

Abstract
Currently, patients with neuroblastoma are classified into risk groups (e.g., according to the Children’s Oncology Group risk-stratification) to guide physicians in the choice of the most appropriate therapy. Despite this careful stratification, the survival rate for patients with high-risk neuroblastoma remains <30%, and it is not possible to predict which of these high-risk patients will survive or succumb to the disease. Therefore, we have performed gene expression profiling using cDNA microarrays containing 42,578 clones and used artificial neural networks to develop an accurate predictor of survival for each individual patient with neuroblastoma. Using principal component analysis we found that neuroblastoma tumors exhibited inherent prognostic specific gene expression profiles. Subsequent artificial neural network-based prognosis prediction using expression levels of all 37,920 good-quality clones achieved 88% accuracy. Moreover, using an artificial neural network-based gene minimization strategy in a separate analysis we identified 19 genes, including 2 prognostic markers reported previously, MYCN and CD44, which correctly predicted outcome for 98% of these patients. In addition, these 19 predictor genes were able to additionally partition Children’s Oncology Group-stratified high-risk patients into two subgroups according to their survival status (P = 0.0005). Our findings provide evidence of a gene expression signature that can predict prognosis independent of currently known risk factors and could assist physicians in the individual management of patients with high-risk neuroblastoma.
INTRODUCTION
Neuroblastoma is the most common solid extracranial tumor of childhood and is derived from the sympathetic nervous system. Patients in North America are currently stratified by the Children’s Oncology Group into high, intermediate, and low risk based on age, tumor staging, Shimada histology, MYCN amplification, and DNA ploidy (1). Patients < 1 year of age or with lower stage diseases (International Neuroblastoma Staging System stages 1 and 2) usually have better outcome than older patients or those with advanced stage diseases (International Neuroblastoma Staging System stages 3 and 4). Certain consistent cytogenetic changes, including gain of 2p24 and 17q and loss of heterozygosity at 1p36 have been associated with a more aggressive phenotype (2, 3). The MYCN gene, located on 2p24, is amplified in ~22% of all neuroblastoma patients (4) and is an independent predictor for poor prognosis, especially for patients >1 year of age. Although other genes, such as TRKA, TRKB, hTERT, BCL-2, caspases, and FYN (4, 5) have been associated with neuroblastoma prognosis, they all lack the predictive power of MYCN and are not used currently in clinical practice. High-risk patients compose ~50% of all neuroblastoma cases; however, despite significant improvement in the therapy of neuroblastoma using neoadjuvant chemotherapy, surgery, and radiation, the death rate for these patients remains at 70% (6). Although the Children’s Oncology Group risk stratification has been carefully developed to take into account the above risk factors, it is primarily used to guide therapy and does not predict which individual patients will be cured from the disease.
DNA microarray technology has been proven to be an efficacious tool to molecularly classify cancers, to predict prognosis, and to identify genes that are potential therapeutic molecular targets (7–12). We have demonstrated previously that the combination of gene expression profiling and artificial neural networks is a powerful method that can accurately diagnose certain pediatric cancers including neuroblastoma (7). In this current study, we used gene expression profiles from cDNA microarrays to predict the outcome and identify an optimal gene set in patients with neuroblastoma using artificial neural networks.
MATERIALS AND METHODS
Tumor Samples
Fifty-six pretreatment primary neuroblastoma tumor samples from 49 neuroblastoma patients with outcome information were obtained retrospectively from three sources presenting between 1992 and 2000 (Table 1). All of the patients were treated according to local or national guidelines that followed similar protocols, which included “wait-and-see” after surgery or combinations of vincristine, doxorubicin, carboplatin, cisplatin, cyclophosphamide, melphalan, and etoposide, depending on the risk factors. All of the samples were anonymized, and our protocol was deemed exempt from the NIH Multiple Project Assurance. Pretreatment tumor samples were snap-frozen in liquid nitrogen after removal. Tumors were diagnosed as neuroblastoma by local centers experienced in the management of these cancers. Patients were divided into two outcome groups: the “good-outcome” group had event-free survival (i.e., neither relapse nor neuroblastoma progression) for at least 3 years (n = 30), and “poor-outcome” died due to neuroblastoma disease (n = 19). The median age for the good-outcome group was 0.9 years (range from 0.1 to 4.6 years) and for the poor-outcome group was 2.8 years (range from 0.8 to 10.5 years; Table 1).
Table 1.
Neuroblastoma samples used in the study and ANN prognostic prediction
| Sample label | Year of diagnosis | Sample source* | Age at diagnosis (yrs) | INSS stage | MYCN amplification status | Shimada histology | COG risk stratification | Years of survival | All 37920 clonesave ANN vote | Top 19 genes ave ANN vote | ANN predicted outcome | Clinical outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NB11 | 1998 | 2 | 0.3 | 1 | NA | F | L | 5.1 | — | 0.02 | A | A |
| NB22 | 1997 | 2 | 0.9 | 4 | NA | F | I | 5.9 | — | 0.02 | A | A |
| NB33 | 1998 | 2 | 1.2 | 4 | NA | F | H | 5.7 | — | 0.01 | A | A |
| NB44 | 1997 | 2 | 1.4 | 4 | NA | F | H | 6.7 | — | 0.02 | A | A |
| NB7 | 1998 | 2 | 1.3 | 1 | NA | — | L | 5.2 | 0.06 | 0.03 | A | A |
| NB14 | 2000 | 2 | 0.9 | 4 | AMP | — | H | 3.2 | 0.18 | 0.05 | A | A |
| NB15 | 1999 | 2 | 0.9 | 2 | NA | — | L | 3.9 | 0.03 | 0.02 | A | A |
| NB18 | 2000 | 2 | 1.8 | 2 | NA | — | L | 1.4 | 0.72 | 0.95 | D | D |
| NB21 | 2000 | 2 | 5.2 | 4 | AMP | — | H | 0.6 | 0.92 | 0.99 | D | D |
| NB27 | 2000 | 2 | 10.5 | 4 | AMP | UF | H | 1.4 | 0.36 | 0.97 | D | D |
| NB291 | 1998 | 2 | 0.3 | 1 | NA | F | L | 5.1 | 0.04 | 0.02 | A | A |
| NB302 | 1997 | 2 | 0.9 | 4 | NA | F | I | 5.9 | 0.05 | 0.01 | A | A |
| NB314 | 1997 | 2 | 1.4 | 4 | NA | F | H | 6.7 | 0.04 | 0.02 | A | A |
| NB323 | 1998 | 2 | 1.2 | 4 | NA | F | H | 5.7 | 0.05 | 0.02 | A | A |
| NB61 | 1997 | 2 | 1.4 | 3 | NA | F | I | 6.3 | 0.22 | 0.2 | A | A |
| NB69 | 1992 | 2 | 4.4 | 4 | NA | — | H | 0.5 | 0.16 | 0.8 | D | D |
| NB75 | 1998 | 2 | 1 | 3 | AMP | F | H | 3 | 0.89 | 0.99 | D | D |
| NB77 | 1994 | 2 | 0.2 | 1 | NA | — | L | 9.7 | 0.07 | 0.01 | A | A |
| NB79 | 1997 | 2 | 2.8 | 4 | AMP | — | H | 1.5 | 0.9 | 0.99 | D | D |
| NB205 | 1995 | 1 | 3.9 | 4 | NA | — | H | 2.3 | 0.52 | 0.84 | D | D |
| NB2075 | 1995 | 1 | 4.4 | 4 | NA | — | H | 3.1 | — | 0.98 | D | D |
| NB208 | 1995 | 1 | 0.8 | 1 | NA | F | L | 4.8 | 0.11 | 0.02 | A | A |
| NB2096 | 1995 | 1 | 1.2 | 4 | NA | UF | H | 1 | — | 0.98 | D | D |
| NB2107 | 1996 | 1 | 2.3 | 4 | NA | UF | H | 1.1 | — | 0.97 | D | D |
| NB216 | 1996 | 1 | 0.6 | 3 | NA | — | I | 6.8 | 0.05 | 0.02 | A | A |
| NB231 | 1998 | 1 | 0.5 | 2 | NA | F | L | 4 | 0.04 | 0.02 | A | A |
| NB237 | 1999 | 1 | 4.1 | 1 | NA | F | L | 3.2 | 0.14 | 0.11 | A | A |
| NB254 | 2000 | 3 | 2.6 | 4 | AMP | — | H | 1.8 | 0.88 | 0.98 | D | D |
| NB255 | 1999 | 3 | 0.5 | 2 | NA | — | L | 4 | 0.71 | 0.24 | A | A |
| NB266 | 1996 | 3 | 2 | 4 | AMP | — | H | 0 | 0.63 | 0.98 | D | D |
| NB2735 | 1995 | 1 | 4.4 | 4 | NA | — | H | 3.1 | 0.84 | 0.96 | D | D |
| NB2756 | 1995 | 1 | 1.2 | 4 | NA | UF | H | 1 | 0.94 | 0.99 | D | D |
| NB2767 | 1996 | 1 | 2.3 | 4 | NA | UF | H | 1.1 | 0.57 | 0.98 | D | D |
| NB278 | 1999 | 1 | 1.7 | 4 | AMP | UF | H | 0.8 | 0.85 | 0.94 | D | D |
| NB283 | 1999 | 1 | 5.5 | 4 | NA | UF | H | 4 | 0.45 | 0.9 | D | D |
| NB8 | 1998 | 2 | 4.6 | 4 | NA | — | H | 1.8 | 0.63 | 0.97 | D | D |
| NB9 | 1996 | 2 | 1.1 | 1 | NA | — | L | 7.1 | 0.04 | 0.02 | A | A |
| NB17 | 2000 | 2 | 1.2 | 1 | NA | F | L | 3.5 | 0.17 | 0.03 | A | A |
| NB24 | 2000 | 2 | 0.6 | 4 | NA | F | I | 3 | 0.08 | 0.03 | A | A |
| NB33 | 1998 | 2 | 1.4 | 1 | NA | F | L | 4.8 | 0.05 | 0.02 | A | A |
| NB34 | 1997 | 2 | 1.2 | 1 | NA | F | L | 5.2 | 0.04 | 0.02 | A | A |
| NB35 | 1997 | 2 | 2.6 | 4 | NA | — | H | 6.5 | 0.13 | 0.07 | A | A |
| NB64 | 1998 | 2 | 0.6 | 4 | NA | — | I | 5.7 | 0.81 | 0.02 | A | A |
| NB72 | 1994 | 2 | 3 | 3 | AMP | — | H | 1 | 0.94 | 0.98 | D | D |
| NB201 | 1994 | 1 | 1.5 | 3 | NA | UF | H | 7.4 | 0.04 | 0.08 | A | A |
| NB206 | 1995 | 1 | 3.3 | 4 | NA | UF | H | 5.8 | 0.82 | 0.96 | D | D |
| NB215 | 1996 | 1 | 1.2 | 3 | NA | F | I | 7.3 | 0.04 | 0.03 | A | A |
| NB220 | 1997 | 1 | 0.4 | 2 | NA | F | L | 6 | 0.06 | 0.04 | A | A |
| NB221 | 1997 | 1 | 0.4 | 1 | NA | F | L | 5.7 | 0.09 | 0.02 | A | A |
| NB232 | 1998 | 1 | 0.1 | 2 | NA | F | L | 4.3 | 0.06 | 0.04 | A | A |
| NB235 | 1999 | 1 | 0.4 | 2 | NA | F | L | 3.2 | 0.15 | 0.03 | A | A |
| NB238 | 1999 | 1 | 1.2 | 1 | NA | F | L | 3 | 0.04 | 0.02 | A | A |
| NB251 | 2000 | 3 | 0.8 | 4 | AMP | — | H | 0.5 | 0.69 | 0.91 | D | D |
| NB265 | 1996 | 3 | 1.8 | 4 | AMP | — | H | 2 | 0.78 | 0.97 | D | D |
| NB269 | 1997 | 3 | 0.8 | 4 | NA | — | I | 5.3 | 0.04 | 0.07 | A | A |
| NB282 | 1999 | 1 | 4.6 | 4 | NA | UF | H | 3.3 | 0.91 | 0.58 | D | A |
NOTE. All samples (except NB1, NB2, NB3, NB4, NB207, NB209, and NB210) were used in the leave-one-out ANN analysis. Samples highlighted in gray are the 21 test samples, and the rest were used for training in the clone optimization procedure. There were 7 replicated samples, marked by the numbers in superscript.
Abbreviations: INSS, International Neuroblastoma Staging System; AMP, amplification; NA, not amplified; F, favorable; −, not known; UF, unfavorable; COG, Children’s Oncology Group; H, high-risk; I, intermediate-risk; L, low-risk; ave ANN vote, average ANN committee votes; ANN prediction, average ANN vote <0.5 = A (alive) and >0.5 = D (dead); A, alive without event; D, deceased due to neuroblastoma disease.
Sample source: 1, Cooperative Human Tissue Network (Ohio); 2, German Cancer Research Center; 3, The Children’s Hospital at Westmead (Australia).
RNA Extraction
Total RNA was extracted according to the published protocols (13). We used an Agilent BioAnalyzer 2100 (Agilent, Palo Alto, CA) to assess the integrity of total RNA from tumors. Total RNA from seven human cancer cell lines (CHP212, RD, HeLa, A204, K562, RDES, and CA46) was pooled in equal portions to constitute a reference RNA, which was used in all of the cDNA microarrray experiments.
RNA Amplification and Labeling of cDNA
mRNA was amplified one round using a modified Eberwine RNA amplification procedure (14). Next, an indirect fluorescent-labeling method was used to label cDNA as described by Hegde et al. (15). In brief, aminoallyl-dUTP (Sigma-Aldrich, St. Louis, MO) was first incorporated into cDNA in a reverse transcription reaction in which amplified antisense RNA was converted into cDNA by Superscript II reverse transcriptase enzyme (Invitrogen, Grand Island, NY) according to the manufacturer’s instructions. Second, unincorporated aminoallyl-dUTP was removed with Qiagen PCR purification kits (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Third, monoreactive-Cye5 or Cye3 dyes (AmershamPharmacia, Piscataway, NJ) were conjugated with the aminoallyl-dUTP on the cDNA. Fluorescent-labeled cDNA was purified with Qiagen PCR purification kits.
Fabrication of cDNA Microarrays, Hybridization, Image Acquisition, and Image Analysis
Sequence-verified cDNA libraries were purchased from Research Genetics (Huntsville, AL), and a total of 42,578 cDNA clones, representing 25,933 unique genes (UniGene clusters; 13,606 known genes and 12,327 unknown expressed sequence tags), were printed on microarrays using a BioRobotics MicroGrid II spotter (Harvard Bioscience, Holliston, MA). Fabrication, hybridization, and washing of microarrays were performed as described by Hegde et al. (15). Images were acquired by an Agilent DNA microarray scanner (Agilent, Palo Alto, CA) and analyzed using the Microarray Suite program as described (16), coded in IPLab (Scanalytics, Fair-fax, VA).
Data Normalization and Filtering
Gene expression ratios between tumor RNA and reference RNA on each microarray were normalized using a pin-based normalization method modified from Chen et al. (16). To include only high-quality data in the analysis, the quality of each individual cDNA spot was calculated according to Chen et al. (17). Next, spots with an average quality across all of the samples < 0.95 were excluded from all of the analyses. There were 37,920 (90.3%) clones that passed this quality filter.
Architecture of Artificial Neural Networks
First, we used principal component analysis and reduced the dimensionality of the data to the top 10 principal components as inputs for artificial neural networks. This procedure reduced the number of variables from 37,920 to 10 to avoid over-fitting the data, which occurs when the number of variables exceeds the number of samples. We used feed-forward resilient back-propagation multilayer perceptron artificial neural networks (coded in Matlab, The Mathworks, Natick, MA) with three layers: an input layer of the top 10 principal components of the data (Fig. 1, A and B) or the gene expression ratios of each cDNA spot (for the minimized gene set, see Fig. 1B); a hidden layer with 3 nodes; and an output layer generating a committee vote that discriminates two classes (i.e., good-and poor-outcome groups). Average artificial neural network committee votes were used to classify samples, and 0.5 was used as the decision boundary for artificial neural network prediction throughout the study. The ideal vote was 0 for the good-outcome group (alive) and 1 for the poor-outcome group (dead). We trained the artificial neural networks using an 8-fold cross-validation scheme in all of the analyses similar to those described previously (7).
Fig. 1.
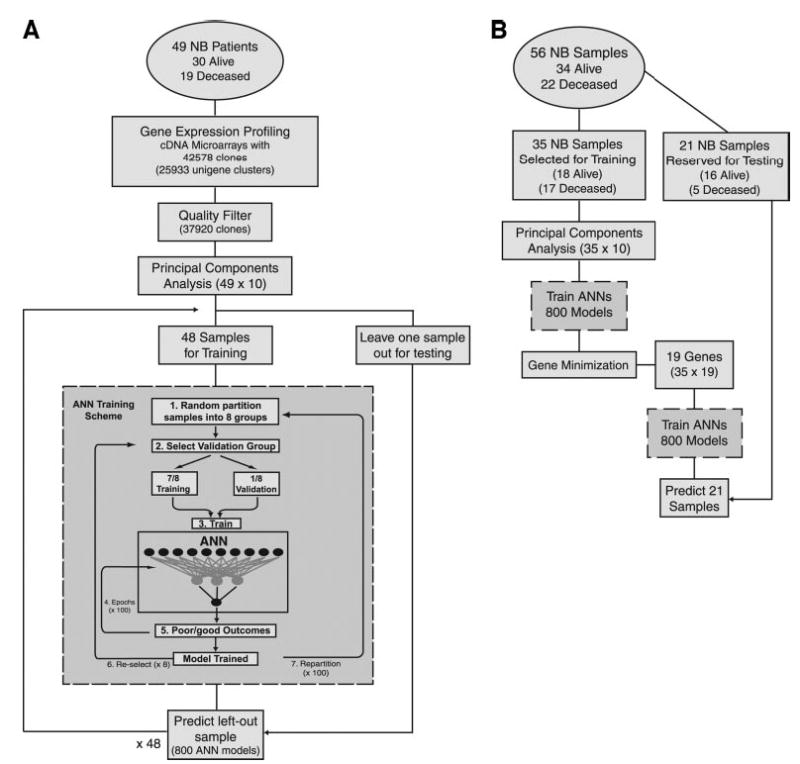
Workflow diagrams. A, workflow for a complete leave-one-out artificial neural network (ANN) analysis using all 37,920 clones. Gene expression profiling was performed on tumors from 49 neuroblastoma (NB) patients (Alive, n = 30; Deceased, n = 19) using cDNA microarrays containing 42,578 clones. After a quality filter, 37,920 clones were used as a data matrix of high quality cDNA measurements for further data analysis. Principal component analysis was used to reduce the dimensionality of the data and reduce noise. The top 10 principal components were used for input to the ANN. One sample was left out as an independent test sample, and the ANNs were trained using the remaining 48 NB samples. ANN training scheme (gray box). 1. All remaining neuroblastoma samples were randomly partitioned into eight groups. 2. One of the eight groups (containing 6 samples) was selected as a validation set, whereas the remaining 7 groups (42 samples) were used to train the network. 3 and 4. The training weights were iteratively adjusted for 100 cycles (epochs). 5. The ANN output (0–1, where 0 = ideal good-outcome and 1 = ideal poor-outcome) was calculated for each sample in the validation set. 6. A different validation set was selected from the same partitioning in 1, and the remaining seven groups were used for training. Steps 2–6 were repeated until each of the eight groups from 1 had been used as a validation set exactly one time. 7. The samples were randomly repartitioned into eight new groups, and steps 2–6 were repeated. Sample partitioning was performed 100 times in total. Thus, steps 1–6 were repeated 100 times. Eight hundred ANN models were, thus, trained and were used to predict the left out test sample. This scheme was repeated for each left out test sample. B, identifying prognostic gene expression signature and outcome prediction. Fifty-six neuroblastoma samples (7 replicates were added to the training group to examine the reproducibility of the results) were partitioned into a training (n = 35) and an independent test (n = 21) set. Principal component analysis was again performed, and ANNs were retrained using the 35 training samples based on the ANN training scheme detailed in the gray box in A. Gene minimization. Each of the input clones was ranked according to its importance to the prediction of ANNs (7). Increasing numbers of the top-ranked clones were used to train ANNs, and the resulting classification error was monitored. The minimal number of clones that yielded the minimal classification error (Fig. 3A) was identified, and the top-ranked clones for each gene were used to retrain the ANNs and predict the 21 test samples without performing a principal component analysis.
Prediction Using a Leave-One-Out Strategy
To test the generalizibility of the artificial neural network approach, we first performed a leave-one-out prediction strategy (Fig. 1A), where we left out each sample (of the 49 unique samples) one time during the training of artificial neural networks and tested it as an independent sample to predict the outcomes with all of the quality-filtered clones (n = 37,920) without additional clone selection.
Identification of Prognostic Signature Using Training and Test Sets
To identify the prognostic genes, we performed a separate artificial neural network analysis using a gene minimization procedure as described by Khan et al. (7). In brief, the 7 replicate samples were placed in the training set, and the remaining samples were then randomly partitioned into training (n = 35) and testing (n = 21) sets. None of the replicate samples were included in the test set to ensure that the selected genes did not bias the prediction outputs of the trained artificial neural networks. The minimal number of clones for outcome prediction was identified using only the training set. Quality-filtered clones were first ranked by determining the sensitivity of prediction of the 35 training samples with respect to a change in the gene expression level of each clone. Then, using increasing numbers of the top artificial neural network-ranked clones, we identified the minimum number of clones that generated minimum prediction errors (Fig. 1B). Where multiple clones represented one gene, we selected the top-ranked clone to obtain a minimal predictor gene set. We recalibrated the artificial neural networks using the expression ratios of these genes with only the training samples (without performing principal component analysis). Finally, we predicted the survival status of the test samples using the trained artificial neural networks (Fig. 1B).
Statistical Analysis for Survival
Survival length was calculated for the 49 unique neuroblastoma patients from date of diagnosis until date of death or last follow-up as appropriate. The probability of survival and significance was calculated using the Kaplan-Meier and Mantel-Haenszel methods, respectively (18, 19). The Cox proportional hazards model (20) was used to determine the hazard ratios and confidence intervals (21) for survival between the dichotomized groups of patients and was used to assess which factors were jointly significant in the association with survival for the 24 high-risk patients (20). The Cox model parameters (bi) were converted to hazard ratios by computing exp(bi), where exp(a) = 2.7183a. The 95% confidence interval for the hazard ratio was computed as [exp-1.96 (biL), exp(biH)] where biL = bi-1.96 [estimated SE (bi)] and biH = bi = [estimated SE (bi)] (21). In this study, the hazard ratio indicates the risk associated with neuroblastoma-caused death while being in a greater-risk category compared with that of being in the lower-risk category. Using the procedure described by Simon and Altman (22), a likelihood ratio test was used to assess for importance of the microarray prediction after adjusting for standard prognostic factors such as MYCN amplification, age, or stage.
RESULTS
Prediction of Outcome Using the Global Expression Profiles of All of the Clones
Visualization of all 56 of the neuroblastoma samples using principal component analysis of all of the quality-filtered 37,920 clones revealed neuroblastoma samples generally grouped according to their clinical outcomes (Fig. 2A), clearly indicating a pre-existent prognostic signature. To demonstrate the generalizability of the artificial neural network approach, we next tested the ability of artificial neural networks to predict prognosis of the 49 unique individuals (excluding 7 replicated samples) with all 37,920 clones using a conservative unbiased leave-one-out prediction strategy (Fig. 1A). We found that the artificial neural networks correctly predicted 16 of 19 poor-outcome and 27 of 30 good-outcome cases (Fig. 2B). This corresponds to a sensitivity of 84% and specificity of 90% for the poor-outcome patients, with a positive predictive value of 84% for the poor- and 90% for the good-outcome patients (Table 2). The Kaplan-Meier curves demonstrated that patients with poor and good gene expression signatures as identified by the artificial neural networks had significantly different survival probabilities (P < 0.0001, see Fig. 2C). The Cox proportional hazard ratio for the risk of death associated with the poor signature was 16.1 (95% confidence interval, 4.6 to 56.9, P < 0.0001), which was higher than those of all of the other risk factors we examined (stage, MYCN amplification, and age) except Shimada histology and was comparable with the Children’s Oncology Group risk stratification (Table 3; Fig. 2D).
Fig. 2.

Predicting the outcomes of neuroblastoma without gene selection. A, plot of the top three principal components (PC) of the 56 neuroblastoma samples using all quality-filtered 37,920 clones demonstrates some separation according to the clinical outcome. Red spheres represent poor-outcome patients, whereas blue spheres represent good-outcome patients. B, artificial neural network voting results for outcome prediction of the 49 unique neuroblastoma patients using 37,920 clones without any additional clone selection in a leave-one-out prediction scheme. (Samples labels: St, stage; NA, MYCN nonamplified; A, MYCN amplified, followed by sample name). Seven replicated samples (NB1, NB2, NB3, NB4, NB207, NB209, and NB210) were excluded for this analysis. Symbols, ANN average committee votes for each sample, whereas the length of the horizontal lines represents the SE. Red triangles, poor-outcome, and blue circles, good-outcome neuroblastomas. Vertical line at 0.5 is the decision boundary for outcome prediction (i.e., good signature < 0.5, poor signature >0.5). C, Kaplan-Meier curves of survival probability for the 49 neuroblastoma patients derived from the results in B. D, Kaplan-Meier curves of survival probability for the 49 neuroblastoma patients using the current Children’s Oncology Group risk stratification.
Table 2.
Performance of ANN prediction
| ANN prediction | Sensitivity (%) poor-outcome | Specificity (%) poor-outcome | Positive predictive value (%) poor-outcome | Positive predictive value (%) good-outcome |
|---|---|---|---|---|
| Leave-one-out with all clones (n = 49) | 84 | 90 | 84 | 90 |
| 19 genes (test samples: n = 21) | 100 | 94 | 83 | 100 |
| 19 genes (n = 49) | 100 | 97 | 95 | 100 |
Table 3.
Univariate proportional hazard analysis for the risk of NB-related death
| Variable | HR | 95% CI | Log-rank P |
|---|---|---|---|
| All NB samples (n = 49) | |||
| All 37920 Clones (poor signature versus good signature) | 16.1 | 4.6 to 56.9 | <0.0001 |
| Top 19 ANN-ranked genes (poor signature versus good signature) | ∞* | <0.0001 | |
| COG risk stratification (high risk versus low and intermediate risk) | 29.7 | 4.0 to 222.9 | <0.0001 |
| COG risk stratification (high and intermediate risk versus low risk) | 13.6 | 1.8 to 101.7 | 0.0009 |
| COG risk stratification (high risk versus low risk) | 23.2 | 3.1 to 175.9 | <0.0001 |
| INSS stage (stage 4 versus stages 1–3) | 7.1 | 2.1 to 24.2 | 0.0003 |
| INSS stage (stage 3 and 4 versus stage 1 and 2) | 13.6 | 1.8 to 101.7 | 0.0009 |
| MYCN status (amplified versus not amplified) | 9.8 | 3.6 to 26.7 | <0.0001 |
| Age (>1 year versus <1 year) | 12.3 | 1.6 to 92.5 | 0.0017 |
| Shimada histology (unfavorable versus favorable) (n = 27) | 19.9 | 2.4 to 166.1 | 0.0001 |
| High-risk samples (n = 24) | |||
| MYCN status (amplified versus not amplified) | 3.5 | 1.2 to 10.0 | 0.01 |
| Top 19 ANN-ranked genes (poor signature versus good signature) | ∞* | 0.0005 | |
| All 37920 clones (poor signature versus good signature) | 5.3 | 1.4 to 19.4 | 0.0067 |
NOTE. The Cox proportional hazards model was used to calculate all HRs and CIs. P values were calculated using the Mantel-Haenszel method.
Abbreviations: NB, neuroblastoma; COG, Children’s Oncology Group; INSS, International Neuroblastoma Staging System; HR, hazard ratio; CI, confidence interval.
These hazard ratios are infinite, because none of the patients predicted to have good-outcome experienced an event (i.e., death).
Identifying Prognostic Gene Expression Signature
To identify the optimal set of genes that results in the minimum classification errors, we performed a gene minimization procedure in a separate artificial neural network analysis using training and test sets as described previously (7). We first randomly partitioned all 56 of the samples into training (n = 35) and testing sets (n = 21) and used only the training set for the gene selection algorithm. We observed that the top 24 artificial neural network-ranked clones resulted in the minimal classification error (Fig. 3A). These 24 clones represented 19 unique genes, and we took the top-ranked clone for each gene and used this as our minimal gene set. When we visualized the overall variance of these genes using principal component analysis on all 56 of the samples we found a clearer separation of the poor- from the good-outcome samples when compared with the principal component analysis for all 37,920 clones (Fig. 3B).
Fig. 3.
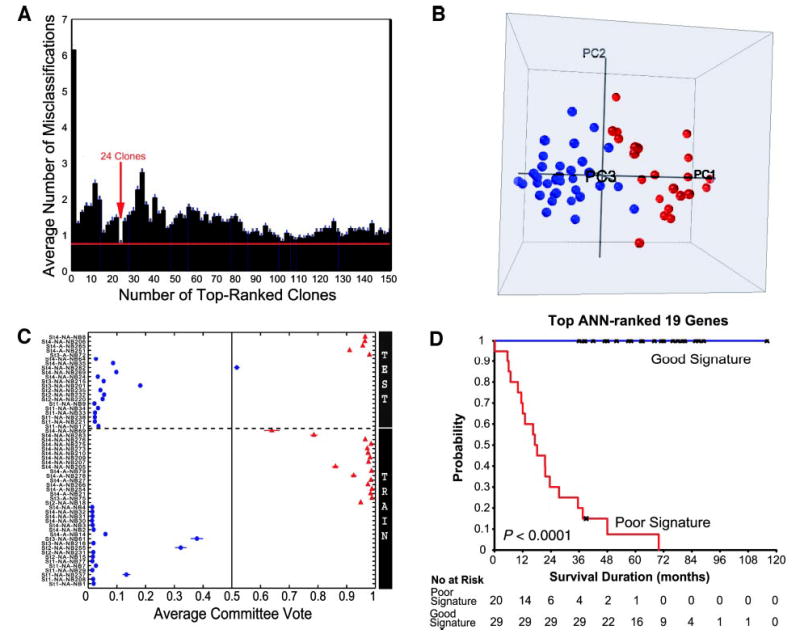
Outcome prediction using top artificial neural network (ANN)-ranked genes. A, clone minimization plot for ANN prediction. ANNs were first trained using the 35 training samples, and all 37,920 clones were ranked according to their importance to the ANN prediction (7). Then ANNs were trained with increasing numbers of the top ANN-ranked clones, and average prediction errors were calculated for the 35 training samples. The minimal number of clones that generated the minimal error rate was 24 (arrow), representing 19 unique genes. Bars, ±SE of number of misclassifications for the validation group during ANN training. B, plot of the top 3 principal components of the 56 neuroblastoma samples using the top 19 genes (duplicated clones of the same gene were removed, and the top-ranked clone for each gene was used in the ANN prediction) demonstrates a clear separation according to the clinical outcome. Red spheres, poor-outcome patients, whereas blue spheres, good-outcome patients. C, ANN committee vote results of the 56 samples using the top 19 ANN-ranked genes. ANNs were retrained using the 35 training samples (including all of the replicated samples) with the top 19 ANN-ranked genes directly without PCA, and these trained models were used to predict the 21 independent test samples. Horizontal dotted line divides the test (above the line) from the training samples. D. Kaplan-Meier curves for survival probability of the 49 patients were derived from the ANN prediction using the 19 genes in C.
We next recalibrated the artificial neural networks with the 35 training samples using the expression ratios for the 19 genes and correctly predicted the outcomes for 5 of 5 poor-outcome and 15 of 16 good-outcome patients in the independent test set, corresponding to a sensitivity of 100% and a specificity of 94% for predicting poor outcome (Fig. 3C; Table 2). The positive predictive values were 83% and 100% for the poor- and good-outcome groups, respectively, for the test samples and 95% and 100% for all of the patients (Table 2). The Kaplan-Meier curves demonstrated that patients with good and poor signatures based on the expression ratios of the 19 genes had significantly different survival probabilities (P < 0.0001, see Fig. 3D). Furthermore, no patients died in the good signature group; thus, the hazard ratio for death risk was infinite (Table 3).
The top 24 artificial neural network-ranked clones represent 19 unique genes including 12 known genes and 7 expressed sequence tags. Four of the known genes, DLK1, ARHI, PRSS3, and SLIT3, were represented by two or more independent cDNA clones (Fig. 4A) and, hence, acted as internal validation for these genes. We also validated all 12 of the known genes by quantified reverse transcription-PCR (data not shown). Nine of the genes were up-regulated and 10 down-regulated in the poor- compared with the good-outcome group (Fig. 4, A and B). To our knowledge, all of the genes, except MYCN and CD44 (23–25), have not been associated previously with neuroblastoma prognosis.
Fig. 4.
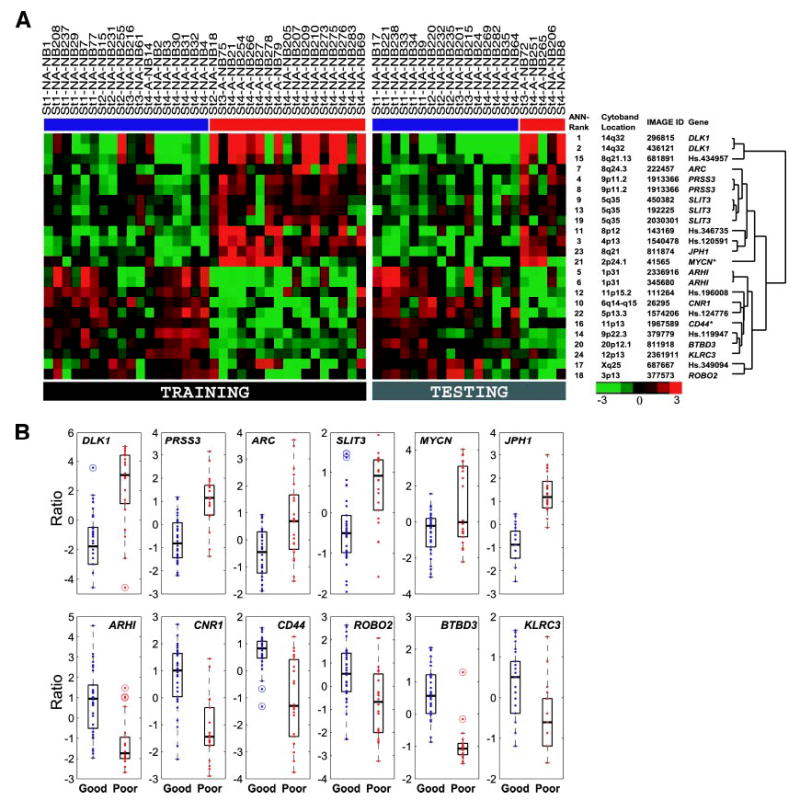
Expression levels of the top 19 ANN-ranked genes in the 56 neuroblastoma samples. A, expression level of each gene was logged (base 2) and mean-centered, and represented by pseudo-colors according to the scale shown on the bottom right. A red color corresponds to up-regulation, and a green color corresponds to down-regulation as compared with the mean. On the right are the artificial neural network (ANN)-ranked order, chromosomal location, IMAGE Ids, gene symbols, and the hierarchical clustering dendrogram. Red bars below the sample labels mark poor-outcome patients, and blue bars below the sample labels mark good-outcome patients. *, genes that have been reported previously to be associated with neuroblastoma prognosis. B, differentially expressed genes in good- and poor-prognostic groups. Box and whisker plots of the mean centered expression levels of the 12 known genes identified in this study. Boxes represent the upper and lower quartiles of the data. The black horizontal line within the box denotes the median. The whiskers extending above and below the box are fixed at 1.5 times the inter-quartile range. Outliers that fall outside the whiskers of the box are plotted as circles with a dot inside.
Outcome Prediction for High-Risk Patients
We next investigated whether the gene expression signatures could predict the survival status of those patients in our study that are currently stratified as high risk (see Table 1). From our 49 patients, 24 were high risk (Table 1). The Kaplan-Meier curves demonstrated that artificial neural networks were able to additionally partition these high-risk patients according to their clinical outcomes using all 37,920 of the quality-filtered clones (P = 0.0067), as well as the top 19 artificial neural network-ranked genes (P = 0.0005; Fig. 5, A and B). As shown in Fig. 5B, the top 19 artificial neural network-ranked genes were able to correctly predict all 5 with good signature as surviving and 18 of 19 with poor signature as dying, suggesting a potential benefit for predicting outcome in these high-risk patients. The hazard ratio was again infinite, because all of the patients that we predicted to have a good outcome survived (Table 3).
Fig. 5.
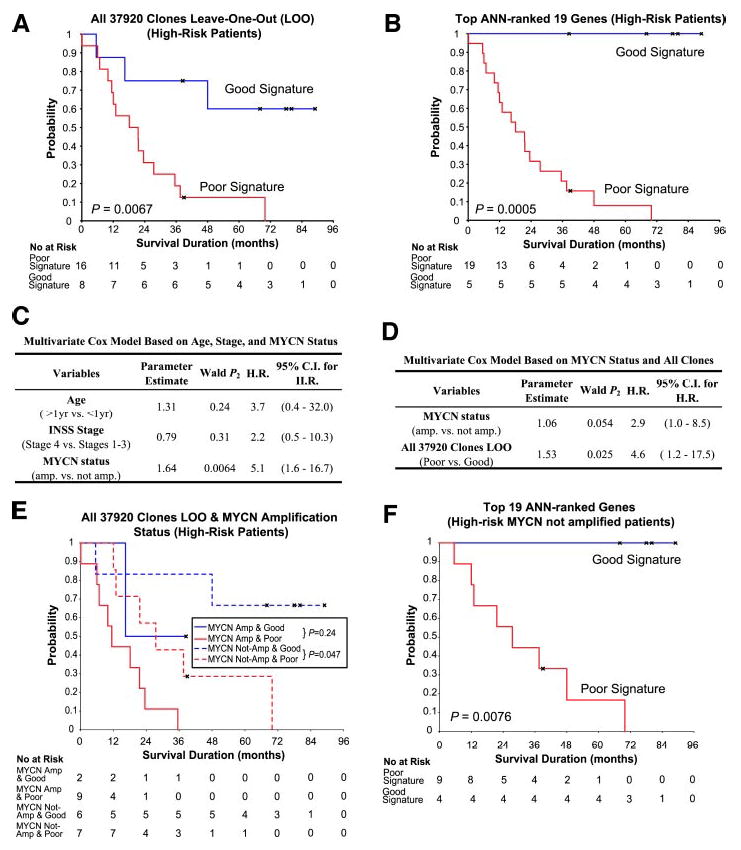
Artificial neural network (ANN)-based outcome prediction for the high-risk group. A, Kaplan-Meier curves of survival probability demonstrated that gene expression profiles without gene selection using all 37,920 clones could additionally separate the high-risk group (n = 24) into two statistically different subgroups according to their outcomes (P = 0.0067). B, Kaplan-Meier curves showed that the top 19 ANN-ranked genes produced statistically significant groupings (P = 0.0005) and were more accurate in predicting the prognosis than all 37,920 clones. C. Multivariate Cox proportional hazards model excluding the ANN prediction showed MYCN is the only statistically significant conventional risk factor in the high-risk patients in our data set (n = 24). (H.R., hazard ratio; C.I., confidence interval). D, multivariate Cox proportional hazards model based on MYCN status and all 37,920 clones ANN prediction. E, Kaplan-Meier curves for survival probability of the high-risk patients (n = 24) based on both MYCN status and the 37,920 clones ANN prediction. ANN-based prediction additionally separates patients without MYCN amplification (P = 0.047). F. Kaplan-Meier curves for survival probability of the MYCN nonamplified high-risk patients (n = 13) using the predictions based on the top 19 genes were statistically significant (P = 0.0076). The 19 genes predicted outcome more accurately than the 37,920 clones. MYCN amplified patients were not considered here, because there was only one amplified patient predicted to have a good-outcome, and, therefore, it was not possible to construct a Kaplan-Meier curve.
To determine whether the gene expression signatures provide additional predictive power over the conventional risk factors, we first created a Cox model using age, stage, and MYCN amplification excluding the artificial neural network prediction results. The model showed that MYCN amplification (P = 0.0064) was the only significant factor (i.e., P < 0.05, see Fig. 5C). Therefore, we built another multivariate model using MYCN amplification and the prediction results based on all 37,920 clones (Fig. 5D). (We used the artificial neural network results based on the 37,920 clones, because there were no deaths in the good signature group using the 19 genes, and in these circumstances it is not possible to create models where the hazard ratios are infinite). Applying the likelihood ratio test, we found that prediction by all of the clones added predictive ability to the model (P = 0.012). Additionally, the Kaplan-Meier curves (Fig. 5, E and F) illustrate that artificial neural network prediction can additionally separate the MYCN nonamplified patients according to their survival status based on either all of the clones (P = 0.047) or in particular the 19 genes (P = 0.0076, see Fig. 5F).
DISCUSSION
We have developed an artificial neural network-based method for predicting the outcome of patients with neuroblastoma using the expression profiles of only 19 genes that provides a significant improvement in prediction over the current known risk factors. Moreover, we found that the most important advantage of our approach was the ability to additionally partition Children’s Oncology Group stratified high-risk patients, in particular those without MYCN amplification, into two subgroups according to their survival status. The ability to predict the outcome of individual patients with high-risk neuroblastoma at initial diagnosis using gene expression signatures has major clinical implications, because ~70% of the patients in this group (~50% of all neuroblastoma patients) succumb to the disease (1). Firstly, patients that are identified to have a poor signature, i.e., predicted to die if given conventional therapy, may directly benefit from the newer therapeutic strategy trials that are currently under investigation by the cooperative study groups such as Children’s Oncology Group. Secondly, because treatment-related death rates have been reported to be as high as 23% (26), it may be possible to design future dose intensity reduction trials to minimize therapy-related morbidity and mortality for the high-risk patients who have a good signature. An example of such a patient in the latter category is NB14 (stage 4, MYCN-amplified) who, despite his high-risk status, experienced event-free survival for >3 years as was predicted by our artificial neural networks. Although the survival rate for patients with Children’s Oncology Group-stratified low-risk disease is 95%, our approach may identify the few patients predicted to have a poor outcome by the artificial neural networks who may benefit from more aggressive therapy. For instance, although case NB18 was classified as low-risk (based on stage 2 and MYCN not amplified), our artificial neural networks predicted this sample as poor-outcome, and this patient died within 1.5 years after diagnosis. These results indicate the potential utility of using our approach for individualized management of patients with cancer. However, they need to be interpreted with some caution in view of the limited number of subjects in our study and some heterogeneity of their treatments, and confirmation is required in larger, prospective trials before these predictor genes are used in the clinic.
Because there was some overlap in the expression levels of the top 19 artificial neural network-ranked genes between the prognostic groups, the prospect of identifying a single gene that can accurately predict outcome is unlikely. Thus, a combinatorial approach using several genes and artificial machine learning algorithms was necessary for accurate outcome prediction. Among these 19 genes, 2 (MYCN and CD44) have been reported to correlate with neuroblastoma prognosis (23–25), thus validating our ability to identify prognostic-specific genes. MYCN amplification is an established marker for high stage and poor outcome (23) and plays a critical role in the aggressive phenotype of neuroblastoma tumors (27, 28). Our analysis confirmed MYCN as an important prognostic marker (ranked 16 of 19); however, the median expression level of this gene was similar in the two groups, in agreement with previous reports that MYCN expression levels are not consistently correlated with survival in patients with nonamplified tumors (29–31). MYCN amplification is currently the only molecular marker used for risk stratification; however, it cannot be used as the sole risk predictor, because only 22% of neuroblastoma patients have this molecular trait.
Of the 19 predictor genes, 8 of the 12 known genes have been reported previously to be expressed in neural tissue. Of these, 5 were up-regulated in the poor-outcome group (DLK1, PRSS3, ARC, SLIT3, and MYCN), and 3 were down-regulated (CNR1, ROBO2, and BTBD3). DLK1 (ranked number 1) is the human homologue of the Drosophila Delta gene and is expressed by neuroblasts in the developing nervous system (32) as well as in neuroblastoma (33, 34). It is a transmembrane protein that activates the Notch signaling pathway, which has been shown to inhibit neuronal differentiation (35). Additionally, ARC (36), MYCN (37), and SLIT3 (38) are also expressed during neural development. The higher expression levels of these genes in the poor-outcome tumors suggest a more aggressive phenotype characterized by a less differentiated state, reminiscent of proliferating and migrating neural crest progenitors. Intriguingly, we observed the up-regulation of the neuron axon repellant gene, SLIT3, with the down-regulation of one of its receptors, ROBO2, in the poor-outcome group suggesting the possibility that these neuroblastoma cells secrete a substrate to repel connecting axons and potentially prevent differentiation. The exact roles and how the interactions of these genes confer an aggressive phenotype in neuroblastoma require more detailed biological studies.
Of additional interest, the ARHI gene, which maps to 1p31, is a maternally imprinted tumor suppressor gene implicated in ovarian and breast cancer (39), possibly through methylation silencing (40), and is among the down-regulated genes for the poor-outcome group. An additional study of its role in tumorigenesis as a potential tumor suppressor gene in neuroblastoma is warranted particularly because of its proximity to the 1p36 region, which is frequently deleted in poor-outcome neuroblastoma patients.
We noted the absence of three prognostic related genes reported previously TRKA, TRKB, and FYN (5, 41, 42), among our 19 genes. Unfortunately, TRKA was not on our microarrays, and TRKB and FYN were not ranked within the top 500 clones by artificial neural networks. At this point, the predictive role of TRKA, TRKB, or FYN is not conclusive, and none are currently used to guide therapy.
In this study we have identified a small subset of 19 predictor genes from a pool of 25,933 unique genes with the majority of these 19 genes showing a >2-fold average differential expression between good- and poor-outcome tumors. This small number of genes can be developed into cost-effective clinical assays for outcome prediction. In addition, the products of 3 genes (DLK1, SLIT3, and PRSS3) are secreted proteins, raising the possibility of using these as serum markers for prognosis.
In this data set, our artificial neural network-based method provided a significant improvement in prediction over the current risk factors in patients with neuroblastoma. Moreover, the most important advantage of our approach was the ability to additionally partition Children’s Oncology Group-stratified high-risk patients, in particular those without MYCN amplification, into two subgroups according to their survival status. These findings merit confirmation on larger, prospective trials. We believe that our approach would allow physicians to tailor therapy for each individual patient according to their molecular profile, with the prospect of improving clinical outcome and survival rates in patients with neuroblastoma.
Footnotes
Note: J. S. Wei, B. T. Greer, and F. Westermann contributed equally to this work.
References
- 1.Brodeur GM, Maris JM. Neuroblastoma. In: PA Pizzo and DG Poplack, editors. Principles and practice of pediatric oncology, 4th ed. Philadelphia: Lippincott-Raven; 2002. p. 895–937.
- 2.Schwab M, Westermann F, Hero B, Berthold F. Neuroblastoma: biology and molecular and chromosomal pathology. Lancet Oncol. 2003;4:472–80. doi: 10.1016/s1470-2045(03)01166-5. [DOI] [PubMed] [Google Scholar]
- 3.Westermann F, Schwab M. Genetic parameters of neuroblastomas. Cancer Lett. 2002;184:127–47. doi: 10.1016/s0304-3835(02)00199-4. [DOI] [PubMed] [Google Scholar]
- 4.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–16. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 5.Berwanger B, Hartmann O, Bergmann E, et al. Loss of a FYN-regulated differentiation and growth arrest pathway in advanced stage neuroblastoma. Cancer Cell. 2002;2:377–86. doi: 10.1016/s1535-6108(02)00179-4. [DOI] [PubMed] [Google Scholar]
- 6.Pearson AD, Philip T. Prognosis of low-risk and high-risk neuroblastoma. In: GM Brodeur, T Sawada, Y Tsuchida, PA Voute, editors. Neuroblastoma, 1st ed. Amsterdam, The Netherlands: Elsevier Science; 2000. p. 555.
- 7.Khan J, Wei JS, Ringner M, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–9. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramaswamy S, Tamayo P, Rifkin R, et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc Natl Acad Sci USA. 2001;98:15149–54. doi: 10.1073/pnas.211566398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature (Lond) 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 10.Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature (Lond) 2002;415:436–42. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 11.Beer DG, Kardia SL, Huang CC, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–24. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 12.Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–7. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 13.Wei JS, Khan J. Purification of total RNA from mammalian cells and tissues. In: D Bowtell, J Sambrook, editors. DNA microarrays: a molecular cloning manual. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2002. p. 110 –9.
- 14.Sotiriou C, Khanna C, Jazaeri AA, Petersen D, Liu ET. Core biopsies can be used to distinguish differences in expression profiling by cDNA microarrays. J Mol Diagn. 2002;4:30–6. doi: 10.1016/S1525-1578(10)60677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hegde P, Qi R, Abernathy K, et al. A concise guide to cDNA microarray analysis. Biotechniques. 2000;29:548–50. doi: 10.2144/00293bi01. 552–4, 556 passim. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Dougherty ER, Bittner ML. Ratio-based decisions and the quantitative analysis of cDNA microarray images. Biomedical Optics. 1997;2:364–74. doi: 10.1117/12.281504. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Kamat V, Dougherty ER, Bittner ML, Meltzer PS, Trent JM. Ratio statistics of gene expression levels and applications to microarray data analysis. Bioinformatics. 2002;18:1207–15. doi: 10.1093/bioinformatics/18.9.1207. [DOI] [PubMed] [Google Scholar]
- 18.Kaplan E, Meier P. Non-parametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- 19.Mantel M. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chem Rep. 1966;50:163–70. [PubMed] [Google Scholar]
- 20.Cox D. Regression models and life tables. J Royal Stat Soc (B) 1972;34:187–202. [Google Scholar]
- 21.Matthews DE, Farewell VT. Using and understanding medical statistics. 3rd ed. Basel: Karger, 1996. p. 150–60.
- 22.Simon R, Altman DG. Statistical aspects of prognostic factor studies in oncology. Br J Cancer. 1994;69:979–85. doi: 10.1038/bjc.1994.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–4. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- 24.Combaret V, Gross N, Lasset C, et al. Clinical relevance of CD44 cell-surface expression and N-myc gene amplification in a multicentric analysis of 121 pediatric neuroblastomas. J Clin Oncol. 1996;14:25–34. doi: 10.1200/JCO.1996.14.1.25. [DOI] [PubMed] [Google Scholar]
- 25.Munchar MJ, Sharifah NA, Jamal R, Looi LM. CD44s expression correlated with the International Neuroblastoma Pathology Classification (Shimada system) for neuroblastic tumours. Pathology. 2003;35:125–9. [PubMed] [Google Scholar]
- 26.Pinkerton CR, Blanc Vincent MP, Bergeron C, Fervers B, Philip T. Induction chemotherapy in metastatic neuroblastoma–does dose influence response? A critical review of published data standards, options and recommendations (SOR) project of the National Federation of French Cancer Centres (FNCLCC) Eur J Cancer. 2000;36:1808–15. doi: 10.1016/s0959-8049(00)00189-1. [DOI] [PubMed] [Google Scholar]
- 27.Schwab M, Ellison J, Busch M, Rosenau W, Varmus HE, Bishop JM. Enhanced expression of the human gene N-myc consequent to amplification of DNA may contribute to malignant progression of neuroblastoma. Proc Natl Acad Sci USA. 1984;81:4940–4. doi: 10.1073/pnas.81.15.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schweigerer L, Breit S, Wenzel A, Tsunamoto K, Ludwig R, Schwab M. Augmented MYCN expression advances the malignant phenotype of human neuroblastoma cells: evidence for induction of autocrine growth factor activity. Cancer Res. 1990;50:4411–6. [PubMed] [Google Scholar]
- 29.Nisen PD, Waber PG, Rich MA, Pierce S, Garvin JR, Jr, Gilbert F, Lanzkowsky P. N-myc oncogene RNA expression in neuroblastoma. J Natl Cancer Inst. 1988;80:1633–7. doi: 10.1093/jnci/80.20.1633. [DOI] [PubMed] [Google Scholar]
- 30.Slavc I, Ellenbogen R, Jung WH, et al. myc gene amplification and expression in primary human neuroblastoma. Cancer Res. 1990;50:1459–63. [PubMed] [Google Scholar]
- 31.Bordow SB, Norris MD, Haber PS, Marshall GM, Haber M. Prognostic significance of MYCN oncogene expression in childhood neuroblastoma. J Clin Oncol. 1998;16:3286–94. doi: 10.1200/JCO.1998.16.10.3286. [DOI] [PubMed] [Google Scholar]
- 32.Bettenhausen B, Hrabe de Angelis M, Simon D, Guenet JL, Gossler A. Transient and restricted expression during mouse embryogenesis of Dll1, a murine gene closely related to Drosophila Delta. Development. 1995;121:2407–18. doi: 10.1242/dev.121.8.2407. [DOI] [PubMed] [Google Scholar]
- 33.Helman LJ, Thiele CJ, Linehan WM, Nelkin BD, Baylin SB, Israel MA. Molecular markers of neuroendocrine development and evidence of environmental regulation. Proc Natl Acad Sci USA. 1987;84:2336–9. doi: 10.1073/pnas.84.8.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Limpt V, Chan A, Caron H, et al. SAGE analysis of neuroblastoma reveals a high expression of the human homologue of the Drosophila Delta gene. Med Pediatr Oncol. 2000;35:554–8. doi: 10.1002/1096-911x(20001201)35:6<554::aid-mpo13>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 35.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science (Wash DC) 1999;284:770–6. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 36.Lyford GL, Yamagata K, Kaufmann WE, et al. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron. 1995;14:433–45. doi: 10.1016/0896-6273(95)90299-6. [DOI] [PubMed] [Google Scholar]
- 37.Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H. Regulation of the neural crest cell fate by N-myc: promotion of ventral migration and neuronal differentiation. Development. 1997;124:1953–62. doi: 10.1242/dev.124.10.1953. [DOI] [PubMed] [Google Scholar]
- 38.Itoh A, Miyabayashi T, Ohno M, Sakano S. Cloning and expressions of three mammalian homologues of Drosophila slit suggest possible roles for Slit in the formation and maintenance of the nervous system. Mol Brain Res. 1998;62:175– 86. doi: 10.1016/s0169-328x(98)00224-1. [DOI] [PubMed] [Google Scholar]
- 39.Yu Y, Xu F, Peng H, et al. NOEY2 (ARHI), an imprinted putative tumor suppressor gene in ovarian and breast carcinomas. Proc Natl Acad Sci USA. 1999;96:214 –9. doi: 10.1073/pnas.96.1.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan J, Luo RZ, Fujii S, et al. Aberrant methylation and silencing of ARHI, an imprinted tumor suppressor gene in which the function is lost in breast cancers. Cancer Res. 2003;63:4174–80. [PubMed] [Google Scholar]
- 41.Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur Gm. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med. 1993;328:847–54. doi: 10.1056/NEJM199303253281205. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto K, Wada RK, Yamashiro JM, Kaplan DR, Thiele CJ. Expression of brain-derived neurotrophic factor and p145TrkB affects survival, differentiation, and invasiveness of human neuroblastoma cells. Cancer Res. 1995;55:1798–806. [PubMed] [Google Scholar]