

Abstract
The tumor necrosis factor alpha (TNF-α) −308 G/A and TNF-β NcO1 polymorphisms have been described to be associated with an increased risk for sepsis in critically ill patients. Functional consequences associated with these polymorphisms remain unclear. We compared the genotype distribution of these TNF polymorphisms with susceptibility to severe sepsis and leukocyte function in blunt trauma patients (n = 70; mean injury severity score, 24 points [range, 4 to 57). Severe sepsis was defined according to the American College of Chest Physicians-Society of Critical Care Medicine consensus conference criteria. Genotyping for the NcO1 polymorphism (alleles TNFB1 and TNFB2) was performed by PCR and digestion of the products with NcO1, and that for the TNF-α −308 G/A polymorphism (alleles TNF1 and TNF2) was performed by real-time PCR. Leukocyte function was assessed by measurement of the production of endotoxin-induced cytokines (TNF-α, interleukin-6 [IL-6], and IL-8) in whole blood. TNF-α, IL-6, and IL-8 were determined by enzyme-linked immunosorbent assay. For the genotypes of the TNF-α −308 G/A polymorphism, differences in the frequency of development of severe sepsis were not detectable. Patients developing severe sepsis after trauma were significantly more likely to posses a homozygous genotype of the TNF-β NcO1 polymorphism. Compared with heterozygotes, the odds ratio for the TNFB2/B2 genotype for the development of severe posttraumatic sepsis was 11 (P = 0.01), and that for the TNFB1/B1 genotype was 13 (P = 0.014). TNF-α −308:TNF-β NcO1 haplotype analysis showed that the TNFB2:TNF2 haplotype is significantly negatively associated with development of severe sepsis. Patients homozygous for the TNFB1 or TNFB2 allele showed a persistently higher cytokine-producing capacity during at least 4 to 8 days after trauma than the heterozygotes. In patients homozygous for the TNF1 allele, a higher TNF-α- and IL-8-producing capacity was found only at day 1 after trauma. Although the TNF-β NcO1 polymorphism appears to be less likely to be causative for development of severe sepsis after trauma, it is thus far the only genetic marker identified which can be used as a relevant risk estimate for severe sepsis in trauma patients immediately after the injury.
Cytokines are integral components of the host's immune response to infection or tissue damage, and, in particular, tumor necrosis factor (TNF) is thought to play a pivotal role (4, 35). Furthermore, an innate profile of cytokine production and a genetic predisposition for fatal infectious diseases has been supposed (3, 42). Subsequently, the TNF gene was considered as a candidate predisposing gene in fatal infectious diseases, and the role of TNF gene polymorphisms in host defense and their influence on cytokine production were studied.
The TNF locus is located on chromosome 6 within the major histocompatibility complex class IV gene cluster. It consists of the functional gene for TNF-α, which is positioned between the TNF-β (lymphotoxin α [LTα]) gene and the LTβ gene. So far at least nine polymorphisms and five microsatellites in the TNF locus have been characterized, and some of them have been found to be strongly associated with certain HLA alleles (33).
Two of these polymorphisms in the human TNF locus have been described to be associated with severe clinical forms of infectious diseases (7, 15, 22, 37), i.e., the NcO1 polymorphism of the TNF-β (LTα) gene and a −308 G/A polymorphism in the TNF-α promoter region. The guanine-to-adenine transition at position −308 in the TNF promoter, which defines the rare allele TNF2 and is strongly associated with the major histocompatibility complex haplotype HLA-A1, B8, DR3, has been reported to influence TNF promoter activity and to be associated with enhanced TNF-α production in vitro and in vivo by some investigators (15, 17, 40, 43), while others could not confirm differences in TNF promoter activity in relation to position −308 (1, 6, 36).
In patients with cerebral malaria and mucocutanous leishmaniasis, the TNF-α −308G/A polymorphism has been shown to be associated with outcome and clinical course of the disease (7, 22). In patients with meningococcal disease (42), the TNF-α −308G/A polymorphism was not associated with outcome, indicating that the significance of the TNF-α −308G/A polymorphism does not in general extend to infectious diseases.
The biallelic restriction fragment length polymorphism in the first intron of the TNF-β gene at positions 1064 to 1069, which is identified by the restriction fragments TNFB1 (5.5 kb) and TNFB2 (10.5 kb) by NcO1 digestion, has been described to be associated with several autoimmune diseases (10, 29, 30).
The TNFB1 allele has been described to be strongly associated with the A1, B8, DR3 haplotype (28). The TNF-β NcO1 polymorphism was found to be correlated with an amino acid variation of the TNF-β sequence at position 26, which is asparagine for the TNFB1 sequence and threonine for the TNFB2 sequence (23). Furthermore, it was described that the TNF-β response of peripheral blood mononuclear cells from TNFB1-homozygous individuals is significantly increased after phytohemagglutinin stimulation, whereas monocytes from TNFB2-homozygous individuals showed a significantly increased interleukin-1β (IL-1β) and TNF-α response after phytohemagglutinin and endotoxin stimulation (23, 25, 29, 30). Recently, it has been shown that the TNF-β NcO1 polymorphism is associated with high mortality in severe sepsis of various origins (37) and with a significantly increased risk for the development of severe sepsis in multiply injured blunt trauma patients (20). Although the TNF-α −308 G/A polymorphism has not been found to be associated with mortality in severe sepsis (36), an association of the rare TNF2 allele with susceptibility and mortality in septic shock patients has been described (24). However, functional consequences associated with these polymorphisms in inflammatory situations such as sepsis or trauma remain unclear. Moreover, until now no human TNF gene polymorphism which is substantially related to TNF production in health and disease states has been identified, and TNF gene expression has been shown to be regulated only in part by transcriptional events (27, 39).
To further compare the roles of these two TNF gene polymorphisms as predisposing markers for severe infectious complications, we studied the genotype distribution of the TNF-α −308 G/A and TNF-β NcO1 polymorphisms in connection with the development of severe sepsis and leukocyte function in blunt trauma patients. In order to exclude influences of cell preparation techniques and to preserve a natural environment, leukocyte function was tested in a whole-blood assay system using the ex vivo cytokine (TNF-α, IL-6, and IL-8) response to endotoxin as a functional parameter (8, 13, 19). Since functional consequences associated with the TNF-α −308 G/A and TNF-β NcO1 polymorphisms are controversial and despite the fact that these polymorphisms are certainly not decisive in the regulation of the IL-6 and IL-8 response to endotoxin, TNF-α, IL-6, and IL-8 were determined because of their biological relevance in inflammatory and septic responses in order to obtain a global measure of the patients' actual reactivity towards an inflammatory challenge.
MATERIALS AND METHODS
Patient evaluation and study protocol.
The protocol used was approved by the local ethics committee. Seventy patients sustaining blunt trauma were considered for entry into the study. Patients were assigned an injury severity score (ISS) (11) by independent evaluators. Exclusion criteria were (i) age of <18 years or >65 years, (ii) admission >8 h after trauma or secondary admission, (iii) penetrating injuries, and (iv) any chronic illness. Injuries of the various body regions (head and neck, face, thorax, abdomen, extremities, and skin) were classified by using the abbreviated injury scale (AIS) (11). The clinical course was monitored prospectively in all patients. Severe sepsis was defined according to the American College of Chest Physicians-Society of Critical Care Medicine consensus conference criteria (2) and was diagnosed not before day 5 after trauma. The demographic data for the patients are given in Table 1. The patients' mean ISS ± standard error of the mean (SEM) was 24 ± 1.3, with the following AIS values of the specific body regions (mean ± SEM): head and neck, 1.4 ± 0.2; face, 0.5 ± 0.1; thorax, 1.6 ± 0.2; abdomen, 1.4 ± 0.2; extremities, 2.6 ± 0.1; and skin, 0.4 ± 0.1.
TABLE 1.
Demographic data for the study population
| Polymorphism and genotype | n | Mean age ± SEM (yr) | % Male | Mean ISS ± SEM (median) | ISS range | % Died |
|---|---|---|---|---|---|---|
| Total | 70 | 37 ± 2 | 65 | 24 ± 1.3 (22) | 4-57 | 8.6 |
| TNF-α −308 G/A | ||||||
| TNF1/TNF1 | 46 | 39 ± 3 | 59 | 24 ± 1.6 (20) | 4-57 | 8 |
| TNF1/TNF2 | 24 | 37 ± 3 | 68 | 23 ± 2.6 (22) | 8-41 | 8 |
| TNF-β NcO1 | ||||||
| TNFB1/TNFB1 | 12 | 47 ± 7 | 83 | 32.5 ± 2.2 (33) | 25-41 | 25 |
| TNFB1/TNFB2 | 27 | 37 ± 2.8 | 62 | 22 ± 1.6 (22) | 8-41 | 4 |
| TNFB2/TNFB2 | 31 | 37 ± 3.4 | 61 | 23 ± 2.6 (22) | 4-57 | 7 |
All patients requiring surgical intervention received standard surgical care and postoperative intensive care unit treatment. Blood samples were drawn within 24 h after admission (designated day 1) and on subsequent days (7:00 to 8:00 p.m. on days 2, 4, 6, 8, and 14 after injury) of hospitalization. Blood (9 ml) was collected in plastic tubes (NH4-heparin tube; Sarsted, Nümbrecht, Germany) along with the routine baseline laboratory work-up and was immediately used for stimulation ex vivo.
Ex vivo endotoxin stimulation of whole blood.
A human whole-blood assay was used as previously described (18). In brief, whole blood mixed 1:1 (vol/vol) with cell culture medium (RPMI 1640 [GibcoBRL, Karlsruhe, Germany], 64 IU of penicillin per ml, 64 μg of streptomycin per ml) was transferred to microtiter plates (Falcon 3072 Microtest III tissue culture tube; Becton Dickinson, Paramus, N.J.). Samples were prepared in duplicate. The mixtures were incubated at 37°C and 5% CO2 with endotoxin (lipopolysaccharide [100 ng/ml] from Salmonella enterica serovar Friedenau [stock solution, 5 mg/ml]) (generously provided by H. Brade, Forschungszentrum Borstel, Borstel, Germany) for 4 h. Control mixtures were incubated without endotoxin. After incubation, the supernatants were separated and stored at −20°C.
Immunologic assays of cytokines. (i) TNF-α.
TNF-α in supernatants of the endotoxin-stimulated blood was measured with an enzyme-linked immunosorbent assay (ELISA). Microtiter plates (Nunc, Wiesbaden, Germany) were coated with a monoclonal anti-human TNF-α antibody (Genzyme, Rüsselheim, Germany). The plates were washed three times and were incubated with blocking buffer (10% fetal calf serum in phosphate-buffered saline) for 1 h. After washing three times, a standard curve was prepared using serial dilutions of human recombinant TNF-α (Genzyme), and standards and samples were added to the plates. After incubation for 2 h, the plates were washed again and a second biotin-labeled TNF-α antibody (Genzyme) was added. After incubation for 1 h, the plates were washed again, followed by a 1-h incubation with horseradish peroxidase-labeled streptavidin (Sigma-Aldrich, München, Germany). All samples and standards were assayed in duplicate. Mean values for each were used in calculating sample concentrations by using a linear regression analysis of the optical density (Dynatech MR 5000 micro-ELISA autoreader; test filter, 405 nm; reference filter, 490 nm) versus TNF-α concentration on the standard curve. For the standard curves of each plate, r2 was 0.98 to 1.0. The lower detection limit was 30 pg/ml.
(ii) IL-6 and -8.
Commercially available ELISA kits were used for determinations of IL-6 and IL-8 (both obtained from Endogen, Freiburg, Germany; lower detection limit, 30 pg/ml) in supernatants of whole-blood cultures according to the manufacturer's instructions.
Genotyping for the TNF-α −308 G/A polymorphism.
Blood samples were drawn, and each patient's genomic DNA was extracted by standard phenol-chloroform extraction (34) from peripheral blood mononuclear cells obtained by Ficoll density gradient centrifugation or from whole blood by using a commercially available DNA isolation kit (QIAmp blood kit; Qiagen, Krefeld, Germany) according to the manufacturers' instructions. One microliter (20 to 80 ng) of genomic DNA was used. Genotyping for the TNF −308 G/A genetic polymorphism was performed in a real-time PCR assay with specific fluorescence-labeled hybridization probes, as reported recently (26). The thermocycler was a LightCycler instrument (Roche Diagnostics, Basel, Switzerland). The primers (sense, 5′-AAGGAAACAGACCACAGACCTG; antisense, 5′-GGTCTTCTGGGCCACTGAC) were used at 0.5 μmol/liter, and the detection probe specific for the G allele (TNF1) (5′-AACCCCGTCCCCATGCC) and the anchor probe (5′-CAAAACCTATTGCCTCCATTTCTTTTGGGGAC) were used at 0.2 μmol/liter. The 10-μl PCR mixture contained 1 μl of reaction buffer. Sixty PCR cycles were run, with one PCR cycle consisting of denaturation at 95°C for 0 s and then 20°C/s, annealing at 57°C for 15 s, and extension at 72°C for 10 s. A sample was classified as TNF −308 genotype AA (TNF2/TNF2), AG (TNF1/TNF2), or GG (TNF1/TNF1) according to the derivative melting curves.
Genotyping for the TNF-β NcO1 restriction fragment length polymorphism.
The genotype of the NcoI restriction fragment length polymorphism of the TNF-β gene was determined by PCR amplification and enzymatic digestion of the products with NcoI as described previously (20). PCR was performed using 1 U of Thermus aquaticus thermostable DNA polymerase (Boehringer, Mannheim, Germany), 0.2 μmol of deoxynucleoside triphosphates, 200 pmol of each primer, 1 or 0.1 μl of chromosomal DNA, 50 mM KCl, 10 mM Tris-HCl, and 1.5 mM MgCl2 (pH 8.3) in a total volume of 100 μl. A 782-bp fragment of the TNF-β gene including the first intron was amplified by 37 cycles of denaturation (30 s at 95°C), primer annealing (30 s at 50°C), and DNA extension (45 s at 72°C) in an automated PCR cycler (GeneAmp PCR system 2400; Perkin-Elmer, Darmstadt, Germany). The following nucleotide sequences were used for PCR amplification (37): 5′CCGTGCTTCGTGCTTTGGACTA3′ and 5′AGAGGGGTGGATGCTTGGGTTC3′. Seventeen microliters of the PCR product was digested with 1 μl of NcoI endonuclease (10 U/μl; Gibco, Eggenstein, Germany) and 2 μl of 50 mM Tris-HCl-10 mM KCl-1 mM dithiothreitol-0.1 mg of bovine serum albumin/ml for 2 h at 37°C. This resulted in complete digestion of the PCR product, since extension of incubation time and addition of fresh enzyme did not change the result. The TNFB1 allele includes a restriction site for NcoI which results in 196- and 586-bp fragments after digestion. Following NcoI digestion, 10-μl portions of digested and undigested samples were electrophoresed in a nonreducing 1.5% agarose gel (160 V, 1 h) and stained with ethidium bromide. The bands were visualized under UV light and digitized with a video camera (VarioCam; Phase, Lübeck, Germany).
Statistical analysis.
If not otherwise mentioned, data are expressed as the mean ± the SEM. The Pearson correlation coefficient, chi-square test, Kruskal-Wallis H test, and Mann-Whitney U test were used to test for significant differences between the groups and were calculated with the SPSS for Windows release 10.0.7 program. A two-tailed P value of <0.05 was considered significant. Statistical significance of the odds ratio (OR) associated with a particular genotype (OR = n1n4/n2n3, where n1 is the number of sepsis patients carrying the genotype, n2 is the number of controls carrying the genotype, and n3 and n4 are the corresponding numbers of sepsis and control patients not carrying the genotype) was calculated by using a χ2 distribution with one degree of freedom (21), where χ2PMH = [(n1-4 − 1)/(n1n4 − n2n3)2]/[(n1 + n2)(n3 + n4)(n1 + n3)(n2 + n4)] must be >3.841 to attain a P value of <0.05. A 95% confidence interval [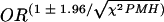 ] excluding the value 1 corresponds to statistical significance.
] excluding the value 1 corresponds to statistical significance.
RESULTS
Allele frequencies and genotype distribution.
The overall allele frequencies were 0.83 for the TNF1 allele and 0.17 for the TNF2 allele of the TNF-α −308 G/A polymorphism and 0.36 for the TNFB1 allele and 0.64 for the TNFB2 allele for the TNF-β NcO1 polymorphism. Twelve patients (17%) were homozygous for the allele TNFB1, 31 patients (44%) were homozygous for the allele TNFB2, and 27 patients (39%) were heterozygous (TNFB1/TNFB2). Forty-six patients (66%) were homozygous for the TNF1 allele of the TNF-α −308 G/A polymorphism, and 24 patients (34%) were heterozygous. No homozygotes were observed for the TNF2 allele of the TNF-α −308 G/A polymorphism (Table 1). A significant association (P < 0.001) was found between the two TNF gene polymorphisms. The distribution of the genotype combinations showed that all TNFB2 homozygotes were TNF1 homozygotes for the TNF-α −308 G/A polymorphism but that the TNF-β NcO1 TNFB2 allele was not always associated with the TNF-α −308 TNF1 allele (Table 2).
TABLE 2.
Association between TNF-α −308 G/A and TNF-β NcO1 genotypes
| TNFβ NcO1 genotype | No. with the following TNFα −308 G/A genotype:
|
|
|---|---|---|
| TNF1/TNF1 | TNF1/TNF2 | |
| TNFB1/TNFB1 | 1 | 11 |
| TNFB1/TNFB2 | 14 | 13 |
| TNFB2/TNFB2 | 31 | 0 |
Ex vivo whole blood cytokine response to endotoxin.
Figure 1 shows the ex vivo cytokine responses of the patients' whole blood to endotoxin for the different genotypes of the TNF-α −308 G/A polymorphism. Significant differences between the TNF1/TNF1 and TNF1/TNF2 genotypes were found on day 1 after trauma for TNF-α and IL-8 production, with a higher cytokine-producing capacity in patients homozygous for the TNF1 allele. For IL-6 production, significant differences between the TNF1/TNF1 and TNF1/TNF2 genotypes were not detectable, although in patients with the TNF1/TNF1 genotype the whole-blood IL-6 response to endotoxin was slightly higher during the 14-day observation period.
FIG. 1.

Ex vivo whole-blood cytokine response to endotoxin in trauma patients carrying the TNF1/TNF1 or TNF1/TNF2 genotype of the TNF-α −308 G/A polymorphism. Values are means ± SEMs. ▪, TNF1/TNF1 genotype (n = 39 to 44); □, TNF1/TNF2 genotype (n = 13 to 19). ∗, P < 0.05. For further details, see Materials and Methods.
The endotoxin-induced cytokine production by the patients' whole blood, depending on the genotype of the TNF-β NcO1 polymorphism, is shown in Fig. 2. Even though statistically significant differences between the genotypes of the TNF-β NcO1 polymorphism were found only on day 2 for TNF-α production, on days 1 to 4 for IL-6 production, and on days 1 and 2 for IL-8 production compared with the heterozygous genotype, patients with a homozygous genotype showed a persistently higher cytokine-producing capacity for at least 4 to 8 days after trauma.
FIG. 2.

Ex vivo whole-blood cytokine response to endotoxin in trauma patients carrying the TNFB1/TNFB1, TNFB1/TNFB2, or TNFB2/TNFB2 genotype of the TNF-β NcO1 polymorphism. Values are means ± SEMs. •, TNFB1/TNFB1 genotype (n = 9 to 12); ▪, TNFB2/TNFB2 genotype (n = 22 to 30); ○, TNFB1/TNFB2 genotype (n = 15 to 24). ∗, P < 0.05 for TNFB1/B2 versus TNFB1/TNFB1; #, P < 0.05 for TNFB1/TNFB2 versus TNFB2/TNFB2. For further details, see Materials and Methods.
Development of severe sepsis.
Fourteen out of 70 patients (20%) developed severe sepsis during the posttraumatic course. The source of infection was predominantly pneumonia. All septic patients had positive blood cultures (41% gram-negative isolates and 59% gram-positive isolates). The first relevant clinical isolates were Enterococcus in two patients, Klebsiella-Enterobacter in one patient, Klebsiella pneumoniae in one patient, Escherichia coli in one patient, Klebsiella oxytoca in one patient, Pseudomonas aeruginosa in one patient, Staphylococcus epidermidis in four patients, and Staphylococcus aureus in four patients. One patient had two isolates.
For the genotypes of the TNF-α −308 G/A polymorphism, differences in the frequency of development of severe sepsis were not detectable (Table 3). In contrast, patients homozygous for the TNFB2 or TNFB1 allele had a significantly higher incidence of severe posttraumatic sepsis than patients with the heterozygous genotype (Table 3). Compared with the heterozygous genotype, the OR for the B2/B2 genotype for the development of severe posttraumatic sepsis was 11 (P = 0.01), and for the homozygous TNFB1 genotype the OR was 13 (P = 0.014). The demographic data for the patients showed that severity of injury, measured using the ISS, was significantly higher in patients homozygous for the TNFB1 allele than in patients with the TNFB2/B2 and TNFB1/B2 genotypes (P < 0.05 by the Kruskal-Wallis H test). This difference was caused by a higher AIS for the body regions head-neck (3.5 ± 0.5 [TNFB1/TNFB1] versus 1.0 ± 0.3 [TNFB2/TNFB2] and 1.3 ± 0.3 [TNFB1/TNFB2]) and extremities (3.0 ± 0.3 [TNFB1/TNFB1] versus 2.8 ± 0.2 [TNFB2/TNFB2] and 2.1 ± 0.3 [TNFB1/TNFB2]). Therefore, the genotype distribution was compared to sepsis development in the subgroup of multiply injured patients (ISS of >15), who carry a high risk for sepsis development. The genotype groups of these patients (n = 58) were homogenous regarding the demographic parameters age, sex, ISS, and AIS for the body regions face, thorax, abdomen, extremities, and skin. Only the AIS of the head-neck region was higher in multiply injured patients homozygous for the TNFB1 allele (3.5 ± 0.5 [mean ± SEM]) than in patients carrying the TNFB2/B2 (1.4 ± 0.5 [mean ± SEM] and TNFB1/B2 (1.5 ± 0.5 [mean ± SEM]) genotypes. Similar to the case for the entire patient population, there were no differences in the frequency of development of severe sepsis between the genotypes of the TNF-α −308 G/A polymorphism, whereas in multiply injured patients the OR for the B2/B2 genotype of the TNF-β NcO1 polymorphism for the development of severe posttraumatic sepsis was 12.6 (P = 0.007), and for the homozygous TNFB1 genotype the OR was 10.5 (P = 0.024), compared with the heterozygous genotype (Table 3). TNF-α −308:TNF-β NcO1 haplotype analysis showed that the TNFB2:−308 A haplotype is significantly negatively associated with sepsis development (Table 4). A significant positive correlation with development of severe sepsis was not found for any of the different haplotypes.
TABLE 3.
Genotype distribution in trauma patients with and without development of severe sepsis
| ISS (no. of patients) | Polymorphism | Genotype | Development of severe sepsis (n)
|
P | OR [95% confidence interval] | |
|---|---|---|---|---|---|---|
| Yes | No | |||||
| 4-57 (70) | TNF-α −308 G/A | TNF1/TNF1 | 10 | 36 | 0.76 | |
| TNF1/TNF2 | 4 | 20 | ||||
| TNF-β NcO1 | TNFB2/TNFB2 | 9 | 22 | 0.78, 0.01 (0.014)a | 11 [1.7-66.7] (13 [1.8-95.5])a | |
| TNFB1/TNFB1 | 4 | 8 | ||||
| TNFB1/TNFB2 | 1 | 26 | ||||
| Homozygousb (TNFB1/TNFB1, TNFB2/TNFB2) | 13 | 30 | 0.008 | 11 [1.92-66.1] | ||
| TNFB1/TNFB2 | 1 | 26 | ||||
| >15 (58) | TNF-α −308 G/A | TNF1/TNF1 | 10 | 28 | 0.59 | |
| TNF1/TNF2 | 4 | 16 | ||||
| TNF-β NcO1 | TNFB2/TNFB2 | 9 | 15 | 0.81, 0.007 (0.024)a | 12.6 [1.97-80.5] | |
| TNFB1/TNFB1 | 4 | 8 | (10.5 [1.3-82.8])a | |||
| TNFB1/TNFB2 | 1 | 21 | ||||
| Homozygous (TNFB1/TNFB1, TNFB2/TNFB2) | 13 | 23 | 0.008 | 11.87 [1.97-71.4] | ||
| TNFB1/TNFB2 | 1 | 21 | ||||
The first value is for the comparison with TNFB1/TNFB1, and the second value is for the comparison with TNFB1/TNFB2. Values in parentheses are for the comparison of the TNFB1/B1 genotype with the TNFB1/B2 genotype.
Patients homozygous for the allele TNFB1 or TNFB2.
TABLE 4.
TNF-α −308:TNF-β NcO1 haplotypes and development of severe sepsis in blunt trauma patients
| Haplotype | No. of patients with the following no. of haplotypesa and with/without development of severe sepsis:
|
rb | Pc | |||
|---|---|---|---|---|---|---|
| 2 | 1 | 0.5 | 0 | |||
| TNFB1:TNF1 | 0/1 | 5/25 | 0/13 | 9/31 | −0.13 | 0.3 |
| TNFB1:TNF2 | 0/0 | 4/11 | 0/13 | 10/46 | 0.048 | 0.69 |
| TNFB2:TNF1 | 9/31 | 1/14 | 0/13 | 4/12 | 0.11 | 0.35 |
| TNFB2:TNF2 | 0/0 | 0/0 | 0/13 | 14/57 | −0.24 | 0.047 |
Haplotypes of heterozygotes were scored as a 0.5 probability for each haplotype.
r, Pearson correlation coefficient for correlation between the number of the patients haplotypes with sepsis development.
P, Level of statistical significance.
DISCUSSION
The TNF-α −308 G/A and the TNF-β NcO1 polymorphisms have both been described to be associated with survival in sepsis or septic shock of various origins (24, 37, 38). Furthermore, we recently reported that the TNFB2/TNFB2 genotype of the TNF-β NcO1 polymorphism is significantly associated with an increased risk for development of severe sepsis in severely injured blunt trauma patients (20). Up to now neither functional consequences associated with these polymorphisms in inflammation nor the relationship of the TNF-α −308 G/A polymorphism to development of severe sepsis and its significance compared with the TNF-β NcO1 polymorphism have been determined for trauma patients.
The determined allele frequency and genotype distribution of the TNF-α −308 G/A and the TNF-β NcO1 polymorphisms were in agreement with the previously described distribution in unrelated and unselected healthy volunteers (6, 12, 16, 23, 24, 28, 30-32). Because of the known low frequency of the TNF2 homozygous genotype of the TNF-α −308 G/A polymorphism (about 5% in a Caucasian population), it is not surprising that no homozygotes for the allele TNF2 were observed in our study population. In line with other reports, we found a tight association between the two polymorphisms (5, 23, 41).
Several polymorphic elements in the TNF gene region as well as the human leukocyte antigen type have been related to TNF production, but the precise location of the genetic elements controlling the TNF response is still unclear. Therefore, we did not expect to detect a strong association between the genotypes of the TNF-α −308 G/A and TNF-β NcO1 polymorphisms and the ex vivo cytokine production in response to endotoxin. Nevertheless, our data indicate that patients with a homozygous genotype of the TNF-β NcO1 polymorphism present with a higher whole-blood cytokine response to endotoxin after trauma, which reaches a level of statistical significance at day 2 for TNF-α, at days 1 to 4 for IL-6, and at days 1 and 2 for IL-8. The differences found for the genotypes of the TNF-α −308 G/A polymorphism at day 1 appear to be caused by the association between the two polymorphisms, with the genotype TNFB2/B2 always associated with the −308 TNF1/TNF1 genotype in our patients.
As expected (20, 37, 41), TNFB2 homozygotes were found to possess a significantly increased risk for the development of severe posttraumatic sepsis, with an OR of 11 (P = 0.01) compared with the heterozygous genotype. Although the small number of TNFB1 homozygous trauma patients makes their risk estimate less accurate, the data indicate that both homozygous genotypes possess a significantly increased susceptibility for the development of severe posttraumatic sepsis compared with the heterozygous genotype.
In contrast to the TNF-β NcO1 polymorphism, the TNF-α −308 G/A polymorphism was not found to be associated with development of severe sepsis in our patients, which is in agreement with previously published data (36, 38, 41). The significant association of the −308GA polymorphism with septic shock susceptibility and/or survival described by some investigators (24, 38) might therefore extend only to the most severe course of infectious disease. On the other hand, several other factors might account for the disparate findings. Besides the relatively small study population in the present and previous studies on the relation of TNF locus polymorphisms to sepsis mortality or susceptibility, differences in distribution of HLA class II alleles based on the ethnic distribution of the study populations also might influence the results. Furthermore, the clinical heterogeneity of patient populations in sepsis studies as well as the possibility that a polymorphism is related to the clinical course and not to the presence or absence of disease should be taken into consideration.
Haplotype analysis indicates that none of the TNF-α −308:TNF-β NcO1 haplotypes is significantly positively associated with development of severe sepsis. On the contrary, the TNFB2:TNF2 haplotype was found to be negatively associated with the development of severe sepsis after blunt trauma. In line with our data, a recent study with a population of community-acquired pneumonia patients (41) showed that TNFB2 homozygous individuals posses a significantly increased risk for the development of septic shock, whereas no association with septic shock development was found for the TNF-α −308 GA polymorphism. Moreover, Waterer et al. (41) described a significant association of the TNFB2:TNF1 haplotype with septic shock susceptibility. Although we could not detect a significant association of the TNFB2:TNF1 haplotype with susceptibility to severe sepsis in our patients, the inverse finding of a negative correlation of the TNFB2:TNF2 haplotype with development of severe sepsis confirms the assumption that the TNF-α −308 locus may exert an influence on sepsis development in trauma patients. In accordance with the weak differences in leukocyte function between the genotypes of the two polymorphisms, the data strengthen the hypothesis that the TNF-β NcO1 polymorphism is a genetic marker associated with so-far-unknown genetic elements responsible for sepsis susceptibility and possibly with regulation of leukocyte function. This would be in line with the finding that patients with development of severe sepsis after severe blunt trauma present with a significantly increased cytokine producing capacity in the early posttraumatic period compared with patients with an uncomplicated recovery (9, 14).
Although it was not an aim of our study to compare mortalities of trauma patients having the different TNF-α −308 G/A and TNF-β NcO1 genotypes, it should be noted that compared with patients with all other genotypes, TNFB1 homozygous trauma patients showed an at least threefold-higher mortality. While this might be a chance observation due to the small number of TNFB1 homozygotes, an increased mortality in these patients cannot be excluded until a larger cohort of patients is studied.
Taken together, in contrast to the TNF-α −308G/A polymorphism, the TNF-β NcO1 polymorphism was found to be significantly associated with the development of severe sepsis in blunt trauma patients. Although the TNF-β NcO1 polymorphism appears less likely to be causative for development of severe sepsis after trauma, it is so far the only genetic marker identified which can be used as a relevant risk estimate for severe sepsis in trauma patients immediately after the injury. Even though knowing the risk for severe sepsis would be without therapeutic consequences today, it may lead to the development of a new therapeutic approach adapted to the genetic background of the individual and to the discovery of causative genetic elements which are involved in the regulation of the immune response to blunt trauma in the future.
REFERENCES
- 1.Abraham, L. J., and K. M. Kroeger. 1999. Impact of the −308 TNF promoter polymorphism on the transcriptional regulation of the TNF gene: relevance to disease. J. Leukoc. Biol. 66:562-566. [DOI] [PubMed] [Google Scholar]
- 2.American College of Chest Physicians-Society of Critical Care Medicine Consensus Conference. 1992. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit. Care Med. 20:864-875. [PubMed] [Google Scholar]
- 3.Beutler, B., and A. Cerami. 1987. Cachectin: more than a tumor necrosis factor. N. Engl. J. Med. 316:379-385. [DOI] [PubMed] [Google Scholar]
- 4.Beutler, B., and G. E. Grau. 1993. Tumor necrosis factor in the pathogenesis of infectious diseases. Crit. Care Med. 21:S423-435. [PubMed] [Google Scholar]
- 5.Bouma, G., J. B. Crusius, M. Oudkerk Pool, J. J. Kolkman, B. M. von Blomberg, P. J. Kostense, M. J. Giphart, G. M. Schreuder, S. G. Meuwissen, and A. S. Pena. 1996. Secretion of tumour necrosis factor alpha and lymphotoxin alpha in relation to polymorphisms in the TNF genes and HLA-DR alleles. Relevance for inflammatory bowel disease. Scand. J. Immunol. 43:456-463. [DOI] [PubMed] [Google Scholar]
- 6.Brinkman, B. M., D. Zuijdeest, E. L. Kaijzel, F. C. Breedveld, and C. L. Verweij. 1996. Relevance of the tumor necrosis factor alpha (TNF alpha) −308 promoter polymorphism in TNF alpha gene regulation. J. Inflamm. 46:32-41. [PubMed] [Google Scholar]
- 7.Cabrera, M., M. A. Shaw, C. Sharples, H. Williams, M. Castes, J. Convit, and J. M. Blackwell. 1995. Polymorphism in tumor necrosis factor genes associated with mucocutanous leishmaniasis. J. Exp. Med. 182:1259-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ertel, W., J.-P. Kremer, J. Kenney, U. Steckholzer, D. Jarrar, O. Trentz, and F. W. Schildberg. 1995. Downregulation of proinflammatory cytokine release in whole blood from septic patients. Blood 85:1341-1347. [PubMed] [Google Scholar]
- 9.Flach, R., M. Majetschak, T. Heukamp, V. Jennissen, S. Flohè, J. Börgermann, U. Obertacke, and F. U. Schade. 1999. Relation of ex vivo stimulated blood cytokine synthesis to posttraumatic sepsis. Cytokine 11:173-178. [DOI] [PubMed] [Google Scholar]
- 10.Fugger, L., N. Morling, P. Ryder, J. Georgsen, B. K. Jakobsen, A. Svejgaard, V. Andersen, P. Oxholm, F. Karup Pedersen, and J. Friis. 1989. Nco1 restriction fragment length polymorphism (RFLP) of the tumor necrosis factor (TNFα) region in four autoimmune diseases. Tissue Antigens 34:17-22. [DOI] [PubMed] [Google Scholar]
- 11.Greenspan, L., B. A. McLellan, and H. Greig. 1985. Abbreviated injury scale and injury severity score: a scoring chart. J. Trauma 25:60-64. [DOI] [PubMed] [Google Scholar]
- 12.Hamann, A., C. Mantzoros, A. Vidal-Puig, and J. S. Flier. 1995. Genetic variability in the TNF-alpha promoter is not associated with type II diabetes mellitus (NIDDM). Biochem. Biophys. Res. Commun. 211:833-839. [DOI] [PubMed] [Google Scholar]
- 13.Haskill, S., C. Hohnson, D. Eierman, S. Becker, and K. Warren. 1988. Adherence induces selective mRNA expression of monocyte mediators and proto-oncogenes. J. Immunol. 140:1690-1694. [PubMed] [Google Scholar]
- 14.Heesen, M., U. Obertacke, F. U. Schade, B. Bloemeke, and M. Majetschak. 2002. The interleukin-6 (IL-6) G(-174)C polymorphism and the ex vivo IL-6 response to endotoxin in severely injured blunt trauma patients. Eur. Cytokine Netw. 13:72-77. [PubMed] [Google Scholar]
- 15.Kroeger, K. M., K. S. Carville, and L. J. Abraham. 1997. The −308 tumor necrosis factor α promotor polymorphism effects transcription. Mol. Immunol. 34:391-399. [DOI] [PubMed] [Google Scholar]
- 16.Laitinen, T., M. L. Lokki, J. Partanen, M. Tulppala, O. Ylikorkala, and S. Koskimies. 1992. Tumor necrosis β gene polymorphism in relation to complotype in couples with spontaneous abortions and in control families. Scand. J. Immunol. 35:131-135. [DOI] [PubMed] [Google Scholar]
- 17.Louis, E., D. Franchimont, A. Piron, Y. Gevaert, N. Schaaf-Lafontaine, S. Roland, P. Mahieu, M. Malaise, D. de Groote, R. Louis, and J. Belaiche. 1998. Tumour necrosis factor (TNF) gene polymorphism influences TNF-alpha production in lipopolysaccharide (LPS)-stimulated whole blood cell culture in healthy humans. Clin. Exp. Immunol. 113:401-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majetschak, M., J. Börgermann, C. Waydhas, U. Obertacke, D. Nast-Kolb, and F. U. Schade. 2000. Diminished endotoxin induced whole blood TNF-α production and its relation to systemic concentrations of interleukin-4, interleukin-10 and transforming growth factor-β1 in multiply injured patients. Crit. Care Med. 28:1847-1854. [DOI] [PubMed] [Google Scholar]
- 19.Majetschak, M., R. Flach, T. Heukamp, V. Jennissen, U. Obertacke, F. Neudeck, K. P. Schmit-Neuerburg, and F. U. Schade. 1997. Regulation of whole blood tumor necrosis factor production upon endotoxin stimulation after severe blunt trauma. J. Trauma 43:880-887. [DOI] [PubMed] [Google Scholar]
- 20.Majetschak, M., S. Flohé, U. Obertacke, J. Schröder, K. Staubach, D. Nast-Kolb, F. U. Schade, and F. Stüber. 1999. Relation of a TNF gene polymorphism to severe sepsis in trauma patients. Ann. Surg. 230:207-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mantel, N., and W. Haenszel. 1959. Statistical aspects of the analysis of data from retrospective studies of disease. J. Natl. Cancer Institute 22:719-748. [PubMed] [Google Scholar]
- 22.McGuire, W., A. V. S. Hill, C. E. M. Allsopp, B. M. Greenwood, and D. Kwiatkowski. 1994. Variation in the TNF-α promotor region associated with susceptibility to cerebral malaria. Nature 371:508-511. [DOI] [PubMed] [Google Scholar]
- 23.Messer, G., U. Spengler, M. C. Jung, G. Honold, K. Blomer, G. R. Pape, G. Riethmüller, and E. H. Weiss. 1991. Polymorphic structure of the tumor necrosis factor (TNF) locus: an Nco1 polymorphism in the first intron of the human TNF-beta gene correlates with a variant amino acid in position 26 and a reduced level of TNF-beta production. J. Exp. Med. 173:209-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mira, J. P., A. Cariou, F. Grall, C. Delclaux, M. R. Losser, F. Heshmati, C. Cheval, M. Monchi, J. L. Teboul, F. Riche, G. Leleu, L. Arbibe, A. Mignon, M. Delpech, and F. Dhainaut. 1999. Association of TNF2, a TNF-alpha promoter polymorphism, with septic shock susceptibility and mortality: a multicenter study. JAMA 282:561-568. [DOI] [PubMed] [Google Scholar]
- 25.Molving, J., F. Pociot, L. Baek, H. Worsaae, L. Dall Wogensen, P. Christensen, L. Staub-Nielsen, T. Mandrup-Poulsen, K. Manogue, and J. Nerup. 1990. Monocyte function in IDDM patients and healthy individuals. Scand. J. Immunol. 31:297-306. [DOI] [PubMed] [Google Scholar]
- 26.Moos, V., M. Rudwaleit, V. Herzog, K. Hohlig, J. Sieper, and B. Müller. 2000. Association of genotypes affecting the expression of interleukin-1beta or interleukin-1 receptor antagonist with osteoarthritis. Arthritis Rheum. 43:2417-2422. [DOI] [PubMed] [Google Scholar]
- 27.Pauli, U. 1994. Control of tumor necrosis factor gene expression. Crit. Rev. Eukaryot. Gene Expr. 4:323-344. [DOI] [PubMed] [Google Scholar]
- 28.Peruccio, D., S. d'Alfonso, I. Borelli, A. Amoroso, G. Mazzola, D. Marsico, M. Bersanti, and P. Richiardi. 1993. Distribution of tumor necrosis factor alleles (Nco1 RFLP) and their relationship to HLA haplotypes in an Italian population. Hum. Hered. 43:103-110. [DOI] [PubMed] [Google Scholar]
- 29.Pociot, F., C. Briant, V. Jongeneel, J. Molvig, H. Worsaae, M. Abbal, M. Thomsen, J. Nerup, and A. Cambon-Thomsen. 1993. Association of tumor necrosis factor (TNF) and class II major histocompatibility complex with the secretion of TNF-alpha and TNF-beta by human mononuclear cells: a possible link to insulin-dependent diabetes mellitus. Eur. J. Immunol. 23:224-231. [DOI] [PubMed] [Google Scholar]
- 30.Pociot, F., J. Molvig, L. Wogensen, H. Worsaae, H. Dalboge, L. Baek, and J. Nerup. 1991. A tumor necrosis factor beta gene polymorphism in relation to monokine secretion and insulin-dependent diabetes mellitus. Scand. J. Immunol. 33:37-49. [DOI] [PubMed] [Google Scholar]
- 31.Pociot, F., S. D'Alfonso, S. Compasso, R. Scorza, and P. M. Richiardi. 1995. Functional analysis of a new polymorphism in the human TNF alpha gene promoter. Scand. J. Immunol. 42:501-504. [DOI] [PubMed] [Google Scholar]
- 32.Rinasco, J., Y. Beraun, A. Nieto, A. Fraile, L. Mataran, E. Pareja, and J. Martin. 1997. Polymorphism at the TNF loci in rheumatoid arthritis. Tissue Antigens 49:74-78. [DOI] [PubMed] [Google Scholar]
- 33.Ruuls, S. R., and J. D. Sedgwick. 1999. Unlinking tumor necrosis factor biology from the major histocompatibility complex: lessons from human genetics and animal models. Am. J. Hum. Genet. 65:294-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed., p. 14-21. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 35.Strieter, R. N., S. L. Kunkel, and R. C. Bone. 1993. Role of tumor necrosis factor alpha in disease states and inflammation. Crit. Care Med. 21:S447-463. [DOI] [PubMed] [Google Scholar]
- 36.Stüber, F., I. A. Udalova, M. Book, L. N. Drutskaya, D. V. Kuprash, R. L. Turetskaya, F. U. Schade, and S. A. Nedospasov. 1996. −308 tumor necrosis factor (TNF) polymorphism is not associated with survival in severe sepsis and is unrelated to lipopolysaccharide inducibility of the human TNF promoter. J. Inflamm. 46:42-50. [PubMed] [Google Scholar]
- 37.Stüber, F., M. Petersen, F. Bokelmann, and F. U. Schade. 1996. A genomic polymorphism within the tumor necrosis factor locus influences plasma tumor necrosis factor-α concentrations and outcome of patients with severe sepsis. Crit. Care Med. 24:381-384. [DOI] [PubMed] [Google Scholar]
- 38.Tang, G. J., S. L. Huang, H. W. Yien, W. S. Chen, C. W. Chi, C. W. Wu, W. Y. Lui, J. H. Chiu, and T. Y. Lee. 2000. Tumor necrosis factor gene polymorphism and septic shock in surgical infection. Crit. Care Med. 28:2733-2736. [DOI] [PubMed] [Google Scholar]
- 39.Trowsdale, J. 1993. Genomic structure and function in the MHC. Trends Genet. 9:117-122. [DOI] [PubMed] [Google Scholar]
- 40.Warzocha, K., P. Ribeiro, J. Bienvenu, P. Roy, C. Charlot, D. Rigal, B. Coiffier, and G. Salles. 1998. Genetic polymorphisms in the tumor necrosis factor locus influence non-Hodgkin's lymphoma outcome. Blood 91:3574-3581. [PubMed] [Google Scholar]
- 41.Waterer, G. W., M. W. Quasney, R. M. Cantor, and R. G. Wunderink. 2001. Septic shock and respiratory failure in community-acquired pneumonia have different TNF polymorphism associations. Am. J. Respir. Crit. Care Med. 163:1599-1604. [DOI] [PubMed] [Google Scholar]
- 42.Westendorp, R. G. J., J. A. M. Langermans, T. W. J. Huizinga, C. L. Verweij, and A. Sturk. 1997. Genetic influence on cytokine production and fatal meningococcal disease. Lancet 349:170-173. [DOI] [PubMed] [Google Scholar]
- 43.Wilson, A. G., J. A. Symons, T. L. McDowell, H. O. McDevitt, and G. W. Duff. 1997. Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc. Natl. Acad. Sci. USA 94:3195-3199. [DOI] [PMC free article] [PubMed] [Google Scholar]