

Abstract
The cytokine tumour necrosis factor (TNF)α is a vital mediator of the innate immune response, and a pleiotropic regulator of cellular function. Its involvement in rheumatoid arthritis is illustrated by the clinical benefits of TNFα blockade. Post-transcriptional regulation (the control of mRNA stability and translation) appears to play a critical role in the regulation of TNFα expression by mitogen activated protein kinase signal transduction pathways and by anti-inflammatory agents. The aim of this article is to review some recent advances in our understanding of these processes, and to speculate on mechanisms of regulation of TNFα and other pro-inflammatory genes.
Keywords: adenosine/uridine-rich element, dexamethasone, inflammation, mRNA stability, translation, tumour necrosis factor α
Full text
Post-transcriptional regulation of gene expression is generally mediated by the 5' or 3' untranslated regions (UTRs) of the mature mRNA, which flank the protein coding sequence. It is assumed that regulation is mediated by proteins that specifically interact with these regions. Much recent attention has focused on adenosine/uridine-rich elements (AREs), particularly repeats of the sequence AUUUA, which are present in the 3' UTRs of many transiently expressed cytokine and growth factor mRNAs, including TNFα, IL 1α, 1β, 6 and 8, IFNγ and granulocyte macrophage colony stimulating factor (GM-CSF) [1,2]. Several AREs have been shown to destabilise heterologous transcripts into which they are inserted [3]. Mammalian TNFα mRNAs possess a 3' UTR of almost 800 nucleotides, within which is a well-conserved ARE containing seven or eight copies of the AUUUA sequence. Among factors known to interact with the TNFα ARE [4,5,6] is tristetraprolin (TTP), an unusual zinc finger protein whose expression is induced by both lipopolysaccharide (LPS) and TNFα itself. In mice deficient in TTP, TNFα protein is overexpressed, and the stability of TNFα mRNA is increased [6]. When coexpressed with a mimic TNFα mRNA, TTP is able to promote shortening of the poly(A) tail, normally an early step in mRNA degradation [7]. This property of TTP is dependent upon a (more or less) intact TNFα ARE. Therefore, TTP may participate in a negative feedback loop whose effect is to limit the magnitude or duration of a burst of TNFα expression by destabilising its mRNA in an ARE-dependent manner.
Mimic transcripts and other 'post-transcriptional reporters' for the study of TNFα gene regulation do not faithfully reproduce all features of endogenous gene expression. A more subtle approach has recently been used by Kollias et al, who precisely deleted the ARE region from the mouse TNFα genomic locus [8]. In the resulting (ΔARE) strain, the dynamics of LPS-stimulated TNFα mRNA accumulation were altered, with a larger, slower and more sustained induction. Altered TNFα mRNA stability was strongly implicated in this change, but not directly demonstrated. Other workers have shown that TNFα mRNA stability is regulated by LPS, and is sensitive to protein kinase inhibitors in myeloid cell lines [9]. These studies should be interpreted with caution because of their dependence upon toxic transcriptional inhibitors like actinomycin D, which may give rise to artefacts.
Previous studies established that the mitogen activated protein kinase (MAPK) p38 regulates TNFα production at the translational level [10,11,12]. Specific inhibitors of p38 blocked TNFα protein synthesis, while downregulating TNFα mRNA by 50% at most. The deletion of the TNFα ARE resulted in the loss of sensitivity to a p38 inhibitor in the mouse strain of Kollias et al [8]. A recent mouse knockout study suggests that the effects of MAPK p38 may be mediated by mitogen activated protein kinase-activated protein kinase 2 (MAPKAPK2), a kinase that is phosphorylated and activated by p38. In the absence of MAPKAPK2 activity, reduced quantities of TNFα protein were synthesised following an LPS challenge, while the induction of TNFα mRNA was unaffected [13]. Together, these findings suggest that p38 regulates TNFα mRNA translation via MAPKAPK2, and by a mechanism involving the TNFα ARE. In contrast, p38 and MAPKAPK2 regulate the stability rather than the translation of IL-6, IL-8, GM-CSF and cyclooxygenase-2 (Cox-2) mRNAs in the human HeLa cell line [14] (Lasa et al, submitted). Regulation of IL-8 and Cox-2 mRNA stability is mediated by 3' AU-rich elements that closely resemble the TNFα ARE. It is significant that these studies do not rely upon pharmacological inhibitors of transcription or of p38, and provide independent confirmation of earlier reports that p38 is able to regulate mRNA stability.
The parallels and differences between TNFα and IL-8 or Cox-2 raise several questions. Do AREs such as those in the TNFα and Cox-2 transcripts regulate translation, stability, or both? What of the p38 signalling pathway? Is it conceivable that distinct mechanisms have evolved for the regulation of both translation and stability by mitogen activated protein kinases, through extremely similar RNA sequence motifs? Perhaps the apparently different consequences of p38 inhibition reflect an underlying common mechanism. It is suggested that both HeLa cells and monocytes/macrophages can label mRNA as 'Not For Use', most probably by means of specific protein-mRNA interactions that involve AREs and are sensitive to p38 (Fig. 1). The immediate consequence of this might be a cessation of translation, with indirect consequences such as mRNA degradation modulated in a species, cell type or transcript specific manner.
Figure 1.
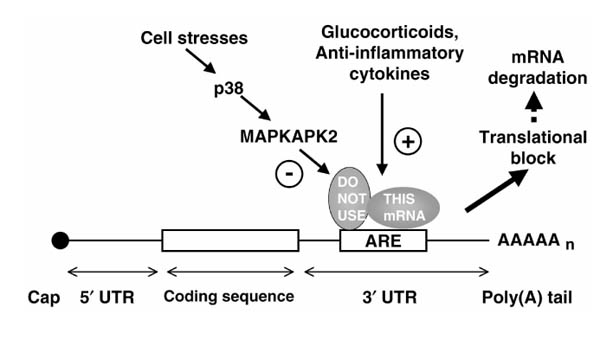
Convergence of regulatory signals at the 3' untranslated region of a pro-inflammatory mRNA. ARE, Adenosine/uridine-rich elements; UTR, untranslated region; MAPKAPK2, mitogen activated protein kinase-activated protein kinase 2.
Studies employing specific inhibitors have implicated MAPK p38 in the post-transcriptional regulation of a number of genes that could be broadly characterised as pro-inflammatory. These include TNFα, IL-6, -8 and -1β, GM-CSF, Cox-2 and the vascular cell adhesion molecule VCAM-1 [12,15,16,17]. In the MAPKAPK2 knockout mouse strain, LPS-induced expression of IL-6 was blocked at both protein and mRNA levels, while expression of both TNFα and IFNγ was blocked at the protein but not the mRNA level [13]. The defective expression of TNFα and IFNγ suggests translational regulation by MAPKAPK2 within both myeloid and T cell lineages.
Dexamethasone, a synthetic glucocorticoid and a potent anti-inflammatory reagent, regulates the expression of TNFα and Cox-2 partly at the post-transcriptional level. Post-transcriptional reporter studies have implicated the TNFα 3' UTR in dexamethasone sensitivity [18], and the deletion of the ARE diminishes the inhibitory effect of dexamethasone upon TNFα gene expression in vivo [8]. In a human pulmonary epithelial cell line, dexamethasone destabilises Cox-2 mRNA by a mechanism that involves shortening of the poly(A) tail, although it has not yet been possible to map the RNA sequences that mediate this effect [19]. Both TNFα and Cox-2 are subject to negative regulation by anti-inflammatory cytokines such as IL-10, which has been shown to regulate the stability of ARE-containing transcripts [20,21], and preliminary evidence indicates that its effects upon TNFα expression may be partly mediated by the TNFα 3' UTR (B. Foxwell, personal communication). The responses of the mouse ΔARE strain to anti-inflammatory cytokines would be interesting to observe.
It is an intriguing possibility that these disparate observations reflect a common mechanism for the post-transcriptional control of several pro-inflammatory genes. In other words, the ARE represents a point of convergence of pro-inflammatory signals (such as MAPK p38 activation) and anti-inflammatory signals (such as activation of IL-10 or glucocorticoid signalling pathways; Fig. 1). In theory, this might allow a cell to modulate its sensitivity to pro- or anti-inflammatory stimuli through changes in the expression of ARE binding factors, and to rapidly and coordinately shut down the expression of several inflammatory mediators by means of phosphorylation-regulated changes in protein-RNA interactions. In the simplest model, all of these regulatory processess could be mediated by a single ARE binding factor; however, in reality, the situation is likely to be rather more complex. One can imagine post-transcriptional regulation involving competition between positive and negative regulatory RNA binding proteins, and other subtleties such as cell-specific differences in the levels of expression of these factors. The assessment of this hypothesis requires the characterisation of putative post-transcriptional regulators, and their functional and physical interactions. It may also be instructive to look for additional targets of suspected regulatory factors such as TTP.
Both the TTP knockout and the ΔARE mouse strains display striking inflammatory pathologies, including an erosive arthritis with features of rheumatoid arthritis (RA) in man [8,22]. Thus, the disruption of either cis-acting (ARE) or trans-acting (TTP) components of negative regulatory control of TNFα synthesis can cause joint disease. One wonders whether 'off phase' or negative regulatory mechanisms for the restraint of TNFα biosynthesis are perturbed in RA. Even if not, post-transcriptional control may represent a target for novel anti- inflammatory therapies. For example, a recent paper reports disease modifying activity of a p38 inhibitor in an animal model of RA, with downregulation of both TNFα and IL-6 expression [23]. In the event of unwanted side effects of intervention with MAPK or glucocorticoid signal transduction pathways, it might be possible to target downstream effectors, the putative RNA binding coordinators of pro-and anti-inflammatory post-transcriptional regulation. It is conceivable that reactivation or reinforcement of 'off phase' regulatory mechanisms may have quite broad anti-inflammatory effects.
References
- Shaw G, Kamen R. A conserved AU sequence from the 3' untranslated region of GM-CSF mRNA mediates selective mRNA degradation. . Cell. 1986;46:659–667. doi: 10.1016/0092-8674(86)90341-7. [DOI] [PubMed] [Google Scholar]
- Caput D, Beutler B, Hartog K, Thayer R, Brown-Shimer S, Cerami A. Identification of a common nucleotide sequence in the 3'-untranslated region of mRNA molecules specifying inflammatory mediators. . Proc Natl Acad Sci USA. 1986;83:1670–1674. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Shyu AB. AU-rich elements: characterization and importance in mRNA degradation. . Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- Sakai K, Kitagawa Y, Hirose G. Binding of neuronal ELAV-like proteins to the uridine-rich sequence in the 3'-untranslated region of tumor necrosis factor-alpha messenger RNA. . FEBS Lett. 1999;446:157–162. doi: 10.1016/s0014-5793(99)00206-9. [DOI] [PubMed] [Google Scholar]
- Gueydan C, Droogmans L, Chalon P, Huez G, Caput D, Kruys V. Identification of TIAR as a protein binding to the translational regulatory AU-rich element of tumor necrosis factor alpha mRNA. . J Biol Chem. 1999;274:2322–2326. doi: 10.1074/jbc.274.4.2322. [DOI] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. . Science. 1998;281:1001–1005. doi: 10.1006/bbrc.2001.4457. [DOI] [PubMed] [Google Scholar]
- Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. . Mol Cell Biol. 1999;19:4311–4323. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. . Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Pitha PM, Shin ML. Poly(A) removal is the kinase-regulated step in tumor necrosis factor mRNA decay. . J Biol Chem. 1992;267:2123–2126. [PubMed] [Google Scholar]
- Prichett W, Hand A, Sheilds J, Dunnington D. Mechanism of action of bicyclic imidazoles defines a translational regulatory pathway for tumor necrosis factor alpha. . J Inflamm. 1995;45:97–105. [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. . Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Young P, McDonnell P, Dunnington D, Hand A, Laydon J, Lee J. Pyridinyl imidazoles inhibit IL-1 and TNF production at the protein level. . Agents Actions. 1993;39:C67–C69. doi: 10.1007/BF01972723. [DOI] [PubMed] [Google Scholar]
- Kotlyarov A, Neininger A, Schubert C, et al. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. . Nat Cell Biol. 1999;1:94–97. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- Winzen R, Kracht M, Ritter B, et al. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. . EMBO J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Kassis S, Kumar S, Badger A, Adams JL. p38 mitogen-activated protein kinase inhibitors - mechanisms and therapeutic potentials. . Pharmacol Ther. 1999;82:389–397. doi: 10.1016/s0163-7258(99)00008-x. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Mori A, Miyata H, Akahane M, Ajisawa Y, Okudaira H. Regulation of interleukin-1 beta-induced interleukin-6 gene expression in human fibroblast-like synoviocytes by p38 mitogen-activated protein kinase. . J Biol Chem. 1998;273:24832–24838. doi: 10.1074/jbc.273.38.24832. [DOI] [PubMed] [Google Scholar]
- Pietersma A, Tilly BC, Gaestel M, et al. p38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. . Biochem Biophys Res Commun. 1997;230:44–48. doi: 10.1006/bbrc.1996.5886. [DOI] [PubMed] [Google Scholar]
- Han J, Thompson P, Beutler B. Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. . J Exp Med. 1990;172:391–394. doi: 10.1084/jem.172.1.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton R, Seybold J, Kuitert LM, Bergmann M, Barnes PJ. Repression of cyclooxygenase-2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post-transcriptional mechanisms involving loss of polyadenylated mRNA. . J Biol Chem. 1998;273:32312–32321. doi: 10.1074/jbc.273.48.32312. [DOI] [PubMed] [Google Scholar]
- Kishore R, Tebo JM, Kolosov M, Hamilton TA. Clustered AU-rich elements are the target of IL-10-mediated mRNA destabilization in mouse macrophages. . J Immunol. 1999;162:2457–2461. [PubMed] [Google Scholar]
- Brown CY, Lagnado CA, Vadas MA, Goodall GJ. Differential regulation of the stability of cytokine mRNAs in lipopolysaccharide-activated blood monocytes in response to interleukin-10. . J Biol Chem. 1996;271:20108–20112. doi: 10.1074/jbc.271.33.20108. [DOI] [PubMed] [Google Scholar]
- Taylor GA, Carballo E, Lee DM, et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. . Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- Badger AM, Griswold DE, Kapadia R, et al. Disease-modifying activity of SB 242235, a selective inhibitor or p38 mitogen-activated protein kinase, in rat adjuvant-induced arthritis. . Arthritis Rheum. 2000;43:175–183. doi: 10.1002/1529-0131(200001)43:1<175::AID-ANR22>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]