

Abstract
We investigated the contribution of the carboxyl terminus region of the β1a subunit of the skeletal dihydropyridine receptor (DHPR) to the mechanism of excitation-contraction (EC) coupling. cDNA-transfected β1 KO myotubes were voltage clamped, and Ca2+ transients were analyzed by confocal fluo-4 fluorescence. A chimera with an amino terminus half of β2a and a carboxyl terminus half of β1a (β2a 1-287/β1a 325-524) recapitulates skeletal-type EC coupling quantitatively and was used to generate truncated variants lacking 7 to 60 residues from the β1a-specific carboxyl terminus (Δ7, Δ21, Δ29, Δ35, and Δ60). Ca2+ transients recovered by the control chimera have a sigmoidal Ca2+ fluorescence (ΔF/F) versus voltage curve with saturation at potentials more positive than +30 mV, independent of external Ca2+ and stimulus duration. In contrast, the amplitude of Ca2+ transients expressed by the truncated variants varied with the duration of the pulse, and for Δ29, Δ35, and Δ60, also varied with external Ca2+ concentration. For Δ7 and Δ21, a 50-ms depolarization produced a sigmoidal ΔF/F versus voltage curve with a lower than control maximum fluorescence. Moreover, for Δ29, Δ35, and Δ60, a 200-ms depolarization increased the maximum fluorescence and changed the shape of the ΔF/F versus voltage curve, from sigmoidal to bell-shaped, with a maximum at ∼+30 mV. The change in voltage dependence, together with the external Ca2+ dependence and additional controls with ryanodine, indicated a loss of skeletal-type EC coupling and the emergence of an EC coupling component triggered by the Ca2+ current. Analyses of d(ΔF/F)/dt showed that the rate of cytosolic Ca2+ increase during the Ca2+ transient was fivefold faster for the control chimera than for the severely truncated variants (Δ29, Δ35, and Δ60) and was consistent with the kinetics of the DHPR Ca2+ current. In summary, absence of the β1a-specific carboxyl terminus (last 29 to 60 residues of the control chimera) results in a loss of the fast component of the Ca2+ transient, bending of the ΔF/F versus voltage curve, and emergence of EC coupling triggered by the Ca2+ current. The studies underscore the essential role of the carboxyl terminus region of the DHPR β1a subunit in fast voltage dependent EC coupling in skeletal myotubes.
INTRODUCTION
The dihydropyridine receptor (DHPR) α1 and β subunits are both essential for Ca2+ channel formation and for triggering excitation-contraction (EC) coupling in skeletal muscle (Tanabe et al., 1988; Gregg et al., 1996). The α1 subunit forms the Ca2+ pore and houses the electrical charges that sense the membrane voltage. α1 belongs to the superfamily of voltage-gated channel proteins with four internal repeats, each composed of six transmembrane segments. The S4 segments form part of a voltage sensor that moves outward in response to membrane depolarization, and this movement is coupled to the opening of the pore (Bezanilla, 2000). β subunits are ∼60 kDa proteins that bind strongly to the cytosolic loop between repeats I and II of α1 via a conserved ∼30-residue β interaction domain (BID) (Pragnell et al., 1994; De Waard et al., 1994). They are structurally related to the MAGUK superfamily of proteins, commonly engaged in membrane signaling (Hanlon et al., 1999). Studies in heterologous cells have shown that β subunits are vital for trafficking the newly synthesized DHPR to the cell surface (Chien et al., 1995; Neuhuber et al., 1998). A mechanism has been proposed where β, by binding to the I-II loop of α1, relieves the inhibitory signal that retains α1 in the endoplasmic reticulum (Bichet et al., 2000). Various β subunits modulate the kinetics of activation and inactivation of the Ca2+ current produced by cardiac and brain pore isoforms (Olcese et al., 1994; Qin et al., 1996; Wei et al., 2000; Berrou et al., 2001) and strengthen the coupling between S4 charge movements and pore opening (Neely et al., 1993; Olcese et al., 1996; Kamp et al., 1996). Furthermore, the skeletal muscle specific β1a subunit (Powers et al., 1992) is required for triggering Ca2+ transients in skeletal muscle (Beurg et al., 1999a), suggesting a role in coupling charge movements to the opening of ryanodine receptor type 1 (RyR1).
In skeletal muscle, charge movements in the DHPR are responsible for activation of RyR1. Yet the Ca2+ current, also activated by the charge movements, is not essential for EC coupling function (Dirksen and Beam, 1999; Ahern et al., 2001a). This signaling mechanism is known as voltage-dependent, or skeletal-type, EC coupling. Its fingerprint characteristics are a sigmoidal relationship between the amplitude of the Ca2+ transient and the magnitude of the depolarization and a saturation of the Ca2+ transient amplitude at large positive potentials (Garcia et al., 1994; Ahern et al., 2001b). In cardiac muscle, the Ca2+ current through the DHPR L-type Ca2+ channel must diffuse to the sarcoplasmic reticulum (SR) to trigger the opening of RyR2. This mechanism is referred to as Ca2+-dependent EC coupling and its main characteristic is a biphasic, bell-shaped relationship between the amplitude of the Ca2+ transient and the magnitude of the depolarization, such that the Ca2+ transient is always proportional to the Ca2+ current (Beuckelmann and Wier, 1988). A change from purely voltage-dependent EC coupling to Ca2+-dependent EC coupling has been described in skeletal dysgenic (α1S-null) myotubes expressing α1C, the cardiac pore isoform, instead of α1S, the endogenous isoform (Tanabe et al., 1990a). This and other related studies (Tanabe et al., 1990b; Garcia et al., 1994) showed that α1S specializes in voltage dependent EC coupling whereas α1C is involved in Ca2+ dependent EC coupling. The α1 isoform alone would thus appear to dictate the intrinsic characteristics of the EC coupling process, at least in the cellular context of the dysgenic myotube.
Sequence comparison between β1a, the major isoform of skeletal muscle, and β2a, primarily expressed in the brain and heart, revealed two conserved central regions amounting to more than half of the total peptide sequence (domains D2 and D4), a nonconserved linker between the two conserved domains (D3), a nonconserved amino terminus (D1), and a nonconserved carboxyl terminus (D5) (Perez-Reyes and Schneider, 1994). We previously tested the participation of D1, D3, and D5 of β1a in the recovery of Ca2+ conductance, charge movements, and EC coupling in β1 KO skeletal myotubes (Beurg et al., 1999b). Deletion of D5 (β1a residues 470-524) drastically reduced the amplitude of voltage-evoked Ca2+ transients without affecting the density of charge movements. Thus the membrane density of DHPR voltage sensors, critical for voltage dependent EC coupling, was not compromised by the carboxyl terminal deletion. On the basis of this result, we implicated D5 in skeletal-type EC coupling. The identification of D5 was also important because it showed that domains of β1a that participate in EC coupling differ from the BID domain present in the D4 region (β1a residues 250-300). Hence it is clear that in the β subunit, the trafficking function controlled by the BID domain (Chien et al., 1995; Bichet et al., 2000), and the EC coupling function have different molecular underpinnings.
In the present study, we further investigated the EC coupling function of D5 in chimeras of β1a and β2a and truncated variants, all having demonstrably intact BID/I-II loop interactions. We show that small truncations of the carboxyl terminus of β1a produce drastic changes in EC coupling, altering it from a purely voltage dependent process to one controlled by voltage and the Ca2+ current. The changes are functionally equivalent to those observed in dysgenic myotubes expressing α1C and are consistent with a view in which the DHPR β subunit, like α1S, is a key structural determinant of skeletal-type EC coupling. Part of this work has been previously published in abstract form (Sheridan et al., 2002a).
MATERIALS AND METHODS
Primary cultures
Cultures of myotubes were prepared from hind limbs of E18 fetuses as described previously (Beurg et al., 1997). Muscles dissected from the fetuses were treated with 0.125% (w/v) trypsin and 0.05% (w/v) pancreatin. After centrifugation, mononucleated cells were resuspended in plating medium containing 78% Dulbeccos's modified Eagle's medium with low glucose, 10% horse serum, 10% fetal bovine serum, and 2% chicken serum extract. Cells were plated on plastic culture dishes coated with gelatin at a density of ∼1 × 104 cells per dish. Cultures were grown at 37°C in 8% CO2 gas. After myoblast fusion (∼6 days), the medium was replaced with fetal bovine serum free medium and CO2 was decreased to 5%.
cDNA transfection
All cDNAs were cloned into the mammalian expression vector pSG5 (Stratagene, CA). cDNA transfection was performed during the myoblast fusion stage with the polyamine LT1 (Panvera). Cells were exposed for 2–3 h to a transfection solution containing LT1 and cDNA at a 5:1 μg ratio. In addition to the cDNA of interest, cells were cotransfected with a plasmid encoding the T-cell protein CD8 used here as a transfection marker. Transfected myotubes expressing CD8 were recognized by surface binding of polystyrene beads coated with a monoclonal antibody specific for an external CD8 epitope (Dynal ASA, Norway). The efficiency of cotransfection of the marker and the cDNA of interest was ∼90%. Whole-cell analysis of Ca2+ currents and Ca2+ transients was performed 3–5 days after transfection.
cDNA constructs
Deletion and chimeric variants were made by polymerase chain reaction (PCR) strategies using cDNAs for mouse β1a (residues 1-524; Genbank accession # NM_031173) and rat β2a (residues 1-604; Genbank accession # M80545). The PCR products were subcloned into the pCR-Blunt vector (Invitrogen Inc., Carlsbad, CA), excised by digestion with AgeI and NotI and cloned into the pSG5 vector in frame with the first 11 residues of the phage T7 gene 10 protein for antibody tagging. All constructs were sequenced twice or more at a campus facility.
β2-β1
This cDNA contains residues 1-287 of β2a fused to the amino terminus of residues 325-524 of β1a and was made by two rounds of PCR as described previously (Beurg et al., 1999b).
β2-β1 Δ7
This cDNA contains residues 1-287 of β2a fused to the amino terminus of residues 325-517 of β1a and was made by PCR using the chimeric β2-β1 cDNA as template. Two primers were used to PCR the 3′ end of the β1a cDNA with the last seven amino acids deleted, B1MluI (5′-tcc aac acg cgt tcc agc ctg gc-3′) and B1Δ7 (5′-tta ggc cac cgc gtc gcc tcc ccg-3′). The PCR product was subcloned into pCR2.1 vector (Invitrogen), excised with MluI and NotI, purified from the gel using QIAquick gel extraction kit (Qiagen) and ligated into the pSG5-T7-β2-β1 chimera via unique MluI and NotI sites. Except for primers, all deletions constructs below were generated by the same strategy.
β2-β1 Δ21
This cDNA contains residues 1-287 of β2a fused to the amino terminus of residues 325-503 of β1a. Primers to PCR the 3′ end of the β1a cDNA with the last 21 amino acids deleted were B1MluI (above) and B1Δ21 (5′-cta cca gaa gct gag att tct cct gag-3′).
β2-β1 Δ29
This cDNA contains residues 1-287 of β2a fused to the amino terminus of residues 325-495 of β1a. Primers to PCR the 3′ end of the β1a cDNA with the last 29 amino acids deleted were B1MluI (above) and B1Δ29 (5′-tta cga ggt gag cac ctg tac ctg g-3′).
β2-β1 Δ35
This cDNA contains residues 1-287 of β2a fused to the amino terminus of residues 325-489 of β1a. Primers to PCR the 3′ end of the β1a cDNA with the last 35 amino acids deleted were B1MluI (above) and B1Δ35 (5′-tat gcg gcc gct tac tgg agg ttg gag acg gg-3′).
β2-β1 Δ60
This cDNA contains residues 1-287 of β2a fused to the amino terminus of residues 325-464 of β1a cDNA and was made as described previously (Beurg et al., 1999b).
β1aΔ278-287
This construct was generated by two steps of PCR and subcloned into the pSG5-T7-tagged expression vector using XhoI and MluI sites. The cDNA encoding β1a residues 1-277 was amplified by S1 primer 5′-gtt ata gga tcc atg gtc cag aag agc ggc-3′ and RBID primer 5′-ctg tca cct cga tgg gcc tca tgg aagg-3′. The cDNA encoding β1a residues 288-300 was amplified by FBID primer 5′-agg ccc atc gag gtg aca gac atg atg-3′ and RMluI primer 5′-agc cag gct gga acg cgt gtt gga g-3′. The two PCR products were purified on QIAquick PCR purification columns (Qiagen, CA), diluted and mixed at a 1:1 weight ratio, and used for a second PCR at 95°C for 1 min, 43°C for 1 min, and 72°C for 1 min for five cycles. Thereafter, primers S1 and RMluI were added to the mixture and the PCR was continued for additional 30 cycles. The amplified PCR product was subcloned into a pCR 2.1 vector, cut with XhoI and MluI, and subcloned into a XhoI/MluI digested pSG5-T7-β1a construct.
α1C (CaV1.2)
A full-length rabbit α1C cDNA encoding residues 1-2171 (Genbank accession # X15539, nucleotide coordinates nt 192 to nt 6707) was fused in frame to the first 11 amino acids of the phage T7 gene 10 protein in the mammalian expression vector pSG5 (Stratagene, CA) using AgeI and NotI cloning sites.
In vitro translation of β subunit cDNAs and GST pull-down experiments
Experiments were carried out essentially as described previously (Pragnell et al., 1994; De Waard et al., 1994; Walker et al., 1998). The α1S I-II loop (α1S residues 335-432) was amplified by PCR from a cDNA template encoding full-length α1S (Genbank accession # M23919), using the forward primer 5′-tca tca gga tcc ggg gaa ttt acc aag gag-3′ that contained a BamHI restriction site and the reverse primer 5′-atg cca gaa ttc tct cga ctt cac cag gtc -3′with an EcoRI restriction site. The PCR product was subcloned into a pGEX-2T vector (Stratagene) and the corresponding protein fragment was fused with the carboxyl terminus of Glutathione S-Transferase (GST). The construct was transformed into the bacterial strain BL21(DE3). Cells were harvested 4 h after isopropul-1-thio-β-d-galactopyranoside induction by incubation in phosphate buffered saline buffer in the presence of protease inhibitors (0.5 mM phenylmethylsulfonyl fluroide and 0.8 mM benzamidine). GST-α1S I-II fusion protein was isolated by incubation with glutathione (GS) Sepharose (Amersham Pharmacia Biotech, NJ) for 1 h on ice. Control and truncated β constructs subcloned into pSG5 vector were in vitro translated using the T7 coupled reticulocyte system (Promega, WI). For radioactivity labeling, 40 μCi of Redivue L-[35S] methionine (Amersham Pharmacia) was used per 50 μl of TNT in vitro translation reaction. For protein-protein binding in vitro, 25 μl of the TNT translated mixture was added to 500 μl binding buffer (50 mM Tris-HCl pH 7.4, 0.1 mM dithiothreitol, 1 mM MgCl2, 100 mM NaCl, 0.05% Tween-20) containing ∼50 μg GST-α1S I-II loop fusion protein immobilized on GS Sepharose beads. The binding reaction was carried out at room temperature for 3 h with agitation and was spun for 30 s in a bench-top centrifuge at 6000 rpm. The pellet containing GS Sepharose beads was washed four times with 1 ml of binding buffer each time. β proteins were released from the GS Sepharose by incubation at 100°C for 3 min in SDS-PAGE loading buffer (50 mM Tris-HCl pH 6.8, 5% β-mercaptoethanol, 2% SDS, 0.1% bromophenol blue, 10% glycerol). The pulled-down β proteins and the in vitro translated β proteins were loaded side by side on a 15% polyacrylamide gel and run for ∼2 h at 36 mA in SDS-PAGE running buffer (25 mM Tris, 250 mM glycine, 0.1% SDS, pH 8.3). The gel was dried and exposed to Kodak X-Omat AR-5 film for 20 h at −70°C.
Immunoblotting
β proteins were translated in vitro using the same conditions described above but without radioactive isotope. A 15 μl of the translation reaction was mixed with 15 μl of twice concentrated SDS-PAGE loading buffer and boiled for 3 min. The 30-μl sample was loaded on a 10% polyacrylamide gel and run for ∼2 h at 36 mA in SDS-PAGE running buffer. The proteins were electrotransferred onto a Hybond ECL nitrocellulose membrane (Amersham Pharmacia) in transfer buffer (48 mM Tris, pH 8.3, 39 mM glycine, 0.037% SDS, 20% methanol) for 4 h at 35 volts and 4°C. The nitrocellulose membrane was blotted with 30 ml of blocking solution (100 mM Tris, pH 7.5, 150 mM NaCl, 0.05% Tween-20, 5% nonfat milk) overnight at room temperature with agitation. The nitrocellulose membrane was blotted for 2 h with 30 ml of blocking solution containing 6 μl of mouse anti-T7 tag monoclonal antibody (Novagen, WI), washed three times with washing buffer (100 mM Tris, pH 7.5, 150 mM NaCl, 0.05% Tween-20), and blotted for 1.5 h with 30 ml of blocking solution containing 6 μl of donkey anti-mouse IgG POD (Boehringer Mannheim). The nitrocellulose membrane was washed four times with washing buffer and revealed with (ECL) Western blotting detection reagents (Amersham Pharmacia). The nitrocellulose membrane was exposed to Hyperfilm ECL high performance chemiluminescence film (Amersham Pharmacia) for 30 s.
Whole cell voltage clamp
Whole-cell recordings were performed as described previously (Strube et al., 1996) with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Effective series resistance was compensated up to the point of amplifier oscillation with the Axopatch circuit. All experiments were performed at room temperature. The external solution was (in mM) 130 TEA methanesulfonate, 10 CaCl2, 1 MgCl2, 10−3 TTX, 10 HEPES titrated with TEA(OH) to pH 7.4. The pipette solution consisted of (in mM) 140 Cs aspartate, 5 MgCl2, 0.1 EGTA (when Ca2+ transients were recorded) or 5 EGTA (when only Ca2+ current was recorded), and 10 MOPS titrated with CsOH to pH 7.2. Patch pipettes had a resistance of 1–1.5 MΩ when filled with the pipette solution.
Confocal fluorescence microscopy
Confocal line scan measurements were performed as described previously (Conklin et al., 1999). Cells were loaded with 4 μM fluo-4 acetoxymethyl ester (Molecular Probes, Eugene, OR) for 60 min at room temperature. All experiments were performed at room temperature. Cells were viewed with an inverted Olympus microscope with a 20× objective (N.A. 0.4) and an Olympus Fluoview confocal attachment (Melville, NY). A 488-nm spectrum line for fluo-4 excitation was provided by a 5-mW Argon laser attenuated to 20% with neutral density filters. Line scans consisted of 1000 lines, each of 512 pixels, acquired at a rate of 2.05 milliseconds per line. Line scans were synchronized to start 100 ms before the onset of the depolarization. The time course of the space-averaged fluorescence intensity change and ΔF/F units were estimated as follows. The pixel intensity in a line scan was transformed into arbitrary units and the mean intensity of each line was obtained by averaging pixels covering the cell exclusively. The mean resting fluorescence intensity (F) corresponds to the mean intensity of each line averaged over the first 50 ms before stimulation and was used as a baseline. The change in mean line intensity above baseline (ΔF) was obtained by subtraction of F (baseline) from the mean line intensity. ΔF was divided by the baseline F for each line in a line scan and was plotted as a function of time. To construct peak Ca2+ fluorescence versus voltage curves, we used the highest ΔF/F line value after the onset of the pulse and up to the termination of the pulse. Image analyses were performed with NIH Image software (National Institutes of Health, Bethesda, MD). To obtain reliable Ca2+ transient versus voltage curves, seven step depolarizations of 50 ms or 200 ms were applied in descending order (from +90 mV to −30 mV) from a holding potential of −40 mV. Between each depolarization, the cell was maintained at the resting potential for 30 s to permit recovery of the resting fluorescence.
Immunostaining
Cells were fixed in 100% methanol and processed for immunostaining as described previously (Gregg et al., 1996) after 4–5 days after transfection. For T7-tagged β constructs, the primary antibody was a mouse monoclonal against the T7 epitope (Novagen) and was used at a dilution of 1:1000. The secondary antibody was a fluorescein conjugated polyclonal goat anti-mouse IgG (Boehringer Mannheim, Indianapolis, IN) and was used at a dilution of 1:1000. For α1C, the primary antibody was an anti-rabbit monoclonal (Chemicon, CA) and was used at a 1:100 dilution. The secondary antibody was a fluorescein conjugated goat anti-rabbit IgG (Chemicon) and was used at a dilution of 1:750. Confocal images of 0.3–0.4 microns per pixel were obtained in the Olympus Fluoview using a 40× oil-immersion objective (N.A. 1.3). Images were Kalman-averaged three times, and the pixel intensity was displayed as 16 levels of gray in reverse. All images were acquired with minimal laser power (6% of maximum 5 mW) and predetermined photomultiplier tube settings to avoid pixel saturation and for accuracy in the comparison of images.
Curve fitting
The voltage dependence of the Ca2+ conductance and the voltage dependence of peak intracellular Ca2+ assayed with the 50-ms depolarization were fitted according to a Boltzmann distribution (Eq. 1) A = Amax/(1 + exp(−(V − V1/2)/k)) where Amax is Gmax or ΔF/Fmax, V1/2 is the potential at which A = Amax/2, and k is the slope factor. The voltage dependence of peak intracellular Ca2+ assayed with a 200-ms depolarization was fit with Eq. 1 for cells expressing the control, Δ7, and Δ21 constructs. All other truncated variants and full-length β2a produced biphasic bell-shaped fluorescence versus voltage curves with varying degrees of curvature. In these cases, curves were fit with the modified Boltzmann distribution (Eq. 2) ΔF/F = (V − ECa)/k′ [(ΔF/Fmax/ (1 + exp((V − V1/2)/k)] where ECa is the Ca2+ equilibrium potential, k′ ∼ −150 mV is a scaling factor, and the other parameters are the same as in Eq. 1. For Δ60, Δ35, and Δ29, a chi-square test indicated that Eq. 2 provided a better fit of the fluorescence versus voltage curve obtained with a 200-ms depolarization. Parameters of a fit of averages of many cells (population average) are shown in figures. The statistics of parameters of the fit of individual cells are shown in Table 1. Student, chi-square, and ANOVA tests were performed with Analyze-it software (Leeds, UK).
TABLE 1.
Boltzmann parameters of β1 KO myotubes expressing carboxyl terminus truncations of the D5 region of β1a
|
G-V
|
ΔF/F-V 50 ms
|
ΔF/F-V 200 ms
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| G(max) (pS/pF) | V1/2 (mV) | k (mV) | ΔF/F(max) | V1/2 (mV) | k (mV) | 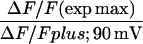 |
ΔF/F(max) | V1/2 (mV) | k (mV) | 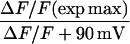 |
|
| β2-β1 | 221 ± 17 | 12 ± 1.4 | 4.2 ± 0.4 | 2.8 ± 0.6 | −1.9 ± 2.8 | 7.8 ± 1.1 | 1.0 ± 0.1 | 2.9 ± 0.4 | −7.4 ± 2.8 | 5.9 ± 1.2 | 1.2 ± 0.1 |
| (20) | (8) | (9) | |||||||||
| Δ7 | 184 ± 34 | 4.7 ± 1.9 | 3.3 ± 0.5 | 1.4 ± 0.3 | 4.3 ± 4.1 | 14 ± 2.2 | 0.9 ± 0.1 | 2.4 ± 0.6 | 4.2 ± 3.5 | 9.7 ± 1.4 | 1.1 ± 0.1 |
| (5) | (10)* | (8) | * | ||||||||
| Δ21 | 170 ± 11 | 8 ± 3 | 4.9 ± 0.7 | 1.5 ± 0.6 | −3.6 ± 2.6 | 9.6 ± 1.1 | 1.1 ± 0.1 | 2.5 ± 0.6 | −1.6 ± 4.0 | 7.8 ± 1.1 | 1.3 ± 0.1 |
| (5) | (9) | (10) | |||||||||
| Δ29 | 132 ± 18 | 10 ± 1.3 | 4.7 ± 0.4 | 0.6 ± 0.2 | 3.2 ± 3.3 | 8.7 ± 1.8 | 1.1 ± 0.2 | 0.8 ± 0.1 | 0.8 ± 1.6 | 4.8 ± 1.0 | 1.5 ± 0.2 |
| (8)* | (6)* | (7)* | |||||||||
| Δ35 | 202 ± 24 | 20 ± 1.4 | 5.4 ± 0.3 | 0.5 ± 0.1 | 10 ± 2.1 | 12 ± 2.9 | 1.3 ± 0.2 | 1.1 ± 0.3 | 6.7 ± 2.5 | 9.2 ± 1.2 | 1.3 ± 0.2 |
| (8) | * | (7)* | * | (8)* | * | ||||||
| Δ60 | 87 ± 10 | 18 ± 1.7 | 5.1 ± 0.3 | 0.4 ± 0.1 | 12 ± 2.1 | 8.2 ± 1.4 | 1.1 ± 0.1 | 0.7 ± 0.1 | 1.4 ± 1.9 | 10 ± 2.3 | 2.2 ± 0.2 |
| (19)* | * | (6)* | * | (6)* | * | * | |||||
| β1a | 195 ± 23 | 15 ± 2.4 | 4.5 ± 0.6 | 2.7 ± 0.5 | −3.2 ± 2.3 | 7.7 ± 0.5 | 1.0 ± 0.1 | 3.4 ± 0.6 | −8.3 ± 1.6 | 5.8 ± 1.1 | 1.2 ± 0.1 |
| (10) | (10) | (10) | |||||||||
| β2a | 184 ± 36 | 11 ± 1 | 5.1 ± 0.3 | 0.9 ± 0.2 | −1.9 ± 3.3 | 7.4 ± 1.5 | 1.5 ± 0.1 | 1.7 ± 0.4 | 5.7 ± 3.1 | 7.6 ± 1.9 | 2.3 ± 0.4 |
| (6) | (10)* | * | (10) | * | * | ||||||
Mean ± SE of Boltzmann parameters fitted to each cell with number of cells in parenthesis. Parameters of fluorescence versus voltage curves are shown for 50-ms and 200-ms depolarizations. Asterisks indicate data in each column compared to control (β2-β1) with 1-way ANOVA significance p < 0.05. G(max), ΔF/F(max), V1/2, and k are parameters of the Boltzmann fit. All data was fit with Eq. 1 except fluorescence data in response to 200 ms in cells expressing Δ29, Δ35, and Δ60 that was fit with Eq. 2. ΔF/F (exp max) corresponds to the experimental maximum ΔF/F for a given fluorescence versus voltage curve. ΔF/F +90 mV corresponds to the experimental ΔF/F at +90 mV.
RESULTS
Based on sequence alignment of several isoforms, domain D5 of mouse β1a comprises the last 50 to 60 residues of the carboxyl terminal end of the subunit (Fig. 1 A). To characterize D5, we serially truncated the carboxyl terminus from back to front and expressed the cDNAs in β1 KO skeletal myotubes that are entirely devoid of EC coupling (Beurg et al., 1997). Truncation mutants were identified by the number of residues missing from the carboxyl terminus end according to the sequence shown in Fig. 1 A. The template for the truncations was the chimera β2a 1-287/β1a 325-524, comprising β2a domains D1, D2, D3, and part of D4 fused to the remaining D4 and D5 of β1a. In β1 KO myotubes, β2a 1-287/β1a 325-524 recovered a high-density L-type Ca2+ current and recapitulated skeletal-type EC coupling quantitatively (Beurg et al., 1999b). For these reasons, and because the chimera eliminates potential contributions of domains D1 and D3 of β1a, it was deemed the ideal control for the present study. All constructs carried a T7 epitope tag at the amino terminus for determining relative levels of protein expression in transfected cells. Fig. 1 B shows confocal anti-T7 immunofluorescence of myotubes expressing the control β2-β1 chimera, the tested truncations, full-length mouse β1a, and a blank consisting of a nontransfected myotube (NT). Cultures were also incubated with anti-CD8 antibody beads, used here as a marker to identify transfected myotubes in voltage clamp experiments. Cells were immunostained 4–5 days after transfection, at the same time as voltage-clamp experiments. At this stage, we found abundant expression of all truncation variants throughout the length and width of the myotube with the exclusion of the cell nuclei. In some myotubes, protein expression was clearly discernable in the submembrane space, away from the perinuclear regions. This suggested that, in such cases, a significant amount of the expressed protein had been routed to peripheral sites. A submembrane staining pattern is significant because in the cultured myotube, EC coupling occurs at peripheral couplings established between the plasma and SR membranes (Takekura et al., 1994). In Fig. 1 B, a preferential peripheral location of the immunostained protein is evident in the myotubes expressing Δ21 or Δ35. Despite these cases, however, we found no evident correlation between the staining pattern and the truncation size. Moreover, submembrane staining, distinct from perinuclear staining, was not evident in all cases. Based on the fluorescence intensity, we estimated that the truncated variants expressed as well as the control chimera or wild-type β1a. Previous studies have shown that the cardiac pore isoform α1C, when transiently transfected in skeletal dysgenic myotubes, expresses Ca2+ currents that contribute to Ca2+ dependent EC coupling (Garcia et al., 1994; Ahern et al., 2000). For this reason we performed controls to determine whether this isoform might be present in the β1 KO myotube due to de novo gene expression. If this were the case, α1C subunits could potentially interact with the transiently expressed β variants and the Ca2+ currents generated by these complexes could contribute to Ca2+ dependent EC coupling. Fig. 1 C shows immunofluorescence of β1 KO myotubes stained with a α1C-specific antibody. We were unable to detect endogenous α1C expression in either nontransfected β1 KO myotubes or in β1 KO myotubes transfected with the cardiac/brain β2a isoform. As a positive control, a rabbit α1C was transiently overexpressed in β1 KO myotubes using the mammalian expression vector pSG5. In this case, we found a robust immunofluorescence signal indicating that exogenous a1C protein was readily detectable by the technique. This result indicated that de novo expression of a1C in the β1 KO myotube was very minor or none at all. In summary, the immunostaining analyses indicated that β constructs with a carboxyl terminus truncation within the D5 region showed abundant expression in β1 KO myotubes and that endogenous expression of α1C was undetectable.
FIGURE 1.

Truncated β variant protein expression and absence of endogenous α1C expression in β1 KO myotubes. (A) Domain D5 of mouse β1a according to a sequence lineup of Perez-Reyes and Schneider (1994). Indicated with numerical coordinates corresponding to the mouse β1a sequence are the carboxyl terminal end residues of the truncated variants: Δ60 (P464), Δ35 (Q489), Δ29 (S495), Δ21 (W503), Δ7 (A517), and β2-β1 control (M524). (B) Confocal immunofluorescence of β1 KO myotubes expressing T7-tagged constructs. Cells were transfected with CD8 cDNA plus the indicated cDNA. β1a corresponds to the full-length mouse cDNA and NT is a nontransfected myotube. Cells were incubated with CD8 antibody beads, fixed, and stained with anti-T7 primary/fluorescein-conjugated secondary antibodies. (C) Confocal immunofluorescence of β1 KO myotubes transfected with CD8 plus α1C (α1C), CD8 plus full-length β2a (β2a), or nontransfected control (NT). Cells were incubated with CD8 antibody beads, fixed, and stained with anti-α1C primary/fluorescein-conjugated secondary antibodies. For (B) and (C), pixel intensity was converted to a 16-level inverted gray scale with high-intensity pixels in black color. Images are 84 × 56 microns and on-focus beads are 4.5 microns in diameter.
To verify the size of the truncated proteins, the cDNAs for the tested β variants were in vitro translated, followed by PAGE analysis on a 10% gel and immunoblotting with anti-T7 antibody (Fig. 2 A). The in vitro translated β1a, β2-β1, and Δ60 proteins migrated with apparent molecular weights of ∼74, 65, and 56 kDa respectively, based on the mobility of five markers in the same gel. The relative mobilities (β1a > β2-β1 > Δ60) are consistent with the theoretical molecular weights predicted from the cDNAs, respectively 58.9, 54.8, and 48.2 kDa. The lower than expected experimental mobility was probably due to the limited resolution of the linear polyacrylamide mini-gel system utilized here for comparative purposes. The mobilities of the rest of the truncated variants (Δ7, Δ21, Δ29, Δ35) fell in a ladder of decreasing molecular weight bracketed by β2-β1 in the low-mobility end and Δ60 in the high-mobility end. We also determined that the truncation of the carboxyl terminus region did not affect the interaction of the β subunit with the pore subunit. If this were to be the case, DHPR surface density would be compromised, given the role of the α1/β interaction in trafficking Ca2+ channels to the cell surface. We used the approach developed for studies of α1/β interactions in neuronal Ca2+ channels (Pragnell et al., 1994; De Waard et al., 1994; Walker et al., 1998). The α1S I-II loop was fused to the carboxyl terminus of GST and the expressed recombinant fusion protein was isolated by affinity binding to GS Sepharose 4B beads. β constructs were translated in vitro in the presence of S35-methione and incubated with GST-(α1S I-II) prebound to GS Sepharose. Strong interactions between β and the I-II loop are recognized by the ability of GS-Sepharose beads to precipitate β bound to the GST-(α1S I-II) fusion protein (Pragnell et al., 1994). Fig. 2 B shows an autoradiogram of a 15% polyacrylamide gel of in vitro translated [35S]methionine-labeled β variants pulled down by recombinant GST- α1S I-II loop fusion protein immobilized on GS Sepharose. Aliquots of the in vitro translated proteins utilized in the pull-down assay were run side by side. All β constructs tested in the present study interacted strongly with the I-II loop, demonstrated by the high intensity of the bands in the pull-down lanes compared to the in vitro translated yields in the right lanes. Furthermore, neither the amount of β variant protein pulled down by the α1S I-II loop nor the in vitro translated yields varied with the truncation length. Controls shown in Fig. 2 C indicated that 1) the GS Sepharose/GST complex without the α1S I-II loop fusion did not pull down full-length β1a nor the truncated variants (not shown); and 2) deletion of essential residues in the BID region of β1a (β1aΔ278-287; De Waard et al., 1994) eliminated the interaction with the I-II loop and thus eliminated the autoradiogram signal. However, both control β1a and β1aΔ278-287 were readily expressed in vitro. This result is consistent with studies in neuronal β isoforms showing that the BID sequence is essential for an interaction of the β subunit with the α1 I-II loop (De Waard et al., 1994). Additional controls shown elsewhere (Cheng et al., 2002) further demonstrated that 3) the β/I-II loop interaction persisted in high salt (0.4 M NaCl) indicating a high affinity; and 4) the β interaction was specific for the I-II loop and not for the other cytosolic loops of α1S. In summary, the GST pull-down experiments demonstrated that partial or total absence of D5 did not affect the ability of the β subunit protein to bind to the α1S I-II loop, consistent with a similar deletion analysis conducted in neuronal subunits (De Waard et al., 1994).
FIGURE 2.

Size of truncated β variants and interaction with the I-II loop of the α1S pore subunit. (A) Immunoblot of a 10% SDS-polyacrylamide gel of in vitro translated β variants. After transfer to nitrocellulose and incubation with anti-T7 antibody, the blot was revealed by high performance chemiluminescence. Indicated are the relative mobilities of two out of five M.W. markers run in the same gel. (B) Autoradiogram of a 15% SDS-polyacrylamide gel of in vitro translated [35S]methionine-labeled β variants pulled down by recombinant GST-α1S I-II loop fusion protein immobilized on gluthatione(GS)-Sepharose. Lanes 1–7 show pellets (pull-downs) after 2 h of incubation of GS-Sepharose/GST-α1S I-II loop and the indicated in vitro translated construct. Lanes 8–14 show equal aliquots of the in vitro translation mixture with each β variant as template. (C) Controls of the pull-down protocol are shown on an autoradiogram of a 15% SDS-polyacrylamide gel of in vitro translated [35S]methionine-labeled β variants. Lane 1 shows absence of β1a pull down by recombinant GST lacking α1S I-II loop fusion. Lane 2 shows β1a pull down by GST-α1S I-II loop fusion. Lane 3 shows absence of pull down of β1aΔ278-287 by recombinant GST-α1S I-II loop fusion. Lanes 4 and 5 show in vitro translated β1a and β1aΔ278-287.
Ca2+ transients are customarily evoked by a brief depolarization, and in our previous studies we used voltage steps with a fixed duration of 50 ms (Beurg et al., 1999a,b). This stimulation time is more than adequate for completion of charge movements in the DHPR (Ahern et al., 2002). However, the slow L-type Ca2+ current also expressed by the DHPR is only partially activated at the end of the 50-ms pulse. To investigate a possible contribution of the Ca2+ current to the EC coupling recovered by the truncated variants, we compared Ca2+ transients activated by depolarizations lasting 50 ms and 200 ms. We used voltage steps in the range of −30 mV to +90 mV from a holding potential of −40 mV to cover both the inward and outward phases of the Ca2+ current. To avoid cell fatigue and to allow for fluorescence recovery, we limited the number of pulses to 14 (seven for each duration) and each pulse was applied every 30 s. Fig. 3 shows the two stimulation protocols in β1 KO myotubes expressing β2-β1 and Δ60, the latter lacking the entire D5 region. The line scan images in color correspond to 103 512-pixel lines stacked vertically from left to right with a total acquisition time (horizontal) of 2.05 s. The time course of the space-averaged fluorescence intensity is shown above each image and the width of the horizontal bar reflects the duration of the pulse. In the control case, Ca2+ transients of nearly identical shape and intensity were obtained with the short and long pulse protocols and the maximum intensity was nearly the same at +30 mV and +90 mV. Both features, namely the absence of pulse duration dependence and persistence at large positive potentials, are hallmarks of skeletal-type EC coupling (Melzer et al., 1986). In contrast, Ca2+ transients activated in the myotube expressing Δ60 were barely detectable with the short pulse, consistent with a previous study (Beurg et al., 1999b). However, in response to the 200-ms depolarization, Ca2+ transients became comparatively larger and behaved in a biphasic manner, increasing from −10 mV to +30 mV and decreasing at more positive potentials. Furthermore, the rate of rise of the fluorescence signal during the depolarization was noticeably slower at all potentials compared to the control case. The decay of the Ca2+ transient after the stimulus was also much slower than in controls, requiring several seconds for the signal to reach resting level. To avoid excessive photobleaching, in Fig. 3 we limited the total line scan time to 2.05 s per pulse. Hence, the decay phase of the Ca2+ transient for the myotube expressing Δ60 is not entirely evident at all potentials. However, it is clear from the line scan images that resting fluorescence levels were consistently reached before each stimulation. It is conceivable that the slow decay kinetics may be due to the much weaker stimulation of the Ca2+ pump produced by the small Ca2+ transients expressed by Δ60. In summary, Ca2+ transients activated by Δ60 had severely altered kinetics and voltage dependence. The protocol with the longer depolarization suggested a direct involvement of the Ca2+ current in the fluorescence signal in the myotube expressing Δ60 but not for the control chimera. This was indicated by the decrease in Ca2+ transient amplitude at positive potentials. Therefore, we further investigated if the Ca2+ current activated by the longer depolarization in myotubes expressing Δ60 was involved in triggering SR Ca2+ release.
FIGURE 3.

Ca2+ transients expressed by β2-β1 and Δ60 revealed by 50 ms and 200-ms depolarizations. Confocal line scan images of fluo-4 fluorescence are shown for a β1 KO myotube expressing β2-β1 (left) and a separate myotube expressing Δ60 (right). For visual reference pseudocolors, in ΔF/F units, are blue <0.2; green <2; and yellow >4. In the images, the total line scan duration (x axis) was 2.05 s and the cell dimension (y axis) was (in microns) 21 for β2-β1 and 30 for Δ60. Traces above the images show the integral of the image fluorescence in ΔF/F units indicated by the vertical bars. Note difference in ΔF/F scale for β2-β1 and Δ60. Myotubes were held at a resting potential of −40 mV and depolarized for 50 ms or 200 ms. The depolarization time is indicated by the horizontal bar under the fluorescence time course.
Ca2+ currents expressed by β2-β1 and Δ60 are shown in Fig. 4 A. The Ca2+ current of nontransfected cells (not shown) was below the limit of detection, and was <20 pA/cell or <0.05 pA/pF for the smallest cells having the lowest capacitative noise. The truncation produced a slight reduction in the activation kinetics and, when currents were corrected for cell size, Δ60 expressed a Ca2+ current density lower than the control chimera (Fig. 4 B). In a previous study (Beurg et al., 1999b), we showed that β2-β1 and Δ60 generated a comparable density of charge movements, hence the reduction in Ca2+ current is probably related to the loss of EC coupling and retrograde coupling rather than due to a reduction in the membrane density of DHPR complexes (Nakai et al., 1996). The Boltzmann parameters of the Ca2+ conductance for all truncations are shown in Table 1, at a resolution of 5 mV in the range of −35 to +80 mV from a holding potential of −40 mV. The most severe reductions in maximum Ca2+ conductance density (Gmax) were observed for Δ29 and Δ60, and for the Ca2+ current recovered by the latter construct, there was also a positive shift in half-activation potential (V1/2) of ∼6 mV. This was also apparent in the current versus voltage relationships shown in Fig. 4 B. The graphs in Fig. 4, C and D, present the voltage dependence of the total ionic charge, in pC/pF, which entered the cell during the 50- and 200-ms depolarizations utilized for triggering Ca2+ transients. Ca2+ currents were obtained concurrent with the cell Ca2+ fluorescence, and the voltage dependence of the latter is described in detail below (see Fig. 7). The ionic charge, which is mostly Ca2+, was obtained by integration of the pulse current with examples at +30 mV provided in the insets of Fig. 4, C and D. Near maximum Ca2+ entry within this series of stimulations occurred at ∼+30 mV, consistent with the voltage dependence of the Ca2+ current measured at a much higher resolution in voltage in Fig. 4 B. For Δ60, Ca2+ entry was minimal with the 50-ms depolarization and increased ∼130-fold with the 200-ms depolarization. Such a large difference in Ca2+ entry produced by the two pulses could explain the pronounced dependence of the Ca2+ transients expressed by Δ60 on pulse duration. Furthermore, the largest Ca2+ transient for Δ60 occurred during the depolarization to +30 mV, a potential that also generated the largest Ca2+ entry. Interestingly, significantly larger Ca2+ entries occurred in myotubes expressing β2-β1 at either 50 or 200 ms. However, the excess of Ca2+ had no obvious effect on the magnitude of the Ca2+ transient (Fig. 3). This result underscores the significant change in EC coupling mechanism produced by the truncation of domain D5. Taken together with the observations in Fig. 3, the data in Fig. 4 suggested that either the Ca2+ current was responsible for the fluorescence change generated in the Δ60-expressing myotube, or the Ca2+ current triggered Ca2+ release from the SR via a Ca2+-induced Ca2+ release mechanism.
FIGURE 4.

Voltage dependence of Ca2+ currents and Ca2+ entry in myotubes expressing β2-β1 and Δ60. (A) Representative Ca2+ currents in β1 KO myotubes expressing the control chimera or Δ60. Cells were depolarized for 200 ms to the indicated potentials from a holding potential of −40 mV. The vertical scale bar is 0.5 nA in both cases. (B) Peak Ca2+ current versus voltage relationship in response to a 500-ms depolarization from a holding potential of −40 mV. (C, D) The voltage dependence of the ionic charge entering the cell calculated by integration of the Ca2+ current during pulses of 50 ms (filled symbols) or 200 ms (open symbols) from a holding potential of −40 mV. Inserts show representative Ca2+ currents in β1 KO myotubes expressing β2-β1 (225 pF) or Δ60 (300 pF). Scale bars are 0.5 nA (vertical) and 50 ms (horizontal) in both cases. Cells were depolarized from −40 mV to +30 mV.
FIGURE 7.

Sigmoidal and bell-shaped voltage dependencies of Ca2+ transients in myotubes expressing truncated β variants. Ca2+ transients in ΔF/F units were measured in β1 KO myotubes expressing the indicated truncated variant using a depolarization of 50 ms (filled symbols) and 200 ms (open symbols) from a holding potential of −40 mV. Ca2+ transients were measured at the end of each depolarization from the time course of the integrated confocal fluo-4 fluorescence. The lines correspond to a Boltzmann fit of the population mean ΔF/F obtained from the number of cells reported in Table 1 for each construct. All fluorescence versus voltage curves were fit with Eq. 1 except those obtained with the 200-ms depolarization in myotubes expressing Δ29, Δ35, and Δ60, which were fit with Eq. 2. Parameters of the fitted lines (ΔF/Fmax in ΔF/F units, V1/2 in mV, and k in mV) are β2-β1: 2.5, −5.5, 8.2 for 50 ms and 2.9, −7.1, 7.8 for 200 ms; Δ7: 1.4, 1.2, 16.4 for 50 ms and 2.4, 0.7, 11.2 for 200 ms; Δ21: 1.5, −7.6, 8.7 for 50 ms and 2.4, −9.4, 6.9 for 200 ms; Δ29: 0.6, 1.2, 11 for 50 ms and 0.9, 2.9, 8 for 200 ms; Δ35: 0.5, 9, 9.6 for 50 ms and 1.1, 10.6, 8.7 for 200 ms; and Δ60: 0.4, 11.8, 6.8 for 50 ms and 0.8, 13.2, 7.1 for 200 ms. Chi-square significance of the fit with Eq.2 versus a fit of the same data with Eq. 1 were respectively as follows: Δ29: p = 0.05 vs. p = 0.085; Δ35: 0.036 vs. 0.078 ; and Δ60: p = 0.023 vs. 0.2. Symbols are the same as in Fig. 9.
We quantified the fluorescence increase due to Ca2+ entering the cell after blocking Ca2+ release form the SR with 0.1 mM ryanodine. To ensure a complete elimination of the fluorescence change due to EC coupling, myotubes were exposed to ryanodine for 30 min. Because whole-cell recordings of the same quality could not be consistently maintained for such a prolonged time, we compared separate control and ryanodine-treated cells within the same batch of transfected cells. The time course of Ca2+ transients of β1 KO myotubes expressing β2-β1 or Δ60 in control external solution and external solution supplemented with ryanodine are shown respectively in Fig. 5, A and B. To maximize the fluorescence signal due to Ca2+ entering the myotube, we used a 200-ms depolarization to +30 mV. Ca2+ currents stimulated by the depolarization are shown below the Ca2+ transients. The treatment with ryanodine drastically reduced the stimulated fluorescence. Further addition of 0.5 mM La3+ and Cd2+ to the external solution completely blocked the Ca2+ current and this intervention eliminated all changes in cell fluorescence (gray traces). In myotubes exposed to ryanodine, we investigated the relationship between the fluorescence increase and the Ca2+ current injected into the cytoplasm during the 200-ms depolarization. Ca2+ current densities expressed by the control and Δ60 constructs were mildly reduced in ryanodine treated cells. However, differences relative to control cells expressing the same constructs were not statistically significant. This can be seen by comparing Ca2+ entry versus voltage curves obtained under the same conditions in Fig. 5, C andD, with their counterparts without ryanodine in Fig. 4, C and D. Thus at +30 mV, Ca2+ entry during the 200-ms depolarization was 0.95 ± 0.08 and 0.27 ± 0.07 pC/pF respectively for β2-β1 and Δ60 expressing myotubes treated with ryanodine, and was 0.98 ± 0.21 and 0.35 ± 0.08 pC/pF respectively for nontreated control myotubes. For the control chimera (Fig. 5 C), the fluorescence change was strictly proportional to the magnitude of the Ca2+ current at each potential in the range of −30 to +90 mV. Consequently, the curve was bell-shaped and centered at approximately +30 mV. At this potential, the contribution of the Ca2+ current expressed by the control chimera was 0.19 ± 0.04 ΔF/F (n = 4) and this value was 15-fold lower than the maximum fluorescence change observed in the presence of a functional SR (2.9 ± 0.4 ΔF/F; n = 9; Table 1). The fluorescence contributed by the Ca2+ current in myotubes expressing Δ60 was barely detectable above the background noise (Fig. 5 D). We attributed this result to the lower Ca2+ current density expressed by this truncated variant, which is evident in the Ca2+ entry versus voltage curve. For Δ60, the fluorescence change at +30 mV was 0.08 ± 0.02 ΔF/F (n = 4), whereas in external solution without ryanodine it was ninefold higher (0.7 ± 0.1 ΔF/F; n = 6; Table 1). These results ruled out a major direct contribution of the Ca2+ current to the fluorescence signal during EC coupling and suggested that Δ60 changed the nature of the EC coupling trigger signal from one controlled by voltage to one involving the Ca2+ current.
FIGURE 5.

Contribution of the Ca2+ current to the cytosolic fluorescence in myotubes expressing β2-β1 and Δ60. (A, B) Representative Ca2+ transients obtained from the integrated confocal line scan fluo-4 fluorescence are shown in two myotubes expressing β2-β1 (A) and two myotubes expressing Δ60 (B). Ca2+ transients were evoked by a 200-ms depolarization to +30 mV from a holding potential of −40 mV. The period of the stimulation is indicated by the horizontal lines. Experiments were conducted in the absence (control) or presence of 0.1 mM ryanodine added to the external solution 30 min before establishing whole cell clamp. Ca2+ current densities in pA/pF are shown in the same cell below the Ca2+ transients. Traces in gray show the complete elimination of the Ca2+ transient and the Ca2+ current after addition of 0.5 mM LaCl3 and 0.5 mM CdCl2 to the bath solution of the same cell. Note changes in ΔF/F and pA/pF scales. (C, D) Voltage dependence of the peak cytosolic Ca2+ increase produced by Ca2+ entry in myotubes exposed to ryanodine. Ca2+ charge was calculated from the running integral of the Ca2+ current in response to 200-ms depolarizations from a holding potential of −40 mV. The peak fluorescence change detected during the depolarization to −30 mV were 0.07 ± 0.01 ΔF/F and 0.06 ± 0.01 ΔF/F respectively for β2-β1 and Δ60, and were due to background noise. For Δ60, the background fluorescence noise at −30 mV and the peak fluorescence change at +30 mV were not significantly different (t-test significance p = 0.14).
The presence of Ca2+ dependent EC coupling in myotubes expressing truncated β variants was further tested by measuring Ca2+ transients in the same cell before and after a rapid change in external Ca2+ concentration. In preliminary experiments, a change in external Ca2+ from the standard 10 mM external Ca2+ to a submillimolar concentration invariably made the pipette seal unstable. However, a change in external Ca2+ from low to high was more successful. This protocol is shown in Fig. 6 for the control chimera, Δ21, Δ29, and Δ60. Myotubes were initially voltage clamped in an otherwise standard external solution but containing 0.5 mM Ca2+ and depolarized to +30 mV for 200 ms. The Ca2+ transient elicited in this condition was acquired concurrently. The myotube was then fast-perfused for 1 s with external solution containing 10 mM Ca2+ before and during the same depolarization and line scan protocols. The perfusion solution was passed through a large-bore pipette placed near the cell, with the pipette connected to a pressurized manifold (ALA Scientific, Westbury, NY). The change in external solution was confirmed by the increase in Ca2+ current shown in Fig. 6 for each cell under the corresponding line scan. Increases in Ca2+ current, indicative of a change in external Ca2+, varied between two- and fivefold for the control construct and Δ21, and between 5- and 10-fold for the most severe trunctations. As expected, Ca2+ transients expressed by the control construct were similar in the low and high external Ca2+ solutions. We found that Ca2+ transients evoked in myotubes expressing Δ7 (see Fig. 8 A) or Δ21 (Fig. 6) were also unaffected by the change in external Ca2+. However, Ca2+ transients in myotubes expressing the more severe truncations such as Δ29 and Δ60 were detectable in the high Ca2+ solution but not in the low Ca2+ solution. The limit of fluorescence change detection, based on microscope settings and the average resting fluo-4 fluorescence, was ∼0.05 ΔF/F units; that is, we could detect a ∼5% change above the cell resting fluorescence. Using a pseudoratiometric equation for estimating the cytosolic free Ca2+ and assuming a resting free Ca2+ of 100 nM (Conklin et al., 1999), the nominal limit of resolution of free Ca2+ change was ∼150 nM. Thus, changes in free Ca2+ concentration under this limit could have taken place in myotubes expressing Δ29, Δ35, or Δ60 when stimulated in 0.5-mM external Ca2+ and would have escaped detection. We noticed that the rise time of the Ca2+ transient was slower when myotubes were subjected to the rapid perfusion protocol compared to myotubes kept in 10 mM external Ca2+ all the time. This was especially obvious for Δ29 and Δ60 but much less for the control chimera and Δ21. It could be that during the exposure to 0.5 mM external Ca2+, which lasted up to 15 min, the SR Ca2+ content was gradually reduced. This might have reduced the Ca2+ sensitivity of SR Ca2+ release which is known to be controlled by the luminal Ca2+ concentration (Lukyanenko et al., 2001). The lower SR Ca2+ load might have also reduced the Ca2+ release rate by reducing the Ca2+ gradient across the SR. Despite these limitations, the protocol was extremely useful for comparative purposes and a summary of this data is presented below (see Fig. 8 A). From these results we concluded that the Ca2+ current contributed in a significant manner to the signal that releases Ca2+ from the SR in myotubes expressing Δ29 and Δ60. However this was not the case for the control chimera and for Δ21-expressing myotubes.
FIGURE 6.
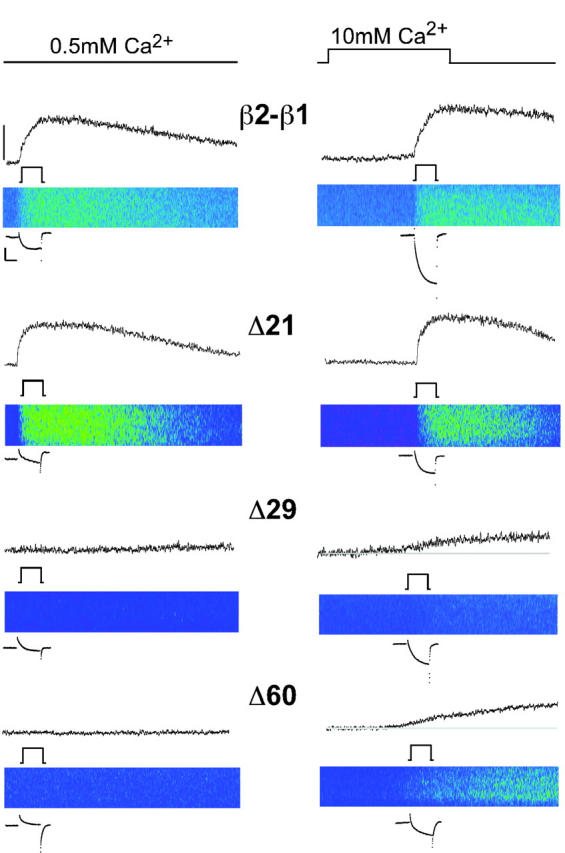
External Ca2+ dependence of Ca2+ transients expressed by truncated β variants. Line scan images and time courses of integrated fluo-4 fluorescence in ΔF/F units are shown for the same β1 KO myotube expressing the indicated variant in external solution with 0.5 mM Ca2+ (left) and 10 mM Ca2+ (right). Ca2+ transients were evoked by a 200-ms depolarization to +30 mV from a holding potential of −40 mV. In the images, the total line scan duration (x axis) was 2.05 s and the cell dimension (y axis) was (in microns) 36 for β2-β1; 25 for Δ21; 34 for Δ29; and 47 for Δ60. For visual reference, the color table was the same used in Fig. 3. Myotubes were initially voltage clamped and stimulated in external solution containing 0.5 mM Ca2+ and later fast-perfused with control external solution for 1 s before and during the same stimulation. The change in external solution and the depolarization are indicated by the line diagrams in black. Ca2+currents are shown before and after the solution change. Scale bars are 0.5 nA and 100 ms. The mean resting fluorescence of myotubes expressing Δ29 and Δ60 is indicated by the gray line. Fluorescence intensity scale denotes 1 ΔF/F for β2-β1, Δ29, and Δ60; and 2 ΔF/F for Δ21.
FIGURE 8.

Ca2+ dependent EC coupling as a function of the truncation length. (A) Histograms of the ratio of Ca2+ transient amplitude at +30 mV obtained as described in Fig. 5 in 0.5 and 10 mM external Ca2+ for the indicated number of cells and β variants. ΔF/F 10 Ca2+/ΔF/F 0.5 Ca2+ = 1 denotes absence of an EC coupling component that depends on Ca2+ entering the cell. (B) Histograms of the ratio ΔF/F exp max/ ΔF/F +90 mV described in the text and Table 1 for the indicated number of cells and β variants. ΔF/F exp max/ ΔF/F +90mV = 1 implies saturation of the Ca2+ transient versus voltage relationship indicative of voltage dependent EC coupling. (C) Correlation of the ΔF/F exp max/ ΔF/F +90mV ratio and the ΔF/F 10 Ca2+/ΔF/F 0.5 Ca2+ ratio for the same variants (correlation coefficient r2 = 0.8).
Ca2+ dependent EC coupling can be distinguished from purely voltage dependent EC coupling by the shape of the Ca2+ fluorescence versus voltage relationship, because the former mechanism follows the voltage dependence of the Ca2+ current. These relationships are shown in Fig. 7 for the control chimera, Δ60, and the four smaller deletions within domain D5, all obtained by integration of the confocal line scan fluorescence. The pulse protocol for obtaining these curves was the same described in Fig. 3. The ΔF/F value reached at the end of the depolarization was plotted as a function of voltage for depolarizations of 50 ms (filled symbols) or 200 ms (open symbols). The duration of the pulse did not affect the sigmoidal voltage dependence of Ca2+ transients expressed by the control construct, in agreement with observations in adult skeletal muscle (Melzer et al., 1986). However, myotubes expressing Δ7 and Δ21 had sigmoidal fluorescence versus voltage curves with a higher maximum fluorescence at large positive potentials (ΔF/Fmax) for the 200-ms than for the 50-ms stimulation. Moreover, as a function of truncation length, we observed a progressive reduction in the ΔF/Fmax produced by the 50-ms stimulation, although the shape of these curves was sigmoidal in all cases. In contrast, fluorescence versus voltage curves generated with the 200-ms pulse showed a significant change in shape, from sigmoidal for Δ7 and Δ21, to bell-shaped for Δ29, Δ35, and Δ60. For the later cases there was an increase in fluorescence in the range of −30 mV to +20 mV, a maximum at ∼+30 mV, and an incremental decline in fluorescence in the range of +40 mV to +90 mV. To quantify these observations, we fitted the curves obtained with the 200-ms depolarization for Δ29, Δ35, and Δ60 with a modified Boltzmann equation that takes into account the Ca2+ equilibrium potential (Eq. 2) while the rest of the curves in Fig. 7 were fitted with a conventional Boltzmann equation (Eq. 1). The fit of the mean fluorescence is indicated by the curve, and chi-square tests of the fit with Eq. 2 are provided in the figure legend. We further gauged the degree of curvature of the fluorescence versus voltage plots at positive potentials by computing the ratio of fluorescence at the experimental maximum and the fluorescence at +90 mV ([ΔF/F exp max]/[ΔF/F +90 mV]). The Boltzmann parameters of the fit of each cell and the fluorescence ratios at 50 ms and 200 ms are shown in Table 1 along with the statistical significance and the number of cells. The [ΔF/F exp max]/[ΔF/F +90 mV] ratio was >1 with ANOVA significance p < 0.05 for the more severe truncations and for full-length β2a when measured with the 200-ms depolarization. However, for the moderate truncations, the ratio was ∼1, within a 10% error. To confirm the basis of the bell-shaped fluorescence versus voltage relationships, we correlated the bending ratio [ΔF/F exp max]/[ΔF/F +90 mV] computed from the 200-ms depolarization with the ratio of Ca2+ transient amplitude at +30 mV in 10-mM and 0.5-mM external Ca2+ using the protocol described in Fig. 6. Fig. 8, A and B, shows histograms of the two indices for all the constructs tested in this study, whereas Fig. 8 C plots one index against the other. We found a clear correlation between these two indices, strongly suggesting that for the most severe truncations, the Ca2+ current contributed to trigger the Ca2+ transient thus explaining the change in the shape of the fluorescence versus voltage curve. In summary, a dependence of the Ca2+ transient amplitude on pulse duration was observed for all the truncated variants. However, only the most severe truncations, namely Δ29, Δ35, and Δ60, had bell-shaped fluorescence versus voltage relationships, consistent with the presence of an EC coupling component triggered by the Ca2+ current. Furthermore, as a function of truncation length, there was a continuous decrease in the maximum amplitude of the Ca2+ transient triggered by the 50-ms depolarization. Because the voltage dependence of the curves measured with the 50-ms depolarization was strictly sigmoidal, this result suggested a progressive loss of voltage-dependent coupling. At the same time, the progressive change in the shape of the plots determined by the 200-ms depolarization, from sigmoidal to biphasic bell-shaped, suggested a progressive gain in Ca2+ current dependent EC coupling.
If the Ca2+ current triggered EC coupling in the myotubes expressing the severely truncated constructs, the rate of rise of the fluorescence signal and the rate of Ca2+ entering the cell should be correlated. Fig. 9 A shows Ca2+ transients and Ca2+ currents produced by a 200-ms depolarization to +30 mV in myotubes expressing the control and Δ60 constructs. As already noted, Ca2+ transients obtained with Δ60 were considerably slower than those obtained with the control chimera. To correlate the time course of the Ca2+ transient and the Ca2+ current, we compared the cumulative sum of the Ca2+ charge during the depolarization (i.e., the running integral of the Ca2+ current) with the fluorescence signal. Because Ca2+ fluorescence was measured in a weakly buffered internal solution containing 0.1 mM EGTA, the cell fluorescence tracked Ca2+ accumulation rather than the rate of Ca2+ release from the SR (Melzer et al., 1984). Hence, we deemed it reasonable to correlate Ca2+ fluorescence with Ca2+ charge accumulation. The digitized data corresponds to the line by line confocal fluorescence in ΔF/F units (gray) and the smooth curve (black) is the time course of the cumulative Ca2+ charge. For the myotube expressing the control chimera, the fluorescence signal increased earlier and faster than the Ca2+ charge. Furthermore, the fluorescence signal saturated before termination of the 200 ms pulse while the Ca2+ charge continued to increase for as long as the pulse was at +30 mV. In contrast, there was a much closer agreement in the kinetics of the two signals in the myotube expressing Δ60. The increase in fluorescence occurred in parallel with Ca2+ charge accumulation and this was especially obvious during the second half of the 200-ms pulse. Thus in cells expressing Δ60, the kinetics of the Ca2+ transient was consistent with the Ca2+ current serving as trigger for SR Ca2+ release. In some myotubes expressing Δ60 we could observe a small fast component of the fluorescence signal at the start of the depolarization which could be interpreted as a residual voltage dependent component. However, this component was not consistently observed. In Fig. 9 B, we compared the rate of fluorescence change during the first 20 ms (dΔF/F/dt) as a function of voltage for myotubes expressing the control construct, the control construct in the presence of 0.1 mM ryanodine (open inverted triangles), and each of the five carboxyl terminal truncations. In control cells, dΔF/F/dt increased with voltage in a sigmoidal manner and reached the highest maximum at potentials more positive than +30 mV. Treatment with ryanodine, reduced the rate of fluorescence change ∼50-fold, consistent with the absence of a functional SR in this group of cells. For a moderate truncation such as Δ7, the maximum rate was statistically indistinguishable from control at all voltages (t-test significance p > 0.1). For the other truncations, the maximum rate at potentials more positive than +30 mV were two- to eightfold lower, and the decrease for Δ21 was less severe than for Δ29, Δ35, and Δ60 (t-test significance p < 0.001 versus control at +90 mV). Hence, the decrease in the rate of cytosolic Ca2+ accumulation was proportional to the truncation length. In summary, truncation of D5 region of β1a eliminated the fast EC coupling typical of skeletal myotubes. Furthermore, the slow EC coupling observed in myotubes expressing Δ60 appears to have its kinetic basis on the slow time course of the skeletal DHPR Ca2+ current.
FIGURE 9.

Initial rate of cytosolic Ca2+ increase during the Ca2+ transient in myotubes expressing truncated β variants. (A) Ca2+ currents and Ca2+ transients in myotubes expressing the control chimera (left) and Δ60 (right) were obtained in response to a 200-ms depolarization from a holding potential of −40 mV to +30 mV in external solution with 10 mM Ca2+. The digitized trace (gray) shows fluo-4 fluorescence in ΔF/F units obtained during the confocal line scan. Each dot corresponds to the mean fluorescence of a single 512-pixel line acquired at a rate of 2.05 ms per line. The black trace shows the cumulative integral of the Ca2+ current (running integral) in fC (pA × ms) superimposed on the fluorescence trace. Actual Ca2+ currents are shown below the trace of fluorescence. (B) The voltage dependence of the initial rate of fluorescence increase is shown for the indicated β variants. dΔF/F/dt was determined from the initial 20 ms of the Ca2+ transient. The lines correspond to a Boltzmann fit (Eq. 1) of the mean dΔF/F/dt. Parameters of the fit were (d(ΔF/F)/dt)max (in ΔF/F units) = 0.08, 0.06, 0.04, 0.02, 0.02, and 0.01; V1/2 (in mV) = −0.6, 8.7, 0, −2.8, 12.3, and 5.7; k (in mV) = 10.2, 13.8, 10.6, 13.9, 10.9, and 6.7; for myotubes expressing β2-β1 (n = 17), Δ7 (n = 18), Δ21 (n = 19), Δ29 (n = 13), Δ35 (n = 15), and Δ60 (n = 12), respectively. Empty inverted triangles (n = 4) correspond to β1 KO myotubes expressing β2-β1 treated with 0.1 mM ryanodine for 30 min before line scan acquisition.
DISCUSSION
The results of the present work show that elimination of carboxyl terminus of the DHPR β1a subunit modifies EC coupling in skeletal myotubes, transforming it from a process controlled by voltage to a much weaker and slower coupling process controlled by the Ca2+ current. As a function of truncation length, we observed an overall decrease in the amplitude of Ca2+ transients with evident changes in the shape of the Ca2+ fluorescence versus voltage relationship and slowing of the kinetics of the Ca2+ transient. For the most severe truncations, we also observed a dependence of Ca2+ transients on external Ca2+. These observations are consistent with the emergence of Ca2+ dependent EC coupling whereby Ca2+ entering the cell via the DHPR induces SR Ca2+ release, presumably by Ca2+ dependent activation of RyR1, although the participation of RyR3 cannot be discarded (Sheridan et al., 2002b). Additionally, there was a continuous decrease in voltage-dependent coupling indicated by the fact that Ca2+ transients activated by the 50-ms depolarization became smaller as a function of truncation length. Finally, the data showed a moderate decrease in Ca2+ conductance of two- to threefold for the most severe Δ29 and Δ60 truncations, whereas the control chimera expressed the same Ca2+ conductance of β1a-transfected myotubes (Table 1). The control β2-β1 chimera and Δ60 were previously shown to express a comparatively large charge movement density of 5.1 ± 0.5 and 4 ± 0.3 fC/pF, respectively (Beurg et al., 1999b). In addition, a truncated β1a lacking 35 residues from the carboxyl terminus also expressed a high charge movement density of 5 ± 0.8 fC/pF (Beurg et al., 1999b). For these reasons, we do not believe that the decrease in Ca2+ conductance observed in the severely truncated β variants may be ultimately explained by a decrease in DHPR surface expression, although this possibility remains to be investigated in detail. Studies in RyR1 KO myotubes show that the bulk of charge movements remain in place after RyR1 is eliminated, however, the Ca2+ current density decreases 5- to 10-fold (Nakai et al., 1996). Thus, expression of the DHPR Ca2+ current appears to be under the specific control of RyR1 by a so-called retrograde signal, from RyR1 to the DHPR. We have recently shown that this retrograde signal is strongly influenced by molecular determinants present on the amino-terminus of β2a (Ahern et al., 2002). Consequently, it is entirely possible that the loss in Ca2+ conductance in the severely truncated variants might represent a loss of retrograde signaling promoted by the truncated β subunit variants. Furthermore, because signals in the retrograde and orthograde directions have closely related structural determinants (Tanabe et al., 1990b; Ahern et al., 2001b), it would be unexpected to lose voltage-dependent EC coupling without losing retrograde enhancement of the Ca2+ current at the same time.
Drastic changes in EC coupling were previously reported when α1S, the skeletal pore isoform, was replaced by α1C, the cardiac pore isoform, in the cellular context of a primary dysgenic α1S-null skeletal myotube (Tanabe et al., 1990a). The Ca2+ fluorescence versus voltage curve expressed by α1C in dysgenic myotubes had a maximum at ∼+30 mV followed by a continuous decrease at more positive potentials. The biphasic nature of this relationship, together with a dependence of Ca2+ transients on external Ca2+, showed that Ca2+-dependent EC coupling could be implemented in skeletal myotubes by a hybrid DHPR composed of a cardiac pore subunit and skeletal nonpore subunits. The observation in α1S-null myotubes gave strong support to the notion that the pore subunit of the skeletal DHPR harbored a unique signal for triggering skeletal EC coupling (Tanabe et al., 1990a). In light of the present data, this concept must be expanded because elimination of the D5 region of β1a, in the context of a skeletal myotube lacking the β1 gene but expressing wild-type α1S, changed the EC coupling trigger signal in a manner similar to the change observed when α1C replaced α1S. Previous studies in β1 KO myotubes depolarized for 50 ms had shown that β2a expressed weak skeletal-type EC coupling (Beurg et al., 1999a). However, the longer stimulus used here demonstrated that a large component of the Ca2+ transient recovered by β2a is activated by the Ca2+ current (see Table 1). The similar outcome of these two sets of experiments, namely α1S replacement by α1C versus β1a replacement by β2a, suggests that the α1S/β1a pair, and not either subunit alone, controls the EC coupling signal in skeletal myotubes.
It could be argued that in skeletal β1 KO myotubes there is reexpression of α1C due to pleotropic effects arising from either the absence of β1a or the reintroduction of exogenous β variants. Thus the Ca2+-dependent EC coupling observed in our expression system might have originated from a DHPR containing the reexpressed α1C isoform. The experimental evidence indicates that this possibility is remote. First, the activation kinetics of the Ca2+ current expressed by Δ60 was even slower than that expressed by the control chimera with bona fide skeletal-type EC coupling, although a complete kinetic analysis has not yet been completed. A similar conclusion was reached recently by comparing the activation kinetics of Ca2+ currents recovered by β2a and β1a in β1 KO and double β1/RyR1 KO myotubes (Ahern et al., 2002). Hence, the truncated variant, as well as β2a, recovered a slow skeletal-type Ca2+ current and not the much faster Ca2+ current expressed by α1C. This result would not be expected if β2a paired up with α1C, because Ca2+ currents generated by the α1C/β2a pair are considerably faster (Kamp et al., 1996). Second, the half-activation potentials of the Ca2+ current recovered by the truncated β variants were +10 to +20 mV (Table 1), whereas the V1/2 expressed by α1C in skeletal myotubes is significantly more negative (Garcia et al., 1994). Third, transfection of β2a in dysgenic α1S-null myotubes does not result in expression of Ca2+ currents (not shown). Therefore, α1S is essential for the β-mediated recovery of DHPR function in skeletal myotubes. Finally, immunofluorescence staining with a primary antibody specific for α1C showed that α1C could not be detected in β1 KO myotubes (Fig. 1 C). For all these reasons, we are rather certain that a DHPR that included α1S was responsible for Ca2+ dependent EC coupling observed in β1 KO myotubes expressing the truncated β variants.
The systematic truncation approach provided clues on at least one structural motif present in the D5 region potentially responsible for the changes in EC coupling. Ca2+-dependent EC coupling was clearly noticeable in the severely truncated Δ29, Δ35, and Δ60 constructs and much less in the Δ21 and Δ7 constructs. This could be inferred from the exaggerated curvature of the fluorescence versus voltage plots for Δ29, Δ35 and Δ60 and by the dependence of the Ca2+ transients generated by these variants on external Ca2+. The D5 region of β1a (Fig. 1 A) contains a quasi-heptad repeat motif, namely L478(n)-V(n+7)-V(n+14)-L(n+22)-L(n+28), that is progressively removed by the Δ21, Δ29, and Δ35 truncations and is absent in Δ60. Heptad repeats are known to be important in protein-protein interactions responsible for gating (McCormack et al., 1991; Garcia et al., 1997). A conserved heptad repeat is present in voltage-gated K+, Na+, and L-type Ca2+ channels in the S4-S5 cytosolic linker immediately after the S4 charges. In the Shaker K+ channel, alanine substitutions in the heptad repeat leads to large positive shifts in the conductance versus voltage curve (McCormack et al., 1991; Bezanilla, 2000). These conductance shifts suggest that the S4 heptad repeat might be critical for the tight coupling that exists in voltage-gated channels between movements of the voltage sensor and the opening of the pore. Perhaps the heptad repeat in the carboxyl terminus of the β subunit could affect the coupling between charge movements in the DHPR voltage sensor and opening of the RYR1 channel. It is also conceivable that the heptad repeat in D5 could interact with a second heptad repeat present in domain D2 of β1a, namely L149(n)-V(n+7)-V(n+14)-L(n+21). This is an interesting possibility because the heptad repeat in D2 is conserved among β subunits, whereas the repeat in D5 is only present in β1a. The voltage dependent coupling mediated by β1a could thus arise from a specific protein folding governed by these two heptad repeats.
One of the important conclusions of the present work is that a DHPR complex with a β subunit having an intact carboxyl terminus is essential for fast skeletal-type EC coupling. Thus we would speculate that molecular determinants in the β subunit are either essential for the physical docking between DHPR tetrads and the tetrameric RyR1 complex or play a critical role in the transmission of the EC coupling signal. However, it must be realized that in closely interacting protein complexes, such as the DHPR and RyR1, these two possibilities are not mutually exclusive (Franzini-Armstrong and Protasiet, 1997). As a function of the truncation length, we observed a progressive loss of voltage-dependent EC coupling and the emergence of Ca2+-dependent EC coupling. Furthermore, for moderate truncations such as Δ7 and Δ21, Ca2+ transients were independent of external Ca2+ (Fig. 8). However, the amplitudes of the Ca2+ transients expressed by these two variants were larger with the 200-ms than with the 50-ms depolarizations (Fig. 7). These results are inherently difficult to explain within the conceptual framework described for EC coupling in skeletal and cardiac cells because pulse length dependence is not an obvious feature of skeletal-type EC coupling and Ca2+ currents had no obvious effects in myotubes expressing Δ7 and Δ21. The intermediate form of EC coupling favored by Δ7 and Δ21, as well as the fully fledged Ca2+-dependent mechanism observed for the more severe truncations, could originate from a progressive weakening of physical docking of the DHPR and RyR1 complexes brought about by a DHPR with a progressively truncated β subunit. The intermediate EC coupling mechanism favored by Δ7 and Δ21 might thus result from the transmission of a voltage signal to RyR1 that is kinetically impaired such that a long stimulus may be necessary to open a significant number of RyR1 channels. This explanation is directly supported by the progressively slower rate of increase of the fluorescence signal observed as a function of β truncation length (Fig. 9 B). A slow voltage dependent activation of RyR1 channels could account for the sigmoidal voltage dependence of Ca2+ transients and the higher ΔF/F max observed with the longer depolarization observed in myotubes expressing Δ7 and Δ21. Clearly, a study of the kinetics of charge movements expressed by Δ7 and Δ21 should shed light on this explanation. Ca2+-dependent EC coupling favored by the more severely truncated Δ29 and Δ60 variants might result from the transmission of a voltage signal to RyR1 that is unable to open a significant number of RyR1 channels due to a severe undocking of the DHPR and RyR1 complexes. Those RyR1 channels not activated by voltage might thus be available for activation by the Ca2+ current. It is also possible that the partial elimination of the carboxyl terminus of the β subunit leads somehow to an increase in the Ca2+ sensitivity of RyR1 and that the stronger Ca2+ transients seen with the 200-ms depolarization were due to excessive Ca2+ feedback on RyR1. Interestingly, spontaneous Ca2+ sparks in β1 KO myotubes were found to be significantly wider and longer in duration than their counterparts in normal or dysgenic (α1S null) myotubes (Conklin et al., 1999). Thus, the properties of clusters of RyR1 channels can be modified by the absence of the β subunit. Finally, there is an alternative explanation to consider brought about by the study of Simon and Hill (1992), demonstrating that voltage activation of four DHPR voltage sensors, presumably one in each DHPR in the tetrad, is required for activation of SR Ca2+ release. Truncated β subunits in each DHPR could disrupt the voltage-sensing activity within the tetrad or disrupt the DHPR tetrad arrangement altogether. In principle, tetrad disruption could account for the slow rate of SR Ca2+ release observed in the present experiments hence, this remains as an attractive possibility. These three mechanisms of EC coupling disruption by truncated β subunits, namely a slowing in the transmission of the voltage signal from the DHPR to RyRl, an increase in RyR1 Ca2+ sensitivity, and/or a disruption of the DHPR tetrad arrangement, deserves close scrutiny in future experiments.
Acknowledgments
This work was supported by National Institutes of Health (AR46448, HL47053) to R. C., Wisconsin Heart Association (predoctoral fellowship) to D. C. S. and C. A. A., and National Institutes of Health (T32 HL07936 fellowship) to C. A. A.
References
- Ahern, C., P. A. Powers, R. G. Gregg, and R. Coronado. 2000. The DHPR beta subunit is essential for cardiac-type excitation-contraction coupling. Biophys. J. 78:426a. (Abstr.) [Google Scholar]
- Ahern, C. A., J. Arikkath, P. Vallejo, C. A. Gurnett, P. A. Powers, K. P. Campbell, and R. Coronado. 2001a. Intramembrane charge movements and excitation-contraction coupling expressed by two-domain fragments of the Ca2+ channel. Proc. Natl. Acad. Sci. USA. 98:6935–6940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern, C. A., D. Bhattacharya, L. Mortenson, and R. Coronado. 2001b. A component of excitation-contraction coupling triggered in the absence of the T671-L690 and L720-Q765 regions of the II-III loop of the dihydropyridine receptor α1S pore subunit. Biophys. J. 81:3294–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahern, C. A., D. C. Sheridan, W. Cheng, L. Mortenson, P. D. Allen, and R. Coronado. 2002. Ca2+ current and charge movements in skeletal myotubes promoted by the β subunit of the dihydropyridine receptor in the absence of ryanodine receptor type 1. Biophys. J. Submitted. [DOI] [PMC free article] [PubMed]
- Berrou, L., G. Bernatchez, and L. Parent. 2001. Molecular determinants of inactivation within the I-II linker of a1E (CaV2.3) calcium channels. Biophys. J. 80:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuckelmann, D. J., and W. G. Wier. 1988. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. J. Physiol. (Lond.). 405:233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurg, M., M. Sukhareva, C. Strube, P. A. Powers, R. G. Gregg, and R. Coronado. 1997. Recovery of Ca2+ current, charge movements, and Ca2+ transients in myotubes deficient in dihydropyridine receptor β1 subunit transfected with β1 cDNA. Biophys. J. 73:807–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurg, M., M. Sukhareva, C. A. Ahern, M. W. Conklin, E. Perez-Reyes, P. A. Powers, R. G. Gregg, and R. Coronado. 1999a. Differential regulation of skeletal muscle L-type Ca2+ current and excitation-contraction coupling by the dihydropyridine receptor β subunit. Biophys. J. 76:1744–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurg, M., C. A. Ahern, P. Vallejo, M. Conklin, P. A. Powers, R. G. Gregg, and R. Coronado. 1999b. Involvement of the carboxy-terminus region of the dihydropyridine receptor β1a subunit in excitation-contraction coupling of skeletal muscle. Biophys. J. 77:2953–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bichet, D., V. Cornet, S. Geib, E. Carlier, S. Volsen, T. Hoshi, Y. Mori, and M. De Waard. 2000. The I–II loop of the Ca2+ channel α1 subunit contains an endoplasmic reticulum retention signal antagonized by the β subunit. Neuron. 25:177–190. [DOI] [PubMed] [Google Scholar]
- Bezanilla, F. 2000. The voltage sensor in voltage-dependent ion channels. Physiol. Rev. 80:555–592. [DOI] [PubMed] [Google Scholar]
- Cheng, W., X. Cao, D. C. Sheridan, and R. Coronado. 2002. A novel α1S/β interaction relevant to excitation-contraction coupling. Biophys. J. 82:78a. (Abstr.) [Google Scholar]
- Chien, A. J., X. L. Zhao, R. E. Shirokov, T. S. Puri, C. F. Chang, K. Sun, E. Rios, and M. M. Hosey. 1995. Roles of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. J. Biol. Chem. 270:30036–30044. [DOI] [PubMed] [Google Scholar]
- Conklin, M. W., P. A. Powers, R. G. Gregg, and R. Coronado. 1999. Ca2+ sparks in embryonic mouse skeletal muscle selectively deficient in dihydropyridine receptor α1S or β1a subunits. Biophys. J. 76:657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard, M., M. Pragnell, and K. P. Campbell. 1994. Ca2+ channel regulation by a conserved β subunit domain. Neuron. 13:495–503. [DOI] [PubMed] [Google Scholar]
- Dirksen, R. T., and K. G. Beam. 1999. Role of calcium permeation in dihydropyridine receptor function. Insights into channel gating and excitation-contraction coupling. J. Gen. Physiol. 114:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong, C., and F. Protasi. 1997. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol. Rev. 77:699–729. [DOI] [PubMed] [Google Scholar]
- Garcia, J., J. Nakai, K. Imoto, and K. G. Beam. 1997. Role of S4 segments and the leucine heptad motif in the activation of an L-type calcium channel. Biophys. J. 72:2515–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, J., T. Tanabe, and K. G. Beam. 1994. Relationship of calcium transients to calcium currents and charge movements in myotubes expressing skeletal and cardiac dihydropyridine receptors. J. Gen. Physiol. 103:125–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg, R. G., A. Messing, C. Strube, M. Beurg, R. Moss, M. Behan, M. Sukhareva, S. Haynes, J. A. Powell, R. Coronado, and P. A. Powers. 1996. Absence of the β subunit (CCHB1) of the skeletal muscle dihydropyridine receptor alters expression of the α1 subunit and eliminates excitation-contraction coupling. Proc. Natl. Acad. Sci. USA. 93:13961–13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon, M. R., N. S. Berrow, A. C. Dolphin, and B. A. Wallace. 1999. Modeling of a voltage-dependent Ca2+ channel β subunit as a basis for understanding its functional properties. FEBS Lett. 445:366–370. [DOI] [PubMed] [Google Scholar]
- Kamp, T., M. T. Perez-Garcia, and E. Marban. 1996. Enhancement of ionic current and charge movement by coexpression of calcium channel β1a subunit with α1C subunit in a human embryonic kidney cell line. J. Physiol. 492:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukyanenko, V., S. Viatchenko-Karpiski, A. Smirnov, T. F. Weisner, and S. Gyorke. 2001. Dynamic regulation of sarcoplasmic reticulum Ca2+ content and release by luminal Ca2+-sensitive leak in rat ventricular myocytes. Biophys. J. 81:785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack, K., M. A. Tanouye, L. E. Iverson, J. W. Lin, M. Ramaswami, T. McCormack, J. T. Campanelli, M. K. Mathew, and B. Rudy. 1991. A role for hydrophobic residues in the voltage-dependent gating of Shaker K+ channels. Proc. Natl. Acad. Sci. USA. 88:2931–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer, W., E. Rios, and M. F. Schneider. 1984. Time course of calcium release and removal in skeletal muscle fibers. Biophys. J. 45:637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer, W., M. F. Schneider, B. J. Simon, and G. Szucs. 1986. Intramembrane charge movement and calcium release in frog skeletal muscle. J. Physiol. 373:481–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai, J., R. T. Dirksen, H. T. Nguyen, I. N. Pessah, K. G. Beam, and P. D. Allen. 1996. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 380:72–75. [DOI] [PubMed] [Google Scholar]
- Nakai, J., N. Sekiguchi, T. A. Rando, P. D. Allen, and K. G. Beam. 1998. Two regions of the ryanodine receptor involved in coupling with L-type Ca2+ channels. J. Biol. Chem. 273:13403–13406. [DOI] [PubMed] [Google Scholar]
- Neuhuber, B., U. Gerster, J. Mittendorfer, H. Glossmann, and B. E. Flucher. 1998. Differential effects of Ca2+ channel β1a and β2a subunits on complex formation with α1S and on current expression in tsA201 cells. J. Biol. Chem. 273:9110–9118. [DOI] [PubMed] [Google Scholar]
- Neely, A., X. Wei, R. Olcese, L. Birnbaumer, and E. Stefani. 1993. Potentiation of the β subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 262:575–578. [DOI] [PubMed] [Google Scholar]
- Olcese, R., N. Qin, T. Schneider, A. Neely, X. Wei, E. Stefani, and L. Birnbaumer. 1994. The amino terminus of a calcium channel β subunit sets rates of channel inactivation independently of the subunit's effect on activation. Neuron. 13:1433–1438. [DOI] [PubMed] [Google Scholar]
- Olcese, R., A. Neely, N. Qin, X. Wei, L. Birnbaumer, and E. Stefani. 1996. Coupling between charge movement and pore opening in vertebrate neuronal α1E calcium channels. J. Physiol. 497:675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Reyes, E., and T. Schneider. 1994. Calcium channels: structure, function, and classification. Drug Dev. Res. 33:295–318. [Google Scholar]
- Powers, P. A., S. Liu, K. Hogan, and R. G. Gregg. 1992. Skeletal muscle and brain isoforms of a beta subunit of human voltage-dependent calcium channels are encoded by a single gene. J. Biol. Chem. 267:22967–22972. [PubMed] [Google Scholar]
- Pragnell, M., M. DeWaard, Y. Mori, T. Tanabe, T. P. Snutch, and K. P. Campbell. 1994. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 368:67–70. [DOI] [PubMed] [Google Scholar]
- Qin, N., R. Olcese, J. Zhou, O. A. Cabello, L. Birnbaumer, and E. Stefani. 1996. Identification of a second region of the β subunit involved in regulation of calcium channel inactivation. Am. J. Physiol. 271:C1539–C1545. [DOI] [PubMed] [Google Scholar]
- Sheridan, D. C., W. Cheng, L. Mortenson, and R. Coronado. 2002a. The C-terminus of the skeletal dihydropyridine β1a subunit determines the rate of rise of the Ca2+ transient. Biophys. J. 82:77a. (Abstr.) [Google Scholar]
- Sheridan, D. C., L. Mortenson, V. Sorrentino, and R. Coronado. 2002b. Ca2+ induced Ca2+ release (CICR) in dyspedic (RyR1 null) skeletal myotubes induced by DHPR β2a. Biophys. J. 82:76a.11751297 [Google Scholar]
- Simon, B. J., and D. A. Hill. 1992. Charge movement and SR calcium release in frog skeletal muscle can be related by a Hodgkin-Huxley model with four gating particles. Biophys. J. 61:1109–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strube, C., M. M. Beurg, P. A. Powers, R. G. Gregg, and R. Coronado. 1996. Reduced Ca2+ current, charge movement, and absence of Ca2+ transients in skeletal muscle deficient in dihydropyridine receptor β1 subunit. Biophys. J. 71:2531–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekura, H., L. Bennett, K. Tanabe, K. G. Beam, and C. Franzini-Armstrong. 1994. Restoration of junctional tetrads in dysgenic myotubes by dihydropyridine receptor cDNA. Biophys. J. 67:793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe, T., K. G. Beam, J. A. Powell, and S. Numa. 1988. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 336:134–139. [DOI] [PubMed] [Google Scholar]
- Tanabe, T., A. Mikami, S. Numa, and K. G. Beam. 1990a. Cardiac-type excitation-contraction coupling in dysgenic skeletal muscle injected with cardiac dihydropyridine receptor cDNA. Nature. 344:451–453. [DOI] [PubMed] [Google Scholar]
- Tanabe, T., K. G. Beam, B. A. Adams, T. Niidome, and S. Numa. 1990b. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 346:567–569. [DOI] [PubMed] [Google Scholar]
- Walker, D., D. Bichet, K. P. Campbell, and M. DeWaard. 1998. A β4 isoform-specific interaction site in the carboxyl-terminal region of the voltage-dependent Ca2+ channel α1A subunit. J. Biol. Chem. 273:2361–2367. [DOI] [PubMed] [Google Scholar]
- Wei, S. K., H. M. Colecraft, C. D. DeMaria, B. Z. Peterson, R. Zhang, T. A. Kohout, T. B. Rogers, and D. T. Yue. 2000. Ca2+ channel modulation by recombinant auxiliary beta subunits expressed in young adult heart cells. Circ. Res. 86:175–184. [DOI] [PubMed] [Google Scholar]