

Abstract
Three pulse echo peak shift and transient grating (TG) measurements on the plant light-harvesting complexes LHCII and CP29 are reported. The LHCII complex is by far the most abundant light-harvesting complex in higher plants and fulfills several important physiological functions such as light-harvesting and photoprotection. Our study is focused on the light-harvesting function of LHCII and the very similar CP29 complex and reveals hitherto unresolved excitation energy transfer processes. All measurements were performed at room temperature using detergent isolated complexes from spinach leaves. Both complexes were excited in their Chl b band at 650 nm and in the blue shoulder of the Chl a band at 670 nm. Exponential fits to the TG and three pulse echo peak shift decay curves were used to estimate the timescales of the observed energy transfer processes. At 650 nm, the TG decay can be described with time constants of 130 fs and 2.2 ps for CP29, and 300 fs and 2.8 ps for LHCII. At 670 nm, the TG shows decay components of 230 fs and 6 ps for LHCII, and 300 fs and 5 ps for CP29. These time constants correspond to well-known energy transfer processes, from Chl b to Chl a for the 650 nm TG and from blue (670 nm) Chl a to red (680 nm) Chl a for the 670 nm TG. The peak shift decay times are entirely different. At 650 nm we find times of 150 fs and 0.5–1 ps for LHCII, and 360 fs and 3 ps for CP29, which we can associate mainly with Chl b ↔ Chl b energy transfer. At 670 nm we find times of 140 fs and 3 ps for LHCII, and 3 ps for CP29, which we can associate with fast (only in LHCII) and slow transfer between relatively blue Chls a or Chl a states. From the occurrence of both fast Chl b ↔ Chl b and fast Chl b → Chl a transfer in CP29, we conclude that at least two mixed binding sites are present in this complex. A detailed comparison of our observed rates with exciton calculations on both CP29 and LHCII provides us with more insight in the location of these mixed sites. Most importantly, for CP29, we find that a Chl b pair must be present in some, but not all, complexes, on sites A3 and B3. For LHCII, the observed rates can best be understood if the same pair, A3 and B3, is involved in both fast Chl b ↔ Chl b and fast Chl a ↔ Chl a transfer. Hence, it is likely that mixed sites also occur in the native LHCII complex. Such flexibility in chlorophyll binding would agree with the general flexibility in aggregation form and xanthophyll binding of the LHCII complex and could be of use for optimizing the role of LHCII under specific circumstances, for example under high-light conditions. Our study is the first to provide spectroscopic evidence for mixed binding sites, as well as the first to show their existence in native complexes.
INTRODUCTION
Green plants contain two types of photosynthetic reaction centers, PSI and PSII, which work in series to split water into hydrogen ions and oxygen molecules and to reduce NADP+ to NADPH (van Grondelle et al., 1994). To collect energy from sunlight photons, these photosynthetic reaction centers are aided by light-harvesting complexes. Specifically, PSII is surrounded by the core antennae CP47 and CP43 and the peripheral antennae LHCII, CP29, CP24, and CP26 (van Amerongen and van Grondelle, 2001; Boekema et al., 1999a, 1999b, 2000). LHCII is by far the most abundant of these antennae at a density of about four LHCII trimers per PSII reaction center (van Amerongen and van Grondelle, 2001). Over the last years, LHCII has been the subject of intense study for several reasons. In the first place, it is of major physiological importance and a total of ≥50% of all chlorophyll on earth is bound to this complex. It is capable of fulfilling several roles, such as light-harvesting for the PSI reaction center, light-harvesting for the PSII reaction center, and protection against excess light damage through nonphotochemical quenching (Ruban et al., 1996; Barzda et al., 2000; van Amerongen and van Grondelle, 2001). The latter two functions are also fulfilled by the minor antennae CP29 and CP26 (Ruban et al., 1996; Bassi et al., 1997). To fulfill these roles LHCII is a relatively flexible complex, which can change at least its xanthophyll content, aggregation state, and phosphorylation state as required. Last but not least, for a long time LHCII was the only plant antenna for which a structural model at reasonably high resolution was available. In its natural form in the membrane, the LHCII antenna appears as a trimeric complex. It is known from biochemical analysis (Kühlbrandt et al., 1994; Croce et al., 1999) that an isolated LHCII trimer contains 21–24 chlorophylls a (Chls a), 15–18 Chls b, and three types of xanthophylls: six luteins, three neoxanthins, and about one violaxanthin.
In 1994, Kühlbrandt et al. (1994) resolved the structure of LHCII to 3.4 Å with the use of electron diffraction on two-dimensional crystals of trimers. In each monomer they found three transmembrane α-helices forming a scaffold for the pigments. For the 12 chlorophylls that are visible, the Chl a/b identity could not be determined. Kühlbrandt therefore used the efficiency of Chl-to-xanthophyll triplet transfer as an argument for a tentative assignment. The seven Chls in the center of the structure in van der Waals contact with the two resolved xanthophylls were taken to be Chls a, the other five were considered as Chls b. The numbering of the Chls in this assignment is still universally used to refer to the corresponding binding sites even though some identities are probably different (see below).
For the minor antenna CP29, no crystal structure is available. From biochemical studies it is known to bind two Chls b and six Chls a (Pascal et al., 1999), and it always appears in monomeric form. The CP29 antenna has a large protein sequence homology (Jansson, 1994; Green and Durnford, 1996; Croce et al., 1996) to LHCII with identical or similar binding sites at eight positions. The identity of the Chls at these sites was originally derived directly from the LHCII assignment of Kühlbrandt (Kühlbrandt et al., 1994). In the last few years, several studies on complexes with mutated binding sites (Remelli et al., 1999; Rogl and Kühlbrandt, 1999; Yang et al., 1999; Bassi et al., 1999; Simonetto et al., 1999) have shed new light on the identity of the Chls for both antennae. The determination of the biochemical and spectral properties of the mutated complexes has led to alternative Chl a/b assignments for LHCII (Remelli et al., 1999; Rogl and Kühlbrandt, 1999; Yang et al., 1999) and to an independent assignment for CP29 (Bassi et al., 1999; Simonetto et al., 1999) (see also van Amerongen and van Grondelle (2001) and references therein).
The structures with the chlorophyll identities as proposed by Bassi and co-workers (Remelli et al., 1999; Bassi et al., 1999; Simonetto et al., 1999) are shown in Figs. 1, a (LHCII) and b (CP29). For both complexes, the same four Chl a sites were found by all authors (Remelli et al., 1999; Rogl and Kühlbrandt, 1999; Yang et al., 1999; Bassi et al., 1999; Simonetto et al., 1999) to be conserved with respect to the original Kühlbrandt assignment, namely the central Chls a at the top: A1, A2, A4, and A5. For CP29, Bassi and co-workers proposed that the other four binding sites, A3, B3, B5, and B6, can bind either Chl a or Chl b, or shortly, that they are mixed sites. For LHCII, the same authors found three mixed sites (Remelli et al., 1999; Bassi, Università de Verona, personal communication, 2001), the sites A3 and B3, like in CP29, and the site A6. The last five sites in LHCII can bind only one type of Chl: B1 binds a Chl a; and B2, B5, B6, and A7 bind Chls b. For CP29, to get the correct Chl a:Chl b ratio, half of the mixed sites will have to bind Chl a, the other half Chl b. For LHCII, the ratio should be two Chls a to one Chl b. Whether this ratio is the same in each single antenna complex or is just an ensemble-averaged property is not known. Kühlbrandt and co-workers, who carried out similar mutagenesis studies on LHCII (Rogl and Kühlbrandt, 1999; Yang et al., 1999; Rogl et al., 2002) propose a different assignment without mixed sites. The mutation studies all involve the in vitro reconstitution of the complexes from a protein and pigment mixture. Arguing that mixed sites are not likely to exist in native complexes (Rogl and Kühlbrandt, 1999; Rogl et al., 2002) Kühlbrand c. s. proposed an assignment for LHCII with Chls a at sites A1, A2, A3, A4, A5, and B3, and Chls b at sites B5 and B6. They did not determine the chlorophyll identities at the sites A6, A7, B1, and B2.
FIGURE 1.


Structural model for (a) LHCII and (b) CP29. Chlorophyll identities are drawn as proposed by Bassi and co-workers (see text), with chlorophylls b in light gray, mixed sites in medium gray, and chlorophylls a in dark gray.
The results from mutation studies described above are mostly in agreement with many studies (Du et al., 1994; Bittner et al., 1994, 1995; Visser et al., 1996; Connelly et al., 1997; Trinkunas et al., 1997; Kleima et al., 1997; Gradinaru et al., 1998a, 1998b, 2000; Hillmann et al., 2001) of the energy transfer dynamics in both complexes, which also have led to questioning of the original Kühlbrandt assignment (Kühlbrandt et al., 1994). In most energy transfer studies, the spectral evolution in time of the Chl Qy band was observed with the pump-probe technique. The Qy transition lies between 640 nm and 660 nm for the Chls b, and between 660 nm and 680 nm for the Chls a. In Fig. 2, the room temperature absorption spectra of both complexes are shown. Although these spectra appear rather featureless, spectroscopy at low temperature (Pascal et al., 1999; Hemelrijk et al., 1992; Nussberger et al., 1994; van Amerongen et al., 1994) has shown that they consist of many different bands. In the Chl b region of CP29, two individual peaks at 638 nm and 649 nm show up in the low-temperature absorption and linear-dichroism (LD) spectra (Pascal et al., 1999). In the Chl b region of LHCII, several distinct features were observed between 648 nm and 652 nm. Unfortunately, the width of the corresponding bands remains larger than their spectral separation at all temperatures (Hemelrijk et al., 1992; Nussberger et al., 1994). The main band of Chl a absorption peaks at 676 nm in both complexes and is thought to correspond to about four to five individual Chls a (Hemelrijk et al., 1992). At low temperature, a lowest state is found at 680 nm (Reddy et al., 1994; Pieper et al., 1999) that corresponds to approximately one pigment. Also, the Chl a band of LHCII has a pronounced shoulder at 670 nm, with an oscillator strength corresponding to two or three pigments. For CP29, no prominent shoulder is observed. Some Chl a absorbing quite far to the blue, at around 663 nm, is present in LHCII (Rogl et al., 2002; Hemelrijk et al., 1992; Nussberger et al., 1994).
FIGURE 2.

Absorption spectra (77 K) of LHCII (solid) and CP29 (dotted) together with spectra of laser pulses at 650 nm (dash-dot) and 670 nm (dashed).
The wide range in excited-state energies introduces a clear directionality in the energy flow in both complexes. The above mentioned pump-probe studies (Bittner et al., 1994, 1995; Visser et al., 1996; Connelly et al., 1997; Trinkunas et al., 1997; Kleima et al., 1997; Gradinaru et al., 1998a, b), two-pulse echo study (Hillmann et al., 2001), and fluorescence study (Du et al., 1994) have identified several downhill energy transfer processes. These can be divided into two categories: Chl b ↔ Chl a transfer and blue Chl a ↔ red Chl a transfer. Additionally, some energy transfer between (almost) isoenergetic pigments, i.e., from Chl b to Chl b (Agarwal et al., 2000), and between main band Chls a (van Amerongen et al., 1994; Savikhin et al., 1994; Du et al., 1994; Gradinaru et al., 1998b) is thought to take place.
Upon excitation of the Chl b pigments, energy transfer to Chl a takes place at a variety of rates that are virtually identical at room temperature (Du et al., 1994; Bittner et al., 1994, 1995; Connelly et al., 1997; Trinkunas et al., 1997), and at cryogenic temperatures (Visser et al., 1996; Kleima et al., 1997; Gradinaru et al., 1998a, 2000). In both LHCII and CP29, fast energy transfer to Chl a is seen to occur with a time constant of ∼200 fs in both complexes. For LHCII, an additional phase of ∼600 fs is also observed (Bittner et al., 1995; Visser et al., 1996; Connelly et al., 1997; Kleima et al., 1997). Much slower energy transfer from Chl b to Chl a, in ∼2 ps for CP29 (Gradinaru et al., 1998a) and in 4–9 ps for LHCII (Visser et al., 1996; Trinkunas et al., 1997; Kleima et al., 1997) also takes place. In CP29, the slow Chl b is the red one absorbing at 649 nm and the fast Chl b is the blue one (Gradinaru et al., 1998a). The red (slow) Chl b transfers to a relatively blue Chl a; the blue (fast) Chl b transfers to main-band Chl a absorbing above 675 nm. For LHCII, the amplitudes of the three lifetimes are distributed more or less as follows: 200 fs (40%), 600 fs (40%), and 4–9 ps (20%). A clear identification of the corresponding pigments is not possible as the Chl b pigments cannot be excited selectively. From simultaneous modeling of steady-state spectroscopy and energy transfer (Visser et al., 1996; Kleima et al., 1997; Iseri et al., 2000), and from comparison of monomers and trimers (Kleima et al., 1997), some information about the involved Chls a was obtained: in the trimer, both fast and slow Chl b → Chl a energy transfer excites mostly main-band Chl a, as well as one or two blue Chls a.
Most pump-probe studies have shown that further equilibration among Chls a occurs after Chl b → Chl a transfer. Studies of relaxation processes upon excitation at several wavelengths in the Chl a band of LHCII were carried out at a temperature of 77 K by Visser et al. (1996), and by Gradinaru et al. (1998b). Gradinaru et al. identified five different transfer processes. The blue-most Chl a pigment, which absorbs at around 663 nm, transfers its energy to main-band Chls a at the rather slow rate of ∼5 ps. The pigments that absorb at around 670 nm transfer to the 676 nm pigments with two very different time constants of 300 fs and 12–20 ps. In room temperature experiments, all based upon Chl b excitation, slow transfer from 670 nm to 676 nm was also observed and found to proceed twice as fast as at low temperature, in ∼6 ps (Bittner et al., 1994, 1995; Connelly et al., 1997). Connelly et al. (1997) also observed a 180 fs relaxation component with some amplitude in the Chl a region. Possibly, all Chl a equilibration is twice faster than at low temperature. Further equilibration of the main-band pigments toward the redmost transition was observed at 77 K by Visser et al. (1996), and Gradinaru et al. (1998b), on two timescales, in ∼450 fs and in 7.5 ps. In CP29, for transfer from 670 nm Chls a to 676 nm Chls a, similar transfer times were seen at 77 K as in LHCII (Gradinaru et al., 1998a). Other Chl a → Chl a transfer processes were not observed. Calculations of Chl a → Chl a transfer in CP29 by Cinque et al. (2000) suggest that subpicosecond equilibration takes place between all of the central Chls a at A1, A2, A4, and A5. For an overview of the processes described above, see the rightmost column of Table 5.
TABLE 5.
Overview of energy transfer times
| This work | Others | |
|---|---|---|
| LHCII | ||
| Chl b ↔ Chl b | 150 fs, 0.5–1 ps | 300 fs, 800 fs* |
| Chl b → Chl a | 300 fs, 5 ps | 180 fs, 600 fs, 4–9 ps†,‡ |
| Chl a ↔ Chl a (670 nm) | 140 fs, 3.6 ps | 350 fs, 3 ps, 6 ps* |
| Chl a → Chl a (670 → 680 nm) | 230 fs, 6 ps | 300 fs, 12–20 ps* 2–5 ps (663 nm exc.) |
| CP29 | ||
| Chl b ↔ Chl b | 360 fs (3 ps ?) | |
| Chl b → Chl a | 130 fs, 2.2 ps | 150–220 fs, 2.2 ps, 10 ps‡ |
| Chl a ↔ Chl a (670 nm) | 3.3 ps | |
| Chl a → Chl a (670 → 680 nm) | 300 fs, 5 ps | 280 fs, 10–13 ps§ |
Energy transfer time constants from this work and from literature.
In the above discussion, all transitions are described as if belonging to a certain pigment. However, calculations (van Amerongen and van Grondelle, 2001; Iseri et al., 2000; Iseri and Gülen, 2001) show that a considerable amount of exciton delocalization occurs in both complexes. This is most strongly the case for the Chls a, for which van Amerongen and van Grondelle (2001) calculate that, in LHCII, excitonic coupling leads to a 50% increase of the width of the total Chl a band. They also find that the Chl a and Chl b band exchange oscillator strength, with the Chl a band increasing 8% in intensity at the expense of the Chl b band. Within the Chl b band, the calculated excitonic coupling can be either strong or weak depending on the specific Chl a/b assignment (van Amerongen and van Grondelle, 2001; Iseri and Gülen, 2001). Altogether, for both complexes, the average number of pigments contributing to a certain transition is usually three or four according to Iseri et al. (2000), and Iseri and Gülen (2001). Nevertheless, in the case of two pigments absorbing at rather different energies, the corresponding exciton states will be mostly localized on one of the pigments.
The situation will be different in the case that transfer between isoenergetic pigments occurs. This type of transfer has not yet been studied very extensively in these antennae for the following reasons. Isoenergetic transfer cannot be observed in isotropic pump-probe experiments, because these rely on the occurrence of spectral changes. A second disadvantage of pump-probe is the need for spectrally narrow pulses that have a long time duration. This complicates the search for fast processes that would correspond to strong coupling. Slow isoenergetic energy transfer between main-band Chls a was studied with the use of polarized pump-probe (Kwa et al., 1992; Savikhin et al., 1994). Anisotropy decay times of ∼5 ps were observed at room temperature for excitation wavelengths of 675 nm and above. Unfortunately, the time resolution of these studies was not sufficient to probe subpicosecond transfer steps. A recent attempt at specific identification of isoenergetic energy transfer in LHCII was done by Agarwal et al. (2000), with the use of the three pulse echo peak shift (3PEPS) technique. They observe a very rapid decay of the peak shift when exciting at 650 nm, which they can only explain if Chl b ↔ Chl b transfer takes place within a few hundred fs. After excitation at 670 nm, they find several peak shift decay times, which they interpret as downhill energy transfer between Chls a. For CP29, no comparable study has yet been done.
In this paper, we describe a 3PEPS study of both LHCII and CP29. Both complexes were excited in the Chl b region at 650 nm and in the Chl a region at 670 nm. By comparing the two complexes, we expect to identify specific transfer processes, in particular between isoenergetic pigments or states. At 650 nm, we hope to observe Chl b ↔ Chl b energy transfer in the 3PEPS decay. The 670 nm peak shift should show whether transfer can also occur between blue Chl a pigments. In addition to 3PEPS, we have used the transient grating (TG) method, which monitors downhill energy transfer. This should enable a good comparison with previous results obtained with the pump-probe technique. On the basis of our results, we also hope to answer the question whether mixed binding sites occur in the native complexes.
The 3PEPS and TG techniques will be described in the Methods section. In the Results section, the data are presented and the main timescales are determined using exponential fits. These timescales form the basis of a qualitative interpretation of the data in the Discussion section, with the focus on Chl a and Chl b energy transfer properties. In a subsequent paper, detailed simulations of the data will be presented.
METHODS
The three pulse echo peak shift technique
The 3PEPS method was first used to study solvation processes in liquids by monitoring the spectral diffusion of a dye solute (see e.g., Joo et al., 1995, 1996a; de Boeij et al., 1995). Later, 3PEPS was applied to several photosynthetic systems with the goal of observing both pigment-protein coupling and interpigment energy transfer (Agarwal et al., 2000; Joo et al., 1996b; Jimenez et al., 1997; Yu et al., 1997; Groot et al., 1998; Salverda et al., 2000; Agarwal et al., 2001). An overview of several of these studies can be found in Salverda and van Grondelle (2001). The 3PEPS technique has been discussed in detail in many previous papers (see e.g., Salverda et al., 2000) and the main principles will be described here.
The configuration of a 3PEPS experiment is shown in Fig. 3. We will describe the principle of this experiment using the impulsive limit. In the 3PEPS experiment, a sequence of three laser pulses, each with a slightly different direction of propagation k1, k2, or k3, is focused onto the sample. A fourth pulse, the photon echo, is emitted in the direction k3 + k2 − k1 at a time τ after the third pulse, with τ the delay between the first two pulses. The time-integrated intensity of the echo is measured as a function of delay τ, with the delay T between the second and third pulse, also known as population time, as a parameter. The distance of the maximum of this signal (further referred to as the echo) from τ = 0, the peak shift, is then determined as a function of T. The decay of the peak shift with T reflects all processes that lead to frequency changes (described as Δω(t)) of those pigments within the spectral window of the pulses that are still excited. This can be explained in the following way.
FIGURE 3.

Configuration of a 3PEPS experiment. The two echoes with wave vectors k3 + k2 − k1 and k3 − k2 + k1 are indicated as B and B′ and are both of equal intensity and have a mirror-symmetric time-dependence on delay time τ between the first and second pulse. The sample must be rotated or flowed as indicated by the curved arrow.
Pulses 1 and 2 generate a frequency-dependent modulation of the excited-state population within the laser bandwidth, a so-called frequency grating, which has a modulation period proportional to 1/τ. During the time T between pulses 2 and 3, the modulation depth and width of the grating produced by the combined effect of the first two pulses will be affected by changes in transition frequencies of the pigments within the laser bandwidth, rather like in a transient hole-burning experiment. The amplitude of the grating also decays, which is due to loss of population, for instance due to energy transfer to pigments absorbing outside the laser window, but this does not affect the peak shift. Because intraband frequency changes efface a fine grating more easily, an increasingly coarse grating is needed to survive when longer T values are used. As a more widely spaced grating corresponds to a smaller delay between pulses 1 and 2, this means that the echo intensity maximum gets closer and closer to zero τ. Thus, the decay of the peak shift as a function of T reflects the loss of correlation, averaged over all pigments, between the excitation frequency at the time of pulse 2 and the frequency at the time of pulse 3. It is intuitively clear that energy transfer among more or less identical pigments of an inhomogeneous distribution leads to an increased rate of disappearance of the frequency grating and thus to a faster peak shift decay. Note that for the case of nonzero pulsewidths, the overlap of the pulses in time, leading to different time ordering, must explicitly be accounted for in the modeling of these experiments. The fluctuating frequency ωi(t) of each pigment i can be described as:
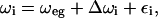 |
(1) |
with ωeg the ensemble average of the transition frequency, Δωi(t) the fluctuation of the frequency of pigment i, and εi its static deviation from average, i.e., the inhomogeneous broadening. The average frequency correlation of the ensemble is then described by the two-point frequency correlation function
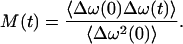 |
(2) |
From M(t) we can calculate the functions R(t, τ, T), which describe the third-order response of the sample medium to an external electric field, such as a light pulse. The third-order polarization P(3) can be calculated by integrating the product of the pulse fields and the response functions over time. All this is described in detail by Mukamel (1995).
The transient grating technique is a special case of the three-pulse echo, with the first two pulses set at τ = 0 and the signal intensity scanned as a function of the population time T. The first two pulses now create a spatial population grating in the sample due to interference between the beams, which intersect at a small angle. The decay of this grating reflects loss of excited-state population from within the laser spectrum. This spatial grating is of course also created in the 3PEPS experiment, but in that experiment the amplitude decay, which is caused by population transfer, is not considered (as mentioned above). Also at nonzero delay between pulses 1 and 2, the phase of this grating will be different for each frequency, leading to canceling contributions.
Experiment
A standard 3PEPS setup was used as described earlier (Joo et al., 1996b). Short (50 fs) pulses at 800 nm were obtained from a Ti: Sapphire oscillator-regenerative amplifier combination (Coherent Mira-SEED and RegA-9050). These were used to pump an infrared Optical Parametric Amplifier (OPA-9850, Coherent) to generate light of the desired wavelength. The output of the OPA was tuned to 1.30 μm or 1.34 μm and frequency doubled to 650 nm or 670 nm for excitation of Chl b or Chl a, respectively. After removal of the remaining infrared light with a cutoff filter, the resulting pulses had a bandwidth of 17–18 nm, a duration of 35–40 fs as measured from their autocorrelation, and an energy of ∼12 nJ at the sample. This energy is divided equally into three parallel beams, which are focused onto a single spot in the sample. All three beams have vertical polarization and can be delayed individually. Two of the beams, referred to as pulse 1 and 2 with wave vectors k1 and k2, travel via retro reflectors on motorized translation stages. The third beam with wave vector k3 travels via a fixed retro reflector and is always last in time. The first two pulses can change role and both echo signals, with wave vectors k3 + k2 − k1 and k3 − k2 + k1 are detected; see the scheme of the beam geometry in Fig. 3. The photon echo (or transient grating) signals are detected by photodiodes. Lock-in amplifiers, connected to a chopper in one of the beams, are used to reduce random noise. The sample cell has a path length of only 200 μm to avoid reabsorption of the generated signal after the focus.
The three pulse echo (3PE) signals are measured at ∼80 values of the population time. These values are distributed unevenly along the full time range, with the emphasis on the early times in a quasi-logarithmic manner. At 670 nm, both for LHCII and for CP29, the echoes are measured up to a population time of 100 ps. At 650 nm, they can only be measured up to T = 10 ps, by which time there are no excitations left on the Chls b. Peak shift values were determined by fitting Gaussians to the two 3PE signals and taking half the time distance between the maxima of these fits. With this fitting method, a small systematic error is introduced in our peak shift curves. At values of the population time below ∼70 fs, all three pulses overlap with each other and this results in the generation of a coherent coupling signal. This coherent artifact peaks at τ = 0 and is detected along with the 3PE, which results in a larger apparent width of the 3PE at the negative side. Fitting Gaussians to these 3PE signals results in a peak shift value that is systematically too small (Larsen et al., 2001). We have checked the effect of this error by comparing with partial Gaussian fits at early delay times. We found that for both complexes and wavelengths, the initial peak shift we use is ∼4 fs too low. Within the pulse overlap region this error rapidly disappears. In the exponential fits described below, this error only affects the amplitude of the fastest component, which should be a few fs larger. This does not affect our main conclusions, which are concerned with the slower components. As the peak shift curves we constructed with partial Gaussians contained more noise, we have based the paper on the curves obtained with full Gaussians.
Transient grating signals are not measured separately, but are constructed from the echo signals by taking the signal intensity at τ = 0 as a function of the population time. This limits the number of time points of each trace to ∼80, just as for the peak shift curves, which is less than usual for TG traces. However, the TG reconstruction does ensure that both 3PEPS and TG are measured under exactly the same sample and setup conditions.
All signals are measured with all three pulses at parallel polarization. This introduces a bias, as the third pulse will now preferentially interact with pigments that were excited by pulses 1 and 2, and with pigments oriented (nearly) parallel to those. This bias is present in almost all previous 3PEPS experiments on photosynthetic systems, and we will not consider it in detail. The main effects are, most likely (see Salverda et al., 2000), a slowing down of the peak shift decay and a speeding up of the transient grating decay, compared to what would result from a magic angle experiment.
The samples are prepared as described previously (Pascal et al., 1999; Peterman et al., 1995). Concentrated solutions of detergent isolated LHCII trimers and CP29 monomers of spinach are thawed and diluted in a buffer with 20 mM HEPES, 0.03% DM (n-dodecyl β-d-maltoside), pH 7.5. The optical density of the used solutions along the cell path depends on sample and wavelength and varied between 0.1 and 0.2, always low enough to avoid reabsorption of the generated signal. To prevent photodamage to the sample, the laser repetition rate is set at 125 kHz and the sample is circulated through a flow cell. The measuring time never exceeded 7 h and the absorption spectrum proved to be the same before and after measuring.
RESULTS
In this section, we will describe the data and present the results of multiexponential fits. The two types of Chl will be treated separately, i.e., we will discuss first the TG and 3PEPS results obtained at 650 nm, then those obtained at 670 nm. LHCII and CP29 data will be considered together.
Excitation at 650 nm
Transient grating
Transient grating curves were constructed from the 3PE signals measured at 650 nm. The resulting traces are shown in Fig. 4 for both LHCII and CP29. The large graph shows the decay up to a population time of 2 ps. The majority of points are concentrated in this interval, so that the fast (sub-ps) dynamics can be determined. The full time range of 0 ps–10 ps is shown in the inset.
FIGURE 4.

Transient grating curves measured at 650 nm for LHCII (solid) and CP29 (dashed). The curves were constructed from the 3PE signals. Both first 2 ps (large graph) and full range up to 10 ps (inset) are shown.
The traces for both complexes share the same general features. In the first few hundred fs, a large decay of 50–70% of the total signal takes place. This fast phase is followed by a slower phase that takes several picoseconds. Within 10 ps, the LHCII transient grating decays to less than 10% of its original value, whereas the final level of the CP29 signal is 25–30%. This difference of ∼15% appears after ∼100 fs and does not change much after that. Despite this difference, the major fact to note about both TG signals is the rapid loss of more than half the amplitude. At 650 nm, Chl a shows considerable absorption at room temperature. However, these directly excited Chls a should also be observable at longer times, so we conclude that for both complexes, mainly Chls b are excited. Nevertheless, the appreciable final level of the CP29 TG suggests that at least in this complex, some Chl a is being monitored in addition to Chl b. After less than 200 fs, the 3PEPS signal, which will be discussed below, is monitoring only a small subset of the initially populated pigments. This subset of (mainly) Chls b is not too close to any Chls a, otherwise they would already have transferred their excitation, but they may be close to other Chls b.
To determine the timescales involved, we have fitted a biexponential decay to the transient grating curves. The parameters describing these fits are listed in Table 1. The faster of the two rates will be contaminated somewhat by coherent coupling artifacts, but they describe mostly population decay. Because the TG intensity scales quadratically with the excited-state population, each decay rate corresponds to an energy transfer rate that is twice as slow. For CP29, this fast energy transfer process takes 2 × 65 fs = 130 fs; for LHCII it is somewhat slower at 2 × 147 fs = 300 fs. LHCII is also slower on the longer timescale, with transfer times of 2 ps for CP29 and 5 ps for LHCII. Both the slow and the fast rates are similar to the well-known Chl b → Chl a transfer rates observed in previous experiments (Du et al., 1994; Bittner et al., 1994, 1995; Visser et al., 1996; Connelly et al., 1997; Trinkunas et al., 1997; Kleima et al., 1997; Gradinaru et al., 1998a, b). Note that, as mentioned in the Introduction, these rates are the same at 77 K and at room temperature. Apparently, the change in shape of the spectra of individual pigments (Zucchelli et al., 1996) does not affect the spectral overlap for the Chl a-Chl b energy gap. The 300 fs rate we observe for LHCII is probably the average of the 150 fs and 600 fs rates, which is also consistent with its large amplitude, 75% of the total signal. For CP29, no room temperature energy transfer study has been reported and the low-temperature rate of ∼220 fs is slower than the one observed here. Probably, this difference can be explained by the better temporal resolution of our study. Altogether, we conclude from the similarity of the transfer rates, that the TG signals measured at 650 nm show Chl b → Chl a energy transfer. Thus, this transfer is seen to remove the population from the spectral window of the laser and we expect that the 3PEPS signal will hardly be contaminated with Chl b → Chl a transfer dynamics.
TABLE 1.
Transient grating at 650 nm
| Parameters | LHCII | CP29 |
|---|---|---|
| A1 | 0.76 | 0.53 |
| τ1 (fs) | 147 | 65 |
| A2 | 0.17 | 0.22 |
| τ2 (ps) | 2.4 | 1.1 |
| Y0 | 0.07 | 0.25 |
Parameters of biexponential fits to transient grating curves measured at 650 nm (with A1, A2 the amplitudes; τ1, τ2 the decay times; and Y0 the final level). Note that population decay times mentioned in the text are taken to be twice the TG intensity decay times listed here.
Three pulse echo peak shift
Peak shift decay curves were constructed from the 3PE signals measured at 650 nm. The resulting traces are shown in Fig. 5 for both LHCII and CP29. The large graph shows the first 2 ps, the complete range of 0–10 ps is shown in the inset.
FIGURE 5.

Peak shift decay curves measured at 650 nm for LHCII (solid) and CP29 (dashed). Both first 2 ps (large graph) and full range up to 10 ps (inset) are shown.
The decays are similar for both complexes with the CP29 signal remaining at a higher level than the LHCII level. Remarkably, however, these 3PEPS signals are quite different from most peak shift decays described in the literature. The initial decay of 3PEPS curves usually leads to a loss of 70% or more of the peak shift in 50 fs or less (see, for example, 3PEPS measurements on LH1 and LH2 (Jimenez et al., 1997)). In Fig. 5, on the other hand, we see that it takes up to 300 fs for LHCII, 500 fs for CP29, to reach such a low level. The peak shift at time zero is ∼20 fs, and after the first 50 fs, the peak shift values are still 15 fs for CP29, 12 fs for LHCII. Obviously, the initial decay has a small amplitude. After this phase, a second, much slower one leads to a peak shift value of a few fs in a time range of 500–1000 fs. A third, yet slower phase leads to the decay of the peak shift to zero within 10 ps. Hence, within this time range all the initially excited pigments transfer their excitation to others.
Apart from a pulse-overlap effect, the fastest phase is generally considered to show the ultrafast redshift due to solvent (i.e., protein) adjustment to the excited state and due to the interference of many high-frequency vibrations of the excited pigment (the Stokes' shift). The small amplitude of this phase is consistent with the fact that at 650 nm, most of the Chl b pigments were excited at the peak or at the red side of their absorption band. For dyes in liquids, Larsen et al. (2001), and Ohta et al. (2001), have shown that the peak shift decay depends on the excitation wavelength, and that the amplitude of the initial phase is the largest at the blue side of the absorption band, due to the excitation of more high-energy vibrations. The fact that for the initial decay, a larger amplitude is observed for LHCII can also be explained: its Chl b absorption is slightly more red on average, with one or two Chls b absorbing around 652 nm (Hemelrijk et al., 1992; Remelli et al., 1999). The small vibrational interference also reflects the absence of a large Chl a contribution. A small role is probably played by Chl a, nevertheless, as the peak shift, at least for CP29, shows a small dip and subsequent increase around T = 20 fs. This could be due to removal of vibrationally relaxed (redshifted) Chl a out of the spectral range of the laser pulse. Afterward, the peak shift only shows leftover Chls b, which have undergone less relaxation and thus have a higher peak shift. Nevertheless, the (partial) rephasing of the interfering vibrations may also explain this peak shift recurrence.
Due to the small amplitude of the initial decay, the slower dynamics can be resolved very well. In experiments with nontransferring pigments (see e.g., B820 (Yu et al., 1997), or blue B800s (Salverda et al., 2000)), only a very fast peak shift decay is seen, therefore we propose that the two slower phases represent energy transfer. To identify the timescales of these transfer processes, a sum of three exponential decays was fit to the peak shift decays. Biexponential fits gave a larger error (a larger χ2) and clearly would not do justice to the observed fast-intermediate-slow decays shown in Fig. 5. Fits with four exponents were also tested, but those led to degeneracy of two of the decay times.
The results of the exponential fits are listed in Table 2. As an example, the measured CP29 peak shift decay and the exponential fit are shown together in Fig. 6. The initial decay is very rapid with a time constant of ∼10 fs for both complexes. Its amplitude was ∼3 fs, which may be artificially small because the dip and recurrence of the peak shift were ignored in this fitting. As said above, this decay is most likely due to a combination of protein reorganization and interference between chlorophyll vibrations.
TABLE 2.
Peak shift at 650 nm
| Parameters | LHCII | CP29 |
|---|---|---|
| A1 (fs) | 6.6 | 3.2 |
| τ1 (fs) | 11 | 11 |
| A2 (fs) | 6.8 | 13 |
| τ2 (fs) | 150 | 360 |
| A3 (fs) | 6.2 | 3.6 |
| τ3 (ps!) | 0.48 | 3.15 |
| Y0 (fs) | 0.9 | 0.1 |
Parameters of 3-exponential fits to 3PEPS decay curves measured at 650 nm (with A1, A2, A3 the amplitudes—also in time units for the peak shift; τ1, τ2, τ3 the decay times; and Y0 the final level).
FIGURE 6.

Measured CP29 peak shift (filled squares) at 650 nm, shown with 3-exponential fit (solid line). Both first 2 ps (large graph) and full range up to 10 ps (inset) are shown. Decay time and amplitudes are detailed in Table 2.
The intermediate decay process, which represents the major part of the peak shift decay, has a time constant of a few hundred fs. In contrast to the decay of the transient grating, the peak shift decay is faster for LHCII than for CP29, with time constants of 145 fs and 360 fs, respectively. This reversal, and the fact that neither time is very similar to the transfer rates measured in transient grating, confirm that completely different processes are monitored by 3PEPS. This leads us to conclude that the 3PEPS measurements show energy transfer between Chls b, in agreement with the conclusion of Agarwal et al. (2000), that transfer among Chls b takes place in LHCII. Definitely, strongly coupled Chl b pairs must be present in both complexes. The assignment of these pairs to binding sites will be discussed in detail in the next section.
The slowest of the three decay processes has a small amplitude of only a few femtoseconds. It should be noted here that the value of 480 fs found for LHCII is rather uncertain. With an equivalent dataset, a value of 1.1 ps was obtained. This transfer time is therefore described from here on as 0.5–1 ps. The slowest transfer times thus found, 3 ps for CP29 and 0.5–1 ps for LHCII, are somewhat different from the transient grating values. Nevertheless, Chl b → Chl a transfer times similar to the 3PEPS decay rates have been observed in pump-probe experiments at both 77 K and room temperature (Visser et al., 1996; Connelly et al., 1997; Kleima et al., 1997; Gradinaru et al., 1998a). Possibly, these rates could be due to transfer from Chls b to Chls a. Excited-state absorption and bleaching in the vibrational wing of Chl a should be visible at 650 nm. However, for LHCII, it can be concluded from the low final value of the TG intensity that this contribution is exceedingly small. In addition, at times less than 1 ps, the Chl a contribution will be significantly smaller than at longer population times. Therefore, we propose that the 0.5–1 ps decay time in the LHCII peak shift represents a second, slower Chl b ↔ Chl b transfer process. For CP29, the Chl a contribution is seen from TG to be much larger, and the decay time of 3 ps is much slower than for LHCII. Therefore, Chl b ↔ Chl a transfer probably contributes to this peak shift decay component.
Excitation at 670 nm
Transient grating
At 670 nm, the 3PE signal was measured up to a population time of 100 ps. Again, transient grating traces were constructed from these signals in the manner described above. In Fig. 7, the TG decay is shown for the first 20 ps, with the range up to 100 ps shown in the inset.
FIGURE 7.

Transient grating curves measured at 670 nm for LHCII (solid) and CP29 (dashed). The curves were constructed from the 3PE signals. Both first 20 ps (large graph) and full range up to 100 ps (inset) are shown.
Just as at 650 nm, the TG decay is similar for both complexes, with the LHCII transient grating being the lower of the two. The difference takes a few picoseconds to develop. Clearly, the first part of the TG decay, until ∼2 ps, is virtually identical for both complexes. This part is responsible for the loss of 60–70% of the total signal amplitude, which shows that most of the excitations are taken away from the spectral maximum of the laser within a few picoseconds. After that, the subsequent decay of the LHCII TG is either faster or has a larger amplitude than that of the CP29 TG. Neither trace shows any decay after ∼20 ps, suggesting that the slow processes occur on a similar timescale in both complexes. The final level is as low as 5% of the initial amplitude for LHCII, and 15–20% for CP29. In isolated antenna complexes, the redmost Chl a states live for several nanoseconds. From the low final level of the curves in Fig. 7, it is therefore evident that this Chl a is hardly visible for the laser pulse applied in this experiment. As CP29 has a less strong blue wing in its Chl a band, the relative contribution at 670 nm of red Chls a will be stronger than for LHCII, given that the optical density is the same at this wavelength. This explains the larger final value for the CP29 transient grating.
To identify the timescales involved, biexponential decays were again fitted to the transient grating curves. The parameters of these fits are given in Table 3. The population decay rates corresponding to these fits are two times slower due to the quadratic nature of the TG signal. To avoid contamination with the rise due to the coherent coupling artifact, datapoints below T = 40 fs were ignored in the fitting. The decay rates for the two complexes are indeed rather similar. The fast process is somewhat faster for LHCII, at 2 × 115 = 230 fs for the population decay time, than for CP29, at 2 × 155 fs = 310 fs. The slow process is a little faster in CP29 with a time constant of 2 × 2.6 = 5 ps, vs. 2 × 3.0 = 6 ps for LHCII. For both decays, the amplitude is somewhat smaller for CP29 than for LHCII, resulting in a larger final value for CP29. For both complexes, the TG signal at 100 ps corresponds to an equilibrated distribution of the Chl a excitations, which will mostly reside on the redder pigments. Whether the redmost pigments are also excited directly or only populated via energy transfer cannot be discerned.
TABLE 3.
Transient grating at 670 nm
| Parameters | LHCII | CP29 |
|---|---|---|
| A1 | 0.68 | 0.53 |
| τ1 (fs) | 116 | 153 |
| A2 | 0.47 | 0.37 |
| τ2 (ps) | 3.0 | 2.6 |
| Y0 | 0.05 | 0.16 |
Parameters of biexponential fits to transient grating curves measured at 670 nm (see Table 1 for definition). Note that population decay times mentioned in the text are taken to be twice the TG intensity decay times listed here.
For excitation at 670 nm, population decay is expected to occur mainly via downhill energy transfer to red Chls a. These red Chls a, absorbing at a wavelength of 676 nm or above, will be referred to as Chl a (680). For fast Chl a (670) → Chl a (680) transfer at 77 K, Gradinaru et al. (1998a, b), find a time constant of ∼300 fs for LHCII and 280 fs for CP29. The 310 fs time we find for CP29 is very close and the difference with our 230 fs time for LHCII is probably due to the limited time resolution of the pump-probe experiment or to the difference in temperature. We conclude therefore that the fast TG decay component at 670 nm indeed represents energy transfer from Chls a absorbing at 670 nm to Chls a absorbing in the main 676 nm band or at longer wavelength.
For slow Chl a (670) → Chl a (680) transfer, a time constant of 5 or 6 ps was observed in pump-probe experiments on LHCII at room temperature (Bittner et al., 1994, 1995; Connelly et al., 1997). Clearly, our 5/6 ps 3PEPS decay component should be attributed to this transfer process. The rate of this particular transfer is clearly temperature dependent as values found from pump-probe on both LHCII and CP29 at 77 K vary between 12 ps and 20 ps (Visser et al., 1996; Kleima et al., 1997; Gradinaru et al., 1998a, b). Probably, within the Chl a band, uphill energy transfer also contributes to the equilibration.
Altogether, it can be assumed that 3PEPS, discussed below, will not be monitoring very red Chls a as these are hardly visible in the transient grating signal.
Three pulse echo peak shift
The constructed 3PEPS curves measured at 670 nm are shown in Fig. 8. As the signal decays very rapidly for both complexes, the large graph shows only the first 2 ps. In the inset, the full 100 ps range is shown. The most remarkable aspect of the 670 nm 3PEPS is the difference with the peak shift at 650 nm, which supports the assumption that the 3PEPS measurement at 650 nm follows almost exclusively Chl b dynamics. At 670 nm, the initial decay is huge and leads to a loss of 70% of the peak shift. Clearly, Chls a are mostly excited to the blue of their absorption maximum by the 670 nm pulse, as should be expected, with the Chl a band peaking at 676 nm for both complexes. Another difference with the 3PEPS measured at 650 nm is the presence of an oscillation, of which the minima are indicated by vertical lines in Fig. 8. This oscillation is almost identical for both complexes. It has a period of ∼90–100 fs, corresponding to a frequency of ∼330–350 cm−1, and persists for ∼600 fs. Most likely, it is due to a single vibrational mode of the excited pigments. A vibrational transition of Chl a with a frequency of 350 cm−1 was observed in several studies on plant complexes with steady-state spectroscopic methods such as hole-burning (Gillie et al., 1989) and fluorescence line narrowing (Peterman et al., 1997; Pieper et al., 1999). According to Small and co-workers (Pieper et al., 1999), this particular vibrational mode has the strongest coupling to the electronic transition. However, the relative strength of such modes can vary a lot depending on the spectroscopic technique used. In pump-probe experiments, no vibrations have ever been observed for either antenna complex. With 3PEPS, a 60 cm−1 oscillation was seen in LHCII by Agarwal et al. (2000), but only when they excited at 650 nm. They did not observe any such feature when exciting Chl a.
FIGURE 8.

Peak shift decay curves measured at 670 nm for LHCII (solid) and CP29 (dashed). Both first 2 ps (large graph) and full range up to 100 ps (inset) are shown.
Another striking feature of the 670 nm curves is that they are qualitatively different for CP29 and LHCII. The CP29 peak shift decays rapidly from 25 fs to 8 fs in a population time range of ∼100 fs. At this point, it shows a very sharp turnoff, after which it decays further on a second, at least 10 times slower timescale to a final level of ∼4 fs at T = 100 ps. The initial peak shift of LHCII is marginally higher at 27 fs, and the final peak shift lower at ∼2 fs. The LHCII peak shift shares the fast initial decay and the picosecond decay. But, more notably, the turnoff is much less sharp. This appears to be due to an intermediate timescale decay that is absent in the CP29 3PEPS. The timescale of this decay is a few hundred fs, which suggests that it corresponds to an energy transfer process. The slow decay components present in the 3PEPS of both complexes are probably also related to energy transfer.
Fits with three exponentials were carried out to identify the energy transfer timescales. The results are listed in Table 4. Although CP29 does not show any intermediate timescale process, its fast decay can be described much better by a sum of two components, of 10 fs and 70 fs, than by a single exponential. This may reflect the contribution of two distinct processes, protein reorganization and interference between vibrations, to this decay. Probably, the fit for LHCII should also have two fast components, and four in total. Unfortunately, the data do not permit a fit with more than three different components. Hence, the rate of 22 fs for LHCII may well reflect a weighted average of two rates. The transfer process, which is present in LHCII only, has a rate of 140 fs, and an amplitude of 7 fs. The rate of this process does not correspond to any of the well-known Chl a (670) → Chl a (680) rates. Of course, this could be due to the fact that such fast Chl a equilibration was not searched for at room temperature. Nevertheless, the decay times observed in the TG decay are remarkably absent from the 670 nm peak shift decay. Therefore, we conclude that the 140 fs transfer process that is seen at 670 nm for LHCII occurs between two blue Chl a states, both contributing to the absorption at 670 nm. Which particular Chls a could be involved will be discussed in the next section. For LHCII, this transfer was not seen before with pump-probe techniques. Agarwal et al. (2000) report a much slower rate of (350 fs)−1 in their 3PEPS experiment, and they attribute this rate to downhill (670) → (680) transfer by referring to pump-probe results in the literature.
TABLE 4.
Peak shift at 670 nm
| Parameters | LHCII | CP29 |
|---|---|---|
| A1 (fs) | 10 | 3.6 |
| τ1 (fs) | 22 | 10 |
| A2 (fs) | 7.6 | 13 |
| τ2 (fs) | 141 | 68 |
| A3 (fs) | 7.4 | 4.0 |
| τ3 (ps!) | 3.6 | 3.3 |
| Y0 (fs) | 1.3 | 4.1 |
Parameters of 3-exponential fits to 3PEPS decay curves measured at 670 nm (see Table 2 for definition).
The slow decay process has a rate of ∼(3 ps)−1 for both complexes. This rate is definitely not comparable to any Chl a (670) → Chl a (680) transfer rate seen in pump-probe studies. It is also absent in the transient grating decay. Consequently, it most likely represents slow energy transfer between Chls a that absorb close to 670 nm. Again, in pump-probe experiments on these complexes, this slow transfer between blue Chls a was not observed either. Probably, its small amplitude makes it very hard to identify, especially as the 670 nm signal will have largely disappeared before it takes place. Agarwal et al. (2000) have probably seen this process in their LHCII 3PEPS, as they do use a 3 ps component in their simulations. Again, they implicitly attribute this component to 670 nm → 680 nm transfer and do not discuss it in detail.
Altogether, we conclude that the 670 nm peak shift shows clear evidence for energy transfer processes between blue Chls a in both LHCII and CP29.
Summarizing the Results section, we have provided an overview of the measured time constants and the transfer processes we attribute them to in Table 5. Energy transfer times known from literature are also included.
DISCUSSION
In this section, the issue of the existence of mixed binding sites will be addressed. Furthermore, we will discuss the assignment of the decay rates to transfer between specific chlorophylls in the structure. This assignment will largely be based on calculations by Iseri and co-workers on both CP29 (Iseri et al., 2000) and LHCII (Iseri and Gülen, 2001). As the calculations indicate that considerable exciton delocalization is present in both complexes, we will frequently refer to states rather than pigments.
By comparing our observed exponential decay rates with the calculations of Iseri, we implicitly equate the decay rates to pairwise transfer steps between two states or pigments, although this is not strictly justified. In fact, observed decay rates show the overall kinetics of the whole complex, which are the effect of many pairwise steps. Nevertheless, for the processes that are of most importance in this work, it is often a matter of only one possible pairwise step that is fast enough. If this step would not occur, the overall kinetics in the complex would be too slow to explain the observations.
Note furthermore that the rates calculated by Iseri for both complexes are all based on the structural LHCII model of Kühlbrandt et al. (1994), either in the directions those authors proposed (designed as 0) or rotated by 90° (designed as 1), apart from small deviations of 5 or 10° that Iseri and Gülen (2001) have included for the Chls b in LHCII. However, the absolute size of the simulated LD spectrum is at least two times smaller than that of the experimental LD spectrum of LHCII (van Amerongen et al., 1994) for any combination of the 0 and 1 permutations, which indicates that these orientations cannot be correct. Thus, the calculated rates should be taken as approximate.
In the papers of Iseri et al. (2000), and Iseri and Gülen (2001), mixed binding sites are assumed to be present in the native complexes. As we will argue below, the measurements presented here largely support this idea. Specifically, Iseri uses the assignments published by Bassi and co-workers (Remelli et al., 1999; Bassi et al., 1999; and Simonetto et al., 1999). For LHCII, this assignment is different from the one presented in Fig. 1 a, with A7 as a fourth mixed site rather than a Chl b. For the moment, we will use the latter assignment and assume that the processes calculated for a Chl a at A7 are irrelevant.
650 nm excitation in CP29
Existence of mixed sites
After excitation at 650 nm, we observe three or four different transfer rates in CP29. From the transient grating decay, we find a fast Chl b → Chl a transfer process with a time constant of 130 fs. From the 3PEPS decay, we find a slower sub-ps transfer that takes 360 fs and that should be energy transfer between two Chls b. Both in TG and 3PEPS, slow components with time constants of 2.2 or 3.2 ps, respectively, are observed, which may or may not be due to the same process. From the above, it can be argued directly that binding sites with both Chl a and Chl b affinity must be present in CP29. We will show this by considering the opposite assumption, namely that the two Chls b of CP29 are always bound at the same two positions. (Note that the following argument is entirely independent of assumptions about the identity of any of the eight binding sites of CP29.) A fast Chl b ↔ Chl a transfer rate is observed, which means that at least one of the two Chls b loses its excitation almost immediately. This means that transfer from this Chl b to the second Chl b will not occur often enough to be observable. With 3PEPS, transfer from the second Chl b to this Chl b cannot be observed as it would be followed by immediate transfer to Chl a, which takes the excitation out of the spectral reach of our pulse. Nevertheless, we do observe a Chl b ↔ Chl b transfer process, which means that two long-lived Chls b must be present. This is in contradiction with the assumption that these Chls b always bind at the same sites. Thus, it must be concluded that mixed binding sites exist in native CP29.
Assignment of Chl b transfer steps
Below, we will discuss the possible site identities and the way the observed transfer processes connect them in more detail. Bassi and co-workers (Remelli et al., 1999; Bassi et al., 1999; Simonetto et al., 1999) propose that CP29 can bind either Chls b or Chls a at the four sites, A3, B3, B5, and B6. For all combinations of Chls a and Chls b at these sites, Iseri et al. (2000) have calculated energy transfer times using the Förster equation.
According to these calculations, fast Chl b ↔ Chl b energy transfer can only take place in complexes with an A3-B3 Chl b pair. Without this pair, only picosecond intra-Chl b processes will occur. For the A3-B3 pair, Iseri et al. (2000) calculate a transfer time of ∼200 fs, which is slightly faster than the 360 fs time we observe. It should be realized that the orientations of the transition dipole moments of the Chls in CP29 have not been determined and that therefore the calculated rates can only be approximate. Moreover, the coupling within the A3-B3 pair is very strong, and Iseri et al. find that the exciton is almost completely delocalized. At nonzero temperature, the exciton will become progressively more localized on one of the two sites, which will be observed as a peak shift decay. The localization rate is not identical to the calculated Förster rate. Nevertheless, the subpicosecond Chl b equilibration will certainly be due to this pair.
The fast Chl b → Chl a transfer rate is significantly different from the fast Chl b → Chl b transfer rate, (130 fs)−1 vs. (360 fs)−1. For the fast transfer from Chl b to Chl a, the A3-B3 pair is also the only candidate, now with a Chl b at one of these sites and a Chl a at the other. To explain the difference in rates, the relative orientation of the b-a pair dipoles must be somewhat different from that of the b-b pair dipoles. A minor problem with the identification of our fast 3PEPS and TG decay components with the A3-B3 pair should be mentioned here. From pump-probe studies at 77 K, it was concluded that fast Chl b → Chl a transfer takes place from a blue Chl b, absorbing at ∼640 nm. Iseri et al. assume that the Chls b at both A3 and B3 absorb at this wavelength and find exciton levels that lie as far to the blue as 636 and 644 nm. However, the fact that the fast Chl b ↔ Chl b transfer is so prominently present in our measurements indicates that their energies may be closer to 650 nm than previously suggested.
For the slow decay component of 3PEPS, with a time constant of 3 ps, several explanations are possible. This process may be slow Chl b ↔ Chl b transfer. Transfer between B5 and B6 as a Chl b pair should take 2 ps, according to Iseri et al. Transfer from this pair to Chls a would be slow enough (5 ps) for the transfer within the pair to be observable at 650 nm. Transfer between other distant Chls b, from those at A3-B3 to those at B5-B6 and vice versa, is too slow to contribute to this component. From the existence of a second Chl b pair at sites B5 and B6, it would follow that all four sites proposed by Bassi and co-workers are mixed sites in native CP29. However, the slow peak shift decay may also be due to Chl b → Chl a energy transfer, as was argued in the Results section. For a Chl a-Chl b pair at the sites B5 and B6, Iseri et al. calculate a Förster transfer rate of 3.5 ps, for both combinations. Also, several other picosecond Chl b → Chl a transfer times are found that may contribute to this component. Therefore, it cannot be concluded with certainty whether only two or all four sites proposed by Bassi and co-workers are mixed in native CP29. Nevertheless, given the existence of any mixed sites in the native complex, we see no reason why these should not be present at all the sites determined in the mutagenesis studies.
650 nm excitation in LHCII
Existence of mixed sites
When exciting Chl b at 650 nm, we observe four different transfer processes in LHCII. From the TG decay, it can be concluded that fast Chl b ↔ Chl a transfer takes place with a time constant of 300 fs, and slow Chl b → Chl a transfer with a time constant of 5 ps. The decay of the peak shift shows that Chl b ↔ Chl b transfer also takes place, both in 150 fs and in 0.5–1 ps. To explain the fast energy transfer between Chls b, a strongly coupled Chl b pair should be present, just like in CP29. Also, this pair should not be too close to any Chls a. To explain the fast TG decay, other Chls b should be present in the complex that do transfer their excitation to Chl a very rapidly. In principle, these different Chls b can all be present in one complex, as LHCII contains five Chls b, and it cannot be concluded directly that mixed binding sites are present. Below, we will discuss the attribution of the above mentioned rates based on the calculations of Iseri and Gülen (2001), which take mixed sites into account. It will become clear that our observations can best be explained if at least some of the sites are indeed mixed.
Assignment of Chl b transfer steps
In LHCII, a pair of Chls b that can transfer excitation to each other can be found at sites B6 and A7 (Bassi, personal communication), independent of the presence of mixed sites. However, the Förster transfer time that Iseri and Gülen calculate for this pair is 500 fs, rather slower than the value of 150 fs we observe. This suggests the presence of a second Chl b pair in some of the complexes. The best candidate would be the A3-B3 pair, for which Iseri and Gülen calculate a Förster transfer time of ∼200 fs just like in CP29. A second possibility is the presence of a Chl b at site A6 in some of the complexes. The transfer between this Chl b and the one at site B6 is calculated to take 300 fs. Along with the B6-A7 transfer, the equilibration in the B6-A7-A6 group may be fast enough to explain the 150 fs peak shift decay component. On the other hand, it is highly unlikely that this group, or the A3-B3 pair, or indeed any second Chl b pair, binds only Chls b in all of the LHCII complexes. In that case, the average Chl b → Chl a transfer time would be very much slower than was observed, both in this work and by many others (Bittner et al., 1994, 1995; Visser et al., 1996; Connelly et al., 1997; Kleima et al., 1997; Gradinaru et al., 1998a, b). Therefore, it seems highly likely that mixed binding sites also exist in native LHCII. Thus, the groups mentioned above can be responsible for fast Chl b ↔ Chl b transfer in some of the complexes, and for fast Chl b → Chl a transfer in the others. Note that neither of these two groups was proposed as an explanation of the observed Chl b ↔ Chl b transfer by Agarwal et al. (2000). This is probably due to the fact that these authors did not consider mutagenesis results. They proposed a Chl b pair at sites A5 and B5, which was wrongly reproduced from Gradinaru et al. (1998b), who suggested that a Chl b pair could be present at sites A6 and B6.
The proposed flexibility in Chl binding would be in line with the character of the LHCII complex, which can adapt itself to many different functions under different light conditions. Specifically, one functional role of a mixed binding site could be the observed quenching of excitation in LHCII aggregates by long-lived Chl a triplets and quenchers formed from these triplets (Barzda et al., 2000). Such long-lived triplets would only occur in Chls a that are not well-connected to the central luteins, for example one at site B3 with a Chl b at site A3. In the case of mixed site occupancy, such a site could bind a Chl b when the quenching effect is not needed, which would be worthwhile as triplets are also a source of damage.
Note that there is a general discrepancy between site identities as proposed by Bassi and co-workers, and the measured Chl b → Chl a energy transfer rates. Both A7 and B5 are always Chls b, according to Bassi and co-workers, and according to the calculations of Iseri and Gülen they always take several picoseconds to transfer their energy to Chl a, whereas measurements indicate that only at most 1.5 of the five Chls b takes this long. With the Chls b at some of the mixed sites, this discrepancy will get larger. Possibly, it can be partly resolved, if the Chl b affinities of the sites A3, B3, and A6 are lower than the 50% proposed by Remelli et al. (1999), and used by Iseri and Gülen (2001), or if A7 sometimes does bind a Chl a. From our measurements, these questions cannot be answered.
Next to the 150 fs transfer process, a slower Chl b ↔ Chl b transfer with a time constant of 0.5–1 ps was observed. Equilibration in the A7-B6-A6 group could very well explain this decay rate, as Iseri and Gülen calculate Förster times of 500 fs for the A7-B6 pair, 300 fs for the A6-B6 pair, with much slower transfer for the A7-A6 pair. The net effect of these three steps would certainly yield a peak shift decay time in the observed range. Even if A6 always binds a Chl a, the A7-B6 transfer would still be visible as a 0.5 ps component.
To the decay components of the TG curve, which correspond to Chl b → Chl a transfer times of 300 fs and 5 ps, any of a large number of Chl b - Chl a combinations can contribute. For example, for the mixed pair at A3-B3, a transfer time of 250 fs is calculated by Iseri and Gülen as well as for the transfer from B2 to A2. Lifetimes of 5–10 ps are found for excitations on the Chl b pair at A3-B3, on B5 and A7, and on A6 and B6 if these are both Chls b. Altogether, the observed rates of Chl b ↔ Chl b and Chl b → Chl a transfer in LHCII can be explained perfectly well.
670 nm excitation in LHCII and CP29
Existence of mixed sites
When exciting at 670 nm, we observe very similar Chl a transfer dynamics in both complexes, and a process which only occurs in LHCII. The LHCII-specific process is observed in the 3PEPS decay as a small component with a time constant of 140 fs. It most likely corresponds to one or more transfer steps between blue Chls a or Chl a states. Both LHCII and CP29 show a slow component in the peak shift decay with a 3 ps time constant, which is probably also due to transfer between blue Chls a. From the transient grating decay, it was determined that transfer from blue to red Chl a states takes place with time constants of a few hundred fs and 5 or 6 ps. From these observations, it cannot be concluded directly that mixed sites are present (other than those identified with the Chl b excitation measurements), as the amount of Chls a in both complexes is too large. Nevertheless, we will compare the above-mentioned rates to the calculations of Iseri et al. (2000), and Iseri and Gülen (2001), which take mixed sites into account. Once again, the best agreement is obtained when some sites are mixed, as we will argue below.
Assignment of Chl a transfer steps
According to the calculations of Iseri and Gülen (2001), excitation transfer in LHCII on a timescale close to 140 fs takes place in two groups of Chls a. Those are the trio A1, A2, and B1, and the A3-B3 pair. For the trio, they find Förster transfer times of 250 fs for transfer from B1 to A1, and 500 fs for transfer from A2 to A1, and vice versa. However, for all three Chls A1, A2, and B1, very low transition energies were determined by Remelli et al. (1999), with wavelengths of 679 nm, 681 nm, and 679 nm, which is also in agreement with excitation energy transfer calculations (van Amerongen and van Grondelle, 2001). Hence, the transfer within this trio is expected to contribute to TG rather than 3PEPS. On the other hand, the 140 fs component can be understood very well if the two sites at A3 and B3 both bind a Chl a in some of the complexes. Based on the difference spectra measured in several mutagenesis studies (Remelli et al., 1999; Rogl and Kühlbrandt, 1999; Rogl et al., 2002), those two are the bluemost Chls a, with transition wavelengths of 663 nm and 665 nm. For a Chl a pair at these sites, Iseri and Gülen (2001) calculate a transfer time of ∼100 fs. Hence, relaxation between these two will be visible as an ultrafast decay component in 670 nm 3PEPS. At the same time, this pairwise step provides the only reasonable explanation for this component, as all other Chl a groups are either too red or too slow. Although the A3-B3 pair was proposed to bind Chls a in all of the complexes (Rogl and Kühlbrandt, 1999; Rogl et al., 2002), we think this is not likely as the overall Chl b → Chl a transfer would be too slow in that case. Thus, this is a strong indication that at least one of these sites must be mixed.
In CP29, sub-ps energy transfer takes place in the A3-B3 pair and in the A1-A2 pair. Neither of these pairs involves only blue Chls a like the A3-B3 pair in LHCII (Bassi et al., 1999; Simonetto et al., 1999). A3 absorbs relatively far to the blue at 668 nm, but B3 absorbs at 674 nm. A1 absorbs at 669 nm, but the site A2 binds the redmost Chl a of the complex with a transition at 680 nm. The lowest exciton level of these pairs will lie quite far to the red, and thus the absence of a detectable component in the CP29 3PEPS is not surprising.
The slow component that is present in both LHCII and CP29 probably also represents transfer between blue Chls a. For LHCII, slow transfer will occur from the isolated Chls a at sites A3, B3, and A6 to the A4-A5 pair and vice versa, taking ∼5 ps (van Amerongen and van Grondelle, 2001; Iseri and Gülen, 2001). All of these Chls are found by Remelli et al. (1999) to absorb fairly close to 670 nm. The transfer within the A4-A5 pair, which takes ∼1 ps, according to Iseri and Gülen, probably also contributes to this component. Transfer from the isolated Chls a to the sites A1, A2, and B1 is also slow, taking as much as 10 ps, but as the latter Chls absorb quite far to the red, this process will not be observed in 3PEPS. In CP29, the rates for transfer from the isolated Chls a A3, B3, B5, and B6 to the A4-A5 pair and vice versa are very similar to the values in LHCII (Iseri et al., 2000). The same is true for the transfer within the A4-A5 pair. Another contribution of the 3 ps component in the CP29 complex may arise from transfer between two Chls a at sites B5 and B6, for which Iseri et al. (2000) calculate a transfer time of 1 ps. Note that such a Chl a pair can only be present if at least one of those two sites is mixed.
With transient grating, both fast and slow downhill energy transfer processes are observed. For red Chl a groups, relaxation from a higher exciton state to the lowest state, which lies close to 680 nm, will be visible as a decay of the transient grating signal at 670 nm. Strongly coupled Chl a groups containing relatively red Chls a are A1-A2-B1 in LHCII and A1-A2 in CP29, which will relax to the lowest level within a few hundred fs (Iseri et al., 2000; Iseri and Gülen, 2001; van Amerongen and van Grondelle, 2001). Picosecond downhill transfer will occur from isolated blue Chls a (e.g., A3, for both complexes) to these red Chl a groups, which subsequently relax rapidly to their lowest exciton level at ∼680 nm.
Altogether, the observed Chl a transfer rates can be explained very well for both complexes. Furthermore, the LHCII dynamics strongly suggest that mixed sites are present at positions A3 and B3.
As a summary of the Discussion section, we have provided block diagrams for both complexes in Fig. 9, which show the determined transfer processes and site identities. Both firm and tentative assignments are included. For processes that are known to occur but that were not observed in this study, no transfer time constants are specified.
FIGURE 9.


Block diagrams of LHCII (a) and CP29 (b), illustrating the determined energy transfer pathways. Only rates determined in our experiments are specified. See text for details about the assignments.
CONCLUDING REMARKS
In this work, we have studied energy transfer between chlorophylls in the plant antenna complexes CP29 and LHCII. The 3PEPS technique was applied to selectively monitor isoenergetic energy transfer. For a comparison with downhill rates under the same experimental conditions, transient grating curves were also determined from these measurements. The chlorophyll b band was excited at 650 nm, and the chlorophyll a band was excited in the blue shoulder at 670 nm. The decay time constants describing these curves were compared with exciton/energy transfer calculations from literature (Iseri et al., 2000; Iseri and Gülen, 2001) to determine which Chls could be involved in the corresponding energy transfer processes.
The transient grating measurements performed at 650 nm show excited-state population decay, probably due to downhill energy transfer from Chls b to Chls a. For CP29, we find population decay times of 130 fs and 2 ps; for LHCII, times of 280 fs and 3 ps. These time constants correspond well with Chl b → Chl a transfer times known from literature, assuming that the fast LHCII rate is a mixture of the two known sub-ps rates. Using 3PEPS, for both complexes, entirely different rates are found at 650 nm. We observe ultrafast, sub-50 fs decay due to electron-phonon coupling and interfering vibrations, followed by two slower components due to energy transfer. The faster of these two processes has a time constant of 150 fs for LHCII, and 360 fs for CP29. We attribute this decay to Chl b ↔ Chl b energy transfer. This leads directly to the conclusion that at least two mixed sites must be present in CP29. Both in CP29 and in LHCII, the A3-B3 pair is the best candidate for such fast Chl b ↔ Chl b energy transfer. We conclude that at least one of these two sites is a mixed site in native CP29. Most likely, this is also the case in LHCII. The slow 650-nm peak shift decay rates are probably also at least partly related to Chl b ↔ Chl b energy transfer. In LHCII, the observed 0.5–1 ps decay time is probably largely due to transfer between the Chls b at B6, A7, and the possible mixed site A6. The slow rate for CP29 (3 ps−1) can also be due to Chl b → Chl a transfer in this complex.
At 670 nm, the transient grating decay again shows almost the same population decay times that were observed in pump-probe studies. Population decay times of 200 fs and 6 ps are determined for LHCII, and times of 300 fs and 5 ps for CP29. These decay times probably reflect energy transfer from blue (670 nm) to red (680 nm) Chl a pigments. Again, the peak shift decay rates are very different from the transient grating rates. Both peak shift curves are dominated by a 20 fs decay component, probably due to interference between many vibrational modes. For CP29, this decay is followed by a slow component with a time constant of 3 ps, whereas for LHCII, an additional decay with a time constant of 150 fs is observed. This decay shows the presence of fast energy transfer between blue Chl a pigments or exciton levels. For such fast transfer, the presence of a Chl a pair at sites A3 and B3 is the best explanation, which suggests that at least one of these sites is mixed in the native LHCII complex. The 3 ps equilibration time observed in both complexes is explained by transfer between distant Chl a molecules, which absorb close to 670 nm. For LHCII, such transfer can occur from B3 or A3 (given that the other site of this pair binds a Chl b), and maybe from A6, to the A4-A5 pair. For CP29, this component may be attributed to transfer between several Chls a, such as from B3 or A3 to the A1-A2 pair, or between B5 and B6, if both sites can bind a Chl a.
Acknowledgments
The authors thank Dr. Andy A. Pascal for repeatedly providing generous batches of CP29 and ing, and Florentine Calkoen for isolating and concentrating enormous amounts of LHCII.
J.M.S. was supported by the Netherlands Organization for Scientific Research via the Board of Earth and Life Sciences, grant 805-02.066. V.N. was supported by a Russian-Dutch Research Cooperation Program (NWO, grant 047.009.014), and by the Russian Foundation for Basic Research, grant 02-04-48779. G.D.S. was supported by a visiting grant from the Institute of Molecular Biological Sciences of the Vrije Universiteit, and B.P.K. by a visiting grant from the COMPAS institute of the Vrije Universiteit.
References
- Agarwal, R., B. P. Krueger, G. D. Scholes, M. Yang, J. Yom, L. Mets, and G. R. Fleming. 2000. Ultrafast energy transfer in LHC-II revealed by three-pulse photon echo peak shift measurements. J. Phys. Chem. B. 104:2908–2918. [Google Scholar]
- Agarwal, R., M. Yang, Q. H. Xu, and G. R. Fleming. 2001. Three pulse photon echo peak shift study of the B800 band of the LH2 complex of Rps. acidophila at room temperature: a coupled master equation and nonlinear optical response function approach. J. Phys. Chem. B. 105:1887–1894. [Google Scholar]
- Barzda, V., M. Vengris, L. Valkunas, R. van Grondelle, and H. van Amerongen. 2000. Generation of fluorescence quenchers from the triplet states of chlorophylls in the major light-harvesting complex II from green plants. Biochemistry. 39:10468–10477. [DOI] [PubMed] [Google Scholar]
- Bassi, R., D. Sandonà, and R. Croce. 1997. Novel aspects of chlorophyll a/b binding proteins. Physiol. Plant. 100:769–779. [Google Scholar]
- Bassi, R., R. Croce, D. Cugini, and D. Sandonà. 1999. Mutational analysis of a higher plant antenna protein provides identification of chromophores bound into multiple sites. Proc. Natl. Acad. Sci. U.S.A. 96:10056–10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner, T., K.-D. Irrgang, G. Renger, and M. R. Wasielewski. 1994. Ultrafast excitation energy transfer and exciton-exciton annihilation processes in isolated light harvesting complexes of photosystem II (LHC II) from spinach. J. Phys. Chem. 98:11821–11826. [Google Scholar]
- Bittner, T., G. P. Wiederrecht, K.-D. Irrgang, G. Renger, and M. R. Wasielewski. 1995. Femtosecond transient absorption spectroscopy on the light-harvesting Chl a/b protein complex of photosystem II at room temperature and 12 K. Chem. Phys. 194:311–322. [Google Scholar]
- Boekema, E. J., H. van Roon, F. Calkoen, R. Bassi, and J. P. Dekker. 1999a. Multiple types of association of photosystem II and its light-harvesting antenna in partially solubilized photosystem II membranes. Biochemistry. 38:2233–2239. [DOI] [PubMed] [Google Scholar]
- Boekema, E. J., H. van Roon, J. F. L. van Breemen, and J. P. Dekker. 1999b. Supramolecular organization of photosystem II and its associated light-harvesting antenna in partially solubilized photosystem II membranes. Eur. J. Biochem. 266:444–452. [DOI] [PubMed] [Google Scholar]
- Boekema, E. J., J. F. L. van Breemen, H. van Roon, and J. P. Dekker. 2000. Arrangement of photosystem II supercomplexes in crystalline macrodomains within the thylakoid membranes of green plants. J. Mol. Biol. 301:1123–1133. [DOI] [PubMed] [Google Scholar]
- Cinque, G., R. Croce, A. R. Holzwarth, and R. Bassi. 2000. Energy transfer among CP29 chlorophylls: calculated Förster rates and experimental transient absorption at room temperature. Biophys. J. 79:1706–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly, J. P., M. G. Müller, M. Hucke, G. Gatzen, C. W. Mullineaux, A. V. Ruban, P. Horton, and A. R. Holzwarth. 1997. Ultrafast spectroscopy of trimeric light-harvesting complex II from higher plants. J. Phys. Chem. B. 101:1902–1909. [Google Scholar]
- Croce, R., J. Breton, and R. Bassi. 1996. Conformational changes induced by phosphorylation in the CP29 subunit of photosystem II. Biochemistry. 36:11142–11148. [DOI] [PubMed] [Google Scholar]
- Croce, R., R. Remelli, C. Varotto, J. Breton, and R. Bassi. 1999. The neoxanthin binding site of the major light harvesting complex (LHCII) from higher plants. FEBS Lett. 456:1–6. [DOI] [PubMed] [Google Scholar]
- de Boeij, W. P., M. S. Psenichnikov, and D. A. Wiersma. 1995. Phase-locked heterodyne-detected stimulated photon echo. A unique tool to study solute-solvent interactions. Chem. Phys. Lett. 238:1–8. [Google Scholar]
- Du, M., X. Xie, L. Mets, and G. R. Fleming. 1994. Direct observation of ultrafast energy transfer processes in light-harvesting complex II. J. Phys. Chem. 98:4736–4741. [Google Scholar]
- Gillie, J. K., G. J. Small, and J. H. Goldbeck. 1989. Nonphotochemical hole burning of the native antenna complex of photosystem I (PSI-200). J. Phys. Chem. 93:1620–1627. [Google Scholar]
- Gradinaru, C. C., A. A. Pascal, F. van Mourik, B. Robert, P. Horton, R. van Grondelle, and H. van Amerongen. 1998a. Ultrafast evolution of the excited states in the chlorophyll a/b complex CP29 from green plants studied by energy-selective pump-probe spectroscopy. Biochemistry. 37:1143–1149. [DOI] [PubMed] [Google Scholar]
- Gradinaru, C. C., S. Özdemir, D. Gülen, I. H. M. van Stokkum, R. van Grondelle, and H. van Amerongen. 1998b. The flow of excitation energy in LHCII monomers: implications for the structural model of the major plant antenna. Biophys. J. 75:3064–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru, C. C., I. H. M. van Stokkum, A. A. Pascal, R. van Grondelle, and H. van Amerongen. 2000. Identifying the pathways of energy transfer between carotenoids and chlorophylls in LHCII and CP29. A multicolor, femtosecond pump-probe study. J. Phys. Chem. B. 104:9330–9342. [Google Scholar]
- Green, B. R., and D. G. Durnford. 1996. The chlorophyll-carotenoid proteins of oxygenic photosynthesis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 47:685–714. [DOI] [PubMed] [Google Scholar]
- Groot, M. L., J.-Y. Yu, R. Agarwal, J. R. Norris, and G. R. Fleming. 1998. Three-pulse photon echo measurements on the accessory pigments in the reaction center of Rhodobacter sphaeroides. J. Phys. Chem. B. 102:5923–5931. [Google Scholar]
- Hemelrijk, P. W., S. L. S. Kwa, R. van Grondelle, and J. P. Dekker. 1992. Spectroscopic properties of LHC-II, the main light-harvesting chlorophyll a/b protein complex from chloroplast membranes. Biochim. Biophys. Acta. 1098:159–166. [Google Scholar]
- Hillmann, F., J. Voigt, H. Redlin, K.-D. Irrgang, and G. Renger. 2001. Optical dephasing in the Light-Harvesting Complex II: a two-pulse photon echo study. J. Phys. Chem. B. 105:8607–8615. [Google Scholar]
- Iseri, E. I., D. Albayrak, and D. Gülen. 2000. Electronic exciton states of the CP29 antenna complex of green plants: a model based on exciton calculations. J. Biol. Phys. 26:321–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseri, E. I., and D. Gülen. 2001. Chlorophyll transition dipole moment orientations and pathways for flow of excitation energy among the chlorophylls of the major plant antenna, LHCII. Eur. Biophys. J. 30:344–353. [DOI] [PubMed] [Google Scholar]
- Jansson, S. 1994. The light-harvesting chlorophyll a/b proteins. Biochim. Biophys. Acta. 1184:1–19. [DOI] [PubMed] [Google Scholar]
- Jimenez, R., F. van Mourik, J.-Y. Yu, and G. R. Fleming. 1997. Three-pulse photon echo measurements on LH1 and LH2 complexes of Rhodobacter sphaeroides: a nonlinear spectroscopic probe of energy transfer. J. Phys. Chem. B. 101:7350–7359. [Google Scholar]
- Joo, T., Y. Jia, and G. R. Fleming. 1995. Ultrafast liquid dynamics studied by 3rd order and 5th order 3-pulse photon echoes. J. Chem. Phys. 102:4063–4068. [Google Scholar]
- Joo, T., Y. Jia, J.-Y. Yu, M. J. Lang, and G. R. Fleming. 1996a. Third-order nonlinear time domain probes of solvation dynamics. J. Chem. Phys. 104:6089–6107. [Google Scholar]
- Joo, T., Y. Jia, J.-Y. Yu, D. M. Jonas, and G. R. Fleming. 1996b. Dynamics in isolated bacterial light harvesting antenna (LH2) of Rhodobacter sphaeroides at room temperature. J. Phys. Chem. 100:2399–2409. [Google Scholar]
- Kleima, F. J., C. C. Gradinaru, F. Calkoen, I. H. M. van Stokkum, R. van Grondelle, and H. van Amerongen. 1997. Energy transfer in LHCII monomers at 77K studied by sub-picosecond transient absorption spectroscopy. Biochemistry. 36:15262–15268. [DOI] [PubMed] [Google Scholar]
- Kühlbrandt, W., D. N. Wang, and Y. Fujiyoshi. 1994. Atomic model of plant light-harvesting complex by electron crystallograpy. Nature. 367:614–621. [DOI] [PubMed] [Google Scholar]
- Kwa, S. L. S., H. van Amerongen, S. Lin, J. P. Dekker, R. van Grondelle, and W. S. Struve. 1992. Ultrafast energy transfer in LHC-II trimers from the Chl a/b light-harvesting antenna of photosystem II. Biochim. Biophys. Acta. 1102:202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, D. S., K. Ohta, Q.-H. Xu, M. Cyrier, and G. R. Fleming. 2001. Influence of intramolecular vibrations in third-order, time-domain resonant spectroscopies. I. Experiments. J. Chem. Phys. 114:8008–8019. [Google Scholar]
- Mukamel, S. 1995. Principles of Nonlinear Optical Spectroscopy. Oxford University Press, New York.
- Nussberger, S., J. P. Dekker, W. Kühlbrandt, B. M. van Bolhuis, R. van Grondelle, and H. van Amerongen. 1994. Spectroscopic characterization of three different monomeric forms of the main chlorophyll a/b binding protein from chloroplast membranes. Biochemistry. 33:14775–14783. [DOI] [PubMed] [Google Scholar]
- Ohta, K., D. S. Larsen, M. Yang, and G. R. Fleming. 2001. Influence of intramolecular vibrations in third-order, time-domain resonant spectroscopies. II. Numerical calculations. J. Chem. Phys. 114:8020–8039. [Google Scholar]
- Pascal, A. A., C. C. Gradinaru, U. Wacker, E. J. G. Peterman, F. Calkoen, K.-D. Irrgang, P. Horton, G. Renger, R. van Grondelle, B. Robert, and H. van Amerongen. 1999. Spectroscopic characterization of the spinach Lhcb4 protein (CP29), a minor light-harvesting complex of Photosystem II. Eur. J. Biochem. 262:817–823. [DOI] [PubMed] [Google Scholar]
- Peterman, E. J. G., F. M. Dukker, R. van Grondelle, and H. van Amerongen. 1995. Chlorophyll a and carotenoid triplet states in light-harvesting complex II of higher plants. Biophys. J. 69:2670–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterman, E. J. G., T. Pullerits, R. van Grondelle, and H. van Amerongen. 1997. Electron-phonon coupling and vibronic fine structure of light-harvesting complex II of green plants: temperature dependent absorption and high-resolution fluorescence spectroscopy. J. Phys. Chem. B. 101:4448–4457. [Google Scholar]
- Pieper, J., J. Voigt, and G. J. Small. 1999. Chlorophyll a Franck-Condon factors and excitation energy transfer. J. Phys. Chem. B. 103:2319–2322. [Google Scholar]
- Reddy, N. R. S., H. van Amerongen, S. L. S. Kwa, R. van Grondelle, and G. J. Small. 1994. Low-energy exciton level structure and dynamics in Light-Harvesting Complex II trimers from the Chl a/b antenna complex of Photosystem II. J. Phys. Chem. 98:4729–4735. [Google Scholar]
- Remelli, R., C. Varotto, D. Sandonà, R. Croce, and R. Bassi. 1999. Chlorophyll binding to monomeric light-harvesting complex. J. Biol. Chem. 274:33510–33521. [DOI] [PubMed] [Google Scholar]
- Rogl, H., and W. Kühlbrandt. 1999. Mutant trimers of light-harvesting complex II exhibit altered pigment content and spectroscopic features. Biochemistry. 38:16214–16222. [DOI] [PubMed] [Google Scholar]
- Rogl, H., R. Schödel, H. Lokstein, W. Kühlbrandt, and A. Schubert. 2002. Assignment of spectral substructures to pigment-binding sites in higher plant light-harvesting complex LHC-II. Biochemistry. 41:2281–2287. [DOI] [PubMed] [Google Scholar]
- Ruban, A. V., A. J. Young, and P. Horton. 1996. Dynamic properties of the minor chlorophyll a/b binding proteins of photosystem II, an in vitro model for photoprotective energy dissipation in the photosynthetic membrane of green plants. Biochemistry. 35:674–678. [DOI] [PubMed] [Google Scholar]
- Salverda, J. M., F. van Mourik, G. van der Zwan, and R. van Grondelle. 2000. Energy transfer in the B800 rings of the peripheral bacterial light-harvesting complexes of Rhodopseudomonas acidophila and Rhodospirillum molischianum studied with photon echo techniques. J. Phys. Chem. B. 104:11395–11408. [Google Scholar]
- Salverda, J. M., and R. van Grondelle. 2001. Primary processes in photosynthesis studied by femtosecond nonlinear spectroscopy. In Femtochemistry. F. C. De Schryver, S. De Feyter, and G. Schweitzer, editors. Wiley-VCH, Weinheim, Germany. 399-415.
- Savikhin, S., H. van Amerongen, S. L. S. Kwa, R. van Grondelle, and W. S. Struve. 1994. Low-temperature energy transfer in LHC-II trimers from the Chl a/b light-harvesting antenna of photosystem II. Biophys. J. 66:1597–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetto, R., M. Crimi, D. Sandonà, R. Croce, G. Cinque, J. Breton, and R. Bassi. 1999. Orientation of chlorophyll transition moments in the higher-plant light-harvesting complex CP29. Biochemistry. 38:12974–12983. [DOI] [PubMed] [Google Scholar]
- Trinkunas, G., J. P. Connelly, M. G. Müller, L. Valkunas, and A. R. Holzwarth. 1997. Model for the excitation dynamics in the light-harvesting complex II from higher plants. J. Phys. Chem. B. 101:7313–7320. [Google Scholar]
- van Amerongen, H., S. L. S. Kwa, B. M. van Bolhuis, and R. van Grondelle. 1994. Polarized fluorescence and absorption of macroscopically aligned light harvesting complex II. Biophys. J. 67:837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen, H., and R. van Grondelle. 2001. Understanding the energy transfer function of LHCII, the major light-harvesting complex of green plants. J. Phys. Chem. B. 105:604–617. [Google Scholar]
- van Grondelle, R., J. P. Dekker, T. Gillbro, and V. Sundström. 1994. Energy transfer in photosynthesis. Biochem. Biophys. Acta. 1187:1–65. [Google Scholar]
- Visser, H. M., F. J. Kleima, I. H. M. van Stokkum, R. van Grondelle, and H. van Amerongen. 1996. Probing the many energy-transfer processes in the photosynthetic light-harvesting complex II at 77 K using energy-selective sub-picosecond transient absorption spectroscopy. J. Chem. Phys. 210:297–312. [Google Scholar]
- Yang, C., K. Kosemund, C. Cornet, and H. Paulsen. 1999. Exchange of pigment-binding amino acids in light-harvesting chlorophyll a/b protein. Biochemistry. 38:16205–16213. [DOI] [PubMed] [Google Scholar]
- Yu, J.-Y., Y. Nagasawa, R. van Grondelle, and G. R. Fleming. 1997. Three pulse echo peak shift measurements on the B820 subunit of LH1 of Rhodospirillum rubrum. Chem. Phys. Lett. 280:404–410. [Google Scholar]
- Zucchelli, G., F. V. Garlaschi, and R. C. Jennings. 1996. Thermal broadening analysis of the light harvesting II absorption spectrum. Biochemistry. 35:16247–16254. [DOI] [PubMed] [Google Scholar]