

Abstract
While remaining extracellular, enteropathogenic Escherichia coli (EPEC) establish direct links with the cytoskeleton of the target epithelial cell leading to the formation of actin-rich pedestals underneath attached bacteria. The translocated adaptor protein Tir forms the transmembrane bridge between the cytoskeleton and the bacterium; the extracellular domain of Tir functions as a receptor for the bacterial adhesin intimin, while the intracellular amino and carboxy termini interact with a number of focal adhesion and other cytoskeletal proteins; and recruitment of some is dependent on phosphorylation of Tyr 474. Using Tir as bait and HeLa cell cDNA library as prey in a yeast two-hybrid screen, we identified cytokeratin 18 as a novel Tir partner protein. Cytokeratin 18 is recruited to the EPEC-induced pedestal and has a direct role in actin accretion and cytoskeleton reorganization. This study is the first to implicate intermediate filaments in microfilament reorganization following EPEC infection.
Introduction
Enteropathogenic (EPEC) and enterohaemorrhagic Escherichia coli (EHEC) are important causes of acute gastroenteritis in humans (Nataro & Kaper, 1998). By adhering to intestinal epithelial cells, EPEC and EHEC subvert cytoskeletal processes to produce attaching and effacing (A/E) lesions, characterized by localized destruction of brush border microvilli and intimate attachment of the extracellular bacteria to the plasma membrane of host epithelial cells (Frankel et al, 1998). The capacity to form A/E lesions is encoded at the locus of the enterocyte effacement (LEE) pathogenicity island (Elliott et al, 1998). The 5′ end of the main coding strand encodes a positive regulator, Ler, and structural components of a type III secretion system (TTSS), commonly found in pathogenic, Gram-negative bacteria (Hueck, 1998). The central part of the LEE encodes the outer membrane adhesin intimin (Elliott et al, 1998) and the translocated intimin receptor (Tir) (Kenny et al, 1997), while the 3′ end encodes additional TTSS structural, translocator and effector proteins.
Tir is delivered to the host cell plasma membrane where it adopts a hairpin topology with a 107-amino-acid external intimin-binding loop and intracellular amino and carboxy termini (Hartland et al, 1999; Kenny, 1999). Following translocation, TirEPEC is phosphorylated at both serine and tyrosine (Y474) residues, a process leading to conformational changes that alter Tir migration through polyacrylamide gels (Kenny, 1999; Warawa & Kenny, 2001). The tyrosine-phosphorylated form of Tir binds the host cell adaptor protein Nck (Gruenheid et al, 2001), which is required for the recruitment of neural Wiskott–Aldrich syndrome protein (N-WASP) and the actin-related protein (Arp)2/3 complex to initiate actin filament assembly and pedestal formation. Tir also binds the focal adhesion proteins talin (Huang et al, 2002) and α-actinin (Goosney et al, 2000). EHEC induces similar reorganization of the cytoskeleton in a Tir tyrosine phosphorylation-independent manner (Goosney et al, 2000). The aim of this study was to identify new host cell Tir partner proteins.
Results
Cytokeratin 18 is a novel Tir partner protein
To identify novel host cell proteins that interact with Tir, we performed a global yeast (strain PJ-69A) two-hybrid screen using Tir as bait (pICC10) and a human cDNA library as prey. Following growth of the co-transformants under conditions that select for protein interactions, prey plasmids were rescued from all emerging clones and retransformed into PJ-69A with or without pICC10. Analysis of the cDNA inserts of plasmids that conferred growth on selective media only in the presence of Tir revealed that the intermediate filament (IF) protein cytokeratin 18 (CK18) is a potential new Tir partner protein.
The function of the nonselective reporter LacZ was assessed in the yeast strains by measuring β-galactosidase activities. While low levels were detected in the host or single plasmid-bearing strains, a 12-fold increase was observed in the strain expressing Tir and CK18 (PJ69-4A pICC10–pICC267) (Fig 1).
Figure 1.
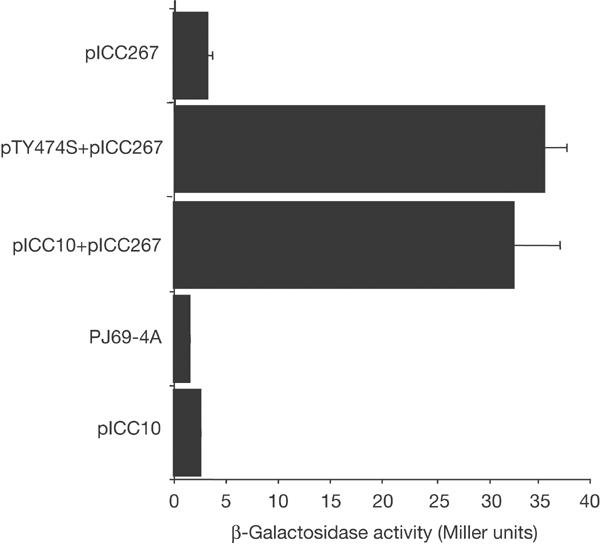
Yeast containing pICC10 (Tir) and pICC267 (CK18) and pTY474S and pICC267 demonstrated an approximately 12.5-fold increase in β-galactosidase activity compared to PJ69-4A alone or single plasmid-bearing strains.
We assessed whether binding of CK18 was Tir tyrosine phosphorylation dependent. PJ69-4A co-transformed with pNC905 (expressing TirY474S) and pICC267 resulted in a 13-fold increase in the level of β-galactosidase Miller units in comparison to single plasmid-bearing strains (Fig 1). These results show that CK18 has the potential to interact with both the phosphorylated and nonphosphorylated Tir forms in the yeast.
Co-immunoprecipitation of CK18 and Tir
To determine whether the observed interaction between Tir and CK18 is relevant to EPEC infection, cytoplasmic extracts of HeLa cells infected with wild-type (WT) EPEC were immunoprecipitated (IP) with mouse anti-CK18 or mouse preimmune serum as a nonspecific control (Fig 2A). As an additional control, cytoplasmic extracts of HeLa cells infected with a tir mutant strain were also IP with the anti-CK18 antiserum. Tir and CK18 were detected in cytoplasmic extracts of WT-infected HeLa cells before IP, while CK18, but not Tir, was observed in extracts of cells infected with the tir mutant (Fig 2A). Only after infection with WT was specific co-IP of Tir with the CK18 antiserum observed. Alternative Tir forms, due to host-mediated phosphorylation (Kenny, 1999; Warawa & Kenny, 2001), were visible in the IP material, implying that CK18 interacts with both the phosphorylated and nonphosphorylated Tir forms in infected cells. In a reciprocal experiment, Tir antiserum was used to IP soluble cytoplasmic and membrane extracts of WT-infected cells. Probing the IP content with antibodies specific for cytoskeletal components revealed that in addition to CK18 (Fig 2B), CK8 was co-IP (Fig 2C) in both soluble cytoplasmic and membrane extracts of WT- infected HeLa cells but not with the tir mutant. The CK18 antibody recognized smaller possible breakdown products in the membrane extracts, but these were not co-precipitated with Tir (Fig 2B). However, the anti-Tir precipitate did include a higher molecular weight form of CK8 detected at approximately 97 kDa by the monoclonal antibody (Fig 2C). The monomeric form of actin (42 kDa) was also co-precipitated with Tir (Fig 2D). In contrast, both α- and β-tubulins were not detected in the IP material (data not shown).
Figure 2.

Co-immunoprecipitation of Tir and CK18. (A) Western blots and IP/western blots of membrane extracts of Δtir- and WT EPEC-infected HeLa cells: lanes 1 and 2 are Δtir- and WT-infected extracts, respectively, detected with anti-CK18; lanes 3–5 are IP/western blots detected with anti-Tir, where lane 3 is a WT-infected extract first IP with preimmune serum and lanes 4 and 5 are Δtir- and WT-infected extracts, respectively, IP with anti-CK18; lanes 6 and 7 are western blots of Δtir- and WT-infected extracts, respectively, detected with anti-Tir. Tir was specifically co-IP with the CK18 antibodies. (B) Western blot of membrane and cytoplasmic extracts of Δtir- and WT EPEC-infected HeLa cells detected with anti-CK18: lanes 1, 2 and 5, 6 represent cytoplasmic extracts, and lanes 3, 4 and 7, 8 are membrane extracts of Δtir (lanes 1, 3, 5 and 7)- and WT (lanes 2, 4, 6 and 8)-infected HeLa cells. Lanes 5–8 were first IP with anti-Tir. CK18 was specifically co-IP with the Tir antiserum. (C) Western blots of membrane and cytoplasmic extracts of Δtir- and WT EPEC-infected HeLa cells detected with anti-CK8. Lanes 1, 3, 5 and 7 represent membrane extracts and lanes 2, 4, 6 and 8 are cytoplasmic extracts of Δtir (lanes 3, 4, 7 and 8)- and WT (lanes 1, 2, 5 and 6)-infected HeLa cells. Lanes 5–8 were first IP with anti-Tir. A higher molecular weight form, at approximately 97 kDa, of CK8 was co-IP with the Tir antiserum. (D) Western blots of membrane and cytoplasmic extracts of Δtir- and WT EPEC-infected HeLa cells detected with anti-actin. Lanes 1, 3, 5 and 7 represent membrane extracts and lanes 2, 4, 6 and 8 are cytoplasmic extracts of Δtir (lanes 3, 4, 7 and 8)- and WT (lanes 1, 2, 5 and 6)-infected HeLa cells. Lanes 5–8 were first IP with anti-Tir. Monomeric actin was specifically co-IP with the Tir antiserum.
Cytokeratins are recruited to the EPEC-induced pedestals
We examined whether CK18 is recruited to the EPEC-induced pedestals. HeLa and intestinal epithelial INT407 cells were infected with WT EPEC and triple stained with either anti-bacterial/CK18/Arp3 or anti-bacterial/CK18/Tir antibodies, and examined by confocal microscopy. In both cell lines, concentrated CK18 was found to be colocalized with Tir and Arp3 beneath adherent EPEC (Fig 3).
Figure 3.
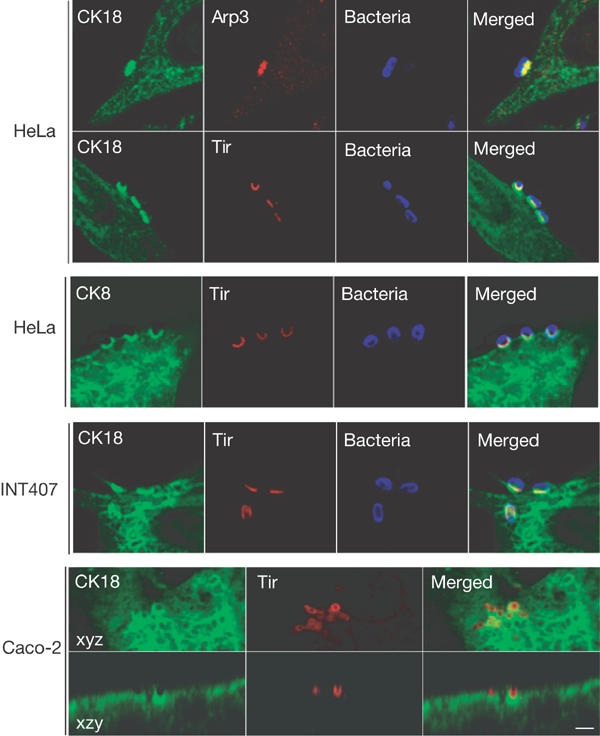
CK18 and CK8 are recruited to EPEC-induced pedestals. Cells were infected with the EPEC strain E2348/69 for 3 h and immunostained for CK18 or CK8 (green), Arp3 or Tir (red), and bacteria (blue). In HeLa cells, CK18 and CK8 are concentrated in EPEC-induced pedestals and show colocalization with Arp3 or Tir. Colocalization of Tir and CK18 in EPEC-induced pedestals was also observed in the human intestinal epithelial INT407 and Caco-2 cell lines. Scale bar corresponds to 2 μm.
We examined recruitment of CK18 to the EPEC-induced pedestal following infection of polarized intestinal Caco-2 brush border cells. Double staining with anti-Tir and anti-CK18 antibodies revealed colocalization underneath attached bacteria (Fig 3). CK8 is a partner protein of CK18 in IF expressed by epithelial cells (Yoon et al, 2001). Moreover, CK8 was co-IP with anti-Tir following EPEC infection (Fig 2). Consistent with these, immunostaining revealed that CK8 is also recruited to the EPEC-induced pedestal (Fig 3).
Tir, tyrosine phosphorylation and recruitment of CK18
We investigated whether recruitment of CK18 to the EPEC-induced pedestal is Tir dependent. HeLa cells were infected with WT and tir mutant strains. Association of CK18 was determined for 100 adhering bacteria, with each infection being repeated in triplicate. CK18 was colocalized with Tir in 87.2% of bacteria following infection with WT (Figs 4, 5). CK18 did not colocalize with adherent bacteria following infection with the tir mutant, but colocalization was restored when the tir mutant was complemented in trans (Figs 4,5). Similarly, no colocalization of CK18 with adherent bacteria was observed following infection of HeLa cells with an espA mutant (TTSS deficient), but the phenotype was restored upon reintroduction of espA on a plasmid (Fig 5). No CK18 was observed in association with adherent bacteria following infection of HeLa cells with EPEC expressing TirY474S (Kenny, 1999) (Fig 4). Importantly, and despite the fact that TirEHEC lacks a Y474 homologue and is not tyrosine phosphorylated, immunostaining of HeLa cells infected with EHEC O157:H7 revealed that CK18 was recruited to the EHEC-induced pedestal (Fig 4).
Figure 4.
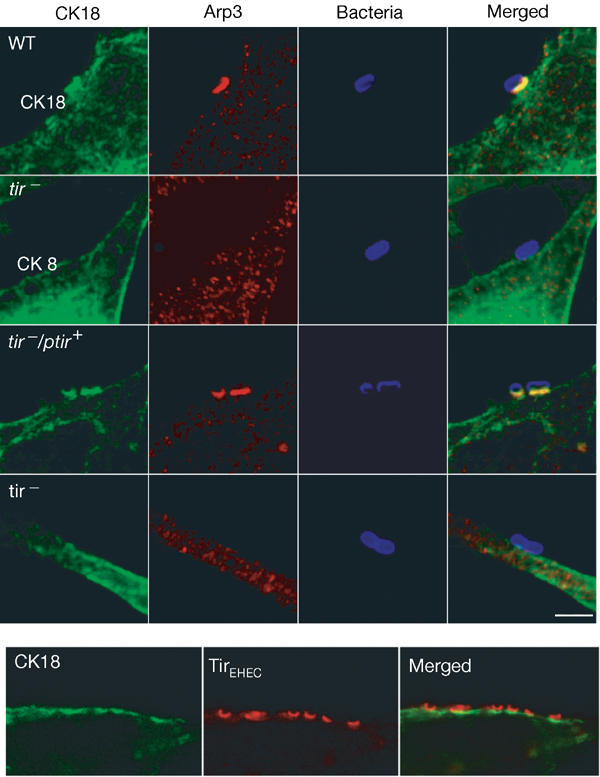
CK18 recruitment to the EPEC pedestal is Tir dependent. HeLa cells were infected with the EPEC strain E2348/69 (WT), tir mutant (tir−), tir mutant complemented in trans (tir−/ptir) or tir mutant complemented in trans with a Tir derivative carrying a Y474S substitution (tir−/ptirY474S). Cells were infected for 3 h and stained for CK18 (green), Arp3 (red) and bacteria (blue). CK18 recruitment in EPEC-induced pedestals was only detected for the WT strain and the complemented tir mutant (tir−/ptir). CK18 is also recruited to, and colocalized with, Tir in the EHEC-induced pedestal. Scale bar corresponds to 2 μm.
Figure 5.

Percentage of EPEC bacteria presenting CK18 at the attachment site in HeLa cells. CK18 was scored for the EPEC strain E2348/69 (WT), the mutant strain UMD872 (espA−), UMD872 complemented in trans (espA−/pespA), the tir EPEC mutant (tir−), the tir EPEC mutant complemented in trans (tir−/ptir) or tir mutant complemented in trans with a Tir derivative carrying a Y474S substitution (TirY474S). The results shown are the means±s.d. of three independent experiments in which a total of 300 bacteria attached to the cell periphery were examined.
CK18 is necessary for A/E lesion formation
To determine the functional significance of recruitment of CK18 to the EPEC-induced pedestal, we used the small interfering RNAs (siRNAs), which provide a sequence-specific, post-transcriptional, gene-silencing mechanism. HeLa cells were transfected with a CK18-specific RNA duplex, infected with WT EPEC, and then triple stained using anti-bacterial/CK18/Arp3 antibodies; Arp3 staining was used as a marker for pedestal formation. Staining for CK18 revealed a transfection efficiency of less than 50%; silencing was confirmed by western blotting (not shown). In transfected cells exhibiting reduced or no CK18 staining, pedestals were observed in 29±1.5% of the adherent bacteria whereas 63±3.6% of adherent bacteria were associated with Arp3 in untransfected cells.
Pedestals formed in transfected cells not completely depleted in CK18 were shorter and the Arp3 staining was less intense (Fig 6). Quantifying fluorescent intensity using confocal microscopy revealed a direct relationship between the intensity of CK18 and Arp3 in the EPEC-induced pedestals; in untransfected cells, the average fluorescent intensity in the pedestal was 108.2 grey value per pixel for Arp3 and 67.6 for CK18 compared with 42.5 and 27.3, respectively, in transfected cells (P<0.001). These results suggest that CK18 is involved in the organization of other cytoskeletal proteins within the actin pedestal and seems to have a major role in the generation of fully formed pedestals.
Figure 6.

Functional role of CK18 in EPEC-induced pedestals. HeLa cells were transfected with CK18 siRNA. At 72 h after transfection, cells were infected for 3 h with the EPEC strain E2348/69 and stained for CK18 (green), Arp3 (red) and bacteria (blue). Transfected cells are indicated with asterisks. CK18 silencing blocked EPEC-induced pedestals (upper panel) or resulted in the formation of small and weakly stained pedestals (lower panel, arrow). Scale bar corresponds to 2 μm.
Discussion
Binding of EPEC and EHEC to epithelial cells is associated with reorganization of host cell cytoskeletal proteins (Goosney et al, 2001). The cytoskeleton of higher eukaryotic cells is composed of three major cytoskeletal systems: actin microfilaments, microtubules and IFs; IFs are considered to be a major contributor to the mechanical integrity of cells and tissues (Fuchs & Cleveland, 1998). In this study, we have shown that the IF protein CK18 interacts with both the phosphorylated and nonphosphorylated forms of Tir. The reason why CK18 was not effectively recruited to the site of bacterial adhesion following infection with EPEC (TirY474S) is not clear, especially as CK18 was recruited to the EHEC-induced pedestal in a Tir phosphorylation-independent manner. However, differences in biological activities between the two Tir forms are well established. It is therefore possible that TirEPEC(Y474S) adopts a conformation not permissive for CK18 binding following membrane targeting, or that other interacting proteins might displace CK18 from this Tir mutant or are needed to stabilize the complex. Nonetheless, the fact that CK18 is recruited to the site of intimate bacterial attachment, that Tir is co-IP with CK18 and that depletion of CK18 abolished A/E lesion formation suggests that the interaction between Tir and CK18 is not only involved in the maintenance of the pedestal but rather has a direct role in its formation.
Although the mechanism through which IFs are involved in actin dynamics is not well defined, our recent finding that Tir, CK18 and 14-3-3 form a complex in EPEC-infected cells (unpublished data) might provide some clues. Indeed, members of the 14-3-3 family represent a group of highly conserved and ubiquitous proteins known to modulate the interactions between proteins involved in fundamental cell signalling processes including cytoskeletal structure (Aitken, 2002).
In studies involving cultured cells and transgenic mouse models, phosphorylation has been shown to regulate cytokeratin filament dynamics, organization and function. Since cytokeratins are substrates for a large number of protein kinases and phosphatases engaged in signal transduction pathways, they are thought to be involved in cell signalling (Paramio & Jorcano, 2002). In this regard, CK8 and CK18, which are the dominant cytokeratins in HeLa cells, have recently been described to affect tumour necrosis factor (TNF) signalling by binding the cytoplasmic domain of TNFR2 and moderating TNF-mediated NFκB and JNK activation (Paramio & Jorcano, 2002). Significantly, it was reported that following infection of T84 cells, EPEC activates NFκB and this activity was dependent on a functional TTSS (Savkovic et al, 1997). Moreover, recent studies have implicated CK8 and CK18 in Salmonella typhimurium cell invasion (Scherer et al, 2000; Carlson et al, 2002). In these studies, the translocator protein SspC (also known as SipC) was shown to bind both IF proteins. Antibody specificity and the staining methods used are likely reasons as to why, in a previous study, Finlay et al (1992) were unable to demonstrate IF recruitment to EPEC pedestals.
The precise role that the association between Tir and CK18 plays during EPEC infection is not known. However, considering that the high molecular weight form of CK8 (representing IF-associated protein) was co-IP with anti-Tir and that CK8 was also recruited to the pedestal, it is possible that CK18 connects Tir directly to IF. Therefore, it appears that Tir bridges the extracellular EPEC and EHEC bacteria with both the microfilament and the IF networks.
Methods
Bacterial strains and plasmids. The bacterial strain and plasmids used in this study are listed in Table 1.
Table 1.
List of strains and plasmids
| Description | Reference | |
|---|---|---|
|
Strains |
|
|
| EPEC E2348/69 |
Wild type |
Levine et al (1978) |
| EPEC tir |
tir mutant |
Kenny et al (1997) |
| UMD872 |
espA mutant |
Kenny et al (1996) |
| EHEC 85-170 |
Wild type |
Tzipori et al (1987) |
|
Plasmids |
|
|
| pACYC-tir deltaBamHI |
Cloned tir |
Kenny et al (2002) |
| Y4-S pBluescript-tir |
Cloned tir Y474S |
Kenny (1999) |
| pMSD2 |
Cloned espA |
Kenny et al (1996) |
| pICC10 |
pGBT9-tir |
Hartland et al (1999) |
| pICC267 |
pGAD-GH-CK18 |
This study |
| pNC905 | pGBT9-tir Y474S | This study |
Preparation of cDNA library and yeast two-hybrid screen. RNA was prepared from HeLa cell cultures. Polyadenylated RNAs were selected using the poly(A) Quik mRNA isolation kit (Stratagene). Oligo(dT)25 (dA/dC/dG) was used to prime first-strand cDNA synthesis by Moloney murine leukaemia virus reverse transcriptase, and second-strand synthesis utilized a mixture of T4DNA polymerase and RNaseH. The cDNAs were ligated to an EcoR1 adapted linker and cloned into pGAD-GH (Clontech). The pGAD-GH cDNA clones were amplified in E. coli SURE (Stratagene) before plasmid DNA was transformed into yeast (PJ69-4A). The bait and prey plasmids were used in the yeast two-hybrid screen as described (Hartland et al, 1999).
Identification of the prey proteins. The cDNA-containing plasmids were rescued from His+ and Ade+ colonies into E. coli HB101. Each of the prey plasmids was then retransformed into the PJ69-4A either on its own (to determine possible self-activation) or with pICC10 (to confirm the results of the general screen).
Activation of the lacZ reporter was assessed by quantification of β-galactosidase activity in cell extracts (Hartland et al, 1999). In each case, activation of lacZ was determined in triplicate for three colonies.
Inserts were sequenced using pGAD-specific forward and reverse primers.
Immunoprecipitation HeLa cells were cultured in DMEM/10% FCS, infected (at a multiplicity of infection of 100) for 3.5 h with EPEC, and mechanically fractionated into membrane and cytoplasmic fractions according to the method of Gauthier et al (2000).
Extracts were precleared with immobilized protein A before IP. Tir and CK18 were IP from EPEC-infected HeLa cell extracts (10–12 mg protein) in RIPA buffer (1% (v/v) Triton X-100; 50 mM Tris–HCl, pH 7.6; 400 μM NaVO4; 100 g ml−1 phenylmethylsulphonyl fluoride; 10 mM leupeptin) with either anti-Tir (Hartland et al, 1999) or anti-CK18 antibody (sc-6259; Santa Cruz) and immobilized protein A (Life Technologies, UK). Western blots were probed with either mouse monoclonal anti-CK18 (ab7797; AbCam) or anti-CK8 (MAB 3414; Chemicon International) or anti-actin (MAB 3128; Chemicon International) and alkaline phosphatase-conjugated secondary antibodies (Sigma).
Immunostaining. HeLa, INT407 and Caco-2 cells were grown and infected with EPEC and EHEC as previously described (Hartland et al, 1999). Cells were fixed in cold methanol for 4 min at −20°C, washed in PBS and immunostained using monoclonal CK8 or CK18 (clone CY-90; Sigma), which reacted with a single band in western blots of whole-cell extracts, and rabbit polyclonal Arp3 or Tir antibodies. Samples were analysed using a confocal laser scanning microscope (LSM510, Zeiss or TCS SP2, Leica).
Small interfering RNAs. siRNA oligonucleotides targeting human CK18 were synthesized and used as described (Harborth et al, 2001). At 72 h after transfection, cells were infected as described above.
Quantification of the fluorescence intensity was determined using confocal microscopy by selecting the more intense area of the attachment site of the bacteria. The results given are representative of two separate experiments performed in which a total of at least 100 pedestals present at the cell periphery were examined.
Acknowledgments
We thank C. Sasakawa for the Arp3 antibodies, B. Kenny for strains and plasmids, and A. Servin for cell lines. This project was supported by the European Union Fifth Framework Quality of Life Program (QLK2-2000-00600) and by the Wellcome Trust. J.G. is supported by a Marie Curie fellowship from the European Commission.
References
- Aitken A (2002) Functional specificity in 14-3-3 isoform interactions through dimer formation and phosphorylation. Chromosome location of mammalian isoforms and variants. Plant Mol Biol 50: 993–1010 [DOI] [PubMed] [Google Scholar]
- Carlson SA, Omary MB, Jones BD (2002) Identification of cytokeratins as accessory mediators of Salmonella entry into eukaryotic cells. Life Sci 70: 1415–1426 [DOI] [PubMed] [Google Scholar]
- Elliott SJ, Wainwright LA, McDaniel TK, Jarvis KG, Deng YK, Lai LC, McNamara BP, Donnenberg MS, Kaper JB (1998) The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol Microbiol 28: 1–4 [DOI] [PubMed] [Google Scholar]
- Finlay BB, Rosenshine I, Donnenberg MS, Kaper JB (1992) Cytoskeletal composition of attaching and effacing lesions associated with enteropathogenic Escherichia coli adherence to HeLa cells. Infect Immun 60: 2541–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel G, Phillips AD, Rosenshine I, Dougan G, Kaper JB, Knutton S (1998) Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol Microbiol 30: 911–921 [DOI] [PubMed] [Google Scholar]
- Fuchs E, Cleveland DW (1998) A structural scaffolding of intermediate filaments in health and disease. Science 279: 514–519 [DOI] [PubMed] [Google Scholar]
- Gauthier A, de Grado M, Finlay BB (2000) Mechanical fractionation reveals structural requirements for enteropathogenic Escherichia coli Tir insertion into host membranes. Infect Immun 68: 4344–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goosney DL, DeVinney R, Pfuetzner RA, Frey EA, Strynadka NC, Finlay BB (2000) Enteropathogenic E. coli translocated intimin receptor, Tir, interacts directly with α-actinin. Curr Biol 10: 735–738 [DOI] [PubMed] [Google Scholar]
- Goosney DL, DeVinney R, Finlay BB (2001) Recruitment of cytoskeletal and signaling proteins to enteropathogenic and enterohemorrhagic Escherichia coli pedestals. Infect Immun 69: 3315–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenheid S, DeVinney R, Bladt F, Goosney D, Gelkop S, Gish GD, Pawson T, Finlay BB (2001) Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nature Cell Biol 3: 856–859 [DOI] [PubMed] [Google Scholar]
- Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci 114: 4557–4565 [DOI] [PubMed] [Google Scholar]
- Hartland EL, Batchelor M, Delahay RM, Hale C, Matthews S, Dougan G, Knutton S, Connerton I, Frankel G (1999) Binding of intimin from enteropathogenic Escherichia coli to Tir and to host cells. Mol Microbiol 32: 151–158 [DOI] [PubMed] [Google Scholar]
- Huang L, Mittal B, Sanger JW, Sanger JM (2002) Host focal adhesion protein domains that bind to the translocated intimin receptor (Tir) of enteropathogenic Escherichia coli (EPEC). Cell Motil Cytoskeleton 54: 255–265 [DOI] [PubMed] [Google Scholar]
- Hueck CJ (1998) Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Res 62: 379–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny B (1999) Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modifications. Mol Microbiol 31: 1229–1241 [DOI] [PubMed] [Google Scholar]
- Kenny B, Lai LC, Finlay BB, Donnenberg MS (1996) EspA, a protein secreted by enteropathogenic Escherichia coli, is required to induce signals in epithelial cells. Mol Microbiol 20: 313–323 [DOI] [PubMed] [Google Scholar]
- Kenny B, DeVinney R, Stein M, Reinscheid DJ, Frey EA, Finlay BB (1997) Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91: 511–520 [DOI] [PubMed] [Google Scholar]
- Kenny B, Ellis S, Leard AD, Warawa J, Mellor H, Jepson MA (2002) Co-ordinate regulation of distinct host cell signalling pathways by multifunctional enteropathogenic Escherichia coli effector molecules. Mol Microbiol 44: 1095–1107 [DOI] [PubMed] [Google Scholar]
- Levine MM, Bergquist EJ, Nalin DR, Waterman DH, Hornick RB, Young CR, Sotman S (1978) Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet i: 119–122 [DOI] [PubMed] [Google Scholar]
- Nataro JP, Kaper JB (1998) Diarrheagenic Escherichia coli. Clin Microbiol Rev 11: 142–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramio JM, Jorcano JL (2002) Beyond structure: do intermediate filaments modulate cell signalling? BioEssays 24: 836–844 [DOI] [PubMed] [Google Scholar]
- Savkovic SD, Koutsouris A, Hecht G (1997) Activation of NF-κB in intestinal epithelial cells by enteropathogenic Escherichia coli. Am J Physiol 273: C1160–C1167 [DOI] [PubMed] [Google Scholar]
- Scherer CA, Cooper E, Miller SI (2000) The Salmonella type III secretion translocon protein SspC is inserted into the epithelial cell plasma membrane upon infection. Mol Microbiol 37: 1133–1145 [DOI] [PubMed] [Google Scholar]
- Tzipori S, Karch H, Wachsmuth KI, Robins-Browne RM, O'Brien AD, Lior H, Cohen ML, Smithers J, Levine MM (1987) Role of a 60-megadalton plasmid and Shiga-like toxins in the pathogenesis of infection caused by enterohemorrhagic Escherichia coli O157:H7 in gnotobiotic piglets. Infect Immun 55: 3117–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warawa J, Kenny B (2001) Phosphoserine modification of the enteropathogenic Escherichia coli Tir molecule is required to trigger conformational changes in Tir and efficient pedestal elongation. Mol Microbiol 42: 1269–1280 [DOI] [PubMed] [Google Scholar]
- Yoon KH, Yoon M, Moir RD, Khuon S, Flitney FW, Goldman RD (2001) Insights into the dynamic properties of keratin intermediate filaments in living epithelial cells. J Cell Biol 153: 503–516 [DOI] [PMC free article] [PubMed] [Google Scholar]