

Abstract
The water-proton spin-lattice relaxation rate constant, 1/T1, was measured as a function of magnetic field strength for several dilute protein solutions. By separating the intermolecular contributions from the intramolecular contributions to the water-proton spin-lattice relaxation, the number of water molecules that bind to the protein for a time long compared with the rotational correlation time may be measured. We find a good correlation between the number of long-lived water molecules and the predictions based on available free volume in the proteins studied. The rotational correlation times of these proteins are larger than predicted by the Stokes-Einstein-Debye (SED) model for a sphere reorienting in a viscous liquid. The discrepancy between experiment and theory is usually attributed to hydration effects increasing the effective radius of the particle. However, the average lifetime of water molecules at the protein interface is far too short to justify such a picture. We suggest that surface roughness may be responsible for the retardation of rotational mobility and find that the SED model provides a reasonable representation of experiment if the radius assumed for the reorienting particle is the arithmetic mean of the crystallographic packing radius and the radius deduced from the effective surface area of the protein.
INTRODUCTION
Although water has been blamed for many mysterious aspects of macromolecular chemistry, several very different experiments have demonstrated that the vast majority of water at the protein surface has short residence times that are practically limited by the diffusion of water away from the surface sites; i.e., in the range of tens to hundreds of picoseconds. However, it is also now clear that there are a few specifically bound water molecules on most proteins that have lifetimes between 0.1 ms and 10 ns. These few water molecule sites affect the nuclear spin relaxation rates of the water protons through protein-water-proton dipolar couplings, which then affect other magnetic resonance observations, most notably, the contrast in nuclear magnetic images (Bryant, 1996a; Bryant, 1996b; Denisov and Halle, 1996; Halle et al., 1999). The same dipolar couplings may affect proton nuclear Overhauser effects in protein solutions and may attenuate signal intensities in high resolution experiments.
The water-protein NMR literature includes complimentary contributions from proton, deuteron, and oxygen-17 spectroscopy. The interpretation of 1H experiments has been somewhat troubled by the effects of labile proton chemical exchange and protein aggregation, but recent instrumental advances that provide both high sensitivity and high resolution (Wagner et al., 1999) permit experiments on dilute protein solutions and high isotope dilutions of protons so that it is possible to isolate the intramolecular contributions to the proton spin-lattice relaxation rate; i.e., the contribution from dipolar interaction between water protons. (Kiihne and Bryant, 2000) Therefore, the proton experiments become practically equivalent in most ways to the more difficult oxygen or deuterium relaxation experiments except that the protons provide a considerable gain in sensitivity. Although the experiments are still technically demanding, the approach provides a means of exploring the number of water molecule binding sites for water molecule lifetimes in the range from 0.1 s to 10 ns. We report here the results of 1H magnetic relaxation dispersion (MRD) experiments for ribonuclease A and cytochrome C, which yield the numbers of long-lived water molecules. We compare the number of bound water molecules with the number predicted based on free volume calculated using structural data obtained from x-ray diffraction, and then examine the dynamical consequences of these bound water molecules on the rotational and translational dynamics of the protein.
Theoretical background
Several studies have shown that the nuclear magnetic relaxation dispersion of 1H, 2H, and 17O relaxation rate of bulk H2O or D2O is caused by a small number of water molecules bound to the protein for a time longer than the rotational correlation time of the protein (Koenig and Schillinger, 1969; Koenig et al., 1975; Halle et al., 1981; Koenig et al., 1993; Denisov and Halle, 1995; Denisov et al., 1995; Koenig, 1995; Bryant, 1996a; Denisov and Halle, 1996; Halle et al., 1999). Indeed, if these bound water molecules exchange with the bulk solvent with a rate that is fast compared to the spin-lattice relaxation rate of the bound water protons, the protein-bound-water-molecule sites act as relaxation sinks for the whole water population. In the case of dilute protein solutions where protein aggregation is minimized, the 1H dispersion in the spin-lattice relaxation rate constant has a Lorentzian shape that permits accurate measurement of the rotational correlation time of the macromolecule, τrot (Kiihne and Bryant, 2000). The observed 1H spin-lattice relaxation rate constant may be written (Denisov and Halle, 1998; Kiihne and Bryant, 2000):
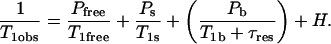 |
(1) |
1/T1free denotes the intramolecular H-H or H-D contributions of the free solvent H2O or HOD molecules and Pfree is essentially unity. The correlation times for reorientation of water in the bulk are short compared with the proton Larmor frequencies over the range studied, 0.01–300 MHz; thus, 1/T1free is independent of magnetic field strength over this range. The second term, 1/T1s, comes from the hundreds of water molecules at the surface of the protein characterized by a probability Ps. These molecules generally have a residence time much shorter than the rotational correlation time of the protein, and the relaxation dispersion for this contribution occurs above the largest magnetic field strength studied. Therefore, these short-lived surface interactions also add to the field independent relaxation rate. Pb is the probability that water molecules are bound to the protein for times the order of or longer than the rotational correlation time, and may be expressed in terms of the number of bound water molecules per protein molecule, Nb, and 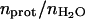 , the ratio of the number of protein molecules to the total number of water molecules:
, the ratio of the number of protein molecules to the total number of water molecules:
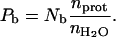 |
(2) |
We have assumed in Eq. 1 that a single spin-lattice relaxation time, T1b, characterizes the bound environment. The mean residence time for water bound on the protein, τres, may be different for different sites on the protein, but its contribution is negligible if 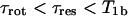 , where τrot is the rotational correlation time of the protein.
, where τrot is the rotational correlation time of the protein.
The last term in Eq. 1, H, represents the contribution from the exchange between protein ionizable groups and the protons of the solvent, which may be expressed as
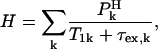 |
(3) |
where the index k runs over all the protein-proton exchange sites occupied with a probability 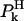 and characterized by a relaxation time,
and characterized by a relaxation time, 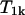 , which is field dependent, and mean residence time,
, which is field dependent, and mean residence time, 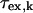 . This contribution is generally a function of temperature and pH; it is often small because the mean residence times for many sites may be long relative to the relaxation times at the site, amide protons for example. The contribution of this term is independent of proton mole fraction and contributes to relaxation in the same way as the intermolecular contribution to 1/T1b, which is discussed below.
. This contribution is generally a function of temperature and pH; it is often small because the mean residence times for many sites may be long relative to the relaxation times at the site, amide protons for example. The contribution of this term is independent of proton mole fraction and contributes to relaxation in the same way as the intermolecular contribution to 1/T1b, which is discussed below.
The bound water rate constant, 1/T1b, may be decomposed as the sum of intermolecular and intramolecular dipolar contributions. In the H2O case, both the intramolecular (water proton-water proton) and intermolecular (water proton-protein proton) contributions are homonuclear and 1/T1b may be written (Abragam, 1961):
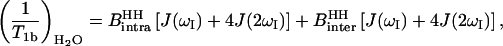 |
(4) |
where 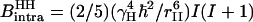 characterizes the strength of the intramolecular dipole-dipole contribution of a proton pair separated by rII, which is 1.58 Å in the water molecule.
characterizes the strength of the intramolecular dipole-dipole contribution of a proton pair separated by rII, which is 1.58 Å in the water molecule. 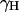 is the magnetogyric ratio of the proton, ℏ the Planck constant divided by 2π, and
is the magnetogyric ratio of the proton, ℏ the Planck constant divided by 2π, and 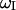 the proton Larmor frequency.
the proton Larmor frequency. 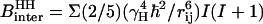 is the intermolecular dipolar proton-proton contribution, which involves several proton-proton contacts characterized by different intermoment distances rij. The minimum separation is determined by the van der Waals contact distance of 2.2 Å but a wide range of interproton distances may contribute.
is the intermolecular dipolar proton-proton contribution, which involves several proton-proton contacts characterized by different intermoment distances rij. The minimum separation is determined by the van der Waals contact distance of 2.2 Å but a wide range of interproton distances may contribute. 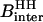 and
and 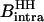 may contain an order parameter,
may contain an order parameter, 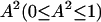 , that may account for partial averaging of the dipolar interactions caused by high frequency motions of the bound water molecules. As previously noted (Denisov and Halle, 1998; Kiihne and Bryant, 2000), these relaxation dispersion experiments in the present field strength range do not provide a characterization of such high frequency motions, i.e., A2. For simplicity we set the order parameter to 1 and make our calculations based on the assumption of rigidly bound water molecules.
, that may account for partial averaging of the dipolar interactions caused by high frequency motions of the bound water molecules. As previously noted (Denisov and Halle, 1998; Kiihne and Bryant, 2000), these relaxation dispersion experiments in the present field strength range do not provide a characterization of such high frequency motions, i.e., A2. For simplicity we set the order parameter to 1 and make our calculations based on the assumption of rigidly bound water molecules. 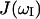 denotes the spectral density of magnetic fluctuations at
denotes the spectral density of magnetic fluctuations at 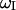 . Assuming a single global correlation time for rotational diffusion, the spectral density function,
. Assuming a single global correlation time for rotational diffusion, the spectral density function, 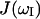 has the Lorentzian form (Abragam, 1961)
has the Lorentzian form (Abragam, 1961)
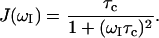 |
(5) |
In the D2O solutions of proteins, the intermolecular contribution to the water 1H spin-lattice relaxation rate comes from the residual HOD protons coupling to protein protons.
The intramolecular contribution to proton relaxation caused by deuterons is heteronuclear and given by
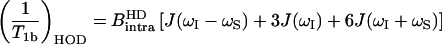 |
(6) |
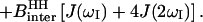 |
where 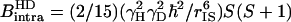 , rIS is the proton-deuteron distance and S=1 for deuterium. In Eq. 4–6 we have neglected the intermolecular interaction between water protons and protein-deuterons because
, rIS is the proton-deuteron distance and S=1 for deuterium. In Eq. 4–6 we have neglected the intermolecular interaction between water protons and protein-deuterons because 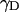 is small compared to
is small compared to 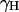 and the protein-deuteron is relatively rare.
and the protein-deuteron is relatively rare.
Both in the H2O and D2O cases, the strength of the intermolecular contribution to the relaxation, 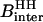 , involves two terms that may be written
, involves two terms that may be written
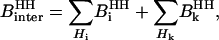 |
(7) |
where 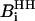 represents the dipolar interaction between a nonexchangeable protein proton,
represents the dipolar interaction between a nonexchangeable protein proton, 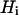 , and a water proton; the sum runs over all the nonexchangeable protein protons.
, and a water proton; the sum runs over all the nonexchangeable protein protons. 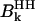 represents the strength of the dipolar interaction between a labile protein proton and a protein-bound water proton, which decreases linearly with the proton mole fraction,
represents the strength of the dipolar interaction between a labile protein proton and a protein-bound water proton, which decreases linearly with the proton mole fraction, 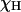 .
.
To estimate the contribution of the second term of Eq. 7, we have used the coordinates based on the crystal structure of ribonuclease A (Wlodawer et al., 1988) to compute the ratio of the dipolar interaction between a fixed water proton and all the nonexchangeable protein protons to that of the same fixed water proton and all the protein protons, labile or not. This calculation demonstrates that ∼10% of the coupling derives from the labile protein protons that are displaced in D2O solutions. In the following we neglect the intermolecular contribution coming from the labile protein-protons and discuss the error introduced by this term in the determination of the number of long-lived bound water molecules later.
At low values of the Larmor frequency, Eqs. 4–6 reduce to
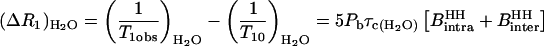 |
(8) |
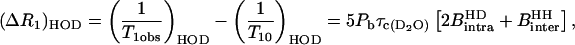 |
(9) |
where 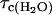 and
and 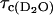 are the rotational correlation times of the protein in H2O and D2O solutions respectively, which are different because the viscosities differ by ∼20%.
are the rotational correlation times of the protein in H2O and D2O solutions respectively, which are different because the viscosities differ by ∼20%.
For both solvents, the intramolecular contribution of the bound water molecules involves a single interspin distance of 1.58 Å; then 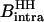 and
and 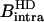 differ by the factor α
differ by the factor α
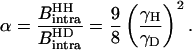 |
(10) |
We then have the coupled equations
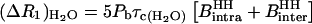 |
(11) |
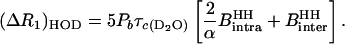 |
(12) |
Let 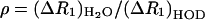 , the ratio of the difference between the high and low field relaxation rate constants in the two solvents, and
, the ratio of the difference between the high and low field relaxation rate constants in the two solvents, and 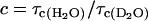 the ratio of the rotational correlation time in the two solvents. Combining Eqs. 8–12, and solving for Pb yields
the ratio of the rotational correlation time in the two solvents. Combining Eqs. 8–12, and solving for Pb yields
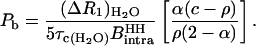 |
(13) |
The numerical value of 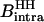 is calculated using 1.58 Å for the interproton distance in the H2O molecule and is equal to 9.7226 109 s−2. Knowing all the other experimentally determined parameters, the number of long-lived bound water molecules is computed using Eq. 2.
is calculated using 1.58 Å for the interproton distance in the H2O molecule and is equal to 9.7226 109 s−2. Knowing all the other experimentally determined parameters, the number of long-lived bound water molecules is computed using Eq. 2.
Aside from measurement noise, there are two sources of error in the calculation of the number of bound water molecules. The first comes from the intermolecular dipoledipole contribution of the labile protein protons, which may cause an overestimate of Nb by 10% at maximum. The second is caused by the uncertainty in the order parameter, A2, set to 1 in the above calculation. The quantity that we compute rigorously from our measurements is 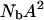 . If
. If 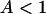 , the number of bound water molecules, Nb, increases as
, the number of bound water molecules, Nb, increases as 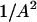 . The reduction of A is caused by restricted high frequency local motions that would, in principle, cause a second relaxation dispersion at very high field strengths. However, if the local correlation time is short, in the range of tens of picoseconds, for example, the contribution to the relaxation rate would be ∼1000 times smaller than that from lower frequency motions. Thus, the primary effect of local motion of a bound water molecule in the binding site is reduction of the low field relaxation rate by the factor, A2.
. The reduction of A is caused by restricted high frequency local motions that would, in principle, cause a second relaxation dispersion at very high field strengths. However, if the local correlation time is short, in the range of tens of picoseconds, for example, the contribution to the relaxation rate would be ∼1000 times smaller than that from lower frequency motions. Thus, the primary effect of local motion of a bound water molecule in the binding site is reduction of the low field relaxation rate by the factor, A2.
EXPERIMENTAL
Bovine pancreas ribonuclease A (R5555), bovine heart cytochrome C (C3131), bovine pancreas α-chymotrypsin (C4129), pepsin (6887), and thermolysin (protease type X P1512) were purchased from Sigma (St Louis, MO) as lyophilized powders. H2O and D2O solutions were made by dissolving the lyophilized proteins in dionized water and deuterium oxide (D, 99.9% Cambridge Isotope Laboratories, Andover, MA) respectively. For α-chymotrypsin and Thermolysin the ionic strength was maintained using 100 mM potassium chloride. Before NMR measurements, the H2O and D2O protein solutions were deoxygenated with a flowing nitrogen stream for one hour, to remove the paramagnetic contribution of the dissolved O2 to the proton relaxation rate.
Deoxygenated samples were sealed in a 5 mm o.d. glass sample tube utilizing a Delrin filler plug and a silicone rubber plug compressed between two threaded components similar to the design reported previously (Wagner et al., 1999). The glass tube is far superior to the Delrin tube because it does not leak oxygen as a function of time and is chemically much more inert. The MRD measurements were made in a dual magnet spectrometer described elsewhere (Wagner et al., 1999). The sample is allowed to achieve equilibrium in the high initial magnetic field, then pneumatically driven to a satellite magnet where it resides for variable relaxation period after which it is pneumatically driven back to the high field magnet where the magnetization is detected. The decay of the magnetization as a function of residence time in the satellite field is fit to an exponential time constant that is the relaxation time constant in the satellite field. The value of the satellite field strength is varied to map the relaxation dispersion over the range of proton Larmor frequencies from 0.01 to 70 MHz. The soak field with a proton Larmor frequency of 300 MHz provides the highest field relaxation rate constant.
RESULTS
Fig. 1 presents the relaxation dispersion curves obtained in H2O and D2O for ribonuclease A, 1.2 mM, and 0.60 mM respectively at pH = 5.3. The two relaxation dispersions are Lorentzian. The fits according to Eq. 6 for the H2O case and Eq. 8 for the D2O case correspond to the solid lines and lead to a correlation time 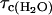 = 3.2 ± 1.3 ns for the H2O solution and
= 3.2 ± 1.3 ns for the H2O solution and 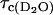 = 4.5 ± 0.7 ns in the D2O case. We note that the viscosity in D2O is larger than that in H2O by 20%, which is only approximately consistent with the larger correlation time found in the D2O solution and is discussed below.
= 4.5 ± 0.7 ns in the D2O case. We note that the viscosity in D2O is larger than that in H2O by 20%, which is only approximately consistent with the larger correlation time found in the D2O solution and is discussed below.
FIGURE 1.


z(Top) The 1H spin-lattice relaxation rate constant for the residual protons, 1/T1, are shown as a function of the magnetic field strength plotted as the proton Larmor frequency for a 0.60 mM solution of ribonuclease A in D2O at ambient laboratory temperature at a pH meter reading of 5.2. (Bottom) The 1H spin-lattice relaxation rate constant, 1/T1, shown as a function of the magnetic field strength plotted as the proton Larmor frequency for 1.2 mM solution of ribonuclease A in H2O at pH 5.2 and ambient laboratory temperature.
The amplitudes of the dispersion in the relaxation rate constant used for the calculation of the number of bound water molecules, 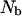 , are
, are 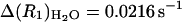 and
and 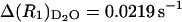 when both data sets are normalized to 0.60 mM concentration of ribonuclease A. Knowing the value of
when both data sets are normalized to 0.60 mM concentration of ribonuclease A. Knowing the value of 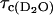 and considering the ratio
and considering the ratio 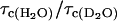 , we find an intramolecular contribution to the relaxation of 30%. According to Eqs. 2 and 15 we find
, we find an intramolecular contribution to the relaxation of 30%. According to Eqs. 2 and 15 we find 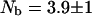 . The neglect of the dipolar contribution from labile protons causes this analysis to over estimate the number of bound water molecules by ∼10%; thus,
. The neglect of the dipolar contribution from labile protons causes this analysis to over estimate the number of bound water molecules by ∼10%; thus, 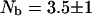 which is not significantly different. Three internal water molecules have been reported based on x-ray diffraction data (Denisov and Halle, 1998). Rashin and coworkers report that there is space in the protein for 2 ± 1 internal water molecules based on calculations of free volume deduced from packing in the reported crystal structure.
which is not significantly different. Three internal water molecules have been reported based on x-ray diffraction data (Denisov and Halle, 1998). Rashin and coworkers report that there is space in the protein for 2 ± 1 internal water molecules based on calculations of free volume deduced from packing in the reported crystal structure.
The value of the rotational correlation time for the water protons associated with ribonuclease A is short compared with expectations based on molecular volume and other measures of the protein reorientation time (Denisov and Halle, 1998). As pointed out by Denisov and Halle, the origin of this apparent discrepancy may derive from the interference between the rotational motion and the exchange of the water from the ribonuclease A binding environments. The water sites for long-lived water molecules on ribonuclease A are on surface pockets or crevasses not buried deeply inside the folded structure. The 17O relaxation dispersion data agree with the proton relaxation dispersion data and imply that the rotational correlation time and the exchange times are of nearly the same size. Because the exchange event is uncorrelated with rotational diffusion, we may write the effective correlation time as
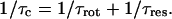 |
(14) |
If we assume that the rotational correlation is 6.6 ns as reported by Denisov and Halle based on deuterium relaxation data, which is also in agreement with the calculation in Table 1 below based on molecular volume, then substitution of the measured rotational correlation time in Eq. 14 yields a value of 6.2 ns for the mean residence time of these bound water molecules. This value is in reasonable agreement with the value of 7.6 ns at 27°C reported by Denisov and Halle (1998). One consequence of the short water-molecule residence times on ribonuclease A is that the deuterium and proton MRD inflections points are not simply related to the solution viscosity. In the deuterium case, the residual water-proton relaxation rate results from the sum of water-proton to protein-proton intermolecular contributions and from the exchange of labile protein protons with the water. The effective correlation time for the first contribution is reduced from that for pure rotation because of the contribution of the short lifetime of the water on the protein according to Eq. 14. For the labile protein proton contribution, the effective correlation time is just the rotational correlation time of the protein. Because the weights of these contributions are different when the proton-proton intramolecular term is added for the bound H2O molecule, the observed MRD inflection frequencies are not simply proportional to the viscosities of the solutions.
TABLE 1.
Correlation time comparison
| Protein | Mw (kDa) | RV (nm) | RS (nm) | Rav (nm) | τth (ns) | τc1 (ns) |
|---|---|---|---|---|---|---|
| α-Chymotrypsin | 25.3 | 1.97 | 2.76 | 2.37 | 13.4 | 13.9 |
| Ribonuclease A | 12.64 | 1.57 | 2.19 | 1.88 | 6.7 | 3.2 |
| Cytochrome C | 12.4 | 1.56 | 2.18 | 1.87 | 6.6 | 6.6 |
| Thermolysin | 34 | 2.18 | 3.84 | 2.62 | 18.2 | 17.0 |
| Pepsin | 35.5 | 2.21 | 3.05 | 2.65 | 18.8 | 18.8 |
| BSA | 68 | 2.74 | 3.84 | 3.29 | 36.3 | 41 |
Comparison between the computed rotational correlation time, 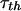 , obtained from the Stokes-Einstein-Debye relation and the experimental rotational correlation time,
, obtained from the Stokes-Einstein-Debye relation and the experimental rotational correlation time, 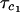 , deduced from nuclear magnetic relaxation dispersion measurements. The radius used for the SED equation is the arithmetic mean of two measures of the effective protein radius. The first, RS, is the effective radius of the sphere that has the same surface area as the surface area of the protein deduced using the methods of Lee and Richards. The second, RV, is the radius deduced from the protein-protein contacts in the protein crystal. We list the molecular mass, Mw, for reference. The experimental value
, deduced from nuclear magnetic relaxation dispersion measurements. The radius used for the SED equation is the arithmetic mean of two measures of the effective protein radius. The first, RS, is the effective radius of the sphere that has the same surface area as the surface area of the protein deduced using the methods of Lee and Richards. The second, RV, is the radius deduced from the protein-protein contacts in the protein crystal. We list the molecular mass, Mw, for reference. The experimental value 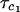 for bovine serum albumin is extracted from previous work (Kiihne and Bryant, 2000). The comparisons are made for H2O solutions with η = 0.01 poise for T = 300 K.
for bovine serum albumin is extracted from previous work (Kiihne and Bryant, 2000). The comparisons are made for H2O solutions with η = 0.01 poise for T = 300 K.
The same experiments and calculations were made for cytochrome C at 0.6 mM and pH = 8. The intramolecular contribution to the relaxation is 64% and we find 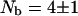 bound water molecules whereas the free volume analysis cited suggests that there should be two internal water molecules (Rashlin et al., 1986). Similar results were obtained for dilute solutions of thermolysin (0.040 mM, pH = 6) and pepsin (0.28 mM, pH = 7). The high field 1H relaxation rate dispersions obtained in H2O solutions yield rotational correlation times
bound water molecules whereas the free volume analysis cited suggests that there should be two internal water molecules (Rashlin et al., 1986). Similar results were obtained for dilute solutions of thermolysin (0.040 mM, pH = 6) and pepsin (0.28 mM, pH = 7). The high field 1H relaxation rate dispersions obtained in H2O solutions yield rotational correlation times 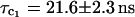 for thermolysin and
for thermolysin and 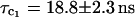 for pepsin. We discuss in the following section the magnitudes of these values in the context of prevalent ideas about protein volume and hydration.
for pepsin. We discuss in the following section the magnitudes of these values in the context of prevalent ideas about protein volume and hydration.
Fig. 2 shows the dispersion of the 1H relaxation rate constant obtained for a H2O solution of α-chymotrypsin 0.3 mM, in 100 mM KCl and pH = 6.9. Contrary to the ribonuclease A and cytochrome C results and those previously obtained in BSA (Kiihne and Bryant, 2000), the dispersion shape is not described by a single Lorentzian function. We have fitted this dispersion curve as the sum of two Lorentzian contributions to obtain the solid line; the correlation times are τc1 = 13.4 ± 1.2 ns and τc2 = 509 ± 112 ns. We attribute 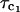 to the rotational correlation time of the monomeric protein. The value of τc2 is large and we attribute that to a low concentration of an impurity with large molecular mass. We note that the low field contributions to the relaxation rates from the particles of different size scale with the ratios of the rotational correlation times. Thus, the observed low field relaxation rate is consistent with only 2.6% of the bound water molecule sites derived from the larger molecule.
to the rotational correlation time of the monomeric protein. The value of τc2 is large and we attribute that to a low concentration of an impurity with large molecular mass. We note that the low field contributions to the relaxation rates from the particles of different size scale with the ratios of the rotational correlation times. Thus, the observed low field relaxation rate is consistent with only 2.6% of the bound water molecule sites derived from the larger molecule.
FIGURE 2.

The 1H spin-lattice relaxation rate constant, 1/T1, as a function of the magnetic field strength plotted as the proton Larmor frequency for a 0.30 mM solution of α-chymotrypsin in H2O at pH 6.9 in 100 mM potassium chloride.
Rotational correlation times and protein hydration
As we show above, the MRD provides a direct report of the rotational motility of a protein as well as a quantitative measure of the number of long-lived water molecules that are associated with the protein. It is really an old (Koenig and Schillinger, 1969) but still remarkable result that the number of long-lived water molecules that hydrate proteins is a very small fraction of the total number of water molecules that are in contact with the protein. The vast majority of the surface contacts between the water and the protein are transient and characterized by short lifetimes, in the range of a few hundreds of picoseconds or shorter (Koenig, 1995; Bryant, 1996; Halle et al., 1999). Recognizing this fact, it is useful to compare the measured rotational correlation times with values predicted based on hydrodynamic theory. Proteins are large molecules that should be appropriate to Stokes-Einstein-Debye theory, which predicts that the rotational correlation time, 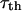 , is proportional to molecular volume and the viscosity, η,
, is proportional to molecular volume and the viscosity, η,
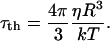 |
(15) |
As commonly noted, the experimental values of τrot for proteins are larger than predicted by this relation and solvation has been blamed for the discrepancy (Yguerabide et al., 1970). A standard approach to understanding the failure of experiments to agree with theory is to ascribe a solvation layer of water molecules to the water-protein interface so that the effective size of the reorientational unit is larger than the volume of the protein presumed to be spherical. However, as the present and other measurements demonstrate, the mean residence time of water at the protein surface is compared with the rotational correlation time of the protein. This fact compromises the model that the hydration layer increases the effective radius of the protein and slows the rotational motion.
An alterative hypothesis is that protein surface roughness retards the rotation of the protein (Garcia de la Torre and Bloomfield, 1981; Denisov and Halle, 1998). Indeed, the protein surface is not a smooth sphere when the different side chains are considered. One approach for measuring this roughness quantitatively is to compare radii computed from the effective surface area with that based on packing volume. The packing volume in a crystal provides a measure of the effective molecular volume from which a radius 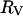 may be computed. Empirical relation between the molecular weight,
may be computed. Empirical relation between the molecular weight, 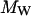 , and the radius
, and the radius 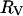 has been offered:
has been offered: 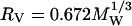 (Richards, 1977). This radius predicts a rotational correlation times that is smaller than that observed experimentally as shown in Table 1. An alternative way to consider the size of the protein is to examine the effective surface area S as considered by Lee and Richards using a probe molecule like water (Lee and Richards, 1971). This surface area may be translated to an effective spherical radius,
(Richards, 1977). This radius predicts a rotational correlation times that is smaller than that observed experimentally as shown in Table 1. An alternative way to consider the size of the protein is to examine the effective surface area S as considered by Lee and Richards using a probe molecule like water (Lee and Richards, 1971). This surface area may be translated to an effective spherical radius, 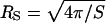 . For the proteins studied here, the value of
. For the proteins studied here, the value of 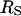 is considerably larger than
is considerably larger than 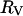 and if
and if 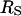 is used in Eq. 15, values of the rotational correlation time much larger than that observed experimentally are obtained.
is used in Eq. 15, values of the rotational correlation time much larger than that observed experimentally are obtained.
Although we have no fundamental or detailed theoretical justification for it, we find that a remarkably simple strategy provides an alternative approach to computing rotational correlation times for globular proteins. If we take the arithmetic mean between 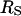 and
and 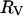 to approximate the reorientational sphere in Eq. 15, reasonable agreement with the experiment is obtained as shown in Table 1. This procedure increases the effective reorientational radius by the factor 1.20 for the proteins listed. Although the concept of a protein as a smooth but enlarged sphere is difficult to defend, an alternative interpretation of this factor is that it represents the effective surface friction coefficient that is different from unity. The essence of the difference between this approach and assuming a bound hydration layer is that it springs from a reasonable physical picture of the macromolecule and avoids the unjustified assumption of ice-like water bound at the surface of the protein.
to approximate the reorientational sphere in Eq. 15, reasonable agreement with the experiment is obtained as shown in Table 1. This procedure increases the effective reorientational radius by the factor 1.20 for the proteins listed. Although the concept of a protein as a smooth but enlarged sphere is difficult to defend, an alternative interpretation of this factor is that it represents the effective surface friction coefficient that is different from unity. The essence of the difference between this approach and assuming a bound hydration layer is that it springs from a reasonable physical picture of the macromolecule and avoids the unjustified assumption of ice-like water bound at the surface of the protein.
References
- Abragam, A. 1961. The Principles of Nuclear Magnetism, Chapter VIII. The Clarendon Press, Oxford.
- Bryant, R. G. 1996a. The dynamics of water-protein interactions. Annu. Rev. Biophys. Biomol. Struct. 25:29–53. [DOI] [PubMed] [Google Scholar]
- Bryant, R. G. 1996b. Magnetization transfer and cross relaxation in tissue. In Encyclopedia of Magnetic Resonance. E. D. M. Grant and R. K. Harris, Editors. John Wiley, New York. 2954–2962.
- Denisov, V. P., and B. Halle. 1995. Protein hydration dynamics in aqueous solution: a comparison of bovine pancreatic trypsin inhibitor and ubiquitin by oxygen-17 spin relaxation dispersion. J. Mol. Biol. 245:682–697. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., and B. Halle. 1996. Protein hydration dynamics in aqueous solution. Faraday Discuss. 103:227–244. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., and B. Halle. 1998. Thermal denaturation of ribonuclease A characterized by water oxygen-17 and deuterium magnetic relaxation dispersion. Biochemistry. 37:9595–9604. [DOI] [PubMed] [Google Scholar]
- Denisov, V. P., B. Halle, J. Peters, and H. D. Horlein. 1995. Residence times of buried water molecules in bovine pancreatic trypsin inhibitor and its G36S mutant. Biochemistry. 34:9046–9051. [DOI] [PubMed] [Google Scholar]
- Garcia de la Torre, J. G., and V. A. Bloomfield. 1981. Hydrodynamic properties of complex, rigid, biological macromolecules: theory and applications. Q. Rev. Biophys. 14:81–139. [DOI] [PubMed] [Google Scholar]
- Halle, B., T. Anderson, S. Forsen, and B. Lindman. 1981. Protein hydration from water oxygen-17 magnetic relaxation. J. Am. Chem. Soc. 103:500–508. [Google Scholar]
- Halle, B., V. P. Denisov, and K. Venu. 1999. Multinuclear relaxation dispersion studies of protein hydration. In Biological Magnetic Resonance, Vol. 17. L. J. Berliner and N. R. Krishna, editors. Klewer Academic/Plenum Press, New York. 419–484.
- Kiihne, S., and R. G. Bryant. 2000. Protein-bound water molecule counting by resolution of (1)H spin- lattice relaxation mechanisms. Biophys. J. 78:2163–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig, S. H. 1995. Classes of hydration sites at protein-water interfaces: the source of contrast in magnetic resonance imaging. Biophys. J. 69:593-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig, S. H., R. D. I. Brown, and R. Ugolini. 1993. A unified view of relaxation in protein solutions and tissue including hydration and magnetization transfer. Magn. Reson. Med. 29:77–83. [DOI] [PubMed] [Google Scholar]
- Koenig, S. H., K. Hallenga, and M. Shporer. 1975. Protein-water interactions studied by solvent proton, deuteron, and oxygen-17 magnetic relaxation. Proc. Natl. Acad. Sci. USA. 72:2667–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig, S. H., and W. E. Schillinger. 1969. Nuclear magnetic relaxation dispersion in protein solutions. I. Apotransferrin. J. Biol. Chem. 244:3283–3289. [PubMed] [Google Scholar]
- Lee, B., and F. M. Richards. 1971. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55:379–400. [DOI] [PubMed] [Google Scholar]
- Rashlin, A. A., M. Iofin, and B. Honig. 1986. Internal cavities and buried waters in globular proteins. Biochemistry. 25:3619–3625. [DOI] [PubMed] [Google Scholar]
- Richards, F. M. 1977. Areas, volumes, packing and protein Structure. Annu. Rev. Biophys. Bioeng. 6:151–176. [DOI] [PubMed] [Google Scholar]
- Wagner, S., T. J. R. Dinesen, T. Rayner, and R. G. Bryant. 1999. Magnetic relaxation dispersion measurements of solute spin probes using a dual magnet system. J. Magn. Reson. 140:172–178. [DOI] [PubMed] [Google Scholar]
- Wlodawer, A., L. A. Svensson, L. Sjoln, and G. L. Gilleeland. 1988. Structure of phosphate free ribonuclease A refined to 1.26 A. Biochemistry. 27:2705–2727. [DOI] [PubMed] [Google Scholar]
- Yguerabide, J., H. Epstein, and L. Stryer. 1970. Segmental flexibility in an antibody molecule. J. Mol. Biol. 51:573–590. [DOI] [PubMed] [Google Scholar]