

Abstract
Because modification of plasma albumin on tyrosine residues generates nitrated albumin (NOA) that may function as a mechanism of nitrogen monoxide clearance from microcirculation, we investigated biochemicaly and morphologically the cell surface binding and the transendothelial transport of NOA. An electron microscopic study was carried out with mouse lungs and hearts perfused in situ with NOA and NOA-Au complexes. The results indicate that NOA-Au can bind to the endothelial cell surface, and its binding can be blocked by albumin plus nitrotyrosine (NO-tyrosine) or abolished by excess NOA. We detected NOA-Au into perivascular spaces as early as 30 sec after the beginning of its perfusion. NOA, unlike native albumin, leaves the vascular lumina via both endothelial caveolae and open junctions. By cross-linking and ligand blotting analysis, we showed that NOA interacted with the same albumin binding proteins of 16–18, 30–32, 60, and 74 kDa as native albumin. ELISA performed on tissue homogenates obtained from the same specimens showed that NOA transport was 2- to 4-fold greater than native albumin. The augmented transendothelial transport of NOA reflects its transcytosis as well as its exit from the microcirculation via open junctions. The increased transport of NOA may serve as an important mechanism that protects a vascular bed against the damaging effects of nitrogen monoxide and peroxynitrite.
Endogenous nitrogen monoxide (°NO) is generated continuously or in bursts as a result of the enzymatic activity of constitutive and inducible forms of NO synthase. The isoforms of NO synthase are haemoproteins with limited tissue-specific distribution, use l-arginine as substrate and molecular oxygen as cosubstrate, and require nicotinamide-adenine-dinucleotide phosphate, flavin adenine-dinucleotide, flavin adenine-mononucleotide, and (6R)-5,6,7,8-tetrahydrobiopterin as cofactors. °NO participates in numerous direct and indirect chemical reactions and regulates a plethora of physiological responses that can be classified as regulatory, protective, or deleterious (1, 2).
Peroxynitrite (ONOO−), released from °NO interaction with the superoxide anion (O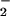 ), nitrates tyrosine residues of proteins, producing NO-tyrosine (3). NO-tyrosine as well as its metabolites have been detected in human urine (4), human tissues, and tissues of animal models of various diseases (5–7). Nitration as a chemical modification can alter protein function and enzymatic activity of different classes of enzymes, and it may interfere with signaling pathways and induce tissue injury (4, 5). Thus, protein nitration may have broad physiological and pathological effects. The reaction of ONOO− with albumin generates a nitrosated albumin as a result of interaction with one cysteine residue (8) and nitrated albumin (NOA) because of six tyrosine residues that are the preferential substrate for ONOO− attack (9). NOA is observed when ONOO− is added to plasma (10), and it appears in the blood and surrounding tissue when ONOO− is injected i.d. (11).
), nitrates tyrosine residues of proteins, producing NO-tyrosine (3). NO-tyrosine as well as its metabolites have been detected in human urine (4), human tissues, and tissues of animal models of various diseases (5–7). Nitration as a chemical modification can alter protein function and enzymatic activity of different classes of enzymes, and it may interfere with signaling pathways and induce tissue injury (4, 5). Thus, protein nitration may have broad physiological and pathological effects. The reaction of ONOO− with albumin generates a nitrosated albumin as a result of interaction with one cysteine residue (8) and nitrated albumin (NOA) because of six tyrosine residues that are the preferential substrate for ONOO− attack (9). NOA is observed when ONOO− is added to plasma (10), and it appears in the blood and surrounding tissue when ONOO− is injected i.d. (11).
Hemoglobin and albumin are among the proteins involved in °NO and ONOO− clearance. Hemoglobin serves as an important antioxidant inside circulating RBC (12), but it is not as effective against externally generated ONOO− (13). Thus, removal of °NO and ONOO− by albumin may function as an important defense mechanism (14).
Although it is known that NOA is present during sepsis (5, 15), little is known about its interactions with endothelial cells and its transendothelial transport. In this study, we investigated the possibility that nitration of albumin can influence its transport across the endothelial barrier and thereby affect its clearance from the circulation. Our results show that NOA is an active species capable of opening interendothelial junctions, thus increasing endothelial permeability. As nitration of circulating albumin has a profound effect in promoting the loss of endothelial barrier, its clearance from circulation may represent a critical mechanism of °NO and ONOO− removal.
Materials and Methods
Animals.
BALB/c mice were used in all surgical procedures carried out in accordance with the guidelines of the university's Animal Care Committee.
Materials.
Reagents and supplies were obtained as follows: PBS, Hanks' balanced solution, dinitrobenzenesulfonic acid, BSA fraction V, tetranitromethane (TNM), bovine IgG, fucoidan, lactalbumin, heparin, polyvinylpyrrolidone (Mr = 40,000), poly-l-glutamic acid, polyethylene glycol (Mr = 17,500), and α1-acidic protein were from Sigma-Aldrich. 3,3′,5,5′-Tetramethylbenzidine was from Kirkegaard & Perry Laboratories. PD-10 desalting columns, pharmalyte pH 3–10, Immobiline Dry Strips pH 4–7 and pH 3–10, peristaltic pump P1, nitrocellulose (NC) membranes (Hybond ECL), and all other reagents and materials for electrophoresis and isoelectrofocusing were from Amersham Pharmacia. The BCA Protein Assay Reagent Kit, disulfosuccinimidyl tartrate, protein A/G, and Super Signal chemiluminiscent detection kit were from Pierce. Rabbit anti-NO-tyrosine IgG was purchased from Upstate Biotechnology, Lake Placid, NY. Rabbit anti-dinitrophenyl (DNP), horseradish peroxidase (HRP)-coupled anti-DNP IgG, affinity-purified goat anti-rabbit IgG HRP-coupled, and HRP-coupled monoclonal IgG fraction anti-rabbit Ab were from Dako, and human lung microvascular endothelial cells (HLMVEC) and the recommended media were from Clonetics, San Diego.
Tracers.
We prepared: (i) monomeric derivatized albumin to obtain dinitrophenylated albumin (DNPA) as described (16), (ii) monomeric NOA obtained either by treating a solution of albumin with TNM (17) or by reacting a blocked albumin solution with ONOO− (18), and (iii) colloidal gold suspensions of 5 nm and 8 nm stabilized with NOA and albumin, which were kept and diluted as described (19). DNPA and NOA were separated from free dinitrobenzenesulfonic acid, free TNM, or secondary products of ONOO− decomposition by gel filtration. The average number of DNP-lysine residues per albumin molecule was estimated as described (20). NOA was obtained by reacting a solution of albumin either with TNM or ONOO−. The concentration of ONOO− was assayed as described (9), and, in both cases, the appearance of absorption maximum at 428 nm was used to monitor formation of NOA, whereas the molar extinction coefficient of 4,100 was used to quantitate the amount of NO-tyrosine residues per albumin molecule.
Perfusion Experiments.
All perfusions used a described protocol (21). Briefly, the thoracic cavity was opened on the median line, and a canula was placed in the aortic arch for heart perfusion or directly into the pulmonary artery for lung perfusion. Oxygenated and warm (37°C) Hanks' solution, to which sodium bicarbonate was added for a final osmolarity of 280 mOs (mHanks'), was perfused at constant pressure of 100 mm Hg, which was kept unchanged throughout all experiments. During perfusion, the lungs were ventilated at 200 breath/min via a canula placed in the trachea (connected to a pressure-controlled ventilator). Perfused lungs and hearts were removed for biochemical assays and fixed in situ for electron microscopic analysis.
To assess whether blood proteins and NO-tyrosine compete with NOA binding, the vascular beds were flushed with mHanks' containing albumin (0.5, 1, 10, and 30 mg/ml) or α1-acidic protein (1 and 5 mg/ml), and/or NO-tyrosine (0.1, 0.5, and 1 M), and then with NOA-Au for the aforementioned time. The effects of high ionic strength were investigated by perfusing first NOA-Au, and then mHanks' containing 0.5 M NaCl. The role of molecular charge in NOA-Au binding was addressed in experiments in which heparin (5 mg/ml), polyglutamic acid (2 mg/ml), or fucoidan (2 mg/ml) was perfused after the tracer. In other control experiments, IgG, lactalbumin, or polyethylene glycol-Au was perfused instead of NOA-Au to assess whether these tracers behaved differently.
Electron Microscopy.
Collected specimens from tissues perfused with tracers and fixed in situ by perfusion with a mixture of freshly prepared 4% formaldehyde + 2.5% glutaraldehyde + 1 mM Ca2+ + 2.5% polyvinylpyrrolidone in 0.1 M Pipes (pH 7.2) were further fixed (1 h on ice) by immersion in a triple fixative (22). The specimens were stained with 7.5% uranyl acetate, dehydrated in increasing concentrations of ethanol and propylene oxide, and embedded in Epon. Sections ≈60 nm thick were counterstained with lead citrate and uranyl acetate and micrographed in a JEOL 1220 transmission electron microscope. The negatives were scanned, and pictures were obtained by using Adobe PHOTOSHOP 7.0 run in an Apple G4 computer.
Tissue and Cell Lysates.
Tissues free of unbound tracers removed from the animals were trimmed of connective tissue, dried, weighed, hand-minced, mixed with lysis buffer (40 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate/1 mM PMSF, in PBS, pH 7.2) at a ratio of 1:10 (wt/vol), and homogenized (3 × 30 sec on ice) by using a Polytron. The same lysis buffer was used for HLMVEC (fourth to sixth passage). The lysates were cleared by centrifugation in a Beckman Optima TLX ultracentrifuge by using the TLA 55 rotor (150,000 × g for 1 h at 4°C), and the supernates were used for biochemical analysis.
Biochemical Procedures.
Protein content.
Protein content was determined by using the BCA kit with BSA as standard.
Denaturing SDS/PAGE and immunoblotting.
Cell or tissue lysates run on 9% or 5–20% SDS/PAGE gels were transferred (2 h, 700 mA, at 4°C) to NC, stained with 2% Ponceau S, dried, and stored. NC membrane strips were blocked with 5% dried milk + 0.05% Tween 20 in PBS, incubated for 2 h with the appropriate dilutions from the first antibody, washed (3 × 15 min), and incubated with diluted HRP-labeled reporter Ab, and the bound antibodies were detected by using the ECL kit.
ELISA.
Anti-DNP HRP-coupled Ab (1:1,000) and the anti-NO-tyrosine Ab (1:5,000) followed by an HRP-coupled monoclonal IgG fraction anti-rabbit Ab (1:10,000) were used to assess the amounts of derivatized tracers transported to the interstitial fluid. The data are expressed as ng of derivatized albumin normalized to mg wet tissue/5 min.
Isoelectric focusing.
The extent of charge modification of albumin after dinitrobenzenesulfonic acid, ONOO−, and TNM treatment was assessed by isoelectric focusing. We used a polyacrylamide slab gel system with a pH range from 3.0 to 10.0 run for 30 min at 150 V and for another 3 h at 200 V.
Cross-linking experiments.
Cross-linking reactions were carried out as described (23) on mechanically removed HLMVEC. The cells were washed (5 × 1 min) and incubated with 2% polyvinylpyrrolidone in PBS for 1 h at room temperature. The pellet was resuspended in 2 ml of 2% polyvinylpyrrolidone in PBS containing 50 μg NOA/ml, incubated 1 h at 4°C, and washed (5 × 5 min), and the final pellet was resuspended in 2 ml of PBS containing disulfosuccinimidyl tartrate (final concentration 1.5 mg/ml). The cross-linked products were solubilized with 4% SDS in PBS and immunoprecipitated with anti-NO-tyrosine Ab (2 μg/ml). After washing (6 × 15 min) the beads were incubated with 0.2 ml of PBS containing 0.015 M Na-metaperiodate, and the supernates were analyzed by SDS/PAGE.
Ligand blotting.
This procedure was performed on lysates of HLMVEC prepared as described above, which was then resolved by SDS/PAGE, and transferred to NC. Strips of NC were blocked with 2 mg/ml bovine IgG in PBS + 0.01 M Ca2+ overnight and probed with NOA-Au and native albumin Au (A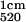 = 1).
= 1).
Data Analysis.
Values are expressed as means ± SEM. The Student's t test was used to examine statistical differences between control and experiments. Statistical significance was set at P values of <0.05.
Results
NOA Properties.
SDS/PAGE and isoelectric focusing used to characterize NOA showed that the derivatization reaction did not significantly affect its molecular weight (Fig. 1A), overall electric charge (Fig. 1B), and antigenic structure (Fig. 1C). Both reagents (TNM and ONOO−) used were effective in modifying albumin as evident by the appearance of NO-tyrosine detected by the anti-NO-tyrosine Ab (Fig. 1D). We calculated that an average of four of nine tyrosine residues were nitrated and available for immunodetection. Moreover, NOA obtained either by treating native albumin with TNM or blocked albumin with ONOO− did not show detectable differences (i.e., the same number of tyrosine residues were modified and there was no significant shift in molecular charge, immunological reactivity, or molecular weight) between the two albumin forms. Based on these observations, we concluded that NOA is a good approximation of native albumin, and thus could be used to address the pathways involved in its transport.
Figure 1.

Biochemical characteristics of NOA. (A) NOA as well as native albumin (A) (5 μg/lane) migrates similarly in a 9% gel. (B and C) Blots of native albumin (A) and NOA (5 μg/lane) probed with anti-albumin antibody (1:1,000) or an anti-NO-tyrosine Ab (1:2,000) show that anti-albumin antibody recognized both forms of albumin (B) whereas anti-NO-tyrosine antibody reacted only with NOA (C). (D) Isoelectrofocusing of native albumin (A) and NOA (5 μg/lane) show that the net electric charge of NOA is similar to A. The ampholytes used were in the range of pH 3 to 10; migration of pI markers and direction of pH gradient are indicated on the right. Results are representative of eight experiments.
Tissue Detection of NOA.
NOA was perfused for 10 min, and its appearance in the final tissue supernatants was determined with anti-NO-tyrosine Ab (Fig. 2). The complex protein profile of tissue supernatants did not show the presence of a protein band corresponding to NOA (Fig. 2A). However, the immunoblots of same supernatants showed the presence of NOA only in the specimens perfused with the tracer (Fig. 2B), an indication of its transendothelial transport. As in the case of native albumin (24), NOA appeared in the final supernatants as early as 30 sec and required <10 min to reach steady state.
Figure 2.

Protein content of final supernatants assessed by electrophoresis and immunoblotting. (A) Final supernatants of tissue perfused with NOA or control specimens (without NOA) at 30 μg total protein/lane, run in a 9% gel, and stained with Coomassie blue. This staining did not show the presence of native albumin or NOA. (B) When the blots were probed with anti-NO-tyrosine Ab (1:2,000), NOA was detected only in the supernatants from tissues perfused with NOA but not in controls. Results are representative of 20 experiments.
Cross-Linking and Ligand Blotting of NOA.
The NOA binding sites (i.e., albumin binding proteins, ABPs) on the endothelial cell surface were cross-linked with disulfosuccinimidyl tartrate and resolved on 5–20% SDS/PAGE gels. Fig. 3A shows the four proteins observed: two major bands at 18 and 32 kDa and two minor bands at 60 and 74 kDa; no band appears if the cross-linker was omitted. Knowing the amount of NOA used in the cross-linking experiments, we estimate that ≈25.3 ± 6.8 μg NOA was bound to 5 mg total endothelial protein (n = 12). In the presence of 500-fold excess albumin, this value decreased to 0.9 ± 0.6 μg NOA per 5 mg total protein; whereas in the presence of the same excess of albumin + 0.5 M NO-tyrosine, the value was 0.4 ± 0.3 μg NO-A per 5 mg total protein (n = 9). The ligand blotting showed only the lower molecular doublets of 18 and 32 kDa (Fig. 3 C and D). These experiments confirmed that NOA binds to the same ABPs as native albumin. Additional faint bands at 14 and 58 kDa already described (25) were observed in some experiments.
Figure 3.

Detection of ABPs by cross-linking and ligand blotting. The electrophoretic pattern, obtained on HLMVEC subjected to the cross-linking protocol, showed four bands: two major bands at 31 and 16 kDa and two minor bands at 58 and 74 kDa (A). The four bands did not appear if the cross-linker was omitted (B). ABPs (16–18 kDa and 30–31 kDa) that appear when NC strips containing lysates of HLMVEC were incubated with NOA-Au (C) or native albumin (A)-Au (D). There are no differences between C and D. Results are representative of 16 experiments.
Quantitation of NOA Transport.
We measured the amount of NOA transported to the interstitial fluid by ELISA carried out on lung and heart tissue supernatants. Fig. 4 shows the results of determinations of NOA transport in comparison with DNPA, a tracer used previously to measure albumin transport (20). We measured a 3- to 4-fold increase in NOA relative to DNPA transport in coronary microvessels (40 mice) and a 2- to 3-fold increase (55 mice) in lung microvessels. The values for DNPA transport are in agreement with values for native albumin (26, 27).
Figure 4.

Quantitation of NOA and albumin transport in heart and lung vascular beds. Monomeric DNPA prepared as described in Materials and Methods was used to measure albumin transport. Under basal conditions an average of ≈280 ± 37 ng albumin/mg wet tissue per 5 min is transported in heart and lung vessels. Perfusion of coronary vascular bed with NOA increased its transport to 790 ± 55 ng NOA/mg wet tissue per 5 min, whereas perfusion of pulmonary vascular bed increased its transport to 410 ± 94 ng NOA/mg wet tissue per 5 min. Results are representative for 40 hearts and 55 lungs.
Binding and Transport of NOA-Au.
The two tracers, NOA-Au and native albumin-Au, were used to visualize at the electron microscopic level their interactions with the continuous endothelium and to identify the cellular pathways involved in their efflux from microvasculature. As early as 30 sec after the beginning of NOA-Au perfusion, particles were found bound to the luminal endothelial plasmalemma proper and to the majority of open caveolae on both sides of endothelial cells (Figs. 5 and 6). The labeling pattern of endothelial plasmalemma and caveolar membranes by NOA-Au (Figs. 5, 6, and 7B) was similar to the decoration described for native albumin-Au (28) (Fig. 5A Inset). No statistical differences were found on comparing the binding densities of the two tracers inside the caveolar cavities (i.e., 12.0 ± 3.2 gold particles for native albumin-Au versus 10.1 ± 2.8 gold particles for NOA-Au). In the case of native albumin-Au, an average of 35% and in the case of NOA-Au an average of 55% of open caveolae were labeled on both sides of endothelial cells (P < 0.005). Diaphragms closing the openings of caveolae and impeding access of tracer particles to the caveolar cavity (Fig. 6B Inset) were much better defined in the lung microvasculature. The tracer particles were transported in large numbers to the perivascular space in both tissues (Figs. 5 and 6).
Figure 5.

Interactions of NOA-Au with murine heart endothelium. (A) Within 5 min of perfusion of murine heart vasculature with NOA-Au, a continuous decoration of plasmalemma proper and open caveolae on both sides of endothelial cells was seen as illustrated in this postcapillary venule. The tracer appeared in the perivascular spaces (pvs) during this period. (Inset) An example of the labeling pattern obtained with native albumin-Au. A more extensive labeling of plasmalemma proper and caveolae is seen with NOA-Au versus native albumin-Au. (Bar: 160 nm, Inset 120 nm.) (B) At the same time point, open interendothelial junctions filled with NOA-Au were seen in capillary endothelial cells. Results are representative of 26 experiments per condition. (Bar: 90 nm.)
Figure 6.

NOA-Au interactions with pulmonary vessels. (A) Tracer labels an open interendothelial junction from of a postcapillary venule. (Inset) Large aggregates of NOA-Au clogging a wide-open interendothelial space. (Bar: 120 nm, Inset 140 nm.) (B) The open interendothelial junctions in pulmonary capillaries are filled with tracer particles from the lumen as well as from transcytotic caveolae, in direct communication with the interendothelial space. (Inset) Illustration that caveolar diaphragms may restrict access of tracer to the caveolar cavity. Results are representative of 18 experiments. (Bar: 100 nm, Inset 140 nm.)
Figure 7.

Competition of NOA-Au binding to the endothelial surface. (A) Only scattered tracer particles were seen in specimens obtained from lung capillaries perfused with NOA or NOA + NO-tyrosine. The few particles inside the cell are illustrative of fluid-phase transcytosis. Results are representative of 10 experiments. (Bar: 80 nm.) (B) Binding and transport of NOA-Au were not altered when 0.5 M NaCl (high ionic strength) or charged species (polyglutamic acid, fucoidan, or heparin) were presented to the vascular endothelia before tracer. Results are representative of 16 experiments. (Bar: 120 nm.) (C) Perfusion of heart or pulmonary microvessels with lactalbumin-Au (a protein with a pI similar with albumin) resulted in the absorption of the gold tracer to the endothelial cell membrane. Note the continuous decoration of the endothelial surface and the fact the junction is not open (Inset) as in the case of NOA-Au. Results are representative of 25 experiments. (Bar: 160 nm, Inset 150 nm.)
The main characteristic of NOA-Au labeling was its presence in opened interendothelial junctions at the level of capillaries (Fig. 5B) and postcapillary and muscular venulae (Fig. 6). NOA-Au often appeared as large aggregates (Fig. 6A Inset) plugging wide-open junctional spaces.
Control Experiments.
The binding and transcytosis of NOA-Au were blocked by native albumin (although some fluid-phase transcytosis still persisted as shown in Fig. 7A). The presence of NO-tyrosine alone had no effect on binding or transcytosis of NOA-Au, whereas the combination albumin + NO-tyrosine abolished its binding and transcytosis (<0.4% of NOA-Au was transported) (Fig. 7A). α1-Acidic protein at 1 or 5 mg/ml did not have an effect on the binding and the transport of NOA (data not shown).
The competition with heparin, polyglutamic acid, fucoidan, or 0.5 M NaCl also had no effect on binding of NOA (Fig. 7B). When lactalbumin-gold complexes (a protein with a pI of 4.5–5.0) were perfused, we observed a uniform decoration of whole endothelial plasmalemma (Fig. 7 C and Inset), in contrast to the labeling by NOA or native albumin.
The NOA-Au particles reached the perivascular space in two ways: transcytosis and opened interendothelial junctions. The observation that NOA-Au particles opened the interendothelial junctions suggests that NO participates in regulating the tightness of endothelial barrier.
Morphometric Analysis.
The cumulated vascular perimeters examined were 1,917.4 μm (i.e., ≈115 μm2) for the heart and 2,234.1 μm (i.e., ≈134 μm2) for the lung, assuming a section thickness of 60 nm. The results of morphometric analysis are presented in Table 1. We observed greater plasmalemma and caveolar labeling with NOA-Au when compared with native albumin-Au (Table 1). The main difference was the labeling by NOA-Au of interendothelial junctional spaces. Native albumin-Au failed to label the interendothelial junctions at any time point, in any experimental condition, or form of albumin used (16, 24, 28–33).
Table 1.
Morphometric analysis of NOA-Au and native albumin-Au interactions with the endothelial surface
| Tracer | Gold particles/μm length of plasmalemma proper
|
Gold particles/μm length of caveolar plasmalemma
|
% of open and labeled caveolae
|
% of open and labeled junctions
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | C | V | A | C | V | A | C | V | A | C | V | |
| A-Au | 3.5 ± 0.7 | 5.7 ± 0.9 | 5.9 ± 1.1 | 8.1 ± 1.7 | 10.5 ± 2.1 | 12.4 ± 2.9 | 27.5 ± 2.3 | 35.6 ± 5.2 | 37.7 ± 4.1 | 0 | 0 | 9.5 ± 3.7 |
| NOA-Au | 9.7 ± 2.2 | 25.2 ± 3.3* | 37.4 ± 4.9* | 8.8 ± 1.9 | 9.7 ± 2.3 | 25.3 ± 4.1* | 34.7 ± 2.2 | 41.1 ± 3.9 | 56.2 ± 7.3* | 0.7 ± 0.2 | 21.4 ± 2.8† | 39.9 ± 5.1† |
Values represent means for aggregated vascular surfaces from lung (134 μm2) and heart (115 μm2). Cumulated vascular perimeters examined from murine lung vascular bed were 337.2 μm for arteries, 862.8 μm for capillaries, and 1,034.1 μm for veins. Cumulated vascular perimeters examined from murine heart vasculature were 237.8 μm for arteries, 773.2 μm for capillaries, and 907.4 μm for veins. From every vascular segment arterial (A), capillary (C), and venous (V), 44 pictures for heart and 69 pictures for lung vasculature were saved as data files and separately analyzed by using the stereology program from Morphometrix, Davis, CA.
P < 0.05.
P < 0.001.
Discussion
Enzymatically produced °NO can be a functional molecule at physiological concentrations, but it may promote tissue injury at high concentrations. The pathophysiological actions of °NO and its congeners are primarily the result of their capacity to covalently modify proteins, and thereby to alter protein function. Thus, the clearance of reactive nitrated species from the circulation is imperative for the maintenance of tissue homeostasis. In the present study, we examined the cellular structures involved in the extraction of NOA as a possible means of clearing °NO and ONOO− from microcirculation. Albumin is nitrated by the direct addition of ONOO− to plasma (10) and it appears at the sites of local administration of ONOO− in human tissues (11). Although the mechanisms of NOA formation are known (5, 11, 34, 35), the cellular and molecular pathways of its clearance remain unclear.
Our morphological and functional data show the pathways and the subcellular structures involved in the removal of NOA from the circulation. As in shown in Fig. 1, we observed no structural differences between NOA and native albumin, and thus we reasoned that the former could be used to describe its interactions with the endothelial surface and to investigate its clearance pathways. We demonstrated by detecting NOA in tissue supernatants of perfused tissues that NOA is transported across the endothelial barrier (Fig. 2). However, we found that its transport was 2- to 4-fold greater than native albumin (Fig. 4). By ligand blotting, we demonstrated that NOA binds to the same low molecular mass ABPs of 16–18 and 30–32 kDa (Fig. 3 C and D) as native albumin (28). When monomeric NOA was used in cross-linking experiments, four surface proteins were detected in the cross-linked complex. The identified bands (18, 32, 60, and 74 kDa), which are also specific ABPs for native albumin (25, 36, 37), were confirmed by our experiments. The interaction of NOA with the four ABPs was inhibited >90% by ≈300-fold excess native albumin and was not affected by up to 1 M NO-tyrosine. As ABPs responsible for the interaction of NOA with the endothelium are not different form the proteins binding native albumin (30), the enhanced transport of NOA cannot be explained by simple differences in binding sites.
We carried out, using NOA-Au and native albumin-Au, extensive electron microscopic surveys of murine lung and heart vascular beds to define the cellular pathways responsible for their transport across the endothelial barrier. Knowing that the continuous endothelium behaves as leaky epithelia with a strong transcytotic component (21, 38, 39), we compared the transport of NOA-Au and native albumin-Au. Our results show that: (i) NOA-Au complex binds to the endothelial cell surface, and it is transported across the endothelial cell bound to the caveolar membrane or in bulk, (ii) NOA-Au complex, unlike other forms of albumin, labels opened interendothelial spaces, and (iii) enhanced transport of NOA-Au is a consequence of open junctions and increased transcytosis occurring both in capillaries and venulae. The binding of NOA was drastically reduced by exogenous albumin but was not affected by NO-tyrosine. The interaction of NOA with the endothelial surface was not electrostatic because competition with high ionic strength (0.5 M NaCl) and charged molecules (heparin, polyglutamic acid, or fucoidan) did not affect its binding. All perfusion experiments showed that NOA interacting specifically with the endothelial surface modified the interendothelial junctions, enabling NOA to enter the wide-open interendothelial space.
Based on previous studies of native (32, 37, 38, 40) or modified albumin (31, 33), we had expected NOA to behave similarly; thus, the finding that NOA induced the opening of interendothelial junctions (range 5–3,000 nm) was unexpected. The opening of capillary interendothelial junctions by NOA resembles the opening of the same junctions observed with vascular endothelial growth factor (41) and platelet-activating factor (42), raising the possibility that the junctional effect may be the result of released NO by these mediators. The opening of interendothelial junctions was a reversible phenomenon. When the vascular beds were washed free of NOA-Au and perfused from 30 to 60 min only with mHanks', the number of opened junctions at the level of capillaries was 1.5 ± 0.7% (versus 21.4% during NOA-Au perfusion), whereas on the venular end this number was 15.7 ± 5.9% (versus 39.9% during NOA-Au perfusion); the value of open junctions in venulae during the recovery phase was similar to the number of open junctions under basal condition (Table 1).
The present data suggest that a possible explanation for the conflicting results reported concerning the role of NO in the control of vascular permeability (43–49) may be the presence of adducts formed by interaction of NO with albumin. Nitrated proteins have a longer half-life (min to days) as compared with °NO or ONOO−. Thus, the magnitude of the increase in endothelial permeability and its duration may critically depend on the concentration of NO-A in the circulation.
In conclusion, we described the cellular pathways and mechanisms involved in the removal of NOA from the vascular lumina. Our results show that the opening of interendothelial junctions induced by NOA is the key determinant of the 2- to 4- fold increases in endothelial permeability; thus, NOA behaves as an active mediator of increased permeability. This increased vascular permeability may be a critical mechanism of clearing °NO or ONOO− and their products from the circulation.
Abbreviations
- NOA
nitrated albumin
- °NO
nitrogen monoxide
- ONOO−
peroxynitrite
- TNM
tetranitromethane
- DNP
dinitrophenyl
- HRP
horseradish peroxidase
- HLMVEC
human lung microvascular endothelial cells
- DNPA
dinitrophenylated albumin
- NC
nitrocellulose
- ABP
albumin binding protein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Moncada S, Palmer R M, Higgs E A. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 2.Grisham M B, Jourd'heuil D, Wink D A. Am J Physiol. 1999;276:G315–G321. doi: 10.1152/ajpgi.1999.276.2.G315. [DOI] [PubMed] [Google Scholar]
- 3.Beckman J S, Beckman T W, Chen P A, Marshall P A, Freeman B A. Proc Natl Acad Sci USA. 1990;87:1620–1626. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohsima H, Friesen M, Brouet I, Bartsch H. Food Chem Tox. 1990;28:647–652. doi: 10.1016/0278-6915(90)90173-k. [DOI] [PubMed] [Google Scholar]
- 5.Ischiropoulos H. Arch Biochem Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- 6.Haddad I Y, Pataki G, Hu P, Galliani C, Beckman J S, Matalon S. J Clin Invest. 1994;94:2407–2413. doi: 10.1172/JCI117607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishiyama S, Hiroe M, Nishikawa T, Abe S, Shimojo T, Ito H, Ozasa S, Yamakawa K, Matsuzaki M, Mohammed M U, et al. Circulation. 1997;95:489–496. doi: 10.1161/01.cir.95.2.489. [DOI] [PubMed] [Google Scholar]
- 8.Radi R, Beckman J S, Bush K M, Freeman B A. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 9.Beckman J S, Chen J, Ischiropoulos H, Crow J P. Methods Enzymol. 1994;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- 10.Sies H, Sharov V S, Klotz L-O, Briviba K. J Biol Chem. 1997;272:27812–27817. doi: 10.1074/jbc.272.44.27812. [DOI] [PubMed] [Google Scholar]
- 11.Greenacre S A B, Evans P, Halliwell B, Brain S D. Biochem Biophys Res Commun. 1999;262:781–786. doi: 10.1006/bbrc.1999.1309. [DOI] [PubMed] [Google Scholar]
- 12.Kondo H, Takahashi M, Niki E. FEBS Lett. 1997;413:236–238. doi: 10.1016/s0014-5793(97)00922-8. [DOI] [PubMed] [Google Scholar]
- 13.Beckman J S. Circ Res. 2001;89:295–299. [PubMed] [Google Scholar]
- 14.Arteel G E, Mostert V, Oubrahim B, Briviba K, Abel J, Sies H. Biol Chem. 1988;379:1202–1205. [PubMed] [Google Scholar]
- 15.Kooy N W, Lewis S J, Royall J A, Ye Y-Z, Kelly D R, Beckman J S. Crit Care Med. 1997;25:812–819. doi: 10.1097/00003246-199705000-00017. [DOI] [PubMed] [Google Scholar]
- 16.Ghitescu L, Bendayan M. J Cell Biol. 1992;117:745–755. doi: 10.1083/jcb.117.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sokolovsky M, Riordan L F, Vallee B L. Biochemistry. 1966;5:3582–3589. doi: 10.1021/bi00875a029. [DOI] [PubMed] [Google Scholar]
- 18.Beckman J S. In: Nitric Oxide: Principles and Actions. Lancaster J, editor. San Diego: Academic; 1995. pp. 1–82. [Google Scholar]
- 19.Predescu D, Palade G E. Am J Physiol. 1993;265:H725–H733. doi: 10.1152/ajpheart.1993.265.2.H725. [DOI] [PubMed] [Google Scholar]
- 20.Predescu D, Horvat R, Predescu S, Palade G E. Proc Natl Acad Sci USA. 1994;91:3014–3018. doi: 10.1073/pnas.91.8.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Predescu S, Predescu D, Palade G E. Am J Physiol. 1997;272:H937–H949. doi: 10.1152/ajpheart.1997.272.2.H937. [DOI] [PubMed] [Google Scholar]
- 22.Simionescu N, Palade G E. J Cell Biol. 1971;50:616–626. doi: 10.1083/jcb.50.3.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Predescu S, Predescu D, Palade G E. Mol Biol Cell. 2001;12:1019–1033. doi: 10.1091/mbc.12.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milici A J, Watrous N E, Stukenbrok H, Palade G E. J Cell Biol. 1987;105:2603–2612. doi: 10.1083/jcb.105.6.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghinea N, Eskenasy M, Simionescu M, Simionescu N. J Biol Chem. 1989;264:4755–4758. [PubMed] [Google Scholar]
- 26.Renkin E M, Joyner W L, Sloop C H, Watson P D. Microvasc Res. 1977;14:191–204. doi: 10.1016/0026-2862(77)90018-8. [DOI] [PubMed] [Google Scholar]
- 27.Ripe B, Kamia A, Folkow B. Acta Physiol Scand. 1979;105:171–187. doi: 10.1111/j.1748-1716.1979.tb06329.x. [DOI] [PubMed] [Google Scholar]
- 28.Ghitescu L, Fixman A, Simionescu M, Simionescu N. J Cell Biol. 1986;102:1304–1311. doi: 10.1083/jcb.102.4.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Handley D A, Chien S. Eur J Cell Biol. 1987;43:163–174. [PubMed] [Google Scholar]
- 30.Simionescu M, Simionescu N. Cell Biol Rev. 1991;25:1–80. [PubMed] [Google Scholar]
- 31.Predescu D, Simionescu M, Simionescu N, Palade G E. J Cell Biol. 1988;107:1729–1733. doi: 10.1083/jcb.107.5.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghitescu L, Desjardines M, Bendayan M. Kidney Int. 1992;42:25–32. doi: 10.1038/ki.1992.256. [DOI] [PubMed] [Google Scholar]
- 33.Galis Z, Ghitescu L, Simionescu M, Simionescu N. Eur J Cell Biol. 1988;47:358–365. [PubMed] [Google Scholar]
- 34.Khan J, Brennan D B, Bradley N, Gao B, Bruckdorfer R, Jacobs M. Biochem J. 1998;330:795–801. doi: 10.1042/bj3300795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alvarez B, Ferrer-Suerta G, Freeman B A, Radi R. J Biol Chem. 1999;274:842–848. doi: 10.1074/jbc.274.2.842. [DOI] [PubMed] [Google Scholar]
- 36.Dobrila L, Antohe F, Heltianu C, Simionescu M. Int Immunol. 1992;4:789–796. doi: 10.1093/intimm/4.7.789. [DOI] [PubMed] [Google Scholar]
- 37.Tiruppathi C, Finnegan A, Malik A B. Proc Natl Acad Sci USA. 1996;93:250–254. doi: 10.1073/pnas.93.1.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palade G E, Milici A J. Mol Mech Cell Growth Diff. 1991;2:223–247. [Google Scholar]
- 39.Lum H, Malik A B. Am J Physiol. 1994;267:L223–L241. doi: 10.1152/ajplung.1994.267.3.L223. [DOI] [PubMed] [Google Scholar]
- 40.Minshall R D, Tiruppathi C, Vogel S M, Niles W D, Gilchrist A, Hamm H E, Malik A B. J Cell Biol. 2000;150:1057–1069. doi: 10.1083/jcb.150.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts W G, Palade G E. J Cell Sci. 1995;108:3269–2379. doi: 10.1242/jcs.108.6.2369. [DOI] [PubMed] [Google Scholar]
- 42.Predescu D, Ihida K, Predescu S, Palade G E. Eur J Cell Biol. 1996;69:86–98. [PubMed] [Google Scholar]
- 43.Oliver J A. J Cell Physiol. 1992;151:506–511. doi: 10.1002/jcp.1041510309. [DOI] [PubMed] [Google Scholar]
- 44.Mayhan W G. Inflammation. 1992;16:295–305. doi: 10.1007/BF00917622. [DOI] [PubMed] [Google Scholar]
- 45.Mayer D J, Huxley V H. Circ Res. 1992;70:382–391. doi: 10.1161/01.res.70.2.382. [DOI] [PubMed] [Google Scholar]
- 46.Kurose I, Kubes P, Wolf R, Anderson D C, Paulson J, Miyasaka M, Granger D N. Circ Res. 1993;73:164–171. doi: 10.1161/01.res.73.1.164. [DOI] [PubMed] [Google Scholar]
- 47.Lippe I T, Stabentheiner A, Holzer P. Eur J Pharmacol. 1993;232:113–120. doi: 10.1016/0014-2999(93)90735-z. [DOI] [PubMed] [Google Scholar]
- 48.Filep J G, Foldes-Filep E, Sirois P. Br J Pharmacol. 1993;108:323–326. doi: 10.1111/j.1476-5381.1993.tb12803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prado R, Watson B D, Kuluz J, Dietrich D. Stroke. 1992;23:1118–1124. doi: 10.1161/01.str.23.8.1118. [DOI] [PubMed] [Google Scholar]