resulted in somewhat lower activation (Fig. 2A). The experiment was repeated with a population of CD8 T cells purified by depletion of CD44 positive cells or by enrichment of cells from RAG−/− animals to even higher purity (>99% TCR+), with identical results (data not shown). This experiment effectively excludes the possibility of presentation of peptide by professional APCs that would have contaminated the culture, in which no class II positive cells were discernible by FACS. Because peptide is bound to the Kb molecule noncovalently, we investigated whether peptide is released from monomers and tetramers in quantities sufficient to evoke activation. Freshly prepared and FPLC-purified monomers loaded with SIINFEKL were diluted in complete media to the highest concentration used in activation assays and concentrated by ultrafiltration in a device with a 10,000 Da cutoff. The material collected in the flow-through contained peptide, in quantities sufficient for activation of CD8 T cells as judged from the up-regulation of CD69 (Fig. 2B) and CD44 (data not shown). The same experiment was performed with a preparation of tetramers, which likewise gave rise to activation by the material in the flow-through fraction (data not shown).
[SIINFEKL] resulted in somewhat lower activation (Fig. 2A). The experiment was repeated with a population of CD8 T cells purified by depletion of CD44 positive cells or by enrichment of cells from RAG−/− animals to even higher purity (>99% TCR+), with identical results (data not shown). This experiment effectively excludes the possibility of presentation of peptide by professional APCs that would have contaminated the culture, in which no class II positive cells were discernible by FACS. Because peptide is bound to the Kb molecule noncovalently, we investigated whether peptide is released from monomers and tetramers in quantities sufficient to evoke activation. Freshly prepared and FPLC-purified monomers loaded with SIINFEKL were diluted in complete media to the highest concentration used in activation assays and concentrated by ultrafiltration in a device with a 10,000 Da cutoff. The material collected in the flow-through contained peptide, in quantities sufficient for activation of CD8 T cells as judged from the up-regulation of CD69 (Fig. 2B) and CD44 (data not shown). The same experiment was performed with a preparation of tetramers, which likewise gave rise to activation by the material in the flow-through fraction (data not shown).
Figure 2.

Peptide released from soluble Kb molecules activates naïve CD8 T cells. (A) Naïve CD8 T cells were incubated with tetramers (Kb4[SIINFEKL]) or monomers (Kb1[SIINFEKL]), loaded with agonist peptide, agonist peptide alone (SIINFEKL), or tetramers loaded with an irrelevant peptide (Kb4[polyS]) for 20 h before staining for activation marker CD69. (B) Naïve CD8 T cells were incubated with freshly prepared monomers (Kb1[SIINFEKL]), or the flow-through fraction of an ultrafiltration of freshly prepared monomers on a concentrator device with a molecular mass cutoff of 10,000 Da (Kb1[SIINFEKL] flow-through). (C) Naïve CD8 T cells were stimulated with tetramers (Kb4) or monomers (Kb1) loaded with agonist peptide, agonist peptide alone (Sfl), or tetramers loaded with an irrelevant peptide (0), for the times indicated. Cells were lysed, immunoprecipitated with an αCD3ζ antibody, and immunoblotted with an αpTyr antibody. (Upper) A longer exposure of the same blot shown (Lower). (D) As in C, but incubated for longer periods of time.
Monomers and Tetramers Evoke Early Activation Signals.
Having established that peptide is sufficient to activate naïve CD8 T cells, we determined the kinetics of activation by peptide in comparison to activation by soluble Kb molecules. Early signaling events were monitored in naïve CD8 T cells incubated with monomers, tetramers, or peptide. Hyperphosphorylation of the CD3ζ chain was detected after 30 sec in cells activated with tetramers and to a much lesser degree in cells activated by monomers after 60 sec (Fig. 2C). Other tyrosine phosphorylated polypeptides were present, and their size identifies them as components of the CD3 complex, namely CD3γ, δ, and ɛ (22). No tyrosine phosphorylation of these proteins was detected in cells incubated with peptide up to 2 min. The apparent increase in immunoreactive material in lane 3 represents increased background activity rather than specific activity and was not observed in duplicate experiments. At 5 min, appearance of the p23 isoform of CD3ζ was noted in cells activated with peptide (Fig. 2D). Association of a 70-kDa phosphoprotein with CD3ζ, likely representing ZAP-70 (23), was seen only after incubation of the cells with tetramers but not monomers or peptide at 1 min (Fig. 2C), resembling the partial signaling patterns reported to be found after nonproductive TCR engagement (24). No tyrosine-phosphorylated polypeptides were detected after incubation with tetramers loaded with an irrelevant peptide. Blots were stripped and reprobed with αTCRζ antibody H146 to verify equal protein loading for all of the lanes (data not shown).
Kb-Negative OT-I CD8 T Cells Display a Naïve Phenotype and Are Responsive to Antigenic Stimuli.
Activation of naïve CD8 T cells by peptide must involve binding of the peptide to Kb molecules displayed on the surface of CD8 T cells. To eliminate the possibility of this mode of T cell activation, we generated class I-deficient CD8 T cells. As expected, CD8 T cells are mostly absent from the periphery of KbDb−/− animals (20), because class I molecules are required to support positive selection. Bone marrow from OT-I transgenic KbDb−/− animals was transferred into irradiated RAG−/− recipients. The recipient provides the class I positive environment that should allow CD8 T cell development in the thymus, but the developing CD8 T cells themselves should then be class I negative. Cells were harvested 8 weeks after the transfer from lymph nodes and spleen and depleted for class I positive (host-derived) cells. Enrichment for CD8 T cells was effectuated by depletion of B, NK, myeloid, CD4 T, and erythroid cells. The resulting preparation was >95% CD8 positive and no Kb-positive cells were detected by FACS (Fig. 3A). Like their class I positive counterparts, class I negative CD8 OT-I T cells displayed a naïve phenotype (Fig. 3 B and C). To assess whether cells that lack endogenous Kb are functionally compromised, Kb-negative and -positive CD8 T cells were stimulated with various agents. The response of class I negative CD8 T cells to phorbol-myristate-acetate/ionomycin, TCR crosslinking, or antigenic peptides presented by APCs is not significantly compromised (Fig. 3 D and E). Activation by tetramers yielded a significantly weaker response than that seen in Kb-positive CD8 T cells, suggesting that part of the response seen in Kb-positive CD8 T cells is due to cell-mediated activation by CD8 T cells themselves. Next, we investigated the effect of costimulation and IL-2. Both αCD28 costimulation and addition of IL-2 enhanced activation of Kb-negative CD8 T cells, yielding maximal responses comparable to those of Kb-positive CD8 T cells, as determined by staining for CD69 (Fig. 3F) and CD44 (Fig. 3G). However, very little activation was seen in response to peptide. Analysis of tyrosine phosphorylation patterns (Fig. 4) revealed that Kb-negative CD8 T cells, as did their Kb-positive counterparts, responded to stimulation with tetramers loaded with agonist peptide but not to tetramers loaded with irrelevant peptide. In Kb-negative and -positive CD8 T cells, monomers evoked the same weak activation patterns, excluding the possibility of an intrinsic inability of Kb-negative CD8 T cells to respond to monomers. Blots were stripped and reprobed with αTCRζ antibody H146 to verify equal protein loading for all of the lanes (data not shown).
Figure 3.

Kb-negative CD8 T cells are functionally normal and depend on costimulation. (A) Kb-negative CD8 T cells were generated by transfer of bone marrow from OT-I KbDb−/− animals into irradiated RAG−/− hosts. Expression of the transgenic TCR is verified by staining for Vα2. Kb-negative CD8 T cells were stained for expression of activation markers CD69 (B) and CD44 (C), revealing a naïve phenotype. Kb-positive (Kb+, black bars) and Kb-negative CD8 T cells (Kb-, white bars) were incubated with PMA/ionomycin, platebound αCD3, DAP-3/Kb cells (APC) loaded with 10−8 M SIINFEKL, and 3 × 10−8 M tetramers (Kb4) loaded with SIINFEKL for 20 h, before staining for activation markers CD69 (D) and CD44 (E). Purified Kb-negative CD8 T cells were incubated with tetramers loaded with agonist peptide (Kb4[SIINFEKL]), agonist peptide alone (SIINFEKL), or tetramers loaded with irrelevant peptide (Kb4[polyS]) for 20 h before staining for activation markers CD69 (F) and CD44 (G). Addition of αCD28 antibody (10 μg/ml) to the culture is indicated, as well as the amount of mIL-2 added.
Figure 4.

Stimulation by soluble Kb molecules induces similar early activation patterns in Kb-negative and -positive CD8 T cells. Patterns of protein tyrosine phosphorylation were examined by immunoprecipitation with an αCD3ζ antibody followed by immunoblotting with an αpTyr antibody. Kb-negative or -positive OT-I CD8 T cells were incubated with agonist-loaded tetramers (Kb4), agonist-loaded monomers (Kb1), or null peptide-loaded tetramers (0) for 2 min before lysis in 1% BRIJ96. (Upper) A longer exposure of the same blot shown (Lower).
Multivalent Engagement of TCRs Is Required for Activation of Naïve Kb-Negative CD8 T Cells.
Having established the requirements for activation of class I negative CD8 T cells by soluble Kb tetramers, we investigated the potency of monomers in this assay. Monomers failed to induce activation above the level induced by peptide alone (Fig. 5 A and B). To explore further the question of valency of the class I ligands required for optimal activation of naïve CD8 T cells, we introduced mixed tetramers, generated with a mixture of agonist- and null peptide-loaded monomers in defined amounts. The purity of mixed tetramers was analyzed by FPLC and native gel electrophoresis. Because mixed tetramers were generated by combining different peptide-loaded monomers with streptavidin, 1 in 16 tetramers produced by mixing three parts of inert monomers and one part of activating monomers will contain two activating arms. This fraction would allow crosslinking of TCRs. Although the intrinsic error in composition of mixed tetramers precludes analysis of the exact number of TCRs to be crosslinked for activation, the drop of activation seen for every active arm replaced with an inert arm is striking (Fig. 5C). The loss of activity is especially obvious when data are plotted as a function of the concentration of agonist-loaded Kb molecules rather than the total number of Kb molecules (Fig. 5D). Stimulation of Kb-positive CD8 T cells with mixed tetramers also revealed a shift of the dose–response curve toward higher concentrations of Kb molecules for every active Kb molecule on tetramers exchanged with an irrelevant Kb molecule (Fig. 5E). However, when the data are plotted as a function of the amount of agonist-loaded Kb molecules, the curves obtained are superimposable, implying that the degree of stimulation depends solely on the amount of active Kb molecules in the assay and not on the valency of ligands (Fig. 5F).
Figure 5.

Multivalent engagement of TCRs is required for activation of naïve Kb-negative CD8 T cells. Kb-negative CD8 T cells were incubated with monomers (Kb1[SIINFEKL]) and tetramers (Kb4[SIINFEKL]) loaded with agonist peptide, agonist peptide alone (SIINFEKL), or tetramers loaded with an irrelevant peptide (Kb4[polyS]) for 20 h, in the presence of 10 μg/ml of αCD28 and 5 units/ml of IL-2, before staining for activation markers CD69 (A) and CD44 (B). (C) Purified Kb-negative CD8 T cells were incubated with tetramers loaded with agonist peptide (Kb4[SIINF]) or irrelevant peptide (Kb4[polyS]), or with mixed tetramers displaying three (Kb4[3SIINF:1polyS]), two (Kb4[2SIINF:2polyS]), or one stimulatory arm (Kb4[1SIINF:3polyS]). After incubation for 20 h in the presence of 10 μg/ml of αCD28 and 5 units/ml of IL-2, cells were stained for activation marker CD69. (D) The same data as in C, but plotted as a function of the concentration of agonist-loaded Kb molecules rather than total Kb molecules. (E) Purified Kb-positive CD8 T cells were incubated with tetramers loaded with agonist peptide (Kb4[SIINF]) or irrelevant peptide (Kb4[polyS]), or with mixed tetramers displaying three (Kb4[3SIINF:1polyS]), two (Kb4[2SIINF:2polyS]), or one stimulatory arm (Kb4[1SIINF:3polyS]). After incubation for 20 h, cells were stained for activation marker CD69. (F) The same data as in E but plotted as a function of the concentration of agonist-loaded Kb molecules rather than total Kb molecules.
Discussion
Soluble class I molecules are widely used as a tool to identify or stimulate antigen-specific CD8 T cells. We show here that the stimulatory properties of soluble class I molecules are obscured by the transfer of peptide onto class I molecules present on the surface of CD8 T cells themselves, resulting in cell-mediated activation. We generated class I negative CD8 T cells to evaluate the requirements for activation by soluble H2-Kb molecules. Monomeric Kb molecules do not activate Kb-negative CD8 T cells and activate only in the presence of endogenous Kb molecules, as does free peptide. We conclude that class I positive CD8 T cells are capable of acquisition and presentation of peptide, presumably by binding of the peptide to class I molecules present on the CD8 T cell surface. It is important to underline that the residual expression of class I molecules in b2m−/− and TAP−/− mice precludes their use for the generation of class I negative CD8 T cells and necessitates the use of KbDb−/− animals.
The capability of CD8 effector T cells to activate and target each other is a matter of record (14, 15), but a similar effect has not been described in naïve cells. On the contrary, it has been claimed that peptide added to naïve (8, 16, 17) or primed (7) CD8 T cells fails to activate them. How can this difference be explained? We show that the response to peptide is slower than the activation evoked by soluble peptide-loaded Kb molecules as assessed by CD3ζ phosphorylation. The study by Daniels and Jameson (8) uses calcium flux, an early event, as readout and reports no activation by peptide, a result concordant with our findings. However, other studies have used stimulation for many hours and have found no activation by peptide. Delon et al. (7) used T cells restricted by Kd, whose affinity for peptide may be adequate to prevent release. We found at least two examples using OT-I TCR transgenic CD8 T cells (16, 17) that fail to show activation by peptide. We cannot explain this discrepancy and can only speculate whether technical issues such as preparation and storage of the peptide might have a detrimental effect on its stimulatory capacity.
Having established that naïve CD8 T cells are capable of activating each other by binding and presentation of peptide, we show that amounts of peptide sufficient to cause stimulation are released from soluble Kb molecules, even if a freshly prepared stock of recombinant Kb molecules is used. Under the conditions of cell culture (37°C for 20 h), peptide release will be more pronounced than under the experimental conditions that we examined.
Purification of the Kb-positive CD8 T cell preparation to >99% of TCR+ with no detectable class II+ cells failed to abolish activation by peptide, whereas purification of Kb-negative CD8 T cells did so almost completely. It is therefore unlikely that contaminating professional APCs are responsible for the observed stimulatory effect of peptides. We repeated these experiments in 4G3 cells, a T cell clone that expresses a TCR specific for SIINFEKL/Kb (15), observing full activation by peptide in the complete absence of APCs (data not shown).
How could such transfer of peptide occur? The source of free peptide could simply be dissociation of the Kb–peptide complex and could be confined essentially to the cell surface. Ligation by the TCR followed by internalization of engaged Kb molecules (25) could play a role, and events that take place intracellularly have not been excluded. Also, a catalytic property of the Kb molecule itself or of the TCR is possible. We refer to this process as transfer of peptide, with no defined mechanism implied at present.
How much of the activation seen after incubation with soluble Kb molecules is due to transfer of peptide? Even when incubated with a vast excess of null peptide, we failed to show complete competition for available binding sites on CD8 T cells stimulated with agonist peptide. At high concentrations of competing null peptide, activation by soluble Kb molecules was decreased, too, presumably due to competition of null peptide for binding to soluble Kb molecules (data not shown). We generated Kb-negative CD8 T cells to examine the stimulatory properties of soluble Kb molecules in the absence of confounding effects caused by transfer of peptide. KbDb−/− mice have virtually no CD8 T cells (20), even when transgenic for a Kb-restricted TCR (26), due to a failure to support positive selection. We therefore transferred bone marrow from KbDb−/− OT-I mice into irradiated Rag−/− mice, so that the class I positive epithelial environment in the thymus would allow positive selection of OT-I CD 8 T cells. The cells generated by using this approach expressed normal levels of TCR and CD8 and displayed a naïve phenotype as assessed by expression of CD69 and CD44. Class I negative CD8 T cells responded normally to various stimuli and showed no major defect in activation. When soluble Kb molecules were used to stimulate them, the response of class I negative CD8 T cells was much weaker than that of Kb-positive CD8 T cells. Because we have demonstrated the ability of peptide to stimulate Kb-positive CD8 T cells, we conclude that a significant part of the response to tetramers seen in Kb-positive CD8 T cells is due to cell-mediated activation, caused by peptide bound to Kb molecules on CD8 T cells. The ability of tetramers to stimulate naïve CD8 cells depended greatly on costimulatory signals and IL-2, in contrast to Wang et al. (18), who found no effect of costimulation on multimer-mediated activation. This discrepancy is easily explained when we consider that a major part of the effect seen in Kb-positive CD8 T cells is due to cell-mediated activation, in which case costimulation is provided by the presenting cells. The unusual aspect, of course, is that here the presenting cell is a CD8 T cell. In Kb-positive CD8 T cells, a significant increase of the activation triggered by tetramers was observed when αCD28 was added to the assay (data not shown), demonstrating that CD8 T cells, as expected, do not provide the full range of costimulation.
Signs of early activation such as calcium flux or CD3ζ phosphorylation do not necessarily imply full activation and acquisition of effector functions. Here, monomers that evoke early signaling events fail to fully activate at 20 h. The phosphorylation patterns observed after stimulation with monomers resemble those described by Reis e Sousa et al. (24) as the result of partial signaling found after nonproductive engagement of the TCR. We did not find phosphorylated ZAP-70 associated with the TCR when using monomers to stimulate CD8 T cells. Using altered peptide ligands, full activation of CD8 T cells has been observed after some delay, even in the absence of a measurable calcium flux and CD3ζ phosphorylation. This led to the suggestion that subthreshold activation events might be summated to yield a fully activating signal (27), although the possibility of peptide release is a concern that remains to be addressed.
When using class I negative CD8 T cells, we find that naïve CD 8 T cells depend on multivalent engagement of their TCR for activation. Using mixed tetramers that allow us to vary the valency of TCR engagement, we find that even dimeric engagement of TCRs leads to much weaker activation than the higher-order multimers, whereas activation of Kb-positive CD8 T cells depended solely on the amount of agonist-loaded Kb molecules in the assay. Delon et al. (7) examined primed CD8 T cells that might be more readily triggered than the naïve CD8 T cells used here. They observed activation by monomers in the presence of CD8, whereas tetramers were necessary for stimulation in its absence. Because incubation with monomers can result in peptide transfer and cell-mediated activation, and given the role of CD8 in forming the complex between presenting and responding CD8 cell, these experiments may require reinterpretation. Sykulev et al. (28) published evidence that cytolytic responses of a 2C T cell clone can be triggered by a single MHC–peptide complex presented on an APC. The average density of ligands in their system was calculated to be three MHC–peptide complexes per APC, a value that after formation of an “immunological synapse” might allow TCR crosslinking in principle. Differences in responsiveness of naïve and activated CD8 T cells may also be of importance.
In summary, we report that peptide is transferred from soluble Kb molecules onto Kb molecules on the surface of CD8 T cells, which in turn present peptide to each other. This effect obscures the stimulation by monomers and tetramers, frequently used in stimulation assays. The use of KbDb−/− CD8 T cells allows us to conclude that multivalent engagement of TCRs is a prerequisite for activation of naïve CD8 T cells. We provide a model to study the effects of activating agents without confounding effects that arise from the capacity of CD8 T cells to present antigenic peptides to each other.
Acknowledgments
We thank Brian Hekking for peptide synthesis and Kristin Hogquist (University of Minnesota) for the donation of OT-I mice. We thank the members of the Ploegh lab for discussing the data, especially Margo Furman and Dina Gould for suggesting the generation of Kb-negative CD8 T cells. We thank H. N. Eisen, J. Chen, and L. J. Stern for helpful discussions. This work was supported by National Institutes of Health grants (to H.L.P.). E.S. is recipient of a fellowship from the Deutsche Forschungsgemeinschaft. N.B. is recipient of a fellowship from the Association pour la Recherche contre le Cancer.
Abbreviations
- TCR
T cell receptor
- APC
antigen-presenting cell
- PE
phycoerythrin
References
- 1.Germain R N, Stefanova I. Annu Rev Immunol. 1999;17:467–522. doi: 10.1146/annurev.immunol.17.1.467. [DOI] [PubMed] [Google Scholar]
- 2.Alam S M, Davies G M, Lin C M, Zal T, Nasholds W, Jameson S C, Hogquist K A, Gascoigne N R J, Travers P J. Immunity. 1999;10:227–237. doi: 10.1016/s1074-7613(00)80023-0. [DOI] [PubMed] [Google Scholar]
- 3.Reich Z, Boniface J J, Lyons D S, Borochow N, Wachtel E J, Davis M M. Science. 1997;387:617–620. doi: 10.1038/42500. [DOI] [PubMed] [Google Scholar]
- 4.Baker B M, Wiley D C. Immunity. 2001;14:681–692. doi: 10.1016/s1074-7613(01)00160-1. [DOI] [PubMed] [Google Scholar]
- 5.Cochran J R, Cameron T O, Stern L J. Immunity. 2000;12:241–250. doi: 10.1016/s1074-7613(00)80177-6. [DOI] [PubMed] [Google Scholar]
- 6.Boniface J J, Rabinowitz J D, Wulfing C, Hampl J, Reich Z, Altman J D, Kantor R M, Beeson C, McConnell H M, Davis M M. Immunity. 1998;9:459–466. doi: 10.1016/s1074-7613(00)80629-9. [DOI] [PubMed] [Google Scholar]
- 7.Delon J, Gregoire C, Malissen B, Darche S, Lemaitre F, Kourilsky P, Abastado J P, Trautmann A. Immunity. 1998;9:467–473. doi: 10.1016/s1074-7613(00)80630-5. [DOI] [PubMed] [Google Scholar]
- 8.Daniels M A, Jameson S C. J Exp Med. 2000;191:335–346. doi: 10.1084/jem.191.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abastado J P, Lone Y C, Casrouge A, Boulot G, Kourilsky P. J Exp Med. 1995;182:439–447. doi: 10.1084/jem.182.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doucey M A, Legler D F, Boucheron N, Cerottini J C, Bron C, Luescher I F. Eur J Immunol. 2001;31:1561–1570. doi: 10.1002/1521-4141(200105)31:5<1561::AID-IMMU1561>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 11.Block M S, Johnson A J, Mendez-Fernandez Y, Pease L R. J Immunol. 2001;167:821–826. doi: 10.4049/jimmunol.167.2.821. [DOI] [PubMed] [Google Scholar]
- 12.Altman J D, Moss P A H, Goulder P J R, Barouch D H, McHeyzer-Williams M G, Bell J I, McMichael A J, Davis M M. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 13.Sykulev Y, Vugmeyster Y, Brunmark A, Ploegh H L, Eisen H N. Immunity. 1998;9:475–483. doi: 10.1016/s1074-7613(00)80631-7. [DOI] [PubMed] [Google Scholar]
- 14.Su M W, Walden P R, Golan D E, Eisen H N. J Immunol. 1993;151:658–667. [PubMed] [Google Scholar]
- 15.Walden P R, Eisen H N. Proc Natl Acad Sci USA. 1990;87:9015–9019. doi: 10.1073/pnas.87.22.9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potter T A, Grebe K, Freiberg B, Kupfer A. Proc Natl Acad Sci USA. 2001;98:12624–1269. doi: 10.1073/pnas.221458898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curtsinger J M, Lins D C, Mescher M F. J Immunol. 1998;160:3236–3243. [PubMed] [Google Scholar]
- 18.Wang B, Maile R, Greenwood R, Collins E J, Frelinger J A. J Immunol. 2000;164:1216–1222. doi: 10.4049/jimmunol.164.3.1216. [DOI] [PubMed] [Google Scholar]
- 19.Hogquist K A, Jameson S C, Heath W R, Howard J L, Bevan M J, Carbone F R. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 20.Vugmeyster Y, Glas R, Perarnau B, Lemonnier F A, Eisen H, Ploegh H. Proc Natl Acad Sci USA. 1998;95:12492–12497. doi: 10.1073/pnas.95.21.12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garboczi D N, Utz U, Ghosh P, Seth A, Kim J, VanTienhoven E A E, Biddison W E, Wiley D C. J Immunol. 1996;157:5403–5410. [PubMed] [Google Scholar]
- 22.Heller M, Goodlett D R, Watts J D, Aebersold R. Electrophoresis. 2000;21:2180–2195. doi: 10.1002/1522-2683(20000601)21:11<2180::AID-ELPS2180>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 23.van Oers N S, Killeen N, Weiss A. Immunity. 1994;1:675–685. doi: 10.1016/1074-7613(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 24.Reis e Sousa C, Levine E H, Germain R N. J Exp Med. 1996;184:149–157. doi: 10.1084/jem.184.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang J F, Yang Y, Sepulveda H, Shi W, Hwang I, Peterson P A, Jackson M R, Sprent J, Cai Z. Science. 1999;286:952–954. doi: 10.1126/science.286.5441.952. [DOI] [PubMed] [Google Scholar]
- 26.Maurice M M, Gould D S, Carroll J, Vugmeyster Y, Ploegh H L. Proc Natl Acad Sci USA. 2001;98:7437–7442. doi: 10.1073/pnas.141143298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosette C, Werlen G, Daniels M A, Holman P O, Alam S M, Travers P J, Gascoigne N R, Palmer E, Jameson S C. Immunity. 2001;15:59–70. doi: 10.1016/s1074-7613(01)00173-x. [DOI] [PubMed] [Google Scholar]
- 28.Sykulev Y, Joo M, Vturina I, Tsomides T J, Eisen H N. Immunity. 1996;4:565–571. doi: 10.1016/s1074-7613(00)80483-5. [DOI] [PubMed] [Google Scholar]