

Abstract
Background
Previous work has suggested that in the liver, adenosine preconditioning is mediated by nitric oxide. Whether the endothelial isoform of nitric oxide synthase plays a part in this mechanism has however not yet been investigated.
Methods
Wistar rats were used (6 in each group) – Groups: (1) sham, (2) ischemia-reperfusion, (3) adenosine + ischemia-reperfusion, (4) endothelial isoform inhibitor + adenosine + ischemia-reperfusion.
Results
Using immunohistochemistry, this study has revealed a decrease in the expression of endothelial nitric oxide synthase following hepatic ischemia-reperfusion. This was prevented by adenosine pre-treatment. When an inhibitor of endothelial nitric oxide synthase was administered prior to adenosine pre-treatment, pre-conditioning did not occur despite normal expression of endothelial nitric oxide synthase.
Conclusions
These findings suggest that adenosine attenuates hepatic injury by preventing the downregulation of endothelial nitric oxide synthase that occurs during ischemia-reperfusion.
Keywords: ischemia, liver, Wistar rats, immunohistochemistry, NG-Nitro-L-arginine
Background
Hepatic surgery may be complicated by liver failure. This is usually a result of poor hepatic reserve and reperfusion injury [1]. The latter is partly due to failure of the hepatic microcirculation. Sinusoidal and Kupffer cell swelling following depletion of adenosine 5'-triphosphate during the ischemic period, increased production of the potent vasoconstrictor endothelin and possibly decreased production of nitric oxide (NO), all contribute to narrowing of the sinusoidal lumen [2].
Nitric oxide (NO), a hydrophobic gaseous molecule, is synthesised from L-arginine by different isoforms of nitric oxide synthase (NOS). Two principal forms of NOS have been described – a constitutive/endothelial form (eNOS) and an inducible form expressed by a number of cells, usually in response to inflammatory mediators. The properties of NO appear to depend on which isoform has contributed to its formation. eNOS, a 135-kDa protein, is dependent on intracellular calcium levels for its activity. It produces a basal level of NO that acts as a vasodilator [3].
Preconditioning refers to a phenomenon in which tissues are rendered resistant to the deleterious effects of ischemia-reperfusion (I-R), either by previous exposure to brief periods of vascular occlusion (ischemic preconditioning) or by the administration of certain chemical agents (pharmacological preconditioning). Of the latter, exogenous adenosine is the most studied and it has been shown to attenuate hepatic reperfusion injury [4-8]. Administration of non-specific inhibitors of NOS reverses this protective effect [4-6]. Adenosine may thus prevent the decrease in NO production believed to occur during early reperfusion. The present study was therefore designed to investigate the effect of exogenous adenosine on eNOS expression during hepatic reperfusion.
Methods
The study was performed using male Wistar rats (6 for each group) weighing 250–300 g. All animals (including controls) were anaesthetised by intraperitoneal administration of 2.7 ml/kg of a mixture of one part midazolam + one part Hypnorm ((1 ml of Hypnorm contains 10 mg fluanisone and 0.315 mg fentanyl citrate)) + two parts sterile water. The rats were placed supine on a heating pad to maintain body temperature between 36.5°C and 37.5°C throughout the procedure. The carotid artery was cannulated using an intravenous cannula (Portex, Hythe, UK, 20 G/32 mm) to allow for continuous blood pressure monitoring (Device Mx2, Lectromed UK, Letchworth, UK) and the infusion of normal saline at 0.5 ml/h. All animals underwent laparotomy. The left lateral and median lobes of the liver were mobilised and delivered through the incision after dividing their suspensory ligaments. An avascular plane was developed between the ventral surface of the liver and the portal vein and hepatic artery supplying these lobes. To induce hepatic ischemia, an atraumatic microvascular clip was applied to occlude the portal vein and hepatic artery just distal to the branches supplying the right lateral lobe for 45 min. After 6 h of reperfusion in the I-R groups, and in the control groups a time period equivalent to the duration of ischemia and reperfusion, blood (0.5 ml) was withdrawn from the arterial cannula for the measurement of liver enzymes. The left lateral and median lobes of the liver were harvested and divided for histological paraffin wax examination and immunohistochemical staining. All animal experimental procedures were carried out in accordance with UK Government legislation (Project Licence Number: 70/4393).
To study whether adenosine could modulate NOS expression in hepatic I-R, the following experimental groups were set up:
Group 1 animals subjected to anaesthesia and sham laparotomy only;
Group 2 animals subjected to 45 minutes of partial hepatic ischemia (as described above), followed by 6 h of reperfusion;
Group 3 animals subjected to hepatic ischemia (as described in Group 2), were pre-treated with adenosine (1 mg/kg) dissolved in bicarbonate-buffered saline (pH 7.4) administered via the carotid artery cannula over a 20 minute period. (Animals enrolled in a group not randomised to receive adenosine were given an equivalent volume of buffer solution at the corresponding time period). Hepatic ischemia was induced immediately following the termination of the adenosine infusion.
Group 4 animals treated with adenosine and subjected to hepatic ischemia (as described in Group 3) were also treated with the relative eNOS inhibitor NG-Nitro-L-arginine (L-NA). This was administered via the carotid cannula at a dose of 10 mg/kg, 3 minutes prior to the administration of adenosine. (Animals in all the other groups received an equivalent volume of buffer solution at the corresponding time period).
Liver transaminases
Heparinised blood samples from each rat were immediately centrifuged at 2,000 g for 10 minutes and the supernatant plasma was stored at -20°C. Aspartate and alanine transaminase levels in plasma were later measured in a clinical chemistry laboratory at Hammersmith Hospital, London, UK, using an Olympus AU600 Analyser (Olympus Optical Co Ltd, Tokyo, Japan), as markers of hepatocellular injury.
H&E staining
Liver sections from each animal were fixed in 10% neutral buffered formalin, dehydrated by passage through graded ethanol series, cleared in xylene and embedded in paraffin blocks. The blocks were 4-μm sectioned and stained with hematoxylin and eosin, according to standard protocols. Sections were evaluated using light microscopy by a blinded member of our research team.
Immunohistochemistry
Liver sections from each animal were fixed in 1% paraformaldehyde in phosphate buffered saline (PBS) for four hours and then transferred into a storage buffer, PBS/sucrose, at 4°C until the cryostat blocks were prepared. Tissue sections (4 μm) were cut on a cryostat and applied to slides. The avidin-biotin complex immunoperoxidase staining system was used. The primary antibody, anti-eNOS monoclonal, was produced in mice and was obtained from Affiniti Research Products Ltd, Exeter UK. The presence of positive staining with the specific antibody was indicated by the development of a reddish-brown stain in the section, as a consequence of using the 3,3'-diamino-benzidine substrate. Each section was reviewed by a blinded member of the research team who noted the presence or absence of staining and its cellular location.
Statistics
The liver enzyme data were log transformed to satisfy the assumptions required to perform parametric tests and are therefore presented as geometric mean and 95% confidence intervals of the mean. Orthogonal contrasts in ANOVA were then used to analyse statistically the difference between any two specified groups.
Results
Liver Transaminases (see figure 1)
Figure 1.

Bar graphs demonstrating the geometric mean and 95% confidence interval for plasma aspartate transaminase and plasma alanine transaminase in each group. [Sham – Group 1 (sham laparotomy); IR only – Group 2 (ischemiareperfusion without prior adenosine therapy); A+IR – Group 3 (ischemiareperfusion with prior adenosine pre-treatment); L-NA+A+IR – Group 4 (ischemiareperfusion with administration of the eNOS inhibitor NG-Nitro-L-arginine prior to adenosine preconditioning).
The concentrations of plasma liver transaminases between the different groups were significantly different at 6 h following the onset of reperfusion (p < 0.001 using ANOVA following logarithmic transformation). Both aspartate and alanine transaminase plasma levels were markedly higher in Group 2 (I-R only) when compared to Group 1 (sham laparotomy) (p < 0.001). The administration of adenosine just prior to the onset of ischemia in Group 3 significantly attenuated the rise in liver enzymes (p < 0.001 for both transaminases when compared with the I-R group). In Group 4, administration of L-NA prior to adenosine pre-treatment resulted in an increase in plasma liver transaminases similar to that seen in the I-R group (p > 0.5 for both transaminases).
H&E staining
On routine histology, liver sections from all the animals in Group 1 (sham) revealed a normal hepatic architecture with no evidence of hepatocyte necrosis. Partial hepatic ischemia followed by reperfusion in Group 2 animals resulted in moderate hepatic injury. This was visualised microscopically as centrilobular hepatocyte necrosis, mitotic figures, trabecular derangement and polymorphonuclear cell infiltrate. In liver sections from rats in Group 3 (A + I-R), there was no evidence of hepatocyte injury except for the occasional appearance of isolated necrotic hepatocytes. Sections from Group 4 (L-NA + A + I-R) were similar to those from Group 2 (I-R only).
Immunohistochemistry (see figure 2)
Figure 2.
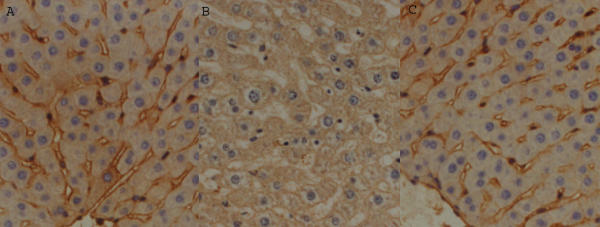
Liver sections, representative of each group, stained with anti-eNOS antibody. Sham and A+IR – very strong staining of sinusoidal endothelial cells; IR only – absence of staining of sinusoidal endothelial cells. [a. Sham – Group 1 (sham laparotomy); b. IR only – Group 2 (ischemia-reperfusion without prior adenosine administration); c. A+IR – Group 3 (ischemia-reperfusion with prior adenosine pre-treatment).]
In all liver samples from Group 1 (sham), staining with the anti-eNOS antibody revealed pronounced staining of all the endothelial cells, i.e. both those lining the blood vessels and the sinusoids. In all specimens from Group 2 (I-R only) staining of sinusoidal endothelial cells in the centrilobular region was absent whilst staining of endothelial cells lining blood vessels and sinusoids in the periphery of the lobule was much less intense than in samples from control animals. Liver sections from all those animals receiving adenosine prior to I-R (Group 3 and 4) had a distribution and intensity of staining with eNOS antibody similar to that in the livers from the control group.
Discussion
The current study has shown that 45 minutes of hepatic ischemia followed by 6 h of reperfusion result in considerable hepatic injury as evidenced by microscopic changes in the liver architecture and release of transaminases. This was associated with a decrease in the normal expression of eNOS within the sinusoidal endothelial cells. This is consistent with previous work suggesting that NO production during the immediate reperfusion period is decreased. Ohmori et al have shown that the administration of NO donors attenuates early hepatic reperfusion injury [9].
In keeping with previous reports from Peralta et al [4,5], these changes were largely prevented by the administration of systemic adenosine prior to the ischemic period. The experimental model in this study, however, differed slightly from that used by the Barcelona group in two main aspects. The left lateral and median lobes, which account for 70% of the rat's liver mass, were rendered ischemic as opposed to the right hepatic lobe (30% of liver mass). The current model is therefore more similar to the clinical situation where the whole liver is rendered ischemic during total vascular exclusion (which is not tolerated in the rat) for liver resections. Outcome measures (hepatic transaminases and histology) were determined at 6 h from the onset of reperfusion as opposed to 90 minutes, since a number of mechanisms, such as neutrophil activation, which contribute to hepatic injury are operative by 6 h but not by 90 minutes [2].
The findings in this study support previous reports that in models of hepatic I-R the protective effect of adenosine seems to be mediated by NO [4,5]. NO is not solely a smooth muscle relaxant. In the last decade NO has been shown to act as a potent anti-inflammatory agent, inhibiting the expression of pro-inflammatory genes [10,11]. This may partly account for its important role in hepatic I-R. We believe that basal production of NO from endogenously expressed eNOS in the sinusoidal cells of the liver may exert an inhibitory effect on the activation of pro-inflammatory transcription factors. During the no-reflow period, downregulation of eNOS results in subnormal NO concentrations. This may lead to activation of pro-inflammatory genes with the subsequent expression of pro-inflammatory cytokines, chemokines and cellular adhesion molecules, all of which are involved in the pathogenesis of hepatic reperfusion injury.
Using immunohistochemical techniques, the current study has revealed that adenosine prevents the downregulation of eNOS. To evaluate whether this finding is part of the mechanism by which adenosine attenuates hepatic I-R injury, a relative eNOS inhibitor was administered prior to adenosine to verify whether the prevention of eNOS downregulation was implicated in this protective mechanism. Despite normal expression of eNOS, the rise in liver enzymes in those animals administered an eNOS inhibitor prior to adenosine was similar to that of those animals not receiving adenosine preconditioning, supporting the hypothesis that the protective effect of adenosine in hepatic I-R is a result of the prevention of eNOS downregulation. We think that adenosine, by preventing the downregulation of eNOS, may prevent the cascade of events resulting from activation of pro-inflammatory nuclear transcription factors, thereby exerting its protective effect. In keeping with this hypothesis, recent experimental work has shown that adenosine prevents the activation of a potent pro-inflammatory nuclear transcription factor when administered prior to cardiac ischemia-reperfusion [12].
Further work is required to study the mechanisms by which adenosine prevents the downregulation of eNOS. Adenosine is a major component of vascular homeostasis, playing an important role in regulating smooth muscle tone acting via cAMP-mediated cascades to induce vascular smooth muscle relaxation [13]. Evidence is accumulating that eNOS expression is regulated in response to shear stress via poorly understood mechanisms [14]. We therefore infer that by altering blood flow through the sinusoids, adenosine may thus, indirectly alter eNOS expression.
This study has shed more light on a mechanism by which hepatic reperfusion injury may be attenuated. By preventing the downregulation of eNOS within the hepatic sinusoidal cells, adenosine has been shown to act as a potent preconditioning agent.
Abbreviations
I-R Ischemia-reperfusion
NO Nitric oxide
(e)NOS (endothelial) nitric oxide synthase
L-NA NG-Nitro-L-arginine
Competing interests
None declared.
Authors' Contributions
Study design: FS-I, RCW, RTM
Experimental animal model FS-I, ITV, RTM
Immunohistochemistry: FS-I, ITV, NAH, RTM
Writing up of manuscript: FS-I
All authors read and approved the final manuscript
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Ferdinand Serracino-Inglott, Email: fsinglott@hotmail.com.
Ioannis T Virlos, Email: itvirlos@hotmail.com.
Nagy A Habib, Email: nagy.habib@ic.ac.uk.
Robin CN Williamson, Email: r.williamson@ic.ac.uk.
Robert T Mathie, Email: rmathie@trusthomeopathy.org.
References
- Delva E, Camus Y, Nordlinger B, et al. Vascular occlusion for liver resections. Operative management and tolerance to ischemia: 142 cases. Ann Surg. 1989;209:211–218. doi: 10.1097/00000658-198902000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serracino-Inglott F, Habib NA, Mathie RT. Hepatic ischemia-reperfusion injury. Am J Surg. 2001;181:160–166. doi: 10.1016/S0002-9610(00)00573-0. [DOI] [PubMed] [Google Scholar]
- Stewart AG, Barker JE, Hickey MJ. Nitric oxide in ischemia-reperfusion injury. In: Grace PA, Mathie RT, editor. Ischaemia-Reperfusion Injury. 1. Oxford: Blackwell Science; 1999. pp. 180–195. [Google Scholar]
- Peralta C, Hotter G, Closa D, Prats N, Xaus C, Gelpí E, et al. The protective role of adenosine in inducing nitric oxide synthesis in rat liver ischemia preconditioning is mediated by activation of adenosine A2 receptors. Hepatology. 1999;29:126–132. doi: 10.1002/hep.510290104. [DOI] [PubMed] [Google Scholar]
- Peralta C, Hotter G, Closa D, Gelpí E, Bulbena O, Roselló-Catafau J. Protective effect of preconditioning on the injury associated to hepatic ischemia-reperfusion in the rat: role of nitric oxide and adenosine. Hepatology. 1997;25:934–937. doi: 10.1002/hep.510250424. [DOI] [PubMed] [Google Scholar]
- Peralta C, Closa D, Hotter G, Gelpí E, Prats N, Roselló-Catafau J. Liver ischemic preconditioning is mediated by the inhibitory action of nitric oxide on endothelin. Biochem Biophys Res Commun. 1996;229:264–270. doi: 10.1006/bbrc.1996.1790. [DOI] [PubMed] [Google Scholar]
- Peralta C, Prats N, Xaus C, Gelpí E, Roselló-Catafau J. Protective effect of liver ischemic preconditioning on liver and lung injury induced by hepatic ischemia-reperfusion in the rat. Hepatology. 1999;30:1481–1489. doi: 10.1002/hep.510300622. [DOI] [PubMed] [Google Scholar]
- Todo S, Zhu Y, Zhang S, Jin MB, Ishizaki N, Tanaka H, et al. Attenuation of ischemic liver injury by augmentation of endogenous adenosine. Transplantation. 1997;63:217–223. doi: 10.1097/00007890-199701270-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori H, Dhar DK, Nakashima Y, et al. Beneficial effects of FK409, a novel nitric oxide donor, on reperfusion injury of rat liver. Transplantation. 1998;66:579–585. doi: 10.1097/00007890-199809150-00005. [DOI] [PubMed] [Google Scholar]
- Peng HB, Libby P, Liao JK. Induction and stabilisation of of IκB-α by nitric oxide mediates inhibition of NFκ-B. J Biol Chem. 1995;270:14214–14219. doi: 10.1074/jbc.270.23.14214. [DOI] [PubMed] [Google Scholar]
- Matthews JR, Botting CH, Panico M, Morris HR, Hay RT. Inhibition of NF-κB DNA binding by nitric oxide. Nucleic Acid Res. 1996;24:2236–2242. doi: 10.1093/nar/24.12.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Ha T, Liu L, Browder W, Kao RL. Adenosine prevents activation of transcriptional factor NF-κB and enhances activator protein-1 binding activity in ischemic rat heart. Surgery. 2000;127:161–169. doi: 10.1067/msy.2000.101582. [DOI] [PubMed] [Google Scholar]
- Stiles GL. Adenosine receptors and beyond: molecular mechanisms of physiological regulation. Clin Res. 1990;38:10–18. [PubMed] [Google Scholar]
- Griffith TM. Shear stress and nitric oxide release: physiological integration of cellular mechanisms, physical forces and flow regulation. In: Mathie RT, Griffith TM, editor. The Haemodynamic Effects of Nitric Oxide. 1. London: Imperial College Press; 1999. pp. 22–51. [Google Scholar]