

Abstract
Neuronal potassium channel subunits of the KCNQ (Kv7) family underlie M-current (IM), and may also underlie the slow potassium current at the node of Ranvier, IKs. IM and IKs are outwardly rectifying currents that regulate excitability of neurons and myelinated axons, respectively. Studies of native IM and heterologously expressed Kv7 subunits suggest that, in vivo, KCNQ channels exist within heterogeneous, multicomponent protein complexes. KCNQ channel properties are regulated by protein phosphorylation, protein-protein interactions, and protein-lipid interactions within such complexes. To better understand the regulation of neuronal KCNQ channels, we searched directly for posttranslational modifications on KCNQ2/KCNQ3 channels in vivo by using mass spectrometry. Here we describe two sites of phosphorylation. One site, specific for KCNQ3, appears functionally silent in electrophysiological assays but is located in a domain previously shown to be important for subunit tetramerization. Mutagenesis and electrophysiological studies of the second site, located in the S4-S5 intracellular loop of all KCNQ subunits, reveal a mechanism of channel inhibition.
Keywords: KCNQ, M-channel, S4-S5 loop, potassium channels, neuromodulation
KCNQ2/KCNQ3 heteromeric channels are voltage-gated potassium channels that limit repetitive firing of neurons. They underlie M-current (1), a slow, noninactivating potassium current named for its modulation by muscarinic agonists (2). Some KCNQ2/KCNQ3 subunits reside in axon initial segments and nodes of Ranvier, where action potentials initiate and propagate (3-5). Thus, potent control over neuronal firing patterns exerted by KCNQ2/KCNQ3 channels reflects both their functionality and their strategic subcellular positioning.
Underscoring this physiological importance, mutations of KCNQ2 and KCNQ3 cause human disorders of neural hyper-excitability, including myokymia (6) and benign familial neonatal convulsions (BFNC) (7-9). Although BFNC is a penetrant, dominantly inherited disorder, some pathogenic mutations reduce channel activity as little as 20% (10). Transgenic mice expressing a dominant-negative KCNQ2 mutation show increased neuronal excitability, hyperactive behavior, and spontaneous epileptic seizures (11).
Regulated inhibition of KCNQ channels by metabotropic neurotransmission underlies an important form of plasticity in vertebrate sympathetic and central neurons (12, 13). PIP2 depletion is the likely mechanism mediating the classic muscarinic inhibition of M-current (14-16), but other mechanisms, including phosphorylation, may contribute to channel modulation by other neurotransmitters (16). Indeed, KCNQ1 and KCNQ2 subunits associate with a variety of kinases and phosphatases via A-kinase anchor proteins (17-20), and mutational studies implicate KCNQ2/KCNQ3 phosphorylation sites for channel regulation by protein kinase A (PKA), protein kinase C (PKC), and src tyrosine kinase (10, 19, 21, 22).
KCNQ2 and KCNQ3 subunits possess large intracellular domains containing many additional potential phosphorylation sites (Fig. 1, refs. 7-9). Here, we take an alternative approach to those previously described. First, we identify sites phosphorylated by endogenous kinases on intact channel proteins in living mammalian cells using mass spectrometry. Then, we examine the physiological consequences of modification at identified sites.
Fig. 1.

Topology of KCNQ2 and KCNQ3 channels. KCNQ2 (843 aa) and KCNQ3 (872 aa) polypeptides are predicted to have six membrane-spanning helices and intracellular N and C termini, like other voltage-gated potassium channels. Stars indicate the positions of the phosphorylation sites identified in this study.
Materials and Methods
DNA Preparation and Site-Directed Mutagenesis. Plasmid cDNAs for human KCNQ2 and KCNQ3, without and with extracellular hemagglutinin (HA) tags (HA-KCNQ2 and HA-KCNQ3) were generously provided by T. J. Jentsch (Zentrum für Molekulare Neuropathobiologie, Hamburg, Germany) (7, 10, 23). Site-directed mutations were incorporated by using PCR with Pfu Turbo DNA Polymerase (Stratagene) and verified by sequencing the channel genes.
Affinity Purification of KCNQ Subunits. Guinea pig anti-KCNQ2 antibodies (17) were crosslinked to Protein A beads (Dynal) using dimethyl pimelimidate (Sigma). Human embryonic kidney 293 (HEK) cells, maintained in DMEM with 10% FBS at 37°C in 5% CO2, were transfected with KCNQ2 and KCNQ3 cDNAs in pcDNA3 in a 1:1 ratio by using FuGENE-6 (Roche), harvested 2 days later in lysis buffer [150 mM NaCl/50 mM Tris·HCl/1% Triton X-100/50 mM NaF/10 mM sodium pyrophosphate/1× Complete protease inhibitor mixture (Roche)/2 mM sodium orthovanadate/100 nM calyculin A/1 mM DTT], kept on ice for 30 min, and centrifuged at 4°C at 20,800 × g for 30 min. The supernatant was transferred to siliconized tubes and incubated overnight at 4°C with 2 μg/ml immobilized KCNQ2 antibodies. Beads were washed four times with lysis buffer, resuspended in SDS sample buffer (Pierce), and heated for 2 min to 85-90°C. After electrophoresis on 7% SDS polyacrylamide gels, proteins were either stained with SYPRO Ruby protein gel stain (Molecular Probes), or subjected to electrotransfer and Western blotting (3, 17, 24). Bands at ≈89 (KCNQ2) and ≈97 kDa (KCNQ3) were excised, minced, washed, dried on a centrifugal concentrator, rehydrated on ice for 10 min in 10 μl of 5 ng/μl sequencing grade trypsin (Promega) in 25 mM NH4HCO3, covered with an additional 25 μl NH4HCO3 solution and incubated overnight at 37°C. Supernatants from the digest reaction and from two subsequent extractions using 50 μl of 50% acetonitrile/1% formic acid were pooled, concentrated to 10 μl, and stored at -80°C until used for mass spectrometry.
Mass Spectrometry. Preliminary analysis was performed by MALDI TOF using an Applied Biosystems Voyager mass spectrometer in reflectron mode. Digest samples (1 μl, estimated 1 pmol of peptides) were further analyzed by nanoflow RPLC (75 μm i.d. × 150 mm long PepMap column; UltiMate, Dionex) coupled to a quadrupole-orthogonal TOF tandem mass spectrometer (MS/MS) (QSTAR XL, Applied Biosystems). Peptides were separated with a 2-35% linear gradient of acetonitrile/0.1% formic acid over 50 min. Eluting peptides were ionized by using a Picotip emitter (New Objective) mounted on the nano-electrospray ion source (Applied Biosystems/MDS Sciex) at an applied voltage of 2 kV. The QSTAR MS was first operated in information-dependent mode, with each full MS scan (1-s acquisition) followed by an MS/MS scan (3-s acquisition) in which the most abundant peptide molecular ion was dynamically selected for collision-induced dissociation. The monoisotopic masses of parent and associated fragment ions, parent ion charge states (z), and ion intensities from the acquired MS/MS spectra were automatically extracted by using the Mascot script for Analyst (www.matrixsciences.com) script and directly submitted for automated database searching for protein identification and characterization using both Mascot and a development version of Protein Prospector (25).
For selected runs, we also manually extracted the monoisotopic masses of all multiply charged ions (signal-to-noise ratio, > 10; n > 1,000 ions) and submitted these masses to Protein Prospector for peptide mass mapping with ms-fit, which compared the submitted list to a calculated set of tryptic peptides (including all possible phosphopeptides) derived from KCNQ2 and KCNQ3 protein sequences. For protein identification, external calibration was used, and mass accuracy of peptide masses was <100 ppm. To achieve better mass accuracy (≈20 ppm) for peptide mass mapping to identify putative phosphopeptides, internal calibration in each LCMS run was carried out by using trypsin autolysis products (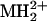 421.7590 and
421.7590 and 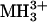 737.3728).
737.3728).
Expression in Xenopus laevis Oocytes. The Ambion mMessage mMachine SP6 kit was used to synthesize capped KCNQ cRNAs from KCNQ cDNAs in pTLN linearized with HpaI. Oocytes were injected with 5 or 10 ng of capped cRNA, and KCNQ2 and KCNQ3 cRNAs were injected in a 1:1 ratio unless otherwise specified.
Electrophysiology. Currents were measured at room temperature in ND96 (96 mM NaCl/2 mM KCl/1 mM MgCl2/1.8 mM CaCl2/5 mM Hepes, pH 7.4) from oocytes 2-3 days after cRNA injection under two-electrode voltage clamp using 0.2-1 MΩ microelectrodes filled with 3 M KCl with a Geneclamp 500B amplifier (Axon Instruments). The voltage-clamp protocol was 0.5-s depolarizing steps from -80 mV in increments of 10 mV up to 60 mV. Recordings were filtered at 1 kHz and acquired and analyzed by using pclamp software (Axon Instruments).
Surface Expression Assays. Surface expression was measured 3-5 days after injection of cRNAs by using mouse monoclonal anti-HA primary (HA.11, Covance), goat horseradish peroxidase (HRP)-conjugated anti-mouse IgG secondary (Jackson ImmunoResearch) antibodies, and the chemiluminescence method described in ref. 26. Results shown are representative of >10 oocytes for each experimental condition.
Results
Affinity Purification of Heterologously Expressed KCNQ2/KCNQ3 Heteromeric Channels. Immunoprecipitated KCNQ2 and KCNQ3 subunits were subjected to SDS gel electrophoresis and protein staining, revealing two major bands, identified by Western analyses as KCNQ2 (≈88 kDa) and KCNQ3 (≈97 kDa), a third prominent band of ≈67 kDa, and numerous minor bands. Protein stains (see Fig. 2A) showed that each two-plate sample yielded about 0.4 μg (≈50 pmol) of KCNQ2 and ≈0.2 μg of KCNQ3. We excised the prominent bands, performed in-gel trypsin digestion, and analyzed the resulting peptide extracts by MALDI TOF and by nano-LC/MS and MS/MS using a hybrid quadrupole-orthogonal TOF MS/MS.
Fig. 2.

Affinity purification of KCNQ2/KCNQ3 channels and identification and electrophysiological characterization of a site of phosphorylation in the C-terminal region of KCNQ3. (A) Protein stained gel showing heterologously expressed, affinity-purified KCNQ2/KCNQ3 channel bands excised for mass spectrometric analysis. Other protein bands identified by Maldi TOF MS are KCNQ2/KCNQ3 oligomers (≈400 kDa) (1), HSP70 (67 kDa) (2), and actin (≈47 kDa) (3). (B) MS/MS spectrum of phosphopeptide representing residues 568-581 of KCNQ3. As shown, collision-induced dissociation spectrum contains doubly charged parent ion (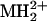 , 798.89) and many b and y series fragment ions (b and y ions are, respectively, the N- or C-terminal fragments produced when the parent peptide is fragmented at peptide bonds). Ions marked
, 798.89) and many b and y series fragment ions (b and y ions are, respectively, the N- or C-terminal fragments produced when the parent peptide is fragmented at peptide bonds). Ions marked 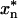 exhibit masses of xn -98 Da, where xn is a bn or yn ion, reflecting the loss of H3PO4, a characteristic signature of phosphopeptides. (C) Schematic of fragment ion coverage in MS/MS spectrum from B. The presence of y6, y4, y7*, and y8* ions localizes the site of phosphorylation to S578 or T579. (D) Wild-type and S578/T579 mutant channels behave similarly. Recordings shown are representative of >15 oocytes from three different batches.
exhibit masses of xn -98 Da, where xn is a bn or yn ion, reflecting the loss of H3PO4, a characteristic signature of phosphopeptides. (C) Schematic of fragment ion coverage in MS/MS spectrum from B. The presence of y6, y4, y7*, and y8* ions localizes the site of phosphorylation to S578 or T579. (D) Wild-type and S578/T579 mutant channels behave similarly. Recordings shown are representative of >15 oocytes from three different batches.
Mass Spectrometry Reveals a Phosphorylation Site on KCNQ3. MALDI analysis confirmed the identity of the KCNQ2 and KCNQ3 bands. The band at ≈67 kDa (Fig. 2 A) was identified as HSP70. This band was seen in all samples of KCNQ2/KCNQ3 transfected cells, although with variable abundance, and was absent from mock immunoprecipitations of untransfected cells, suggesting that at least some KCNQ2 polypeptides were associated with HSP70.
LC/MS/MS analysis of the KCNQ2 or KCNQ3 digests also located a phosphopeptide of KCNQ3, within a C-terminal region important for subunit interactions (the subunit interaction domain), subsequently confirmed by MS/MS sequencing (Fig. 2 B and C, see Fig. 1 for location of sites) (27). This candidate phosphopeptide contained three potential phosphoacceptor residues; however, analysis of the MS/MS spectrum and careful manual examination of all peptides detected during the LC/MS run revealed only a singly phosphorylated peptide that could have arisen from phosphorylation of S578 or T579.
To test the functional effect of this KCNQ3 phosphorylation, we recorded currents from wild-type or mutant channels. A priori, channels heterologously expressed may or may not be substantially phosphorylated at any particular site; the abundance of a phosphopeptide is one of many factors affecting ion intensity observed in the MS. However, comparison of wild-type channels with alanine (abolishing the possibility of phosphorylation) and aspartate or glutamate (to mimic the phosphorylated state) mutants will help identify effects attributable to modulation via phosphorylation.
As previously shown (1, 10), oocytes coinjected with wild-type KCNQ2 and KCNQ3 cRNAs gave slowly activating outward currents upon depolarization (Fig. 2D). Mutation of S578 or T579 of KCNQ3 to alanine, or both residues together to alanine, had no detectable effects on functional expression, voltage-dependence, or activation kinetics of heteromeric KCNQ2/KCNQ3 channels. Mutating either residue to aspartate also resulted in heteromeric currents that were indistinguishable from the wild-type currents (Fig. 2D). Thus, phosphorylation of this C-terminal region did not significantly impact channel assembly or function.
Evidence for Phosphorylation in the S4-S5 Loop of KCNQ2/KCNQ3 Channels. To identify additional phosphopeptides from KCNQ2 or KCNQ3 digests, we analyzed LC/MS spectra carefully and extracted all multiply charged ions for mass mapping. ms-fit results revealed evidence of a potential phosphorylation at a threonine in the S4-S5 loop of KCNQ2/KCNQ3 (Fig. 3 A and B), within a sequence highly conserved among KCNQ family members (Fig. 3C). Although the phosphopeptide was never abundant enough to be selected for LC/MS/MS, several overlapping peptides were found that included this phosphorylation site, strongly suggesting that the S4-S5 loop threonine is phosphorylated. This was the only phosphorylation site we identified in the intracellular loops between the transmembrane helices (see Fig. 1). Because the S4-S5 loop has been shown to influence channel conductance and gating (28, 29), we hypothesized that the addition of a phosphate group here, with its negative charge, could modulate the activity of the channel.
Fig. 3.

Analysis of a potential phosphorylation site in the S4-S5 loop. (A) A set of overlapping peptide mass matches implicates a conserved threonine in the S4-S5 loop as a site of phosphorylation. The observed and calculated monoisotopic (MH+) masses of the KCNQ subunit peptides, the error δ (in parts per million), the peptide sequence, and position in each subunit are indicated. (B) MS spectrum showing low intensity triply charged ion with m/z (mass normalized by charge) of 534.91 atomic mass units (amu), detected in tryptic digest of KCNQ2. This corresponds to monoisotopic mass of 1602.714 amu, a close match for the residues 208-219 of KCNQ2 if Thr-217 is phosphorylated. (C) The KCNQ S4-S5 loop sequence is highly conserved. All family members contain a threonine corresponding to the phosphorylated threonines, Thr-217 of KCNQ2 and Thr-246 of KCNQ3, and surrounding basic residues.
Mimicking the Dephosphorylated State of the S4-S5 Loop Threonine Results in Functional Channels. Therefore, we generated mutants in which the S4-S5 loop threonine was replaced by alanine to prevent the possibility of phosphorylation at this site. The heteromeric KCNQ2_T217A/KCNQ3_T246A mutant channels gave similar amounts of current as wild-type channels, although the mutant channels activated at more depolarized potentials (Fig. 4 A and B). A right shift of the voltage-dependence of conductance of ≈20 mV was confirmed by tail current measurements in high-potassium solution (Fig. 6, which is published as supporting information on the PNAS web site). Similar effects of S4-S5 loop mutations on voltage dependence have been reported for other voltage-gated potassium channels, such as for Shaker (28) and KCNQ1 (29).
Fig. 4.

Physiological characterization of S4-S5 loop mutants. (A) Both mutant and wild-type channels generate large currents upon depolarization, with similar activation kinetics. (B) Current-voltage relation for KCNQ2_T217A/KCNQ3_T246A, KCNQ2_T217D/KCNQ3_T246D, wild-type KCNQ, and endogenous channels. KCNQ2_T217D/KCNQ3_T246D channels are nonfunctional. Results shown are representative of more than five batches of oocytes, with n ≥ 5 oocytes per condition per batch. Error bars represent SEM.
Replacement of the S4-S5 Loop Phosphorylation Site in both KCNQ2 and KCNQ3 With Acidic Residues Abolishes KCNQ2/KCNQ3 Currents Without Reducing Channel Protein Expression. To explore the effects of phosphorylation, we substituted the threonines in the S4-S5 loops of the heteromeric channel, Thr-217 of KCNQ2 and Thr-246 of KCNQ3, with aspartate to mimic the negatively charged phosphate group. Coexpression of KCNQ2_T217D and KCNQ3_T246D cRNAs gave no current above the level of endogenous currents (Fig. 4B). Replacing the S4-S5 loop threonine with glutamate also eliminated functional channel expression (data not shown), suggesting that charge, and not size, of the residue at this position is responsible for the loss of channel function. Western blots showed no differences in steady-state protein levels for mutant and wild-type subunits (Fig. 7, which is published as supporting information on the PNAS web site), indicating that there was no major change in the balance of degradation and synthesis of the mutant subunits.
KCNQ2_T217D and KCNQ3_T246D Mutations Do Not Impair Tetramerization and Surface Trafficking. To determine whether mutant channels with acidic residues replacing the S4-S5 loop threonine appear on the cell membrane, we used immunochemiluminescence to quantify surface expression of channels extracellularly tagged with a HA-epitope (23, 26). HA-KCNQ2_T217D homomeric channels trafficked to the plasma membrane at least as well as HA-KCNQ2 (wild-type) channels (Fig. 5Ai). Moreover, cells expressing HA-KCNQ2_T217D/KCNQ3_T246D (which gave no current, see Fig. 4B) usually yielded higher surface expression than seen with HA-KCNQ2/KCNQ3. Although coexpression of KCNQ2 and KCNQ3 gave 10-fold larger currents than expression of KCNQ2 alone (Fig. 4B), levels of HA-KCNQ2 subunits detected at the membrane were not altered by coexpression of KCNQ3 subunits (Fig. 5Ai), contrary to previous reports (23, 27). Homomeric KCNQ2 channels represent a substantial component of the somatic M-current recorded from sympathetic neurons during early development (30). KCNQ2 subunits are also expressed without associated KCNQ3 subunits in axons (3, 17). Our study provides further evidence for functional expression of homomeric KCNQ2 channels.
Fig. 5.

The loss of channel activity exhibited by S4-S5 loop aspartate mutants is not caused by reduced subunit protein expression or impaired surface trafficking. (A) Quantification of surface channels using a whole-oocyte chemiluminescence assay (see Materials and Methods). Mean surface expression levels are shown in absolute luminescence units (LU, Ai) or relative LU (RLU), where values have been normalized to KCNQ2/HA-KCNQ3 (Aii) or KCNQ2_T217D/HA-KCNQ3_T246D (Aiii) levels. Error bars represent standard error of the mean. (Ai) Surface expression levels of wild-type and mutant HA-KCNQ2 are comparable, regardless of whether KCNQ3 or KCNQ3_T246D subunits are coexpressed. (A ii and iii) Compared to the background level of surface expression of HA-KCNQ3 or HA-KCNQ3_T246D when expressed alone, KCNQ2 coexpression with HA-KCNQ3 greatly increased the surface luminescence (fold-increase = 60.7 ± 16.4; Aii) as did coexpression of KCNQ2_T217D with HA-KCNQ3_T246D (fold-increase = 59.7 ± 11.5) and coexpression of wild-type KCNQ2 with HA-KCNQ3_T246D (fold-increase = 70.6 ± 12.8). (B) The KCNQ3_T246D mutant forms functional heteromeric channels with KCNQ2. Although expression of KCNQ3 or KCNQ3_T246D (data not shown) alone did not result in current above endogenous levels, coexpression of KCNQ2 and wild-type KCNQ3 (n = 8) or KCNQ3_T246D (n = 5) resulted in larger average currents than expression of KCNQ2 alone (n = 5).
When expressed alone, HA-KCNQ3 was hardly detectable on the surface; but, when coexpressed with KCNQ2, the surface expression of HA-KCNQ3 was greatly increased (fold increase = 60.7 ± 16.4), strongly suggesting that the HA-tagged channels on the cell surface are heteromers (Fig. 5Aii) (23). To learn whether KCNQ2_T217D and KCNQ3_T246D subunits form heterotetramers and traffic to the plasma membrane, we expressed H A-KCNQ3_T246D alone or together with KCNQ2_T217D. Coexpression of the two mutant subunits also increased surface channel expression (fold increase = 59.7 ± 11.5), indicating that aspartate substitution of the threonine in the S4-S5 loop did not interfere with channel assembly and surface expression.
We also tested whether the KCNQ2 and KCNQ3 S4-S5 loop aspartate mutant subunits coassemble with wild-type subunits. Coexpression of KCNQ3_T246D and KCNQ2 resulted in currents that were larger than KCNQ2 expression alone, but smaller than the currents in cells coexpressing KCNQ2 and KCNQ3 (Fig. 5B). However, expression of KCNQ2_T217D with KCNQ3 gave no detectable current (data not shown). Thus, the aspartate mutation in the highly conserved S4-S5 loop of KCNQ2 appears to exert much stronger inhibitory effects on channel function.
Discussion
Ion channel function can be modulated by reversible covalent modifications such as phosphorylation. Phosphorylation sites identified by metabolic labeling experiments, mutational analysis, and electrophysiology (31) concern a small subset of kinases, such as the serine/threonine kinases PKA and PKC and a few tyrosine kinases (32-34). Given that >500 distinct kinase genes are expressed in mammals (35), our knowledge about phosphorylation within ion channel complexes is very incomplete. This consideration provides incentive for the development of novel approaches for identifying phosphorylation sites. Mass spectrometry complements traditional approaches because sites of phosphorylation can be identified directly and in vivo (36). Here we used mass spectrometry to identify sites of phosphorylation on channels formed by KCNQ2 and KCNQ3 potassium channel subunits. Because these channels contribute to control of neuronal excitability and are critical for nervous system health, understanding the factors affecting KCNQ2/KCNQ3 channel activity may reveal targets for therapeutic intervention.
We used MALDI TOF and electrospray ionization/quadrupole-orthogonal TOF mass spectrometers to analyze heterologously expressed KCNQ2/KCNQ3 proteins. A substantial portion of the channel polypeptide was represented in the high signal intensity (highly abundant) ions detected by LS/MS and automatically sequenced by data-dependent MS/MS, demonstrating that the subunit peptides were the principal constituents of the bands analyzed. However, some regions of the KCNQ polypeptides were not covered by the set of high-intensity ions, and thus, full analysis of the material required consideration of lower intensity species. Although this analysis was facilitated by the large dynamic range and low noise of the instrument, inclusion of the entire dynamic range raised the potential for contamination by peptides derived from lower abundance background proteins and made comprehensive analysis more difficult. Because our approach demanded complete coverage of the polypeptides, heterologous expression was necessary to isolate sufficient molar amounts of protein for analysis. Mass spectrometry allowed localization of sites, but did not reveal the stoichiometry of phosphorylation. Such methodological issues will likely be common to other studies using mass spectrometry to identify sites of phosphorylation in low-abundance membrane proteins, and represent areas for ongoing method development.
This study revealed evidence for phosphorylation at a residue conserved among all KCNQ subunits in the S4-S5 loop, and at one of two KCNQ3 residues, S578 and T579, near the domain for tetramerization and subunit interaction in the C terminus. Our studies do not rule out the possibility that other sites are also involved in regulation of KCNQ2/KCNQ3 channels. Although our studies did not reveal any significant effect of phosphorylation at the C-terminal site on channel function, phosphorylation there may be involved in regulating other aspects of KCNQ function in vivo, such as interactions with other proteins or targeting channels to specific sites in neurons (37, 38). Although the C-terminal phosphorylation site is near a calmodulin-binding consensus sequence (39), we have been unable to detect an interaction between KCNQ3 and calmodulin (S. Shivakumar and E.C.C., unpublished observations) and calmodulin does not alter KCNQ3 channel function (40), so it is unlikely that this phosphorylation modulates calmodulin binding. However, it is possible that functional consequences of channel phosphorylation only become apparent in supramolecular complexes of the channel, as shown with PKA phosphorylation of Kv4.2 (41).
Multiple ions detected by LC/MS suggested an additional site of phosphorylation at a conserved threonine residue in the S4-S5 loop of the KCNQ2/KCNQ3 heteromeric channel (Fig. 1). Because of the important contribution of the S4-S5 loop to voltage-gated channel function (42), we hypothesized that the addition of a negatively charged phosphate moiety to the S4-S5 linker could provide a robust means of regulation.
To test this hypothesis, the phosphorylation site in the S4-S5 loop was mutated to mimic the phosphorylated and dephosphorylated states. Mutations mimicking the dephosphorylated state gave rise to channels functioning similarly to wild-type channels. Our results differ from those reported by Hoshi et al. (19), who observed that KCNQ2_T217A homomeric channels yielded half the normal current level. However, heteromeric channels with mutations that mimic the phosphorylated state in KCNQ2 did not conduct current, despite their robust surface expression. In addition, heteromeric channels with mutations that mimic the phosphorylated site in KCNQ3 yielded significantly smaller currents than wild-type channels. Thus, our mutagenesis studies predict that phosphorylation of the threonine in the S4-S5 loop of KCNQ2/KCNQ3 channels drastically decreases the channel activity. Given that a mere 25% reduction in KCNQ2/KCNQ3 channel activity is sufficient to cause seizures in newborns with the autosomal dominant disorder benign familial neonatal convulsions (10), this phosphorylation is a powerful modulation. However, the responsible kinase has remained elusive. Given the basic nature of the surrounding residues, the most obvious candidates are PKA, PKC, and CaMKII. PKA effects on channel activity have been attributed to another site (10). PKC and CaMKII activation altered channel activity, but neither required the phosphorylation of the S4-S5 loop site for their actions (T.S.S., unpublished results). Moreover, complete muscarinic suppression occurs even when the S4-S5 loop site cannot be phosphorylated (Fig. 8, which is published as supporting information on the PNAS web site). Thus, whereas the predicted effect of phosphorylation at this site is similar to the effect of muscarinic suppression on the M-current, which results in an increase in neuronal excitability (2), phosphorylation of the threonine in the S4-S5 loop of KCNQ2/KCNQ3 channels represents a different channel regulation that could increase repetitive firing. Phosphorylation of the S4-S5 loop could affect voltage-gating by preventing movement of the voltage-sensor. It could also uncouple the movement of the sensor from the gate, as suggested by mutagenesis studies and the recently solved Kv1.2 crystal structure (42-44).
Many other voltage-gated potassium channels contain putative phosphorylation sites in the S4-S5 loop that are surrounded by basic residues (28, 45). Consistent with our mass spectrometry results that the S4-S5 loops of KCNQ channels are phosphorylated, the potential phosphorylation site of the S4-S5 loop is also exposed in the Kv1.2 channel structure (42). Our studies raise the possibility that posttranslational modification of the S4-S5 loop of voltage-gated potassium channels may be a potent means of regulating the activity of voltage-gated potassium channels.
Supplementary Material
Acknowledgments
We thank the members of the University of California, San Francisco, Mass Spectrometry Facility and A. L. Burlingame for generous support and helpful discussions; T. J. Jentsch for kindly providing KCNQ2 and KCNQ3 clones; M. Harris, T. Cheng, and A. Shenoy for assistance with mutagenesis; and members of the Jan laboratory for encouragement and advice. This work was supported by National Institute of Neurological Disorders and Stroke Grant R21 NS42100 (to E.C.C.), National Center for Research Resources Grants RR14606 and RR01614 (to L.H. and A. L. Burlingame), and by National Institute of Mental Health Grant R01 MH65334 (to L.Y.J.). L.Y.J. and Y.N.J. are investigators of the Howard Hughes Medical Institute. E.C.C. is a recipient of a Miles Family Fund Independent Clinician-Scholar Award.
Author contributions: T.S.S., Y.N.J., L.Y.J., and E.C.C. designed research; T.S.S., L.H., and E.C.C. performed research; L.H. and E.C.C. contributed new reagents/analytic tools; T.S.S., L.H., and E.C.C. analyzed data; and T.S.S., L.Y.J., and E.C.C. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: HA, hemagglutinin; MS/MS, tandem MS.
References
- 1.Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., Dixon, J. E. & McKinnon, D. (1998) Science 282, 1890-1893. [DOI] [PubMed] [Google Scholar]
- 2.Brown, D. A. & Adams, P. R. (1980) Nature 283, 673-676. [DOI] [PubMed] [Google Scholar]
- 3.Devaux, J. J., Kleopa, K. A., Cooper, E. C. & Scherer, S. S. (2004) J Neurosci. 24, 1236-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roper, J. & Schwarz, J. R. (1989) J. Physiol. 416, 93-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarz, J. R. & Vogel, W. (1971) Pflugers Arch. 330, 61-73. [DOI] [PubMed] [Google Scholar]
- 6.Dedek, K., Kunath, B., Kananura, C., Reuner, U., Jentsch, T. J. & Steinlein, O. K. (2001) Proc. Natl. Acad. Sci. USA 98, 12272-12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biervert, C., Schroeder, B. C., Kubisch, C., Berkovic, S. F., Propping, P., Jentsch, T. J. & Steinlein, O. K. (1998) Science 279, 403-406. [DOI] [PubMed] [Google Scholar]
- 8.Charlier, C., Singh, N. A., Ryan, S. G., Lewis, T. B., Reus, B. E., Leach, R. J. & Leppert, M. (1998) Nat. Genet. 18, 53-55. [DOI] [PubMed] [Google Scholar]
- 9.Singh, N. A., Charlier, C., Stauffer, D., DuPont, B. R., Leach, R. J., Melis, R., Ronen, G. M., Bjerre, I., Quattlebaum, T., Murphy, J. V., et al. (1998) Nat. Genet. 18, 25-29. [DOI] [PubMed] [Google Scholar]
- 10.Schroeder, B. C., Kubisch, C., Stein, V. & Jentsch, T. J. (1998) Nature 396, 687-690. [DOI] [PubMed] [Google Scholar]
- 11.Peters, H. C., Hu, H., Pongs, O., Storm, J. F. & Isbrandt, D. (2005) Nat. Neurosci. 8, 51-60. [DOI] [PubMed] [Google Scholar]
- 12.Marrion, N. V. (1997) Annu. Rev. Physiol. 59, 483-504. [DOI] [PubMed] [Google Scholar]
- 13.Robbins, J. (2001) Pharmacol. Ther. 90, 1-19. [DOI] [PubMed] [Google Scholar]
- 14.Zhang, H., Craciun, L. C., Mirshahi, T., Rohacs, T., Lopes, C. M., Jin, T. & Logothetis, D. E. (2003) Neuron 37, 963-975. [DOI] [PubMed] [Google Scholar]
- 15.Suh, B. C., Horowitz, L. F., Hirdes, W., Mackie, K. & Hille, B. (2004) J. Gen. Physiol. 123, 663-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winks, J. S., Hughes, S., Filippov, A. K., Tatulian, L., Abogadie, F. C., Brown, D. A. & Marsh, S. J. (2005) J. Neurosci. 25, 3400-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper, E. C., Aldape, K. D., Abosch, A., Barbaro, N. M., Berger, M. S., Peacock, W. S., Jan, Y. N. & Jan, L. Y. (2000) Proc. Natl. Acad. Sci. USA 97, 4914-4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marx, S. O., Kurokawa, J., Reiken, S., Motoike, H., D'Armiento, J., Marks, A. R. & Kass, R. S. (2002) Science 295, 496-499. [DOI] [PubMed] [Google Scholar]
- 19.Hoshi, N., Zhang, J. S., Omaki, M., Takeuchi, T., Yokoyama, S., Wanaverbecq, N., Langeberg, L. K., Yoneda, Y., Scott, J. D., Brown, D. A. & Higashida, H. (2003) Nat. Neurosci. 6, 564-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurokawa, J., Motoike, H. K., Rao, J. & Kass, R. S. (2004) Proc. Natl. Acad. Sci. USA 101, 16374-16378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gamper, N., Stockand, J. D. & Shapiro, M. S. (2003) J. Neurosci. 23, 84-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, Y., Langlais, P., Gamper, N., Liu, F. & Shapiro, M. S. (2004) J. Biol. Chem. 279, 45399-45407. [DOI] [PubMed] [Google Scholar]
- 23.Schwake, M., Pusch, M., Kharkovets, T. & Jentsch, T. J. (2000) J. Biol. Chem. 275, 13343-13348. [DOI] [PubMed] [Google Scholar]
- 24.Cooper, E. C., Harrington, E., Jan, Y. N. & Jan, L. Y. (2001) J. Neurosci. 21, 9529-9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang, L., Jacob, R. J., Pegg, S. C., Baldwin, M. A., Wang, C. C., Burlingame, A. L. & Babbitt, P. C. (2001) J. Biol. Chem. 276, 28327-28339. [DOI] [PubMed] [Google Scholar]
- 26.Zerangue, N., Schwappach, B., Jan, Y. N. & Jan, L. Y. (1999) Neuron 22, 537-548. [DOI] [PubMed] [Google Scholar]
- 27.Schwake, M., Jentsch, T. J. & Friedrich, T. (2003) EMBO Rep. 4, 76-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isacoff, E. Y., Jan, Y. N. & Jan, L. Y. (1991) Nature 353, 86-90. [DOI] [PubMed] [Google Scholar]
- 29.Franqueza, L., Lin, M., Shen, J., Splawski, I., Keating, M. T. & Sanguinetti, M. C. (1999) J. Biol. Chem. 274, 21063-21070. [DOI] [PubMed] [Google Scholar]
- 30.Hadley, J. K., Passmore, G. M., Tatulian, L., Al-Qatari, M., Ye, F., Wickenden, A. D. & Brown, D. A. (2003) J. Neurosci. 23, 5012-5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moss, S. J., Smart, T. G., Blackstone, C. D. & Huganir, R. L. (1992) Science 257, 661-665. [DOI] [PubMed] [Google Scholar]
- 32.Raymond, L. A., Blackstone, C. D. & Huganir, R. L. (1993) Trends Neurosci. 16, 147-153. [DOI] [PubMed] [Google Scholar]
- 33.Jonas, E. A. & Kaczmarek, L. K. (1996) Curr. Opin. Neurobiol. 6, 318-323. [DOI] [PubMed] [Google Scholar]
- 34.Davis, M. J., Wu, X., Nurkiewicz, T. R., Kawasaki, J., Gui, P., Hill, M. A. & Wilson, E. (2001) Am. J. Physiol. 281, H1835-H1862. [DOI] [PubMed] [Google Scholar]
- 35.Johnson, S. A. & Hunter, T. (2005) Nat. Methods 2, 17-25. [DOI] [PubMed] [Google Scholar]
- 36.Mann, M., Ong, S. E., Gronborg, M., Steen, H., Jensen, O. N. & Pandey, A. (2002) Trends Biotechnol. 20, 261-268. [DOI] [PubMed] [Google Scholar]
- 37.Varga, A. W., Anderson, A. E., Adams, J. P., Vogel, H. & Sweatt, J. D. (2000) Learn. Mem. 7, 321-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Misonou, H. & Trimmer, J. S. (2004) Crit. Rev. Biochem. Mol. Biol. 39, 125-145. [DOI] [PubMed] [Google Scholar]
- 39.Yus-Najera, E., Santana-Castro, I. & Villarroel, A. (2002) J. Biol. Chem. 277, 28545-28553. [DOI] [PubMed] [Google Scholar]
- 40.Gamper, N., Li, Y. & Shapiro, M. S. (2005) Mol. Biol. Cell 16, 3538-3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schrader, L. A., Anderson, A. E., Mayne, A., Pfaffinger, P. J. & Sweatt, J. D. (2002) J. Neurosci. 22, 10123-10133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long, S. B., Campbell, E. B. & Mackinnon, R. (2005) Science 309, 903-908. [DOI] [PubMed] [Google Scholar]
- 43.Lu, Z., Klem, A. M. & Ramu, Y. (2002) J. Gen. Physiol. 120, 663-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piper, D. R., Hinz, W. A., Tallurri, C. K., Sanguinetti, M. C. & Tristani-Firouzi, M. (2005) J. Biol. Chem. 280, 7206-7217. [DOI] [PubMed] [Google Scholar]
- 45.Chandy, K. G. & Gutman, G. A. (1995) in Ligand- and Voltage-Gated ion Channels, ed. North, R. A. (CRC Press, Boca Raton, FL), pp. 1-71.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.