

Abstract
Soluble methane monooxygenase (sMMO) of Methylosinus trichosporium OB3b is a three-component oxygenase that catalyses the O2- and NAD(P)H-dependent oxygenation of methane and numerous other substrates. Despite substantial interest in the use of genetic techniques to study the mechanism of sMMO and manipulate its substrate specificity, directed mutagenesis of active-site residues was previously impossible because no suitable heterologous expression system had been found for expression in a highly active form of the hydroxylase component, which is an (αβγ)2 complex containing the binuclear iron active site. A homologous expression system that enabled the expression of recombinant wild-type sMMO in a derivative of M. trichosporium OB3b from which the chromosomal copy of the sMMO-encoding operon had been partially deleted was previously reported. Here we report substantial development of this method to produce a system for the facile construction and expression of mutants of the hydroxylase component of sMMO. This new system has been used to investigate the functions of Cys 151 and Thr 213 of the α subunit, which are the only nonligating protonated side chains in the hydrophobic active site. Both residues were found to be critical for the stability and/or activity of sMMO, but neither was essential for oxygenation reactions. The T213S mutant was purified to >98% homogeneity. It had the same iron content as the wild type and had 72% wild-type activity toward toluene but only 17% wild-type activity toward propene; thus, its substrate profile was significantly altered. With these results, we have demonstrated proof of the principle for protein engineering of this uniquely versatile enzyme.
Methane monooxygenases (MMOs) are unique among known catalytic systems in their ability to force the unreactive hydrocarbon methane to react with molecular oxygen under ambient conditions to yield methanol as the sole oxygenation product (8). MMOs are found in methanotrophs, which are bacteria that appear to be ubiquitous in the environment (39) and can grow with methane as the sole source of carbon and energy. MMOs catalyze the first step in the methane oxidation pathway (17). Methanotrophs such as Methylosinus trichosporium OB3b can produce two forms of MMO, the membrane-associated or particulate form (pMMO) and the soluble form (sMMO) (47). sMMO also has many potential applications in synthetic organic chemistry and bioremediation because it can cooxidize a very wide range of adventitious substrates, including alkanes, alkenes, alcohols, ethers, alicyclics, and aromatics (7, 48) and chlorinated organic compounds, such as the pollutant trichloroethylene (11).
sMMO catalyzes the NAD(P)H-dependent oxygenation of methane and other substrates (X) in the following reaction:
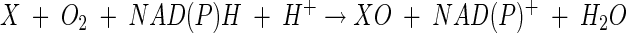 |
The sMMO enzyme complex has three components. (i) The 250-kDa hydroxylase has an (αβγ)2 structure, in which each α subunit contains a μ-(hydr)oxo-bridged binuclear iron center that is the site of oxygen and substrate activation. (ii) The 37-kDa reductase has FAD and Fe2S2 prosthetic groups and supplies reducing equivalents from NAD(P)H to the hydroxylase. (iii) Protein B, or the coupling-gating protein, is a 15-kDa polypeptide that has no prosthetic groups, binds to the hydroxylase, and is necessary for efficient sMMO catalysis (13, 23, 28).
Despite much interest and speculation and the availability of high-resolution X-ray crystal structures for the sMMO hydroxylases from two methanotrophs (M. trichosporium OB3b [9] and Methylococcus capsulatus Bath [41, 42]), there have been no experimental studies of the roles played by individual amino acids in the hydroxylase in determining the unique catalytic properties of sMMO. Until fairly recently, the problem has been that a suitable genetically tractable expression system, a prerequisite for protein engineering, was not available for the sMMO hydroxylase. Previous studies found that this component was inactive when expressed in Escherichia coli (50). The first reports of expression of the genes encoding the sMMO gene cluster of M. trichosporium in a heterologous host were by Wood and colleagues, who demonstrated the degradation of trichloroethylene by sMMO expressed in Pseudomonas putida, Agrobacterium tumefaciens, and Rhizobium meliloti (19, 20). We subsequently described a homologous expression system for the wild-type sMMO hydroxylase, in which recombinant (wild-type) sMMO genes were expressed, yielding highly active sMMO in a derivative of M. trichosporium known as mutant F (26). Mutant F could be propagated with methane as the growth substrate because at a high copper-to-biomass ratio, it oxidized methane with pMMO. When a plasmid-encoded copy of the sMMO operon (5) was introduced by means of conjugation and the resulting strain was cultured at a low copper-to-biomass ratio, pMMO became down-regulated and recombinant sMMO was expressed from its natural copper-repressed promoter (26). Thus, we proved that, in principle, a homologous expression system such as this could be used for the expression of mutant sMMO genes.
In order to test the practicability of such a mutagenesis regimen, Cys 151 and Thr 213 in the α subunit of the sMMO hydroxylase were chosen as targets for the initial mutagenesis experiments. The binuclear iron center is ligated by four Glu and two His residues and three solvent molecules, which lie in a solvent-accessible cavity lined by hydrophobic residues. The proximity of this cavity to the site of oxygen activation (9), together with molecular docking studies (14), identifies it as the most likely site for the binding of methane and other substrates. Cys 151 and Thr 213 are the only aminoacyl residues possessing protonated side chains that are not involved in ligating the binuclear iron center in this cavity (Fig. 1); thus, they are candidates for involvement in a number of processes, including delivery of protons to the binuclear iron center (9, 41), radical chemistry required for substrate oxygenation (10, 31), stabilization of the protein structure surrounding the active site (41), and intersubunit interactions (42). Thr 213 is conserved in all known homologous binuclear iron center monooxygenases (21, 53), whereas the amino acid at the position equivalent to Cys 151 in sMMO correlates with the function of the enzymes. All known sMMOs have cysteine here, monooxygenases that naturally epoxygenate alkenes have glutamate or aspartate (i.e., a carboxyl side chain) (43, 53), monooxygenases that perform ring hydroxylations of aromatic compounds have glutamine (21), and ribonucleotide reductases have tyrosine (46). Also, in the R2 subunit of class I ribonucleotide reductase, which is also homologous to sMMO (31), O2-dependent one-electron oxidation of Tyr 122 (equivalent to Cys 151 in sMMO) produces a stable tyrosyl radical that initiates the radical-dependent reduction of ribonucleotides at the active site of the R1 subunit of this enzyme (46).
FIG. 1.

Active site of the sMMO hydroxylase based on the X-ray crystal structure (9), showing the positions of the mutated residues. The iron atoms Fe-1 and Fe-2, which constitute the binuclear iron center, are ligated by four Glu and two His residues and three solvent molecules (dark grey). They lie in a solvent-accessible cavity lined by hydrophobic residues (light grey). The side-chain Oγ and Sγ moieties of the mutated residues (Cys 151 and Thr 213) are the only nonligating protonated side-chain groups within this cavity and are shown in black.
Here we report substantial improvement of the homologous expression system, resulting in a new vector system for mutagenesis of the α subunit of sMMO and expression of the mutants as part of a functional sMMO enzyme complex. Subsequent construction and analysis of Cys 151 and Thr 213 mutants have confirmed the practicability of the system and given the first direct experimental evidence for the importance of these residues in the catalytic properties of sMMO.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The plasmids and M. trichosporium strains used in this study are described in Table 1. Antibiotics, when necessary, were used at the following concentrations: ampicillin, 50 to 100 μg ml−1; streptomycin, 20 μg ml−1; spectinomycin, 20 μg ml−1; and kanamycin, 10 to 25 μg ml−1. E. coli was cultivated at 37°C in Luria-Bertani broth or on Luria-Bertani agar plates (44). Strains of M. trichosporium were grown on methane in continuous fermentor cultures as described previously (26) but with the following modifications. The total amounts of added copper in the nitrate minimal salts medium used for fermentor growth were 0.l mg of CuSO4 · 5H2O liter−1 during the initial batch phase of growth and 0 or 0.1 mg liter−1 in the subsequent continuous phase, as indicated in each experiment. It was found that antibiotics seriously reduced recombinant wild-type or mutant sMMO specific activity during continuous cultures, and so such cultures were grown in the presence of antibiotics (spectinomycin, 20 μg ml−1; streptomycin, 20 μg ml−1; and kanamycin, 10 μg ml−1) only during the initial batch phase of growth. Genetic analysis confirmed that the cells retained the recombinant sMMO genes. Since the maximum growth rate that could be sustained by cells expressing mutant sMMOs was 0.02 h−1, this dilution rate was used for all strains to ensure comparability of results.
TABLE 1.
Plasmids and M. trichosporium strains
| Plasmid or strain | Description | Source or reference |
|---|---|---|
| Plasmids | ||
| pBluescript KS(−) | Cloning vector, 3.0 kb; AprlacZ′ | Stratagene |
| pMTL22 | Cloning vector, 2.4 kb; AprlacZ′ | 6 |
| pMTL24 | Cloning vector, 2.4 kb; AprlacZ′ | 6 |
| pUC18 | Cloning vector, 2.7 kb; AprlacZ′ | 49 |
| pGEX-2T | GST fusion expression vector, 4.9 kb; Apr | Amersham-Pharmacia |
| pJB3Km1 | Broad-host-range cloning vector, 6.1 kb; Mob+ Apr KnrlacZ′ | 1 |
| pHM2 | pHM1 containing a 10-kb KpnI fragment carrying the sMMO operon from M. trichosporium OB3b; Spr Smr | 26 |
| pHP45Ω | Plasmid carrying the Ω cassette, 4.3 kb; Apr Spr Smr | 37 |
| pNPB101a | pUC18 carrying the 1.0-kb XbaI-NdeI fragment of pSJH1a, including the 5′ portion of mmoX | This study |
| pSJH1a | End-filled 5.6-kb MluI-SmaI fragment of pHM1, including the sMMO operon, cloned into the SmaI site of pBluescript KS in the direction opposite that of lacZ′ | This study |
| pSJH2 | pGEX-2T carrying a 0.5-kb BamHI-EcoRI insert containing the mmoB gene of M. trichosporium OB3b; Apr | This study |
| pTJS140 | Broad-host-range cloning vector, 7.5 kb; Mob+ Apr Spr SmrlacZ′ | This study |
| pTJS141 | pTJS140 carrying the 2.0-kb SacI fragment of pSJH1a, including mmoX, in the direction opposite that of lacZ′; Apr Spr Smr | This study |
| pTJS142 | pTJS140 carrying the 2.0-kb SacI fragment of pSJH1a, including mmoX, in the same direction as lacZ′; Apr Spr Smr | This study |
| pTJS170 | pMTL22 carrying the 5.4-kb HindIII-NdeI fragment of pHM2; Apr | This study |
| pTJS171 | pJB3Km1 carrying the 5.6-kb insert of pSJH1a cloned with BamHI and KpnI; Apr Knr | This study |
| pTJS172 | pMTL22 carrying the 4.4-kb HindIII-BamHI fragment derived from pTJS170 by PCR; Apr | This study |
| pTJS173 | 4.4-kb BglII-BamHI fragment of pTJS172 cloned into the BamHI site of pTJS172 such that the two parts of the insert have the same relative orientation as in pHM2; Apr | This study |
| pTJS175a | 10-kb KpnI insert of pTJS173 cloned into pTJS140 in the same direction as lacZ′; Apr Spr Smr | This study |
| pTJS176a | 10-kb KpnI insert of pTJS173 cloned into pMTL24; Apr | This study |
| Strains | ||
| OB3b | Wild-type, sMMO-positive strain | Laboratory stock |
| Mutant F | Knr sMMO-negative double-crossover mutant of OB3b | 26 |
| TJSHM1a | Mutant F into which pTJS175 had been introduced by conjugation | This study |
Derivatives containing mutations of the mmoX gene were also constructed and identified by appending the mutation to the name of the plasmid or strain, e.g., TJSHM1. C151E is TJSHM1 carrying the C151E mutation in mmoX.
Cloning and DNA analytical methods.
Manipulation of plasmids was performed with E. coli INVαF′ (Invitrogen) or XL-10 Gold (Stratagene) as the host. Purification of plasmid DNA from E. coli and in vitro manipulation of DNA for cloning were performed as described by Sambrook et al. (44), except when stated otherwise. For the construction of large (>12-kb) plasmids, transformation was effected by using SoloPack Gold supercompetent cells (Stratagene). PCRs were performed with Taq (Gibco) or Pfu Turbo (Stratagene) proofreading DNA polymerase under the conditions recommended by the manufacturer. When necessary, cloned PCR products were checked for unwanted mutations by cycle sequencing with a Dye Terminator kit (Applied Biosystems). For expression of the genes encoding the hydroxylase of sMMO, plasmids were electrotransformed (50) into E. coli S17-1 (45) and then introduced by conjugation (26) into M. trichosporium mutant F. Chromosomal DNA was purified from strains of M. trichosporium as described previously (32). The presence of freely replicating plasmids in M. trichosporium strains was tested by back transformation into E. coli (25).
Construction of plasmids.
pTJS140, the vector used for shuttling sMMO genes from E. coli to M. trichosporium mutant F, was constructed as follows. A 1,376-bp fragment carrying the Smr Spr gene was amplified from pHP45Ω (36, 37) by means of PCR with Taq polymerase and primers 5′-CAG CAA CTC GAG CAA TGC CTG ACG ATG CGT GGA GAC C-3′ and 5′-AGA GGT CTC GAG CGT CAT CAC CGA AAC GCG CGA G-3′ (XhoI sites are underlined), digested with XhoI, and cloned into the unique XhoI site of pJB3Km1 (1). Transformants in E. coli INVαF′ were selected on the basis of resistance to streptomycin and spectinomycin.
All subclones of the sMMO operon and flanking sequences were derived from pHM2 (26) (Table 1). The construction of pTJS141, pTJS142, and pTJS175, which were used for the introduction of sMMO genes into M. trichosporium mutant F, is shown in Fig. 2. During this procedure, the 4.4-kb region upstream from the sMMO operon was amplified by PCR from pTJS170 with the M13 forward sequencing primer (3′-GTA AAA CGA CGG CCA GT-5′) and the oligonucleotide 5′-G ACG CGT GGA TCC GAT CGT CGT ATG GCG ATG C-3′ (BamHI site is underlined). The cloned PCR product contained none of the sMMO structural genes; therefore, verification of its sequence was unnecessary.
FIG. 2.

Construction of plasmids. All manipulations shown were performed with E. coli as the host; at each stage, ampicillin, to which all the plasmids conferred resistance, was used for selection. The antibiotic resistance markers and the origin of conjugative transfer (oriT) and the broad-host-range replicon (ori-RK2) from plasmid RK2 (1) are shown only for plasmids pTJS141, pTJS142, and pTJS175, which were used for introducing sMMO genes into M. trichosporium mutant F. Sequences of primers and PCR conditions are stated in Materials and Methods. The diagrams are not drawn to scale.
pNPB101, the subclone containing the 5′ portion of mmoX, which was used as the target DNA for mutagenesis, was constructed by cloning the smaller (1.0-kb) XbaI-NdeI fragment of pSJH1a (Fig. 2) into pUC18. pSJH2, the construct used for the expression of the glutathione S-transferase (GST)-protein B fusion, was constructed by amplifying the mmoB gene from pSJH1a with primers 5′-G ATC GGA TCC ATG TCC AGC GCT CAT AAC G-3′ (BamHI site is underlined) and 5′-G ATC GAA TTC CGA TCA GAT GTC GGT CAG-3′ (EcoRI site is underlined) and cloning it into pGEX-2T (Amersham-Pharmacia) by using BamHI and EcoRI.
Site-directed mutagenesis.
The mutagenesis of mmoX was performed by means of the four-primer overlap extension PCR method (18) with pNPB101 as the template. The upstream and downstream external primers, which were the same for all mutagenesis experiments, were the M13 reverse sequencing primer (5′-CAG GAA ACA GCT ATG AC-3′) and the oligonucleotide 5′-CC GTT CGC CAT ATG ACG CAG CTC GTC-3′ (NdeI site is underlined), respectively. The complementary pairs of mutagenic oligonucleotides specific to each mutation are described in Table 2. These were designed also to introduce a change in restriction pattern that made no additional change to the encoded amino acid sequence, so that the presence of the mutation could be easily confirmed by restriction analysis during subsequent manipulations. The PCR-amplified 1.0-kb mutant gene fragments, containing the first 736 bp of mmoX and incorporating the desired mutations, were cloned into pUC18 with NdeI and XbaI, thus producing a mutated derivative of pNPB101. At this stage, the sequences of the PCR-derived mutant fragments were verified.
TABLE 2.
Oligonucleotides used to mutate mmoXa
| Mutation | Oligonucleotide (5′-3′) |
|---|---|
| C151E | Novel StuI site |
| C ACG CAT CAA GAG GCC TTC ATC AAT C | |
| Glu 151 | |
| C151Y | Novel RsaI site |
| C ACG CAT CAGTAC GCC TTC ATC AAT C | |
| Tyr 151 | |
| T213A | PvuII site removed |
| GCT CGT CGG CGA AGC CTG CTT CGC GAA TCC GCT CAT C | |
| Ala 213 | |
| PvuII site removed | |
| CGG ATT CGC GAA GCA GGC TTC GCC GAC GAG CTG CAG A | |
| Ala 213 | |
| T213S | PvuII site removed |
| GCT CGT CGG CGA AGC CTG CTT CTC GAA TCC GCT CAT C | |
| Ser 213 | |
| PvuII site removed | |
| CGG ATT CGA GAA GCA GGC TTC GCC GAC GAG CTG CAG A | |
| Ser 213 |
Mutations are shown in bold type. Codons encoding altered amino acids are underlined; changes to the restriction pattern associated with each mutation are shown by a line above the sequence. The pairs of mutagenic primers used to create each mutation at amino acid 151 were complementary, and so only the forward primer is shown. The pairs of mutagenic primers used to make changes at amino acid 213 spanned slightly different regions of the gene; both are shown for each mutation, the forward primer first.
Expression of mutant sMMOs.
The 1.0-kb mutant gene fragments were excised from the pNPB101 derivative with NdeI and BamHI before being united with the rest of the sMMO operon by ligation with the larger NdeI-BamHI fragment of pTJS176 (Fig. 2). The reconstructed 10-kb fragments, including the whole sMMO operon with the desired mutations within mmoX, were then excised from the pTJS176 derivative with KpnI and cloned into the broad-host-range vector pTJS140. The correct orientation of the sMMO operon (with the sMMO genes in the same orientation as the interrupted lacZ′ gene) was confirmed by restriction analysis with BamHI, and then the plasmid (a mutant of pTJS175) was introduced into M. trichosporium mutant F by conjugation. The changes in restriction patterns associated with the mutations were used to confirm the presence of the mutant mmoX genes in the exconjugants by restriction analysis of a 1,183-bp fragment of mmoX, amplified by PCR from whole cells or purified chromosomal DNA, with primers 5′-CTG TCA GGA GGA ACA AGC-3′ and 5′-CCG AGA CGG CCG ATC CAG-3′. Chromosomal integration of plasmids in the exconjugants was assessed by means of Southern blot analysis of HindIII-, PstI-, SalI-, and PvuII-digested chromosomal DNA with the 1.2-kb XhoI fragment of mmoX that was deleted from the chromosome of M. trichosporium mutant F as the probe. Similar digests of chromosomal DNA from wild-type M. trichosporium and mutant F, together with digests of plasmid pHM2, were used as controls.
Purification of the sMMO hydroxylase and reductase from M. trichosporium.
The hydroxylase and reductase components of sMMO were purified from strains of M. trichosporium by a method developed from published protocols (12, 35) as follows. Hydroxylase and reductase activities of fractions were determined by propene oxygenation assays in the presence of an excess of the other sMMO components. A culture sample (20 to 30 liters) was collected from a continuous fermentor, and the cells were harvested (8,900 × g, room temperature, in a continuous centrifuge) and resuspended in the minimum volume of 25 mM 3-(N-morpholino)propanesulfonic acid (MOPS)-NaOH (pH 7.0) (buffer A). The suspension was diluted with 350 ml of buffer A containing 5 mM dithiothreitol and 1 mM benzamidine (buffer B), and the cells were washed by centrifugation (14,500 × g, 4°C, 15 min) and resuspended in 150 ml of buffer B. DNase I (Sigma-Aldrich) was added to 20 μg ml−1, and the cells were broken by two passages though a cell disrupter (Constant Systems, Warwick, United Kingdom; 142 MPa, 4°C). The broken cell suspension was centrifuged (48,000 × g, 90 min, 4°C), and the supernatant (the soluble extract) was stirred with DEAE-Sepharose CL-6B anion-exchange matrix (Amersham-Pharmacia; 80 ml of settled bed volume in buffer B); the matrix was washed with buffer B by centrifugation (2,750 × g, 10 min, 4°C) and resuspension until the washings were colorless. The matrix was subsequently resuspended in buffer B (to 150 ml), packed into a column (3 by 12.5 cm), and washed with two column volumes of buffer B. Proteins were eluted with a linear gradient of 0 to 0.4 M NaCl in buffer B. The hydroxylase-containing fractions, which peaked at 0.12 M NaCl, were concentrated to 2 ml by ultrafiltration with an XM50, 50-kDa molecular mass cutoff membrane (Millipore-Amicon). The reductase-containing fractions, which peaked at 0.35 M NaCl, were similarly pooled and concentrated, except that the cutoff of the ultrafiltration membrane was 10 kDa.
The hydroxylase was further purified by using a Superdex 200 gel filtration column (2.5 by 60 cm; Amersham-Pharmacia) with buffer A. This step was followed by anion-exchange chromatography with a Mono Q HR16/10 anion-exchange column (Amersham-Pharmacia) from which the pure hydroxylase was eluted with a linear gradient of 0.05 to 0.25 M NaCl in buffer A.
The reductase-containing fractions were further purified by using a Superdex 75 (Amersham-Pharmacia) column (26 mm by 60 cm) with buffer A containing dithiothreitol (5 mM). The reductase is light sensitive, and so unnecessary exposure of reductase-containing samples to light was avoided at all stages.
Purification of protein B.
The protein B used during this study was an enzymatically active fusion in which the GST affinity tag was attached to the N terminus of protein B from M. trichosporium OB3b. The fusion protein was expressed in E. coli AD202 carrying pSJH2, purified by affinity chromatography (24), and used without cleavage of the affinity tag.
Enzyme assays.
The colorimetric naphthalene oxidation assay was used to detect sMMO activity in whole cells in liquid culture samples (3) or on agar plates (2). The sensitivity of sMMO to inactivation by acetylene was determined by prior incubation of plates for 30 min at 30°C in a 1:1 (vol/vol) acetylene-air atmosphere. Except when otherwise stated, quantitative assays of sMMO activity were done with mutant or wild-type hydroxylase (8 μM) and GST-protein B fusion and reductase (each at 16 μM) in buffer A. Propene oxygenation activity was assayed at 30°C with shaking in a final volume of 100 μl in 2-ml sealed reaction vials. One milliliter of headspace gas was removed and replaced with 1 ml of propene, and the reaction was initiated by the addition of NADH (to 10 mM). Epoxypropane formation was measured after 3 min of incubation by gas chromatography (GC) analysis of liquid-phase samples as described previously (15). Assays with toluene as the substrate were performed similarly, except that all three sMMO components were used at 8 μM, no headspace gas was removed, toluene (to a 10 mM liquid-phase concentration) was added in place of propene, and the reaction time was 30 min. The products (benzyl alcohol and p-cresol) were quantified by reference to standard solutions by using a Philips PU 4500 GC apparatus fitted with a flame ionization detector and a BP10 capillary column (SGE, Milton Keynes, Buckinghamshire, United Kingdom; internal diameter, 25 m by 0.33 mm; film thickness, 0.5 μm). Samples (1 μl) were introduced by splitless injection. The injector and detector temperatures were 240 and 260°C, respectively. The linear flow rate of the carrier gas (N2) was 40 cm s−1; the column temperature was 100°C for the first 5 min after injection, followed by a temperature ramp at 8°C min−1 up to 260°C. Methane oxidation rates were estimated from the increase in the rate of oxygen uptake upon the addition of NADH (to 1 mM) and 0.04 volume of methane-saturated water (giving a methane concentration of about 57 μM). Oxygen uptake was measured at 30°C with a Rank Brothers (Cambridge, United Kingdom) model 10 digital Clark-type oxygen electrode calibrated with air-saturated water, presuming an oxygen concentration at 30°C of 235 μM (40).
SDS-PAGE and MS.
Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) was performed with the buffer system of Laemmli (22). Proteins were visualized by staining with Coomassie blue. Mass analysis of purified mutant and wild-type sMMO hydroxylases was performed by means of electrospray ionization mass spectrometry (MS) with a Quattro II QhQ tandem mass spectrometer (Micromass) (4).
RESULTS
Development of a system for the expression of mutant hydroxylase genes.
In order to facilitate the expression of recombinant sMMO hydroxylases in M. trichosporium mutant F, vector pTJS140 was constructed as described in Materials and Methods. pTJS140 had the following properties: Apr and Spr Smr markers for selection in E. coli and M. trichosporium, respectively; broad-host-range replicon and origin of conjugative transfer from plasmid RK2; and multiple cloning site with unique KpnI and SacI sites positioned within the lacZ′ gene to allow blue-white selection of recombinant clones.
In M. trichosporium mutant F, only mmoX, the gene encoding the α subunit of the sMMO hydroxylase, is inactivated (27); thus, it was initially thought that it would be possible to express recombinant wild-type or mutant hydroxylases in mutant F by providing a plasmid-encoded copy of mmoX alone. Surprisingly, however, pTJS141 and pTJS142 (Fig. 2), which are clones of mmoX and its presumed promoter region in opposite orientations in pTJS140, did not restore sMMO activity when introduced into M. trichosporium mutant F. In light of these results, it was reasoned that the simplest way to obtain the expression of sMMOs that contained mutant MmoX derivatives was to construct plasmids to allow the cloning of mutant mmoX genes into the 10-kb fragment containing the sMMO-encoding operon and flanking sequences, which was known to direct the functional expression of the sMMO complex in M. trichosporium mutant F (26). Plasmid pTJS176 was therefore constructed (Fig. 2) in order to provide a system to allow facile cloning of mutagenized copies of mmoX into this 10-kb fragment.
Evaluation of the modified expression system.
The efficiency of the modified expression system was tested by using it to express wild-type sMMO. The modified 10-kb DNA fragment containing the wild-type sMMO operon was cloned into pTJS140 to give pTJS175, and then pTJS175 was introduced by conjugation (26) into M. trichosporium mutant F to give strain TJSHM1. TJSHM1 oxygenated naphthalene, as determined by the naphthalene plate test (2, 3), whereas the parental strain, M. trichosporium mutant F, did not. Since sMMO is the only enzyme synthesized by M. trichosporium that oxygenates naphthalene, pTJS175 was thus shown to be an effective plasmid for obtaining the expression of recombinant sMMO in M. trichosporium mutant F.
Flask-grown liquid batch cultures (50 to 1,000 ml) of TJSHM1 did not show reproducible sMMO expression. This was probably because recombinant sMMO was expressed from its natural promoter, which is controlled by the concentration of available copper in the medium. At high copper-to-biomass ratios, M. trichosporium grows with copper-dependent pMMO, and sMMO is repressed. It is only at low copper-to-biomass ratios that sMMO is induced (29, 47); thus, the irreproducibility of sMMO expression in batch-grown liquid cultures probably resulted from the difficulty in precisely controlling cell density and the concentration of available copper. In order to control the growth conditions more carefully, TJSHM1 was grown on methane in a continuous fermentor culture with no added copper as described in Materials and Methods. During the continuous phase of growth, the culture reached an optical density at 600 nm of 6.0, and 20 liters of this culture yielded 45 mg of >98% pure sMMO hydroxylase. This recombinant enzyme was confirmed to be fully active wild-type sMMO hydroxylase by comparison of its specific activity for propene oxygenation (260 nmol min−1 mg of protein−1) to the almost identical activity (244 nmol min−1 mg of protein−1) of the hydroxylase purified from wild-type M. trichosporium OB3b.
Mutagenesis and construction of strains for expression of the mutant proteins.
Cys 151 of sMMO was converted to glutamate (as in alkene monooxygenase of Rhodococcus rhodochrous B-276 [43]) and tyrosine (as in ribonucleotide reductase [46]). The function of the side chain of Thr 213 was investigated by mutation to serine (preserving the Oγ group at position 213) and alanine (removing the Oγ functionality at position 213).
All four mutations of the mmoX gene, which encodes the α subunit of the sMMO hydroxylase, were constructed and cloned into the rest of the sMMO operon and then introduced by means of conjugation into M. trichosporium mutant F for homologous expression of the mutant hydroxylases along with the other components of the sMMO enzyme complex. Attempts to transform E. coli with total DNA prepared from the exconjugants yielded no Apr progeny, suggesting the absence of freely replicating plasmids. Southern blot analysis of chromosomal DNA purified from the exconjugants confirmed that the plasmid in each strain had been incorporated into the interrupted copy of the sMMO operon, presumably by homologous recombination (data not shown). It was confirmed that the exconjugants contained the desired mutations by restriction analysis of the recombinant mmoX genes amplified by PCR, exploiting the changes in restriction patterns that were introduced along with each mutation (data not shown).
Analysis of the Cys 151 mutants.
The C151E and C151Y mutant methanotroph strains were tested for sMMO activity by the semiquantitative naphthalene plate test and for the sensitivity of sMMO activity to inactivation by acetylene, which is a turnover-dependent irreversible inhibitor of the sMMO hydroxylase (38) (Table 3). The lack of naphthalene oxygenation activity observed in the C151Y mutant strain (TJSHM1.C151Y) was concluded to be due to inactivity or instability of the mutant hydroxylase (or, conceivably, its mRNA) and not due to genetic rearrangement of the strain, because TJSHM1.C151Y contained the desired mutant mmoX gene, as shown by restriction analysis after PCR, and because a further 63 C151Y exconjugants that were screened for naphthalene oxygenation activity were all negative.
TABLE 3.
Properties of recombinant sMMO-expressing strains
| sMMO-expressing strain (in TJSHM1 expression system) | Activity with naphthalene | Inhibition by acetylene | Added Cu requirement (mg of CuSO4 · 5H2O liter−1) | Culture OD600 during continuous growtha |
|---|---|---|---|---|
| Wild type | Yes | Yes | 0 | 6 |
| C151E | Yes | Yes | 0.1 | 9 |
| C151Y | No | NAb | 0.1 | 9 |
| T213A | Diminished | Yes | 0.1 | 8 |
| T213S | Yes | Yes | 0 | 4 |
Strains that could not be maintained without copper during continuous culturing were grown in the presence of 0.1 mg of CuSO4·5H2O liter−1.
NA, not applicable.
TJSHM1.C151E and TJSHM1.C151Y were grown in continuous fermentor cultures (Table 3) with methane as the growth substrate in order to investigate whether the mutant hydroxylases could be purified. Although both strains grew (initially with pMMO until copper was depleted from the culture medium), the sMMO subunits could not be detected by SDS-PAGE after cell breakage (data not shown); this result suggested that the mutant hydroxylases were synthesized at low levels or were unstable. The TJSHM1.C151E culture had strong positive naphthalene test results, showing the presence of active sMMO in the intact cells, but all MMO activity was lost upon cell breakage. The addition of the serine protease inhibitor phenylmethylsulfonyl fluoride (to 0.5 mM) to the breakage buffer, either with or without the normal protease inhibitor benzamidine (1 mM), failed to stabilize the mutant sMMO in the cell extract long enough for it to be assayed. Likewise, the iron-containing breakage buffer used by Fox et al. (12) did not stabilize the mutant hydroxylase.
The inability of either mutant to sustain a growth rate of 0.02 h−1 in the absence of 0.1 mg of added CuSO4 · 5H2O liter−1 (Table 3) suggested that the mutant strains were unable to oxidize the growth substrate methane with (the mutant) sMMOs. This result cannot, however, be taken as proof that the TJSHM1.C151E and TJSHM1.C151Y mutant hydroxylases had diminished activity toward methane, because their abundance within intact cells was not known.
Whole-cell analysis of Thr 213 mutants and purification of the T213S mutant hydroxylase.
The whole-cell properties of the T213S and T213A mutant strains, together with their characteristics during continuous cultivation, are shown in Table 3. Upon preparation of a soluble extract from T213A mutant cells, the hydroxylase subunits could be detected by SDS-PAGE at only a low level (data not shown), which was found to be insufficient to permit purification by the method used for the wild-type hydroxylase. However, the soluble extract from the T213S mutant strain contained amounts of all three sMMO hydroxylase subunits comparable to those in recombinant wild-type strain TJSHM1, as determined by SDS-PAGE (data not shown). The T213S mutant hydroxylase was successfully purified by the same method as that used for the wild type to yield 140 mg of >98% pure protein from 30 liters of culture.
Characterization of the T213S mutant hydroxylase.
The characteristics of the T213S mutant hydroxylase are shown in Table 4. The mutant hydroxylase behaved in a manner identical to that of the wild type during ion-exchange and gel filtration chromatography, and the identical iron contents strongly suggested that it was fully folded and that the binuclear iron active center was intact. As was observed previously with the hydroxylase from M. capsulatus (Bath) (4), the α subunits of the M. trichosporium wild-type and mutant hydroxylases underwent fragmentation before or during electrospray ionization MS analysis. The largest molecular ions detected with the mutant and the wild type were assigned to amino acids 9 to 526 of the α subunit. For the mutant, this assignment corresponded to a mass of 59,124.1 Da, which represented a 13.6-Da decrease relative to the wild-type sequence, almost exactly equal to the 14-Da decrease predicted for a Thr → Ser mutation.
TABLE 4.
Properties of T213S mutant of sMMO
| Property | Value for:
|
|
|---|---|---|
| Wild type | T213S | |
| Iron content (mol of Fe per mol of 250-kDa hydroxylase)a | 2.74 ± 0.05 | 2.63 ± 0.04 |
| Activity (nmol min−1 mg of hydroxylase−1) with: | ||
| Propeneb | 244 ± 7 | 43 ± 3 |
| Tolueneb | 7.80 ± 0.83 | 5.6 ± 0.49 |
| Methanea,c | 235 ± 22 | 169 ± 27 |
| Product distribution with toluene substrate (mol of benzyl alcohol/mol of p-cresol)b | 2.6 ± 0.8 | 2.8 ± 0.6 |
Measurements were conducted in duplicate; values are shown as the mean and standard deviation.
Derived from GC quantitation of the product(s). Measurements were performed at least three times each; values are shown as the mean and standard deviation.
Derived from substrate-stimulated increase in oxygen uptake as described in Materials and Methods.
DISCUSSION
The low abundance of three out of the four mutant enzymes constructed during this study, including both mutants with changes at position 151, limits the conclusions that can be drawn about the functions of Cys 151 and Thr 213. At least one firm conclusion can, however, be made: neither residue is essential for oxygenase activity. This finding has important consequences for the role of Cys 151, which was a candidate for involvement in active-site radical chemistry because it occupied a position equivalent to that of the radical-forming Tyr 122 of ribonucleotide reductase (9, 31). Activation of the inert C—H bond of methane must involve homolytic cleavage (to produce radical intermediates), heterolytic cleavage (to produce a methyl cation that can be stabilized by coordination to iron) (16), or a concerted mechanism of bond breakage and formation (52). Free radicals have been observed during sMMO catalysis by use of radical trap agents (51), but whether they are an essential part of the catalytic process or merely the result of side reactions is unresolved (8). In this study, it was observed that substitution of Cys 151 with glutamate preserved the activity of the hydroxylase toward naphthalene. A catalytically essential free radical at Glu 151 is therefore very unlikely, because a glutamyl radical would decarboxylate spontaneously (30). It remains to be seen whether essential radicals form elsewhere in the active site or whether a radical at Cys 151 is required only for oxygenation of methane and other inert substrates.
Intriguingly, the phenotypes of the strains expressing the mutant sMMOs support the correlation between the function of the binuclear iron active site and the amino acid at position 151 and its equivalents. Mutation of Cys 151 to Glu, as in alkene monooxygenase of R. rhodochrous B-276 (43), preserves oxygenase activity, whereas mutation of Cys 151 to Tyr, as in ribonucleotide reductase (46) (which is not a monooxygenase), abolishes it. In a study with toluene 4-monooxygenase, the equivalent residue, Gln 141, was mutated to cysteine (as in sMMO), preserving monooxygenase activity (33).
A study of the role of the conserved threonine (Thr 201) in toluene 4-monooxygenase found that mutation to Ser, Ala, or Gly had almost no effect on stability, catalytic activity, or coupling efficiency (34). The results presented here suggest an important role for the equivalent Thr 213 in sMMO because the T213A mutant, which lacks the side-chain HOγ functional group, had diminished stability compared to the wild type and the T213S mutant. The Oγ group of Thr 213 is closely associated with the binuclear iron center, being 6.3 Å from Fe-1 and 6.8 Å from Fe-2 at the binuclear iron center (Fig. 1) (9); thus, it is reasonable that the interaction between this Oγ group and the binuclear iron center is important in determining the effect of Thr 213 on the stability of the enzyme. The most important outcome from the mutagenesis at position 213 was that the T213S mutant, which exhibited a modest but significant alteration in substrate specificity, could be easily expressed and purified by using the homologous expression system. Hence, we have provided proof of the principle for a system for the expression and purification of active mutants of sMMO with altered catalytic properties. This expression system can also be exploited to improve the catalytic utility of sMMO in the same way that Wood and coworkers have recently carried out directed evolution studies using DNA shuffling of the related di-iron center monooxygenase toluene ortho-monooxygenase (4a). They demonstrated that random in vitro protein engineering could be used to enhance the catalytic utility of this large multisubunit monooxygenase. This type of approach, together with the capacity to produce recombinant enzymes using low-cost methane and methanol feedstocks, may open new avenues for industrial exploitation of sMMO.
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council through grant 88/B07030 (to H.D. and J.C.M.), by the European Union through TMR grant (Iron-Oxygen Protein Network) FMRXCT98-0207 (to H.D.), and by European Commission grant QLK3-2000-01528 (to J.C.M.).
We are grateful to Svein Valla for the gift of plasmid pJB3Km1, to Steve Harris for constructing plasmids pSJH1a and pSJH2, to Suki Balendra for preparing the GST-protein B fusion, and to Wendy Foxall for assistance with MS.
REFERENCES
- 1.Blatny, J. M., T. Brautaset, H. C. Winther-Larsen, K. Haugan, and S. Valla. 1997. Construction and use of a versatile set of broad-host-range cloning and expression vectors based on the RK2 replicon. Appl. Environ. Microbiol. 63:370-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bodrossy, L., J. C. Murrell, H. Dalton, M. Kalman, L. G. Puskas, and K. L. Kovacs. 1995. Heat-tolerant methanotrophic bacteria from the hot-water effluent of a natural-gas field. Appl. Environ. Microbiol. 61:3549-3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brusseau, G. A., H.-C. Tsien, R. S. Hanson, and L. P. Wackett. 1990. Optimization of trichloroethylene oxidation by methanotrophs and the use of a colorimetric assay to detect soluble methane mono-oxygenase activity. Biodegradation 1:19-29. [DOI] [PubMed] [Google Scholar]
- 4.Buzy, A., A. L. Millar, V. Legros, P. C. Wilkins, H. Dalton, and K. R. Jennings. 1998. The hydroxylase component of soluble methane monooxygenase from Methylococcus capsulatus (Bath) exists in several forms as shown by electrospray-ionisation mass spectrometry. Eur. J. Biochem. 254:206-209. [DOI] [PubMed] [Google Scholar]
- 4a.Canada, K. A., S. Iwashita, H. Shim, and T. K. Wood. 2002. Directed evolution of toluene ortho-monooxygenase for enhanced 1-naphthol synthesis and chlorinated ethene degradation. J. Bacteriol. 184:344-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cardy, D. L. N., V. Laidler, G. P. C. Salmond, and J. C. Murrell. 1991. Molecular analysis of the methane monooxygenase (MMO) gene-cluster of Methylosinus trichosporium OB3b. Mol. Microbiol. 5:335-342. [DOI] [PubMed] [Google Scholar]
- 6.Chambers, S. P., S. E. Prior, D. A. Barstow, and N. P. Minton. 1988. The pMTL nic-cloning vectors. 1. Improved pUC polylinker regions to facilitate the use of sonicated DNA for nucleotide sequencing. Gene 68:139-149. [DOI] [PubMed] [Google Scholar]
- 7.Colby, J., D. I. Stirling, and H. Dalton. 1977. The soluble methane monooxygenase of Methylococcus capsulatus (Bath): its ability to oxygenate n-alkanes, n-alkenes, ethers, and alicyclic, aromatic and heterocyclic compounds. Biochem. J. 165:395-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deeth, R. J., and H. Dalton. 1998. Methane activation by methane monooxygenase: free radicals, Fe-C bonding, substrate-dependent pathways and the role of the regulatory protein. J. Biol. Inorg. Chem. 3:302-306. [Google Scholar]
- 9.Elango, N., R. Radmakrishnan, W. A. Froland, B. J. Wallar, C. A. Earhart, J. D. Lipscomb, and D. H. Ohlendorf. 1997. Crystal structure of the hydroxylase component of methane monooxygenase from Methylosinus trichosporium OB3b. Protein Sci. 6:556-568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feig, A. L., and S. J. Lippard. 1994. Reactions of nonheme iron (II) centers with dioxygen in biology and chemistry. Chem. Rev. 94:759-805. [Google Scholar]
- 11.Fox, B. G., J. Bourneman, L. Wackett, and J. D. Lipscomb. 1990. Haloalkene oxidation by the soluble methane monooxygenase from Methylosinus trichosporium OB3b: mechanistic and environmental implications. Biochemistry 29:6419-6427. [DOI] [PubMed] [Google Scholar]
- 12.Fox, B. G., W. A. Froland, J. E. Dege, and J. D. Lipscomb. 1989. Methane monooxygenase from Methylosinus trichosporium OB3b: purification and properties of a three-component system with high specific activity from a type II methanotroph. J. Biol. Chem. 264:10023-10033. [PubMed] [Google Scholar]
- 13.Fox, B. G., Y. Liu, J. E. Dege, and J. D. Lipscomb. 1991. Complex formation between the protein components of methane monooxygenase from Methylosinus trichosporium OB3b. J. Biol. Chem. 266:540-550. [PubMed] [Google Scholar]
- 14.George, A. R., P. C. Wilkins, and H. Dalton. 1996. A computational investigation of the possible substrate binding sites in the hydroxylase of soluble methane monooxygenase. J. Mol. Catal. B 2:103-113. [Google Scholar]
- 15.Green, J., and H. Dalton. 1986. Steady-state kinetic-analysis of soluble methane monooxygenase from Methylococcus capsulatus (Bath). Biochem. J. 236:155-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green, J., and H. Dalton. 1989. Substrate specificity of soluble methane monooxygenase—mechanistic implications. J. Biol. Chem. 264:17698-17703. [PubMed] [Google Scholar]
- 17.Hanson, R. S., and T. E. Hanson. 1996. Methanotrophic bacteria. Microbiol. Rev. 60:439-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, and R. L. Pease. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain-reaction. Gene 77:51-59. [DOI] [PubMed] [Google Scholar]
- 19.Jahng, D., and T. K. Wood. 1994. Trichloroethylene and chloroform degradation by a recombinant pseudomonad expressing soluble methane monooxygenase from Methylosinus trichosporium OB3b. Appl. Environ. Microbiol. 60:2473-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jahng, D., K. S. Kim, R. S. Hanson, and T. K. Wood. 1996. Optimization of trichloroethylene degradation using soluble methane monooxygenase of Methylosinus trichosporium OB3b expressed in recombinant bacteria. Biotechnol. Bioeng. 51:349-359. [DOI] [PubMed] [Google Scholar]
- 21.Johnson, G. R., and R. H. Olsen. 1995. Nucleotide sequence analysis of genes encoding a toluene benzene-2-monooxygenase from Pseudomonas sp. strain JS150. Appl. Environ. Microbiol. 61:3336-3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 23.Lipscomb, J. D. 1994. Biochemistry of the soluble methane monooxygenase. Annu. Rev. Microbiol. 48:371-399. [DOI] [PubMed] [Google Scholar]
- 24.Lloyd, J. S., A. Bhambra, J. C. Murrell, and H. Dalton. 1997. Inactivation of the regulatory protein B of soluble methane monooxygenase from Methylococcus capsulatus (Bath) by proteolysis can be overcome by a Gly to Gln modification. Eur. J. Biochem. 248:72-79. [DOI] [PubMed] [Google Scholar]
- 25.Lloyd, J. S., P. De Marco, H. Dalton, and J. C. Murrell. 1999. Heterologous expression of soluble methane monooxygenase genes in methanotrophs containing only particulate methane monooxygenase. Arch. Microbiol. 171:364-370. [DOI] [PubMed] [Google Scholar]
- 26.Lloyd, J. S., R. Finch, H. Dalton, and J. C. Murrell. 1999. Homologous expression of soluble methane monooxygenase genes in Methylosinus trichosporium OB3b. Microbiology 145:461-470. [DOI] [PubMed] [Google Scholar]
- 27.Martin, H., and J. C. Murrell. 1995. Methane monooxygenase mutants from Methylosinus trichosporium constructed by marker-exchange mutagenesis. FEMS Microbiol. Lett. 127:243-248. [Google Scholar]
- 28.Murrell, J. C., B. Gilbert, and I. R. McDonald. 2000. Molecular biology and regulation of methane monooxygenase. Arch. Microbiol. 173:325-332. [DOI] [PubMed] [Google Scholar]
- 29.Nielsen, A. K., K. Gerdes, and J. C. Murrell. 1997. Copper-dependent reciprocal transcriptional regulation of methane monooxygenase genes in Methylococcus capsulatus and Methylosinus trichosporium. Mol. Microbiol. 25:399-409. [DOI] [PubMed] [Google Scholar]
- 30.Nonhebel, D. C., J. M. Tedder, and J. C. Walton. 1979. Radicals. Cambridge University Press, Cambridge, United Kingdom.
- 31.Nordlund, P., H. Dalton, and H. Ecklund. 1992. The active-site structure of methane monooxygenase is closely related to the binuclear iron center of ribonucleotide reductase. FEBS Lett. 307:257-262. [DOI] [PubMed] [Google Scholar]
- 32.Oakley, C. J., and J. C. Murrell. 1988. nifH genes in the obligate methane oxidising bacteria. FEMS Microbiol. Lett. 49:53-57. [Google Scholar]
- 33.Pikus, J. D., J. M. Studts, K. McClay, R. J. Steffan, and B. G. Fox. 1997. Changes in the regiospecificity of aromatic hydroxylation produced by active site engineering in the diiron enzyme toluene 4-monooxygenase. Biochemistry 36:9283-9289. [DOI] [PubMed] [Google Scholar]
- 34.Pikus, J. D., K. H. Mitchell, J. M. Studts, K. McClay, R. J. Steffan, and B. G. Fox. 2000. Threonine 201 in the diiron enzyme toluene 4-monooxygenase is not required for catalysis. Biochemistry 39:791-799. [DOI] [PubMed] [Google Scholar]
- 35.Pilkington, S. J., and H. Dalton. 1990. Soluble methane monooxygenase from Methylococcus capsulatus Bath. Methods Enzymol. 188:181-190. [Google Scholar]
- 36.Prentki, P., A. Binda, and A. Epstein. 1991. Plasmid vectors for selecting IS1-promoted deletions in cloned DNA: sequence analysis of the omega interposon. Gene 103:17-23. [DOI] [PubMed] [Google Scholar]
- 37.Prentki, P., and H. M. Krisch. 1984. In vitro insertional mutagenesis with a selectable DNA fragment. Gene 29:303-313. [DOI] [PubMed] [Google Scholar]
- 38.Prior, S. D., and H. Dalton. 1985. Acetylene as a suicide substrate and active-site probe for methane monooxygenase from Methylococcus capsulatus (Bath). FEMS Microbiol. Lett. 29:501-509. [Google Scholar]
- 39.Reeburgh, W. S., S. C. Whalen, and M. J. Alperin. 1993. The role of methylotrophy in the global methane budget, p. 1-14. In J. C. Murrell and D. P. Kelly (ed.), Microbial growth on C1 compounds. Intercept, Andover, United Kingdom.
- 40.Robinson, J., and J. M. Cooper. 1970. Method of determining oxygen concentrations in biological media, suitable for calibration of the oxygen electrode. Anal. Biochem. 33:390-399. [DOI] [PubMed] [Google Scholar]
- 41.Rosenzweig, A. C., C. A. Frederick, S. J. Lippard, and P. Nordlund. 1993. Crystal structure of a bacterial nonheme iron hydroxylase that catalyzes the biological oxidation of methane. Nature 366:537-543. [DOI] [PubMed] [Google Scholar]
- 42.Rosenzweig, A. C., P. Nordlund, P. M. Takahara, C. A. Frederick, and S. J. Lippard. 1995. Geometry of the soluble methane monooxygenase catalytic diiron center in two oxidation states. Chem. Biol. 2:409-418. [PubMed] [Google Scholar]
- 43.Saeki, H., and K. Furuhashi. 1994. Cloning and characterisation of the Nocardia corallina B-276 gene cluster encoding alkene monooxygenase. J. Ferment. Bioeng. 78:399-406. [Google Scholar]
- 44.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 45.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilization system for in vivo genetic engineering-transposon mutagenesis in Gram-negative bacteria. Bio/Technology 1:784-791. [Google Scholar]
- 46.Sjöberg, B.-M. 1997. Ribonucleotide reductases—a group of enzymes with different metallosites and a similar reaction mechanism. Struct. Bonding 88:139-173. [Google Scholar]
- 47.Stanley, S. H., S. D. Prior, D. J. Leak, and H. Dalton. 1983. Copper stress underlies the fundamental change in intracellular location of methane monooxygenase in methane-oxidizing organisms—studies in batch and continuous cultures. Biotechnol. Lett. 5:487-492. [Google Scholar]
- 48.Stirling, D. I., and H. Dalton. 1979. Purification of the methane monooxygenase from extracts of Methylosinus trichosporium OB3b and evidence for its similarity to the enzyme from Methylococcus capsulatus (Bath). Eur. J. Biochem. 96:205-212. [DOI] [PubMed] [Google Scholar]
- 49.Vieira, J., and J. Messing. 1982. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene 19:259-268. [DOI] [PubMed] [Google Scholar]
- 50.West, C. A., G. P. C. Salmond, H. Dalton, and J. C. Murrell. 1992. Functional expression in Escherichia coli of protein B and protein C from soluble methane monooxygenase of Methylococcus capsulatus (Bath). J. Gen. Microbiol. 138:1301-1307. [DOI] [PubMed] [Google Scholar]
- 51.Wilkins, P. C., H. Dalton, I. D. Podmore, N. Deighton, and M. C. R. Symonds. 1992. Biological methane activation involves the intermediacy of carbon-centered radicals. Eur. J. Biochem. 210:67-72. [DOI] [PubMed] [Google Scholar]
- 52.Yoshizawa, K., T. Yamabe, and R. Hoffmann. 1997. Possible intermediates for the conversion of methane to methanol on dinuclear iron centers of methane monooxygenase models. New J. Chem. 21:151-161. [Google Scholar]
- 53.Zhou, N.-Y., A. Jenkins, C. K. N. C. K. Chion, and D. J. Leak. 1999. The alkene monooxygenase from Xanthobacter strain Py2 is closely related to aromatic monooxygenases and catalyzes aromatic monohydroxylation of benzene, toluene, and phenol. Appl. Environ. Microbiol. 65:1589-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]