

Abstract
KATP channels are comprised of a pore-forming protein, Kir6.x, and the sulfonylurea receptor, SURx. Interaction of adenine nucleotides with Kir6.2 positively charged amino acids such as K185 and R201 on the C-terminus causes channel closure. Substitution of these amino acids with other positively charged residues had small effects on inhibition by adenine nucleotide, while substitution with neutral or negative residues had major effects, suggesting electrostatic interactions between Kir6.2 positive charges and adenine nucleotide negative phosphate groups. Furthermore, R201 mutation decreased channel sensitivity to ATP, ADP, and AMP to a similar extent, but K185 mutation decreased primarily ATP and ADP sensitivity, leaving the AMP sensitivity relatively unaffected. Thus, channel inhibition by ATP may involve interaction of the α-phosphate with R201 and interaction of the β-phosphate with K185. In addition, decreased open probability due to rundown or sulfonylureas caused an increase in ATP sensitivity in the K185 mutant, but not in the R201 mutant. Thus, the β-phosphate may bind in a state-independent fashion to K185 to destabilize channel openings, while R201 interacts with the α-phosphate to stabilize a channel closed configuration. Substitution of R192 on the C-terminus and R50 on the N-terminus with different charged residues also affected ATP sensitivity. Based on these results a structural scheme is proposed, which includes features of other recently published models.
INTRODUCTION
KATP channels are comprised of a pore-forming protein, Kir6.x, and the sulfonylurea receptor, SURx. When expressed alone, Kir6.2 forms functional channels that are inhibited by adenine nucleotides. Coexpression of SUR1 with Kir6.2 confers MgADP stimulation as well as sulfonylurea sensitivity and increases channel open probability (Po) (Tucker et al., 1997; John et al., 1998).
Using mutational analysis of Kir6.2, Tucker et al. (1998) identified several residues that affect ATP sensitivity. Notably two of these mutants, K185Q and R50G, did so without changing the Po in the absence of ATP, suggesting that the decrease in ATP sensitivity was due to a decrease in ATP binding affinity. In 1999 Reimann et al., reported that substitution of K185 with another positive residue had little effect on ATP sensitivity, while mutation to negatively charged residues dramatically reduced ATP sensitivity. Based on this observation, they suggested that ATP binding to Kir6.2 may involve positively charged residues interacting with the negatively charged phosphate groups of ATP, although allosteric effects that decrease ATP binding indirectly were not ruled out.
More recently, in a systematic approach. Shyng et al. (2000) mutated all positively charged residues on Kir6.2 COOH-terminus and showed that R201 and K185 were the only residues that control ATP binding in that region. However, in their study mutations, R176A, R177A, R192A, R206A, K222A, R301A, and R314A did not yield current in the absence of PIP2 and their sensitivity to ATP could not be assessed. In contrast, we have recently shown that R176C and R177C could yield functional channels in the absence of PIP2 and they did not contribute to ATP binding, but interacted instead with SUR1 to confer MgADP-dependent activation (John et al., 2001). In this study we also showed that mutations of the positively charged residues R301C and R314C could yield currents, but again they did not affect significantly ATP sensitivity. On this basis it may be concluded that ATP binds to the C-terminus in a region delimited by K185 and R201.
Contributions of the Kir6.2 N terminus to ATP binding have also been investigated. In 1998 Tucker et al. showed that mutation of R50 reduced dramatically channel sensitivity to ATP, while mutation of K47 and K39 had little effect. In addition, Koster et al., (1999) showed that truncation of up to 30 amino acids of the N-terminus had no direct effect on ATP binding. Outside of these two regions of the N- and C-termini, neutral residues such as G334 have been identified that affect ATP sensitivity dramatically (Drain et al., 1998). It remains to be demonstrated whether this residue interacts with the phosphate groups or the adenosine moiety of adenine nucleotides.
Two processes have been suggested to account for ATP-dependent channel closure. One involves preferential binding of ATP to a closed state and stabilization of the channel in this configuration (Trapp et al., 1998; Fan and Makielski, 1999; Enkvetchakul et al., 2000), which explains why ATP sensitivity increases as Po decreases. The other involves ATP binding to the open channel and destabilization of this configuration (Trapp et al., 1998; Fan and Makielski, 1999; Li et al., 2002), accounting for the decrease in channel open time caused by ATP. In the present study, we have further investigated the adenine nucleotide sensitivity of KATP channels. First we reexamined the N- and C-termini to identify the positively charged residues that affect ATP sensitivity without affecting channel Po in the absence of ATP. Next, we investigated how the nature of the charge at these positions affected the sensitivity to ADP and AMP as well as ATP, to provide insight into whether charged interactions occurred between the negatively charged phosphate groups of ATP (α, β, or γ) and the positively charged residues and, if so, which of the phosphate groups interacted with which residues. Finally, we investigated how mutation of these residues affected the relationship between adenine nucleotide sensitivity and Po. Based on the findings, we propose a model detailing how the individual phosphate groups of ATP bind, in state-dependent and state-independent fashions, to specific positively charged residues on the N- and C-termini of Kir6.2. In generating our final scheme, we incorporated recent structural information about other related K channels (Cukras et al. 2002; Jiang et al., 2002).
MATERIAL AND METHODS
The techniques for cDNA expression and patch clamp recording have been recently described in detail (John et al., 1998) and are only briefly outlined here.
Molecular biology and cDNA expression in HEK293 cells
In most cases, HEK293 cells were transfected with cDNA for Kir6.2 mutants linked to green fluorescent protein (GFP) at the C-terminus (Kir6.2-GFP) so that insertion of the constructs into the plasma membrane could be followed. Our previous findings showed that linkage to GFP did not affect the kinetics or adenine nucleotide sensitivity of wild-type Kir6.2+SUR1 channels (John et al., 1998). The results presented here now show that linking GFP to wild-type Kir6.2 or the Kir6.2K185Q mutant does not affect their sensitivity to ATP, ADP and AMP as previously reported for wild-type and K185Q mutant not linked to GFP (Reimann et al., 1999; Koster et al., 1999). These observations validate the use of GFP to identify the cells with high levels of channel insertion into the plasma membrane. All wild-type cDNAs were subcloned into the vector pCDNA3amp (Invitrogen, Carlsbad, CA). cDNAs used to make the GFP chimeras were subcloned into the pEGFP vector (Clontech, Palo Alto, CA). Both vectors use the CMV promoter. Single site Kir6.2 mutations were constructed using the QuikChange technique from Stratagene (La Jolla, CA). All mutants were in the Kir6.2-GFP backbone. The R201A mutant was kindly provided by Dr. C. Nichols (University of St Louis, MO) and subcloned into the Kir6.2-GFP backbone. The transfections were carried out using the calcium phosphate precipitation method (Graham and van der Eb, 1973). Expression of proteins linked to GFP was detected as early as 12 h after transfection. Patch clamp experiments were started ∼30 h after transfection. HEK293 cells were cultured in DMEM high glucose medium supplemented with 10% (v/v) fetal calf serum, penicillin (100 units/ml), streptomycin (100 units/ml), and 2mM glutamine and divided once a week by treatment with trypsin.
Patch clamp methods
Currents were recorded in HEK293 cells using the inside-out patch clamp configuration, with the pipette solution containing (in mM): 140 KCl, 10 NaCl, 1.1 MgCl2, 10 HEPES and pH adjusted to 7.2 with KOH. The bath solution consisted of 140 KCl, 10 NaCl, 1.1 MgCl2, 10 HEPES, 5 EGTA, and 0.5 CaCl2, with the pH adjusted to 7.2 with KOH. Adenine nucleotides were added directly to the bath. With AMP the highest concentration used was 20 mM, thus, with some mutants such as R201A that exhibited AMP sensitivity around this concentration, the IC50 was deduced by fitting data obtained with concentrations lower than 20 mM using a Hill coefficient of 1. Data obtained with wild-type and other mutant channels indicate that the Hill coefficient deduced from dose-response curve fitting is similar to that obtained with ATP and ADP and is often close to 1. In this study we use removal of Mg2+ and addition of the sulfonylurea glibenclamide to decrease channel activity and investigate the effects of Po on adenine nucleotide sensitivity. Inasmuch as the activity of Kir6.2 alone is insensitive to sulfonylureas and does not spontaneously run-down, the present experiments were performed using Kir6.2 coexpressed with SUR1.
The data, filtered at 2 kHz with an 8-pole Bessel filter, was recorded with a List EPC 7 (Darmstadt, Germany) patch clamp amplifier and recorded on videotape at a fixed frequency of 44 kHz after digitization with a digital audio processor. For analysis, the data was sampled at a rate of 5.5 kHz.
RESULTS
Identification of the positively charged residues in the N- and C-terminus involved in ATP binding
We have neutralized all positively charged residues on the N-terminus between residues 30 and the beginning of transmembrane domain M1, and on the C-terminus between the end of the transmembrane domain M2 and residue 222. Our data illustrated in Fig. 1 confirm previous findings with R50, K185, and R201, and further suggest that R192 also affect ATP binding. Thus, the ATP binding site on the C-terminus is confined between K185 and R201, and the ATP binding site on the N-terminus may be confined to a region near R50.
FIGURE 1.

Effect on ATP sensitivity of mutating positively charged residues in the N- and C-terminus. Wild-type or mutant Kir6.2 were coexpressed with SUR1 in HEK293 cells. Inside-out patches were excised in the absence of Mg2+ and the sensitivity to ATP was measured after rundown. (In many instances, increasing ATP concentrations was applied up to three times to reach the maximum increase in ATP sensitivity with rundown.) For each mutant, the ATP sensitivity was assayed at least in three different patches. The error bars illustrate standard deviation of the mean. The dashed line illustrates the IC50 for wild-type Kir6.2+SUR1 channels. The * sign indicates mutant channels that were not inserted in the membrane. The + sign indicates mutant channels that were inserted in the membrane, but did not yield current.
The mutant channels R34C/G, K38C/G, and R206C did not yield currents when coexpressed with SUR1. However, when linked to GFP, R206C + SUR1, but not R34C/G + SUR1 or K38C/G + SUR1, yielded fluorescent plasma membrane labeling. These results indicate that R34C/G and K38C/G mutations prevented channel insertion into the plasma membrane, suggesting that these residues may play a role in assembly of functional channels. In contrast, R206C was inserted into the plasma membrane without yielding current, indicating that this residue may be important for channel gating.
Interaction of positively charged residues R50, K185, R192, and R201 with adenine nucleotide phosphate groups
Based on extensive mutation of K185 in Kir6.2, it has been suggested that negatively charged phosphate groups of adenine nucleotides may interact with positively charged residues on Kir6.2 (Reimann et al., 1999; Shyng et al., 2000). To assess whether such electrostatic interactions take place between the phosphate groups and the other Kir6.2 positive residues that have been shown to affect ATP sensitivity (R50, R192, and R201), we substituted these charges with other positive, neutral, and negative amino acids. The data in Table 1 show that mutations at R192 and R201 behave like K185, with positively charged substitutions having little effect on ATP sensitivity and neutral or negatively charged substitutions having major effects. On the other hand, R50K has little effect on ATP sensitivity, but R50E also exhibits ATP sensitivity very similar to wild-type. These results are discussed later.
TABLE 1.
ATP sensitivity of three positively charged residues mutated with positive, neutral, and negative amino acids
| Residue | Amino acid | ATP IC50 (μM) |
|---|---|---|
| Wt | 12.5±3 | |
| R201 | A | 820±175 |
| K | 140±54 | |
| E | 1415±280 | |
| R192 | C | 150±27 |
| K | 55±25 | |
| E | 225±50 | |
| R50 | G | 1450±245 |
| K | 92.5±12.5 | |
| E | 22.5±7.5 |
To gain insight into the roles of specific adenine nucleotide phosphate groups, we compared channel inhibition by ATP (with α-, β-, and γ-phosphates), ADP (with α- and β-phosphates), and AMP (with only the α-phosphate). These experiments were performed after channel rundown was induced by removal of Mg2+, in which case the adenine nucleotide sensitivity was maximum and no longer subject to variability due to change in channel Po as a result of rundown.
When Kir6.2 was expressed alone, inhibition by ADP and AMP was respectively threefold and 110-fold less potent than inhibition by ATP (ATP IC50 = 15 μM, ADP IC50 = 325 μM, and AMP IC50 = 12,500 μM). In addition, adenosine alone has almost no inhibitory effect (data not shown). These data indicate that adenine nucleotide-evoked inhibition primarily involves interaction of the α- and β-phosphates of adenine nucleotides with Kir6.2. As shown in Fig. 2, when Kir6.2 was co-expressed with SUR1 and after rundown in the absence of Mg2+, the channel sensitivity to the three adenine nucleotide was higher than with Kir6.2 alone, and the inhibitory efficacy of ADP was reduced compared to ATP. Thus, the ratio of ADP IC50/ATP IC50 increased from 3 in Kir6.2 alone to 9 in Kir6.2 + SUR1. In comparison, the ratio of AMP IC50/ATP IC50 did not vary significantly and remained near 120–140. This lesser inhibition by ADP in the presence of SUR1 may be explained by a stimulatory effect of ADP, even though Mg2+ was absent.
FIGURE 2.

Adenine nucleotide sensitivity of wild-type Kir6.2+SUR1 channels. (A, B, and C) Inward currents measured in the absence of Mg2+ were progressively suppressed by increasing concentrations of ATP, ADP, and AMP with the IC50 near 12 μM, 120 μM, and 1.85 mM, respectively. On average, ADP was 8- to 10-fold less potent than ATP in blocking the channel, and AMP 140-fold less potent. (D) Plot of ATP (•), ADP (○), and AMP (▴) sensitivity for Kir6.2+SUR1 in the absence of Mg2+. The data points were fit with the equation y = 1/(1 + (k/[ATP])n), which yielded k = 12.5 μM for ATP (n = 5), k = 115 μM (n = 5) for ADP, and k = 1.85 mM for AMP (n = 5). The three Hill coefficients were close to unity.
Similar studies comparing sensitivity to ATP, ADP, and AMP were performed with the Kir6.2 mutants R50G, K185Q, R192C, and R210A coexpressed with SUR1. Fig. 3 and Fig. 4 show representative data obtained with K185Q and R192C mutations, respectively. Both mutations decreased ATP and ADP sensitivity, ∼35-fold for K185Q and 12-fold for R192C, but the K185Q mutation decreased the AMP sensitivity by only 2.5-fold as compared to wild-type channel. Thus the K185Q mutation decreased primarily the sensitivity to ATP and ADP and had comparatively minor effects on AMP sensitivity. In contrast, the R192C mutation decreased the inhibitory effect of all three adenine nucleotides with similar potency.
FIGURE 3.

Adenine nucleotide sensitivity of the Kir6.2 mutant K185Q+SUR1. (A, B, and C) Inward currents measured in the absence of Mg2+ were progressively suppressed by increasing concentrations of ATP, ADP, and AMP with the IC50 near 440 μM, 3.8 mM, and 4.5 mM, respectively. On average ADP was eightfold less potent and AMP 10-fold less potent than ATP at blocking the channel. As compared to wild-type channels, the ATP and ADP sensitivity decreased by ∼30-fold, while the AMP sensitivity was only 2.5-fold lower. (D) Plot of channel ATP (•), ADP (○), and AMP (▴) sensitivity for Kir6.2+SUR1 in the absence of Mg2+. Fit of the data points yielded k = 0.44 mM for ATP (n = 3), k = 3.8 mM (n = 3) for ADP, and k = 4.5 mM for AMP (n = 3). The three Hill coefficients were close to unity.
FIGURE 4.

Adenine nucleotide sensitivity of the Kir6.2 mutant R192C+UR1. (A, B, and C) Inward currents measured in the absence of Mg2+ were suppressed by increasing concentrations of ATP, ADP, and AMP with the IC50 near 150 μM, 2 mM, and 18 mM, respectively. On average, ADP was 13-fold less potent and AMP 120-fold less potent than ATP at blocking the channel. Compared with wild-type channels, the ATP, AMP, and ADP sensitivity of the mutant decreased by ∼10-fold. (D) Plot of channel ATP (•), ADP (○), and AMP (▴) sensitivity for Kir6.2+SUR1 in the absence of Mg2+. Fit of the data points yielded k = 150 μM for ATP (n = 3), k = 2.1 mM (n = 3) for ADP, and k = 18 mM for AMP (n = 3). The estimated Hill coefficients were close to unity for ATP and ADP data points. The AMP data were fitted using a Hill coefficient of 1.
These data are summarized in Fig. 5, which also shows that R50G behaved similarly to K185Q. In this case ATP and ADP sensitivity was reduced ∼120- to 150-fold compared to wild-type channels, while the AMP sensitivity was decreased only 10-fold. These results indicate that R50G, like K185Q, affects primarily ATP and ADP sensitivity, while having less effect on AMP sensitivity. Based on these data, we postulate that R50 and K185 interact primarily with the β-phosphate of ATP and ADP.
FIGURE 5.

Adenine nucleotide sensitivity of R50G, K185Q, R192C, and R201A mutants coexpressed with SUR1, compared with wild-type Kir6.2+SUR1 channels. As already shown with wild-type channels, the ADP sensitivity was ninefold lower than ATP sensitivity and AMP 100-fold lower. As compared to wild-type, K185Q mutation decreased the ATP and ADP sensitivity by 35-fold, but had minimal effect on AMP sensitivity. In contrast, the R192C mutation had similar effects on ATP, ADP, and AMP sensitivities and caused a 12- to 15-fold decrease. R50G behaved like K185Q and decreased the ATP and ADP sensitivity by ∼120- to 140-fold, but the AMP sensitivity decreased by only 11-fold. R201A behaved like R192C and decreased the sensitivity to all three nucleotide. The sensitivity to ATP and ADP decreased by 60-fold. The AMP sensitivity with R201A+SUR1 could not be measured accurately, but 20 mM AMP caused 15% channel inhibition. Thus, assuming a Hill coefficient of 1, the IC50 for channel inhibition by AMP would be close to 100 mM and the AMP sensitivity decreased by a similar amount (60-fold).
Fig. 5 also shows that the R201A mutation decreased the sensitivity to all three adenine nucleotides to a similar extent, by ∼60- to 70-fold. In this case, the inhibition by AMP could not be directly measured and was deduced from fitting data obtained with AMP concentrations up to 20 mM (see Methods). The similar effects of the R201 and R192 mutations on the sensitivity to ATP, ADP, and AMP may be explain assuming that these two residues interact with the α-phosphate group common to all three adenine nucleotides.
ATP sensitivity increases as Po decreases in the K185Q, R50G, and R192C mutants similar to wild-type Kir6.2
We recently reported that KATP channel inhibition by the sulfonylurea glibenclamide caused a six- to eightfold increase in ATP sensitivity (John et al., 2001). Because glibenclamide promotes channel closing, such a shift may result from preferential binding of ATP to a closed state of the channel (Trapp et al., 1998; Fan and Makielski, 1999; Enkvetchakul et al., 2000). To assess whether any of the positively charged residues that control ATP binding also involved state-dependent ATP binding, we investigated the effects of decreasing channel Po with glibenclamide on ATP sensitivity. In these experiments, inside-out patches from cells coexpressing wild-type or mutant Kir6.2 with SUR1 were excised in a bath solution containing 200 μM MgADP to promote a high initial Po. The channel sensitivity to adenine nucleotides was assessed immediately after removal of MgADP and then again after inhibition by glibenclamide. Upon removal of MgADP, channel activity remained constant in most cases for at least 10–15 min and thus allowed for measurements of ATP sensitivity before rundown. Upon addition of glibenclamide, channel activity decreased within 30 s and ATP sensitivity was then reassessed.
As we previously reported (John et al., 2001), decreasing Po with glibenclamide (and also spontaneous or Ca2+-induced rundown) caused a dramatic increase in ATP sensitivity in wild-type Kir6.2, with the IC50 for inhibition by ATP decreasing ∼sixfold, from ≈75 μM to ≈12 μM. The Kir6.2 mutant K185Q exhibited a low ATP sensitivity before glibenclamide with an IC50 close to 2 mM. Addition of 200 nM glibenclamide caused on average a 40% decrease in channel activity, close to that observed with wild-type channels (45%). As with wild-type channels, the decrease in channel activity evoked by glibenclamide caused a five- to sixfold increase in ATP sensitivity (Fig. 6 B). This increase was similar to that observed during spontaneous rundown of K185Q + SUR1 channels in the absence of Mg2+ (Fig. 6 D). In addition, glibenclamide-induced decrease in channel activity was associated with loss of MgADP-dependent reactivation. These findings indicate that both glibenclamide and spontaneous rundown promote functional uncoupling of SUR1 from K185Q, as we have previously shown for wild-type Kir6.2 (Ribalet et al., 2000).
FIGURE 6.
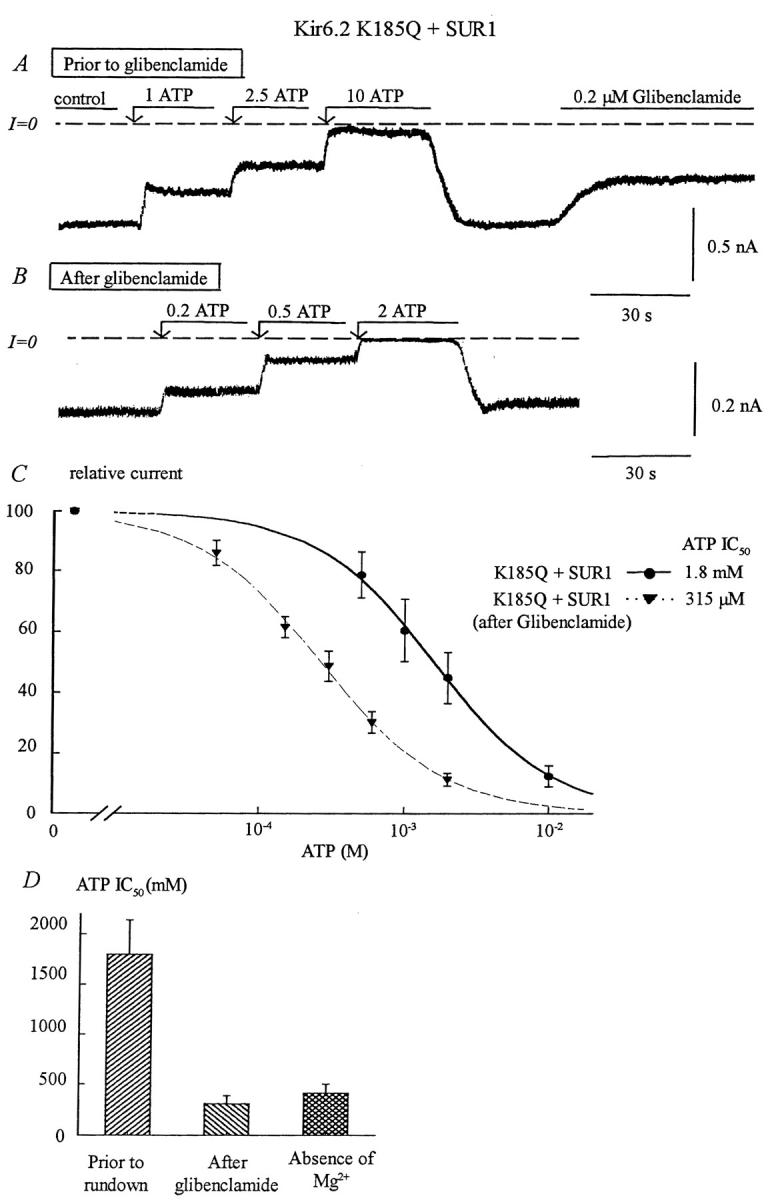
Effects of glibenclamide and rundown on ATP sensitivity of the Kir6.2 mutant K185Q + SUR1. (A) ATP sensitivity in an inside-out patch with K185Q + SUR1. Inward currents were progressively suppressed by increasing ATP concentrations, with the IC50 near 1.8 mM. Glibenclamide (0.2 μM) was then added after removal of ATP. In this series of experiments, patches with low ATP sensitivity were selected to better show the effect of glibenclamide on ATP sensitivity. (B) After glibenclamide the ATP sensitivity increased, the IC50 for inhibition by ATP shifted from 1.8 mM, before glibenclamide, to near 300 μM after glibenclamide. (C) Plot of channel ATP sensitivity for Kir6.2K185Q + SUR1 before (•) and after (▾) glibenclamide. In this case, fit of the data points yielded k = 1.8 mM and n = 1.15 before glibenclamide (n = 5) and k = 315 μM and n = 1.05 after glibenclamide (n = 5). (D) Shows average IC50 values before and after glibenclamide and compares the latter with values obtained after prolonged channel rundown. This graph indicates that glibenclamide and rundown cause similar increases in ATP sensitivity.
Similar findings were obtained with R192C + SUR1 channels. Glibenclamide had a potent inhibitory effect (>50% inhibition), which was accompanied by an eightfold increase in ATP sensitivity (Fig. 7). The dramatic reduction in R192C + SUR1 channel activity after glibenclamide-induced functional uncoupling of R192C and SUR1 indicates that the Po of R192C is low, consistent with our finding that R192C expressed without SUR1 had a Po < 0.2 in the absence of ATP (data not shown). These observations suggest that the decreased ATP sensitivity in this mutant is due to a decrease in ATP binding affinity rather than an inherently high Po.
FIGURE 7.

Effects of glibenclamide on the ATP sensitivity of the Kir6.2 mutants R50G, K185Q, R192C, and R201A mutants coexpressed with SUR1, compared with wild-type channels. As already shown, with wild-type channels, rundown evoked by the sulfonylurea glibenclamide induced a six- to eightfold increase in ATP sensitivity. The bar graph indicates that similar shifts were observed with R50G, K185Q, R192C, and K185Q/R192C mutants. In contrast, glibenclamide caused dramatic decrease in channel activity with R201A and K185Q/R201A mutants but did not induce a shift in ATP sensitivity, suggesting that the mechanism responsible for stabilization of the closed channel configuration is impaired by R201A mutation. The solid and dashed lines illustrate the wild-type channel ATP sensitivity before and after glibenclamide, respectively.
Data in Fig. 7 show that for R50G+SUR1 channels, upon patch excision and before rundown, the IC50 for ATP inhibition was close to 12.5 mM. Glibenclamide blocked R50G + SUR1 activity by 30% on average and, as with the K185Q and R192C mutations, the ATP sensitivity increased by ∼8.5-fold.
In summary, the K185Q, R192C, and R50G mutants, which are all implicated in regulating ATP binding to Kir6.2, all showed a potent increase in ATP sensitivity as Po decreased in response to glibenclamide. Thus, the K185, R192, or R50 residues do not appear to be responsible for preferential binding of ATP to a closed state of the channel. Furthermore, comparison of IC50 for channel inhibition by ATP in the absence of Mg2+ (Fig. 1) and after glibenclamide (Fig. 7) indicates that decrease in Po evoked by spontaneous rundown and sulfonylureas caused a very similar increase in ATP sensitivity.
ATP sensitivity is unaffected by decreased Po in R201A + SUR1
Fig. 8 B illustrates that glibenclamide (200 nM) had a potent inhibitory effect on R201A + SUR1. Even though the extent of inhibition, which averaged 85%, was much greater than in the wild-type Kir6.2 (40%), there was no significant shift in ATP sensitivity (Fig. 8, C and D). These results implicate R201 as a residue in wild-type Kir6.2 that binds ATP preferentially in the closed channel configuration to stabilize the channel in that state.
FIGURE 8.

Effects of glibenclamide on the ATP sensitivity of the Kir6.2 mutant R201A+SUR1. (A) ATP sensitivity in a representative inside-out patch with R201A+SUR1. Inward currents were progressively suppressed by increasing ATP concentrations, with the IC50 near 1 mM. MgADP (400 μM) was then added to the patch in the presence of 1 mM ATP to illustrate functional coupling between Kir6.2R201A and SUR1. (B) Glibenclamide (0.2 μM) had a potent inhibitory effect. (C) After glibenclamide, the ATP sensitivity remained almost the same with the IC50 for ATP inhibition near 0.9 mM. (D) Plot of channel ATP sensitivity for Kir6.2R201A + SUR1 before (•) and after (▾) glibenclamide. Fit of the data points yielded k = 1050 μM and n = 1.03 before glibenclamide (n = 3) and k = 930 μM and n = 1.04 after glibenclamide (n = 3).
Furthermore, Fig. 8 A shows that, before glibenclamide, addition of MgADP caused a 10% reactivation of the ATP-inhibited channels, indicating that functional coupling between the mutant Kir6.2 subunit and SUR1 remained intact (see John et al., 2001). As with the wild-type channel, glibenclamide blocked reactivation by MgADP in the R201A mutant (data not shown), indicating that the sulfonylurea induced functional uncoupling of R201A and SUR1 similar to the wild-type channel (John et al., 2001).
ATP sensitivity of the double mutants K185Q/R192C and K185Q/R201A + SUR1 before and after glibenclamide
Fig. 7 summarizes data obtained with the double Kir6.2 mutant K185Q/R192C coexpressed with SUR1. The ATP sensitivity of the double mutant was further reduced compared to the single mutants K185Q and R192C (IC50 of 4.7 mM vs. 1.8 mM and 1.1 mM, respectively). Glibenclamide had a potent inhibitory effect averaging 63% inhibition accompanied by a 7.5-fold increase in sensitivity to ATP. These additive effects of the single mutations K185Q and R192C are consistent with K185 and R192 being part of the same mechanism that binds ATP in a state-independent manner to induce channel closure.
The results obtained with the K185Q/R201A double mutant, on the other hand, were different. As shown in Fig. 7, channel inhibition by ATP was almost completely suppressed. Glibenclamide blocked channel activity almost completely (data not shown), but failed to increase ATP sensitivity. These observations indicate that the mechanisms of state-independent ATP binding to K185 and closed state-dependent ATP binding to R201 function synergistically to cause channel inhibition.
Effects of decreased channel Po on ATP, ADP, and AMP sensitivities
Fig. 9 shows that upon patch excision and before rundown, the IC50 for inhibition of wild-type Kir6.2 + SUR1 by ADP was close to 850 μM. After inhibition by glibenclamide, ADP sensitivity increased and the IC50 reached ∼110 μM, an eightfold increase similar to ATP. This observation suggests that the sensitization process is common to both ADP and ATP, and therefore does not require the γ-phosphate group of ATP. Data obtained with AMP (Fig. 9, D–F) implicates the α-phosphate group as a key element in the sensitization process, because upon patch excision and before rundown, the sensitivity to AMP was close to 10 mM and after glibenclamide increased to 1.75 mM. This sixfold increase was not significantly different from that observed with ATP and ADP, supporting the hypothesis that the α-phosphate group common to all three adenine nucleotides is responsible for the closed state-dependent ATP binding conferring stabilization of the closed channel.
FIGURE 9.

Effects of glibenclamide on the ADP and AMP sensitivity of wild-type Kir6.2 + SUR1. The left and right sides of the figure show that glibenclamide induces an increase in sensitivity of wild-type Kir6.2+SUR1 to both ADP and AMP, respectively. (A) Inward currents were suppressed by increasing concentrations of ADP, with the IC50 near 1 mM. Glibenclamide (200 nM) was then added after removal of ATP. (B) After glibenclamide, the ADP sensitivity increased, with the IC50 near 100 μM. (C) Plot of channel ADP sensitivity for Kir6.2+SUR1 before (•) and after (▾) glibenclamide. In this case, fit of the data points yielded k = 850 μM before glibenclamide (n = 5) and k = 105 μM after glibenclamide for (▾) (n = 5). (D) Inward currents were suppressed by increasing concentrations of AMP, with the IC50 near 10 mM. (E) After glibenclamide, the AMP sensitivity increased with the IC50 near 2 mM. (F) Plot of channel AMP sensitivity for Kir6.2+SUR1 before (○) and after (□) glibenclamide. In this case, fit of the data points yielded k = 9.5 mM before glibenclamide (n = 5) and k = 1.8 mM after glibenclamide (n = 5).
Further evidence that the α-phosphate of adenine nucleotides preferentially binds to and stabilizes the closed state of the channel was obtained with the R201A mutant coexpressed with SUR1. As with ATP, the R201A mutation caused a similar six- to eight-fold decrease in ADP sensitivity (the IC50 for inhibition by ADP being 6.7 ± 2 mM for R201A as compared to 850 μM for wild-type channels), but decrease in channel Po due to glibenclamide had negligible effects on sensitivity to ADP. After the decrease in channel activity, the IC50 for inhibition by ADP was 5.8 ± 1.4 mM. In this mutant it was not possible to estimate the sensitivity to AMP after glibenclamide, because this mutant's AMP sensitivity was almost nonexistent. These results strongly implicate R201 as a residue to which the α-phosphate binds preferentially when the channel is in a closed state.
DISCUSSION
Identification of positively charged residues on N- and C-terminus of Kir6.2 that interact with ATP
Mutation of all positively charged residues on the N- and C-terminus of Kir6.2 have enabled us to clarify the role that these two regions play in the interaction of the protein with ATP. Data summarized in Fig. 1 confirm that both the N-terminus and the C-terminus contribute to the binding of ATP. Specifically, neutralization of R50 in the N-terminus and K185 and R201 in the C-terminus, which were previously shown not to affect Po in the absence of ATP (Tucker et al., 1998; Shyng et al., 2000), caused a marked reduction in ATP sensitivity. In addition, we showed that neutralization of R192 had similar effects in reducing the sensitivity of Kir6.2 to ATP. In agreement with Shyng et al. (2000), we also found that mutation of positively charged residues outside of the K185–R201 region had no significant effect on ATP sensitivity, suggesting that adenine nucleotides interact specifically with the K185–R201 region on the C-terminus. The analogous systematic screening in the N-terminus indicated that R50 is the only positively charged residue in this domain that affects strongly ATP binding. Moreover, the most novel aspects of the present study derive from the investigation of the effects of mutations of these key residues on ADP and AMP sensitivity, in addition to ATP sensitivity, at both high and low Po. The findings provide insights into interaction between specific phosphate groups of these adenine nucleotides with specific positively charged residues in Kir6.2, as well as their dependence on the channel state.
Binding of the negatively charged phosphate groups of adenine nucleotides to positively charged residues on Kir6.2 has been proposed to account for channel inhibition (Shyng et al., 2000). In support of this hypothesis, it has been shown that the modulation of ATP sensitivity by K185 involves charge-charge interaction. Mutation of this residue to another positive charge had little effect on ATP sensitivity, while substitution with a negative charge dramatically suppressed ATP sensitivity, and neutral charges had intermediate effects (Reimann et al., 1999). We have carried out similar charge reversal experiments with R201, R50, and R192 to test whether these residues exhibited similar characteristics. Our data (Table 1) indicate that, similar to K185, both R201 and R192 control ATP binding via electrostatic interaction with the negative phosphate groups. A word of caution should be given, however, regarding R192. Using multiple sequence alignments to deduce a secondary structure of Kir6.2 COOH tail, Cukras et al., (2002) have recently suggested that R192 was not part of the ATP binding domain, but was positioned instead near R176 to interact with PIP2. If this prediction was to be correct, then R192 would not interact with the phosphate groups and the mutations would only have indirect effects. The results of R50 mutations are more ambiguous. Although mutation of R50 to another positive residue (R50K) only moderately affected ATP sensitivity, mutation to a negative residue (R50E), which should have disrupted a charge-charge interaction, had little effect on ATP sensitivity. In contrast, the neutral residue (R50C) caused the largest reduction on ATP sensitivity, suggesting that steric factors may play a role for the interaction of R50 with adenine nucleotides. Further studies will therefore be required to address this point and assess the role of a negative charge at this position, for instance a negative residue may mimic a bound phosphate group. For the present discussion we will treat R50 like the other positive residues that control ATP binding, keeping in mind that R50 may have additional effects.
Interaction of specific adenine nucleotide phosphate groups with positively charged residues in Kir6.2
When Kir6.2 is expressed without SUR1, ATP is three times more effective at suppressing channel activity than ADP, with IC50s of ∼100 μM and ∼300 μM, respectively (John et al., 1998). In contrast, the inhibitory effect of AMP is at least 100-fold less than ATP (IC50 close to 10 mM) (Tucker et al., 1998) and adenosine has almost no inhibitory effect, even though its binding to Kir6.2 is required for adenine nucleotide specificity (Reimann et al., 1999). These observations suggest that channel inhibition by adenine nucleotides involves primarily binding of the α- and β-phosphate groups of adenine nucleotides. To establish which positively charged residues interacted specifically with the β-phosphate group, we looked for mutations that affected ATP and ADP sensitivity without changing AMP sensitivity. To find which residues interacted solely with the α-phosphate group, we identified mutations that affected the sensitivity to all three adenine nucleotides. Our data show that R50G and K185Q mutations had dramatic effects on ATP and ADP sensitivity, with little effect on AMP sensitivity, implicating these two residues in charge-charge interaction with the β-phosphate group of adenine nucleotides. In contrast, the R192C and R201A mutations affected the sensitivity to all three adenine nucleotides to similar extent, implicating charge-charge interaction with the α-phosphate group. Based on these data, however, we cannot rule out that R192 and R201 bind to the adenosine moiety rather than (or in combination with) the α-phosphate group. However, this seems unlikely, because if these residues interact with the adenosine moiety alone rather than the phosphate group, then a loss of adenine nucleotide specificity would be expected.
Upon coexpressing SUR1 with Kir6.2, the ratio between the IC50 for inhibition by ADP and ATP increases, with ADP becoming 8–10 times less efficient than ATP at closing the channel, compared to a threefold difference in the absence of SUR1. This increased ratio has been attributed to the stimulatory interaction of MgADP with SUR1. However, an increased ratio is also observed in the absence of Mg2+ or after exposure to glibenclamide to functionally uncouple SUR1 from Kir6.2. Furthermore, coexpression of SUR1 with Kir6.2 had little effect on the potency of inhibition by AMP relative to ATP, with AMP remaining 100- to 150-fold less potent. Thus, there may be a residual ADP-dependent interaction between SUR1 and Kir6.2 even in the absence of Mg2+ or after functional uncoupling by glibenclamide. We also considered whether the increased ATP sensitivity of Kir6.2 + SUR as compared to Kir6.2 alone could be due to PIP2. However, the available data whereby PIP2 decreases channel sensitivity to adenine nucleotides and Kir6.2 + SUR1 is more sensitive to the phospholipid than Kir6.2 alone (Ribalet et al., 2000) does not support this hypothesis.
State-dependent block of KATP channels by adenine nucleotides
To account for the increased ATP binding affinity associated with decreased Po, it has been suggested that channel inhibition by ATP involves preferential binding of ATP to a closed state (Trapp et al., 1998; Fan and Makielski, 1999; Enkvetchakul et al., 2000). However, based on the finding that ATP shortens channel open time as well as prolonging closed time, it has also been proposed that channel closure by ATP may involve ATP binding to the open channel configuration with subsequent destabilization of this state (Trapp et al., 1998; Fan and Makielski, 1999; Li et al., 2002). To investigate whether ATP binding to the positively charged residues R50, K185, R192, and R201 showed state-dependent features, we examined the relationship between Po and adenine nucleotide sensitivity in these mutants, using either spontaneous rundown in the absence of Mg2+ or glibenclamide-evoked channel closure to decrease Po. In cases in which both methods of decreasing Po were compared (e.g., Fig. 6 D), the results were similar. However we often used glibenclamide rather than Mg2+ removal to obtain more accurate result quantitation, due to the long time required to reach steady-state rundown after Mg2+ removal. We found that with the R50G, K185Q, and R192C mutants, decreasing Po led to a substantial increase in sensitivity to ATP, comparable to that in wild-type channels. In all three cases, the ATP sensitivity increased ∼10-fold. In contrast, with the R201A mutation, the very large decrease in channel activity (80%) evoked by glibenclamide was not associated with increased ATP sensitivity. Thus R50, K185, and R192 appear to be involved in state-independent binding of ATP, whereas R201C is involved in preferential binding of ATP to a closed state.
A structural model for Kir6.2 channel regulation by ATP
Combining the data on the relative nucleotide sensitivity and the relationship between Po and ATP sensitivity in these mutants, we generated the structural scheme illustrated in Fig. 10, based on the model recently proposed by Jiang et al. (2002), for K channels. As typical of ligand-gated K channels, Kir6.2 has four identical subunits. Each subunit has two transmembrane domains and the last one, M2, forms the inner helix lining the pore. Tilting of the lower portion of this helix around G165 is thought to be responsible for channel gating. With ligand-gated channels, tilting of this lower region is directly controlled by the transverse movement of a cytosolic regulatory domain, referred to as the gating ring, which pulls the pore into the open configuration upon ligand binding. In the absence of SUR, the channel formed by Kir6.2 alone has a low Po (0.1), even in the absence of ATP and is thus in a closed relaxed state (not shown). However, when coexpressed with SUR and in the absence of ATP, the Po increases to 0.4–0.6 (Fig. 10 A). As previously suggested, the stimulatory effect of SUR is due in part to the MgADP-dependent interaction of SUR nucleotide binding folds with the Kir6.2 positive residues R176/R177 (John et al., 2001). Thus, analogous to the scheme for the Ca-gated channel where channel opening results from binding of the ligand to the regulatory domain (Jiang et al., 2002), we hypothesize that in the absence of SUR, KATP channels favor the closed state. In the presence of SUR, however, channels favor the open state due to a side pull involving interaction of SUR with positive residues (R176/R177) right at the cytoplasmic junction of the gate and of the regulatory domain. In the open channel configuration (Fig. 10 B) ATP binds to the regulatory domain. Based on our experimental results, we postulate that this interaction involves binding of adenine nucleotide β-phosphate group to a domain formed by R50 and K185. Because R50 is on the N-terminus (not shown) and K185 is on the C-terminus, it follows that the N- and C-termini may closely interact to form a pocket (see also structural model by Cukras et al., 2002) where the β-phosphate group binds, facilitating channel closure. The assignment of R50 and K185 to interaction with the β-phosphate of ATP is based on the finding that neutralization of either residue markedly decreased ATP and ADP sensitivity but had little effect on AMP sensitivity. Thus, with Kir6.2 adenine nucleotide binding to the gating ring translates into destabilization of the open state and thus channel closure (Fig. 10, B→C). Our data obtained with R210A mutation suggests that the α-phosphate group of adenine nucleotides interacts with R201. This interaction occurs preferentially when the channel is in a closed state, thereby stabilizing the closed state configuration. In our scheme, this last state, D, suggests a mechanism whereby in the open configuration, R201 is not accessible to interaction with the α-phosphate, but upon channel closure, R201 binds to this phosphate to stabilize the closed state.
FIGURE 10.

Model for interaction of the α- and β-phosphate groups of adenine nucleotides with positively charged residues on Kir6.2 N- and C-terminus. In the channel open configuration (A), the β-phosphate group interacts with both K185 on the C tail and R50 on the N-terminus (B). Binding of adenine nucleotide forces the open channel toward closure (C). Once the channel is closed, R201 interacts with the α-phosphate group to stabilize the closed channel configuration (D). The schematized structure is derived from a recent model for K channels (Jiang et al., 2002).
Acknowledgments
This work was supported by grants R37 HL60025 and SCOR in Sudden Cardiac Death P50 HL52319 from the National Institutes of Health; Laubisch and Kawata Endowments to J.N.W.; and by a grant-in-aid from the American Heart Association (Western States Affiliate) to S.A.J.
References
- Cukras, C. A., I. Jeliazkova, and C. Nichols. 2002. Structural and functional determinants of conserved lipid interaction domains of inward rectifying Kir6.2 channels. J. Gen. Physiol. 119:581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain, P., L. Li, and J. Wang. 1998. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore forming subunit. Proc. Natl. Acad. Sci. USA. 95:13953–13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul, D., G. Loussouarn, E. Makhina, S. L. Shyng, and C. G. Nichols. 2000. The kinetic and physical basis of KATP channel gating: toward a unified molecular understanding. Biophys. J. 78:2334–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, Z., and J. C. Makielski. 1999. Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K+ channel. A molecular probe for the mechanism of ATP-sensitive inhibition. J. Gen. Physiol. 114:251–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, F. L., and A. J. van der Eb.1973. Transformation of rat cells by DNA of human adenovirus 5. Virology. 52:456–467. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chalt, and R. MacKinnon. 2002. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- John, S. A., J. R. Monck, J. N. Weiss, and B. Ribalet. 1998. The sulfonylurea receptor SUR1 regulates ATP-sensitive mouse Kir6.2 K channels linked to the green fluorescent protein in human embryonic kidney cells (HEK 293). J. Physiol. 510 (Pt 2):333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John, S. A., J. N. Weiss, and B. Ribalet. 2001. Regulation of cloned ATP-sensitive K channels by adenine nucleotides and sulfonylureas: Interactions between SUR1 and positively charged domains on Kir6.2. J. Gen. Physiol. 118:391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster, J. C., Q. Sha, S. L. Shyng, and C. G. Nichols. 1999. ATP inhibition of KATP channels: Control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J. Physiol. (Lond.). 515:19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L., X. Geng, and P. Drain. 2002. Open state destabilization by ATP occupancy is mechanism speeding burst exit underlying KATP channel inhibition by ATP. J. Gen. Physiol. 119:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribalet, B., S. A. John, and J. N. Weiss. 2000. Regulation of cloned ATP-sensitive K channels by phosphorylation, MgADP, and phosphatidylinositol bisphosphate (PIP2): A study of channel rundown and reactivation. J. Gen. Physiol. 116:391–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann, F., T. J. Ryder, S. J. Tucker, and F. M. Ashcroft. 1999. The role of lysine 185 in the Kir6.2 subunit of the ATP-sensitive channel in channel inhibition by ATP. J. Physiol. 520 (Pt 3):661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S. L., C. A. Cukras, J. Hardwood, and C. G. Nichols. 2000. Structural determinant of PIP2 regulation of inward rectifier KATP channels. J. Gen. Physiol. 116:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp, S., P. Proks, S. J. Tucker, and F. M. Ashcroft. 1998. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J. Gen. Physiol. 112:333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, S. J., F. M. Gribble, C. Zhao, S. Trapp, and F. M. Ashcroft. 1997. Truncation of Kir6.2 produces ATP-sensitive K channels in the absence of the sulfonylurea receptor. Nature. 387:179–182. [DOI] [PubMed] [Google Scholar]
- Tucker, S. J., F. M. Gribble, P. Proks, S. Trapp, T. J. Ryder, H. Haug, F. Reimann, and F. M. Ashcroft. 1998. Molecular determinant of KATP channel inhibition by ATP. EMBO J. 17:3290–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]