

Abstract
We report here the first direct measurements of changes in protein hydration triggered by a functional binding. This task is achieved by weighing hemoglobin (Hb) and myoglobin films exposed to an atmosphere of 98% relative humidity during oxygenation. The binding of the first oxygen molecules to Hb tetramer triggers a change in protein conformation, which increases binding affinity to the remaining empty sites giving rise to the appearance of cooperative phenomena. Although crystallographic data have evidenced that this structural change increases the protein water-accessible surface area, isobaric osmotic stress experiments in aqueous cosolutions have shown that water binding is linked to Hb oxygenation. Now we show that the differential hydration between fully oxygenated and fully deoxygenated states of these proteins, determined by weighing protein films with a quartz crystal microbalance, agree with the ones determined by osmotic stress in aqueous cosolutions, from the linkage between protein oxygen affinity and water activity. The agreements prove that the changes in water activity brought about by adding osmolytes to the buffer solution shift biochemical equilibrium in proportion to the number of water molecules associated with the reaction. The concomitant kinetics of oxygen and of water binding to Hb have been also determined. The data show that the binding of water molecules to the extra protein surface exposed on the transition from the low-affinity T to the high-affinity R conformations of hemoglobin is the rate-limiting step of Hb cooperative reaction. This evidences that water binding is a crucial step on the allosteric mechanism regulating cooperative interactions, and suggests the possibility that environmental water activity might be engaged in the kinetic control of some important reactions in vivo.
INTRODUCTION
Water molecules play an active role in the regulation of protein and enzymatic reactivity. This action occurs because of the energies associated with the solvation/desolvation of macromolecular surfaces that change solvent accessibility upon reaction (Colombo et al., 1992; Eisenberg and McLachlan, 1986). Hence, part of the integral free energy change determining the equilibrium and kinetic properties of biochemical reactions comes from the differential hydration between products and reactants. This partial contribution, i.e., the free energy due to hydration changes, accounts for the work of transfer water molecules from the bulk plus that of binding to the new exposed hydration sites and vice versa. Because of that, the reaction equilibrium position is affected by changes in the bathing water activity, aw, in proportion to the differential hydration between products and reactants (Colombo et al., 1992; Tanford, 1969). Although this knowledge has been available since Tanford's original analysis of linkage effects of aw solution on biochemical equilibrium (Tanford, 1969), only recently aw solution was recognized as a practical thermodynamic variable for the experimental determination of hydration changes regulating biochemical reactions in aqueous solution (Colombo et al., 1992; Kornblatt and Hoa, 1990; Parsegian et al., 1995). The proposal of osmotic stress strategy has greatly contributed to that. In this strategy, the equilibrium of the reaction is displaced by the independent change in aw solution achieved by the addition of osmolytes to the buffer solution, and the Δnw associated with the reaction is determined using Wyman linkage equations (Colombo et al., 1992; Parsegian et al., 1995). Osmolytes, such as sugars and polyols, are used in these experiments inasmuch as most often these solutes are preferentially excluded from macromolecular surfaces (Gekko and Timasheff, 1981), avoiding direct binding interactions between the aw depressant and the reacting species (Colombo et al., 1992; Kornblatt and Hoa 1990; Parsegian et al., 1995). An important finding, which first supported the use of aw as a variable for the determination of the reactive changes in macromolecular hydration, was the demonstration that the hydration change associated with hemoglobin (Hb) oxygenation determined by osmotic stress finds agreement with the computed differences in water-accessible surface areas computed from the crystallographic structures of deoxy-Hb and oxy-Hb (Colombo et al., 1992).
Since its proposal, the osmotic stress strategy has been used to probe water binding associated with many different biochemical reactions. After the original studies of water effects on allosteric regulation (Colombo et al., 1992) and on the electron transfer reaction in the cytocrome-c oxidase system (Kornblatt and Hoa, 1990), changes in hydration have been observed to be associated with cooperative binding to a dimeric Hb (Royer et al., 1996) and to aspartate transcarbamylase (LiCata and Allewell, 1998); substrate binding to enzymes (Swaminathan et al., 1998); protein and drug binding to DNA (Lynch and Sligar, 2000; Ruggiero Neto and Colombo, 2000; Sidorova and Rau, 2000; Vossen et al., 1997); DNA helix transition (Preisler et al., 1995; Ruggiero Neto et al., 2001); and antibody to antigen association (Xavier et al., 1997), to name but a few representative examples. These and many other osmotic stress studies have accumulated evidence for the role of water activity in regulating the thermodynamics of biochemical reactions via differential hydration. Some of these studies, alike in Hb, have also shown agreement between osmotic stress measurements of Δnw and that derived from crystallographic information. Nonetheless, the thermodynamic interpretation of osmolyte effects on biochemical equilibrium in terms of changes in water binding has been the target of recent criticism (Davis-Searles et al., 2001; Timasheff, 1998).
The presumed weakness of the osmotic stress strategy is that it probes the contribution of water binding to biochemical equilibrium indirectly, via solute effects on the chemical potential of water in solution. A direct experimental determination of the hydration changes associated with a biochemical reaction, which could be critically compared with osmotic stress results, is certainly required. Such measurements would not only contribute to solve interpretative disagreements about the physical causes of solute effects on macromolecular conformational stability in cosolutions, but also would provide further information on the energetic and mechanistic contribution of hydration to protein function.
In this work we have investigated the role of water on Hb and myoglobin (Mb) function using a quartz crystal microbalance (QCM) (Kennerly, 1969) to measure oxygen-linked changes in protein hydration. Previous osmotic stress studies have shown that human Hb oxygenation in salt solutions is followed by the binding of ∼72 water molecules (Colombo and Bonilla-Rodriguez 1996; Colombo et al., 1992), whereas in salt-free buffers only ∼25 water molecules follow the transition from deoxy-Hb to oxy-Hb (Colombo and Seixas, 1999). Contrary to that, Mb does not change hydration upon oxygenation (Colombo et al., 1992). These studies have also suggested that deoxy-Hb changes allosteric state upon anion binding, and that ∼45–50 water molecules bind to deoxy-Hb upon chloride release (Colombo and Seixas, 1999). Now we have used a QCM to weigh the mass of Hb and Mb in films equilibrated with a constant water vapor activity, and the mass changes induced by O2 uptake. The equilibrium measurements of Hb and Mb hydration at different states of ligation confirm the earlier osmotic stress results. This gives unprecedented support to the current interpretation of osmotic stress experiments in terms of water differential binding. Although the focus of our analysis will be on the kinetics of protein hydration, we note that the results presented in this work suggest a reevaluation of the potential physical causes for the enhanced stability of protein and enzymes in cosolutions of natural osmolytes (Yancey et al., 1982). This issue has been a matter of recent debate (Parsegian et al., 2000) and even of strong disagreements (Davis-Searles et al., 2001; Timasheff 1998). Our direct measurements shall solve some of these disagreements. We also present the first measurements of kinetic changes in protein hydration triggered by functional binding. These data reveal that water binding is coupled to the rate-limiting step of Hb cooperative reaction, evidencing that water molecules contribute to the allosteric mechanism of Hb oxygenation through binding to the R-like transition state of the protein. Thus we present evidence that water molecules play a role on the kinetic mechanism of protein allosteric regulation.
MATERIALS AND METHODS
HbA0 was purified chromatographically from human blood, stripped of organic and inorganic ions by several passages through an Amberlite MB-1 column, concentrated to ∼8 mM Hb by centrifugation, and stored in liquid N2 until use (Colombo and Bonilla-Rodriguez, 1996). Hb samples consist in a solution ∼7 mM protein, with or without 200 mM NaCl, 10 mM Hepes (N-[2-hydroxyethyl] piperazine-N′-[2-ethane sulfonic acid]), pH 7. Ferro-Mb was obtained by anaerobic reduction of sperm whale metmyoglobin (Sigma) with sodium dithionite, which is removed by size exclusion chromatography. Mb samples consist in a solution ∼5 mM in the same buffer as Hb. All chemicals were from Sigma Chemical, St. Louis, MO.
The hydration of Hb and Mb films was determined with a QCM. The resonance frequency f of the quartz crystal resonator plate (AT-cut) is extremely sensitive to changes on the mass, Δm, of films deposited on the area A of the crystal electrodes. The change on the mass deposited on the quartz crystal with area A and density ρq causes a change in frequency 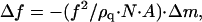 where f is the resonance frequency of the clean crystal and N a constant. For the quartz crystals used in this work (Cristales Argentinos, S.A.), f = 6000 kHz, A = 1.266 cm2, N = 167 cm kHz, and ρq = 2.65g cm−3, which determines a sensitivity of 20 ng per Hz. The QCM was mounted into the closed cell (Fig. 1). The cell is positioned in a Cary 3E UV-Vis spectrophotometer, and the quartz crystal connected to a resonator and to a frequency meter that displays frequency to 1 Hz. The films were equilibrated with an inert atmosphere of nitrogen at 98% relative humidity (RH) until the protein is fully deoxygenated and its hydration equilibrated with the vapor phase, as judged by optical and frequency measurements. This process takes at least 4 h. After equilibration, air at 98% RH is admitted into the sample cell and the protein spectra between 450 and 700 nm and the quartz crystal frequency are taken simultaneously until the protein is fully oxygenated and hydrated. During this transient, water vapor activity is kept constant throughout the system (vapor and film phases) by the saturated K2SO4 solution deposited on the bottom of the closed cell. Hence, the small amount of extra water molecules sorbed by Hb films during oxygenation is counterbalanced by the saturated solution buffering aw in the vapor phase. After measurements, the Hb film is dried for at least 12 h over P2O5 and the mass of the dried sample determined. The protein oxygen saturation, foxy, was evaluated from the optical absorbance at 576 nm, and the intrinsic hydration, m, taking the difference between the mass of the film at 98% RH and that of the fully dried film. m is given in moles of water/mol of protein. Measurements were made at room temperature.
where f is the resonance frequency of the clean crystal and N a constant. For the quartz crystals used in this work (Cristales Argentinos, S.A.), f = 6000 kHz, A = 1.266 cm2, N = 167 cm kHz, and ρq = 2.65g cm−3, which determines a sensitivity of 20 ng per Hz. The QCM was mounted into the closed cell (Fig. 1). The cell is positioned in a Cary 3E UV-Vis spectrophotometer, and the quartz crystal connected to a resonator and to a frequency meter that displays frequency to 1 Hz. The films were equilibrated with an inert atmosphere of nitrogen at 98% relative humidity (RH) until the protein is fully deoxygenated and its hydration equilibrated with the vapor phase, as judged by optical and frequency measurements. This process takes at least 4 h. After equilibration, air at 98% RH is admitted into the sample cell and the protein spectra between 450 and 700 nm and the quartz crystal frequency are taken simultaneously until the protein is fully oxygenated and hydrated. During this transient, water vapor activity is kept constant throughout the system (vapor and film phases) by the saturated K2SO4 solution deposited on the bottom of the closed cell. Hence, the small amount of extra water molecules sorbed by Hb films during oxygenation is counterbalanced by the saturated solution buffering aw in the vapor phase. After measurements, the Hb film is dried for at least 12 h over P2O5 and the mass of the dried sample determined. The protein oxygen saturation, foxy, was evaluated from the optical absorbance at 576 nm, and the intrinsic hydration, m, taking the difference between the mass of the film at 98% RH and that of the fully dried film. m is given in moles of water/mol of protein. Measurements were made at room temperature.
FIGURE 1.

Scheme of the closed optical cell containing a quartz crystal microbalance. Films of Hb and Mb are made spreading <10 μl of solution samples on a glass slide and on the quartz crystal, which are immediately arranged into the closed cell containing a small volume of saturated solution of K2SO4, producing an atmosphere of constant relative humidity (RH) of 98%. The Hb films deposited on one glass slide and on the faces of the crystal are first fully equilibrated with an N2 atmosphere of constant RH of 98%. After equilibration, air at 98% RH is admitted into the cell, and the optical spectrum and the crystal frequency are measured as a function of time to determine simultaneously oxygen and water bind to Hb.
RESULTS
Fig. 1 portrays the closed cell used to weight the hydration of Hb and Mb at constant 98% RH and varied degree of O2 saturation. Initially thin films of fully deoxygenated protein are cast on the surface of the quartz crystal resonator of the microbalance for weighing (Kennerly, 1969), and on the optical glass slide for optical measurements of O2 saturation. After equilibration with the anaerobic wet atmosphere, moist air at the same RH is admitted into the chamber, and the time course of the frequency and of the spectral changes of the protein films are simultaneously recorded. As shown in Fig. 2, the increase in Hb oxygen saturation—reported by the increase on the absorption peaks at 540 and 576 nm (Fig. 2 b)—is accompanied by the decrease on the frequency of the Hb-coated crystal resonator (Fig. 2 a). This frequency change, converted in mass units, shows that the mass difference between oxy-Hb and deoxy-Hb exceeds that due to O2 uptake. Five isosbestic points specific of the spectral conversion between deoxy- and oxygenated Hb (Klinger and Ackers 1998) are observed in Fig. 2 b. This assures that no other Hb specie, such as met-Hb or hemicrome (Colombo and Sanches, 1990), are formed in the course of the experiment. Consequently, the excess mass change of Hb films must be attributed to the O2-induced change in protein hydration.
FIGURE 2.

Time evolution of (a), the resonant frequency change of the quartz crystal plat covered with a thin Hb film, and (b), the spectra of Hb film, prepared from a 7 mM Hb/heme in 10 m/M Hepes, 200mM NaCl, pH 7.0. The films were initially equilibrated in an N2 atmosphere at 98% RH. Data collection starts when oxygen at 98% RH is admitted into the closed cell. The increase of the absorption intensities at 540 and 576 nm and the decrease of that at 550 nm with time report the kinetics of O2 uptake by the protein. The decrease in frequency reports the increase on the hydration mass of the film. The lines connecting these points are the best fit of an exponential growth function to the data.
To explore the water binding under situations in which is known the existence or nonexistence of conformational changes, we have also carried out experiments with Hb films prepared from salt-free and from 200-mM NaCl buffer solutions, and with Mb. Such experiments are shown in Fig. 3, where representative plots of the kinetics of water and of oxygen binding to Hb and Mb are displayed. Fig. 3, a and b, show, respectively, data for Hb in the presence and in the absence of NaCl. Fig. 3 c shows data for Mb. The fractional O2 saturation foxy computed from the optical density at 576 nm is displayed in the left coordinate. The right coordinate display m, the mass difference between the fully dehydrated and the hydrated protein computed from the resonant frequency of the QCM (Kennerly, 1969). The m values are given in relative units (moles of H2O/mol of protein) without correction for the contribution of O2 uptake.
FIGURE 3.
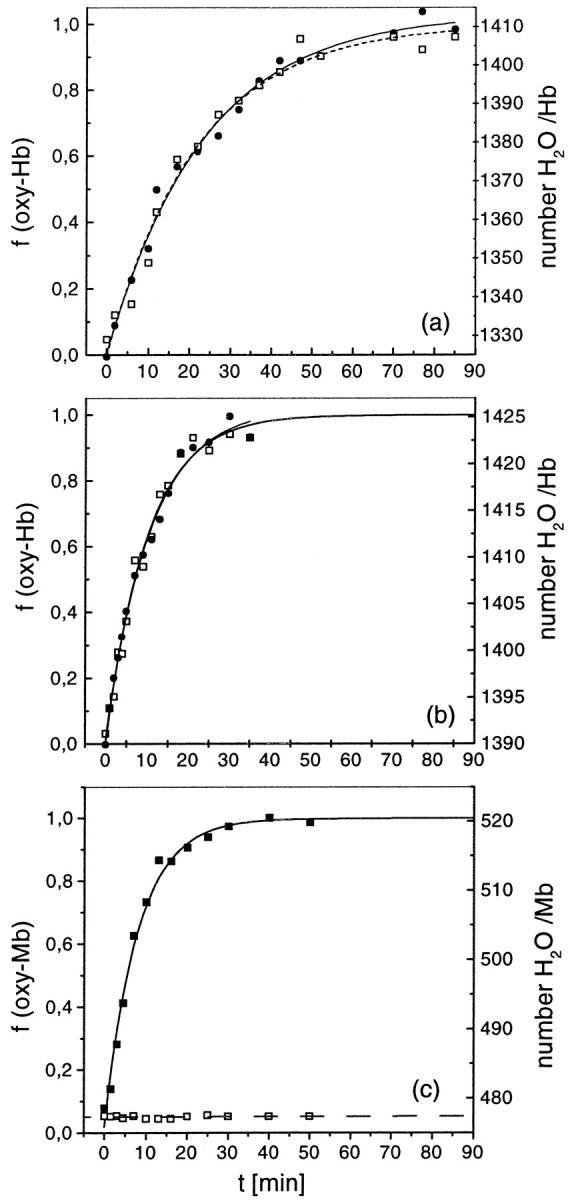
Kinetic of oxygen binding (foxy) and water binding (m) to protein films: (a), prepared from Hb solutions containing 200 mM NaCl; (b), Hb without NaCl; and (c), Mb. The O2-fractional saturation (•) is shown in the left axis, and the Hb gain in mass due to O2-binding, m (□), is shown in the right axis. m is expressed in units of number of water molecules bound per Hb tetramer at 98% RH. The lines connecting the points are the best fit of an exponential growth function to the data, and show that both binding processes follow a kinetic of first order. Fitting parameters are given in Table 1.
The end values of hydration seen in these plots (Fig. 3, a–c) measure the hydration of fully deoxygenated and of fully oxygenated protein states equilibrated at 98% RH. Table 1 summarizes the differential hydration between these states of Hb (and of Mb), 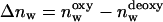 , as determined averaging different experiments. These results are given in units of moles of water per mol of protein. For comparison, the Δnw values previously determined in cosolutions are also tabulated (Colombo and Bonilla-Rodriguez, 1996; Colombo et al., 1992; Colombo and Seixas, 1999), showing the agreement between the two methods. Strikingly, the Δnw of Hb oxygenation weighed in the presence of salt is also in agreement with that determined from the analysis of density measurements of Hb in alcohol-water cosolutions (Bulone et al., 1991). As seen, the binding of O2 to Mb does not cause any perceivable change in protein hydration, whereas the hydration change of Hb in the presence of NaCl is about three times larger than that measured in the absence of the salt.
, as determined averaging different experiments. These results are given in units of moles of water per mol of protein. For comparison, the Δnw values previously determined in cosolutions are also tabulated (Colombo and Bonilla-Rodriguez, 1996; Colombo et al., 1992; Colombo and Seixas, 1999), showing the agreement between the two methods. Strikingly, the Δnw of Hb oxygenation weighed in the presence of salt is also in agreement with that determined from the analysis of density measurements of Hb in alcohol-water cosolutions (Bulone et al., 1991). As seen, the binding of O2 to Mb does not cause any perceivable change in protein hydration, whereas the hydration change of Hb in the presence of NaCl is about three times larger than that measured in the absence of the salt.
TABLE 1.
Best nonlinear fit parameters of the kinetic of O2 and H2O binding to Hb films, and weighed difference in hydration between fully deoxygenated and fully oxygenated Hb and Mb in equilibrium with 98% RH
| Protein films | τfoxy (min)* | τΔm (min)† | Δnw‡ | Δnw§ |
|---|---|---|---|---|
| Hb with NaCl | 22.2 ± 2.0 | 23.4 ± 2 | 80.1 ± 2.6 | 72 ± 2.0 |
| Hb without NaCl | 10.0 ± 1.0 | 10.7 ± 0.8 | 27.4 ± 1.0 | 25 ± 2.0 |
| Mb | 6.9 ± 0.8 | 0.0 | 0.0 | 0.0 |
τfoxy and †τΔm are the best fitted first order rate constant for oxygen and water binding to Hb to the data shown in Fig. 3, a and b, respectively.
Δnw of Hb oxygenation determined in this work by weighing oxygenated and deoxygenated protein films equilibrated with a moist atmosphere at 98% RH. These values are averaged of four–six independent experiments.
Δnw of Hb oxygenation measured in cosolutions. Data taken from Colombo and Bonilla-Rodriguez 1996; Colombo et al., 1992; Colombo and Seixas, 1999.
Different from Hb, the binding of oxygen or chloride to Mb does not lead to changes in protein conformation. Consistently, the hydration of the Mb molecule, both in the film and in cosolutions, is unaffected by O2 uptake. On the other hand, the present measurements confirm that the hydration change associated with Hb oxygenation is larger in the presence than in the absence of NaCl (Table 1) (Colombo and Seixas, 1999). Taken together, the measurements of Δnw of Hb oxygenation, in the film and in solution, exemplify how the determinations of changes in macromolecular hydration associated with biochemical reactions can be used as a probe of structural/function relationships.
In their analysis of the influence of anions on Δnw of Hb oxygenation in solution, Colombo and Seixas (1999) had originally concluded that deoxy-Hb coexist in two distinct allosteric states depending whether or not anions are bound: one T state of low-affinity/low-hydration for Hb complex to anions, and another unforeseen state of intermediate-affinity/intermediate-hydration, which is typical of deoxy-Hb free of anions bound (Colombo and Seixas 1999). In the absence of crystallographic evidence for the structure of this intermediate affinity state of deoxy-Hb, Colombo and Seixas had defined it, operationally, as a P state. Recent crystallographic studies have revealed, however, that although the increase in O2-affinity and the increase in protein hydration accompanies the release of anions from deoxy-Hb, the T quaternary arrangement of the tetramer is preserved (Seixas et al., 1999). The existence of two T states of different O2 affinities, predicted by Δnw measurements, has been confirmed more recently, by oxygen-binding experiments with Hb encapsulated in silica gel, which prevents the O2-induced T to R quaternary transition of Hb (Bruno et al., 2001; Shibayama and Saigo, 2001).
Now, the experiments shown in Fig. 3, a and b, proves directly that the anion-induced changes in Δnw of Hb oxygenation is associated with the transition between two T-like states induced by anion release, from the low-affinity/low-hydration to the intermediate-affinity/intermediate-hydration state. As compared with Δnw values determined in osmotic stress experiments (Colombo and Seixas, 1999), the agreement is quantitative, i.e., deoxy-Hb in the absence of NaCl binds ∼50 extra water molecules more than deoxy-Hb complex with chloride. This again demonstrates that Δnw accounts reliably for the structural changes of Hb both in the film as well as in solution. Moreover, it strongly evidences that the structural characteristics of Hb are not altered on the transfer from aqueous solution to the wet film state. This conclusion is consistent with a recent study on the effect of crystal water content, set by controlled atmosphere vapor pressure, on the x-ray crystal structures of human oxy-Hb and deoxy-Hb. It is shown that the structure of deoxy-Hb crystals equilibrated with an RH below 93% changes conformation but preserves the quaternary T arrangement, whereas the structure of the oxy-Hb is unchanged by change in crystal water content (Biswal and Vijayan, 2002). It is worth reminding that the measurements reported here were carried out at 98% RH, above the RH-producing alterations on the T conformation in the crystal. Alike in solution, the determination of changes in Hb hydration upon O2-binding by weighing reports the change in protein conformation between oxy- and deoxy-Hb. These measurements do not resolve which quaternary conformation oxy-Hb assumes in solution, R or R2 (or even some intermediate between these). These different quaternary conformations have been observed in crystals of fully ligated human Hb grown at different salt and pH conditions (Mueser et al., 2000) and in a human variant (Smith et al., 1991). But it is still unknown which quaternary conformation of ligated Hb, R, R2, or a mixture, prevails in solution. It is interesting to note that the computed water-accessible surface area of oxy-R human Hb is practically the same as that of R2 conformation, suggesting that hydration measurements would hardly distinguish then (unpublished results). Even though we cannot say which quaternary arrangements of Hb prevails, the finding that Δnw Hb oxygenation is unchanged upon transfer from solution to the wet film evidences that the allosteric changes controlling Hb cooperative binding shall be the same in both physical states. This allows us to consider the kinetics of water binding reported in Fig. 3 as a parallel measurement of the kinetics of Hb conformational changes induced by O2 uptake. Therefore, we have analyzed the O2-linked kinetics of water binding to Hb in order to elucidate the role of water binding on the kinetic mechanism of allosteric regulation.
All kinetic data shown in Fig. 3, a–c, are well described by an exponential growth function, as shown by the continuous lines through the experimental points. The best-fitting parameters are also given in Table 1. Alike in solution, the kinetics of O2 uptake by Mb monomer in the film is, as expected, bimolecular. Strikingly, the kinetics of oxygen and of water binding to Hb tetramers in the wet film are, at each salt condition, coincident and follow an apparent first order reaction as if both reactions were bimolecular. This contrasts with the kinetics of Hb oxygenation in solution, where the rate of O2-binding increases stepwise with the progress of the reaction (Antonini and Brunori, 1971). In solution, the concomitant increase on the rate of Hb oxygenation with saturation is due to the very fast displacement of the allosteric equilibrium from the T toward the R state, which binds the ligand with the faster rate (Gibson, 1999). Although the transfer from the solution to the wet film state does not prevent the O2-induced quaternary change of Hb structure, the rates of O2 uptake by Hb and Mb decrease by least 5–6 orders of magnitude. Similarly, the rate of the T to R transition as reported by the rate of hydration is also strongly depressed in the wet film.
The significant decrease on the rates of Hb and Mb oxygenation can be attributed to looser flexibility of the hydrated proteins in the film (Rupley et al., 1983). At 98% RH, the hydration of Hb and Mb species are within 0.36–0.40 g H2O/g protein. These values are very similar to the hydration values measured in aqueous solution (Kuntz and Kauzmann, 1974). Nevertheless, the lack of external layers of water surrounding the protein in the film appears to hinder hydrogen-bonding exchange required for fast anharmonic fluctuations in the protein moiety. As a consequence, the rate of O2-binding to Mb monomer and to Hb tetramer decreases. First, because the structural relaxation of the amino acid residues lining the O2-binding path to the buried heme within monomers is dumped. This is the only cause decreasing the rate of O2 association to Mb, a true bimolecular process. Comparatively to Mb, the rate of O2-binding to Hb is further slowed down (Fig. 3) by the rate of the quaternary change.
The intrinsic rates of tertiary and of quaternary structural changes triggered by O2-binding to Hb are determinants of the observed rates of water binding measured in this work. The monoexponential kinetics of water binding to Hb implicates either a), that the tertiary and quaternary changes in protein structure occur at nearly the same rate, or b), that the rate of the T to R quaternary transition is much slower than the direct and reverse rates of tertiary rearrangement. Any of these two possibilities explain why the fitted kinetics amplitude equals the total difference in hydration between fully oxygenated Hb and fully deoxygenated Hb. Therefore, the measured rates of water binding to Hb probes directly kT–R, the rate of T to R transition. If the intrinsic rates of O2-binding to the T and to the R states of Hb are slower than kT–R, as it appears from comparing the apparent rates of Hb reaction with that of Mb oxygenation, then the quaternary change is the rate-limiting barrier for Hb reaction. This suggests that the transition state is R-like.
The energetic characteristics of the transition state of Hb oxygenation has been analyzed on the framework of the transition state theory (Szabo, 1978). On this background, Szabo first predicted a linear relationship between the free energy of the transition state of Hb oxygenation (ΔG±) and the free energy change between T and R states (ΔGT–R). Considering this, and the kinetic behavior of Hb oxygenation in solution, several workers have already proposed that the transition state of Hb oxygenation is, alike found in this work, R-like (Goldbeck et al., 2001; Henry et al., 1997; Szabo, 1978). The kinetics of O2-linked changes in Hb hydration reported here appears to give structural confirmation of these energy-based predictions.
To further characterize the structure of the transition state, we have replotted the data shown in Fig. 3, a and b, and computed the increment in protein hydration per O2 molecule bound from the slope of the linear plots of protein hydration (m) versus O2 partial saturation (foxy) (Fig. 4). The slopes of these plots report that the binding of oxygen is always associated with a full change in protein hydration. Accordingly, Hb oxygenation only proceeds to completion in the R state. The absence of changes in protein hydration on the true bimolecular association of O2 with Mb is consistent with this conclusion. It also indicates that the rates of O2 and H2O association are much faster than the T–R transition in the film, which implies that at each point of the transient, the protein is practically in equilibrium with the oxygen and vapor activities in the vapor phase.
FIGURE 4.

The change in Hb and Mb (inset) hydration in an atmosphere of 98% RH, m, due to the change in oxygen partial saturation, foxy. These data were replotted from the data shown in Fig. 2: (▪) for Hb films prepared from solutions containing 200 mM NaCl, (•) Hb without NaCl, and Mb (♦). The solid lines are the best linear fit to the data, whose slopes are measurements of the increase in Hb hydration induced by oxygen binding. They equal to (▪) 76.7 ± 4.6 (r = 0.979) and (•) 29.7 ± 1.4 (r = 0.987) water molecules per Hb. Mb (♦) do not show changes in hydration during the oxygen binding.
The simultaneous kinetics of water and oxygen binding to Hb can now be outlined within the Monod-Wyman-Changeux two-state allosteric model (Henry et al., 1997; Monod et al., 1965). In this framework, the kinetics of Hb oxygenation are determined by the intrinsic rates of ligand association and dissociation to the T state, kT and k−T, and to the R state, kR and k−R, and by the rate of the allosteric transition, kT–R (Gibson, 1999; Henry et al., 1997). In solution, kT–R is faster than the rates of O2 association to the T and R states. Thus the binding of oxygen to the T state, which occurs at slower rate because kT < kR, displace the equilibrium toward the fast reacting R state. This explains the stepwise increase on the rate of O2 association to the Hb tetramer observed in solution (Gibson, 1999). Contrary to that, in the wet film, kT–R is strongly dumped, and all other rates became faster than it. Combining this with the fact that the rates of O2 dissociation from Hb, k−T and k−R, are much faster than the direct rates (Gibson, 1999; Henry et al., 1997), we arrive to the following kinetics mechanism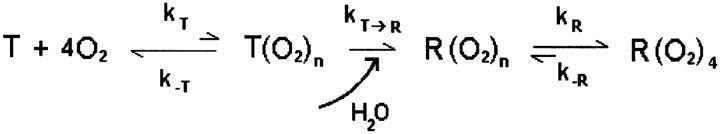 which accounts for the bimolecular kinetics of Hb oxygenation observed in the film state when kT–R is rate-limiting. This mechanism highlights the role of water binding on the stabilization of the R-like, partially ligated transition state of Hb, R(O2)n, where the subscript n < 4. Once this occurs, Hb cooperativity is expressed by speeding up the reaction to completion. Otherwise, the reaction keeps pushed toward the fully unligated T state. Hence, during the transition, the only species significantly populated in the Hb film state are the fully unligated T and the fully ligated R states. This kinetic behavior is obviously distinct from that observed in solution or in vivo conditions. In the liquid environment, kT–R is fast and cooperativity is observed on the stepwise increase on the rates of O2-association. In the wet film, kT–R is slow and cooperativity is observed on the increase on the rate of O2 association on the transition from the T to R-like transition state. Nevertheless, the concomitant measurements of water and oxygen binding to Hb and Mb films evidence water binding in the mechanism of allosteric regulation, and highlights the role of solvation energies to cooperativity.
which accounts for the bimolecular kinetics of Hb oxygenation observed in the film state when kT–R is rate-limiting. This mechanism highlights the role of water binding on the stabilization of the R-like, partially ligated transition state of Hb, R(O2)n, where the subscript n < 4. Once this occurs, Hb cooperativity is expressed by speeding up the reaction to completion. Otherwise, the reaction keeps pushed toward the fully unligated T state. Hence, during the transition, the only species significantly populated in the Hb film state are the fully unligated T and the fully ligated R states. This kinetic behavior is obviously distinct from that observed in solution or in vivo conditions. In the liquid environment, kT–R is fast and cooperativity is observed on the stepwise increase on the rates of O2-association. In the wet film, kT–R is slow and cooperativity is observed on the increase on the rate of O2 association on the transition from the T to R-like transition state. Nevertheless, the concomitant measurements of water and oxygen binding to Hb and Mb films evidence water binding in the mechanism of allosteric regulation, and highlights the role of solvation energies to cooperativity.
We find that the binding of extra water molecules to Hb surface is required to stabilize the high affinity R-like transition state of the protein for the oxygen binding reaction to proceed cooperatively to completion. It is likely that the functional changes in macromolecular hydration associated with many other allosteric proteins and with ligand binding to enzymes and to DNA molecules could be regulated by a similar kinetic mechanism as observed in this work. Significant evidence for that has been recently found in in vitro studies on the effect of solution water activity on the kinetics of protein binding to DNA (Lynch and Sligar, 2000; Sidorova and Rau, 2000) and on the kinetics of Hb oxygenation in cosolutions (Goldbeck et al., 2001). Taken together, these results suggest that changes in cellular osmolality and water activity may play an eventual active role in in vivo processes via a kinetic regulation of some important biological reactions.
Acknowledgments
We thank M. Cornelio, V.A. Parsegian, D.C. Rau, T.S. Grigera, and S.A. Grigera for careful reading of the manuscript.
This work was partially supported by the Brazilian agencies: Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo a Pesquisa da UNESP; and by the following Argentinian agencies: Consejo Nacional de Investigaciones Científicas y Tecnológicas (CONICET: and Agencia Nacional de Promoción Científica y Tecnológica. A.G.S is fellow Comision de Investigaciones Científicas Provincia de Buenos Aires and of Universidad Nacional de La Plata. J.R.G. is a member of the “Carrera de Investigador” of CONICET. M.F.C. is a Researcher Fellow of CNPq.
References
- Antonini, E., and M. Brunori. 1971. Hemoglobin and myoglobin in their reactions with ligands. North-Holland Pub. Co., Amsterdam.
- Biswal, B. K., and M. Vijayan. 2002. Structures of human oxy- and deoxyhaemoglobin at different levels of humidity: variability in the T state. Acta Crystallogr. D Biol. Crystallogr. 58:1155–1161. [DOI] [PubMed] [Google Scholar]
- Bruno, S., M. Bonaccio, S. Bettati, C. Rivetti, C. Viappiani, S. Abbruzzetti, and A. Mozzarelli. 2001. High and low oxygen affinity conformations of T state hemoglobin. Protein Sci. 10:2401–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulone D., I. D. Donato, M. B. Palmavittorelli, M. U. Palma. 1991. Density, structural lifetime, and entropy of H-bond cages promoted by monohydric alcohols in normal and supercooled water. J. Chem. Phys. 94:6816–6826. [Google Scholar]
- Colombo, M. F., and G. O. Bonilla-Rodriguez. 1996. The water effect on allosteric regulation of hemoglobin probed in water/glucose and water/glycine solutions. J. Biol. Chem. 271:4895–4899. [DOI] [PubMed] [Google Scholar]
- Colombo, M. F., D. C. Rau, and V. A. Parsegian. 1992. Protein solvation in allosteric regulation: a water effect on hemoglobin. Science. 256:655–659. [DOI] [PubMed] [Google Scholar]
- Colombo, M. F., and R. Sanches. 1990. Hydration-dependent conformational states of hemoglobin. Equilibrium and kinetic behavior. Biophys. Chem. 36:33–39. [DOI] [PubMed] [Google Scholar]
- Colombo, M. F., and F. A. Seixas. 1999. Novel allosteric conformation of human HB revealed by the hydration and anion effects on O(2) binding. Biochemistry. 38:11741–11748. [DOI] [PubMed] [Google Scholar]
- Davis-Searles, P. R., A. J. Saunders, D. A. Erie, D. J. Winzor, and G. J. Pielak. 2001. Interpreting the effects of small uncharged solutes on protein- folding equilibria. Annu. Rev. Biophys. Biomol. Struct. 30:271–306. [DOI] [PubMed] [Google Scholar]
- Eisenberg, D., and A. D. McLachlan. 1986. Solvation energy in protein folding and binding. Nature. 319:199–203. [DOI] [PubMed] [Google Scholar]
- Gekko, K., and S. N. Timasheff. 1981. Mechanism of protein stabilization by glycerol-preferential hydration in glycerol-water mixtures. Biochemistry. 20:4667–4676. [DOI] [PubMed] [Google Scholar]
- Gibson, Q. H. 1999. Kinetics of oxygen binding to hemoglobin A. Biochemistry. 38:5191–5199. [DOI] [PubMed] [Google Scholar]
- Goldbeck, R. A., S. J. Paquette, and D. S. Kliger. 2001. The effect of water on the rate of conformational change in protein allostery. Biophys. J. 81:2919–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, E. R., C. M. Jones, J. Hofrichter, and W. A. Eaton. 1997. Can a two-state MWC allosteric model explain hemoglobin kinetics? Biochemistry. 36:6511–6528. [DOI] [PubMed] [Google Scholar]
- Kennerly, M. G. 1969. A technique for the measurement of the water adsorption of small amount of hygroscopic materials. Polymer. 10:833–840. [Google Scholar]
- Klinger, A. L., and G. K. Ackers. 1998. Analysis of spectra from multiwavelength oxygen-binding studies of mixed metal hybrid hemoglobins. Methods Enzymol. 295:190–207. [DOI] [PubMed] [Google Scholar]
- Kornblatt, J. A., and G. H. Hoa. 1990. A nontraditional role for water in the cytochrome c oxidase reaction. Biochemistry. 29:9370–9376. [DOI] [PubMed] [Google Scholar]
- Kuntz, I. D., Jr., and W. Kauzmann. 1974. Hydration of proteins and polypeptides. Adv. Protein Chem. 28:239–345. [DOI] [PubMed] [Google Scholar]
- LiCata, V. J., and N. M. Allewell. 1998. Measuring hydration changes of proteins in solution: applications of osmotic stress and structure-based calculations. Methods Enzymol. 295:42–62. [DOI] [PubMed] [Google Scholar]
- Lynch, T. W., and S. G. Sligar. 2000. Macromolecular hydration changes associated with BamHI binding and catalysis. J. Biol. Chem. 275:30561–30565. [DOI] [PubMed] [Google Scholar]
- Monod, J., J. Wyman, and J. P. Changeux. 1965. On nature of allosteric transitions—a plausible model. J. Mol. Biol. 12:88–118. [DOI] [PubMed] [Google Scholar]
- Mueser, T. C., P. H. Rogers, and A. Arnone. 2000. Interface sliding as illustrated by the multiple quaternary structures of liganded hemoglobin. Biochemistry. 39:15353–15364. [DOI] [PubMed] [Google Scholar]
- Parsegian, V. A., R. P. Rand, and D. C. Rau. 1995. Macromolecules and water: probing with osmotic stress. Methods Enzymol. 259:43–94. [DOI] [PubMed] [Google Scholar]
- Parsegian, V. A., R. P. Rand, and D. C. Rau. 2000. Osmotic stress, crowding, preferential hydration, and binding: A comparison of perspectives. Proc. Natl. Acad. Sci. USA. 97:3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preisler, R. S., H. H. Chen, M. F. Colombo, Y. Choe, B. J. Short, Jr., and D. C. Rau. 1995. The B form to Z form transition of poly(dG-m5dC) is sensitive to neutral solutes through an osmotic stress. Biochemistry. 34:14400–14407. [DOI] [PubMed] [Google Scholar]
- Royer, W. E., Jr., A. Pardanani, Q. H. Gibson, E. S. Peterson, and J. M. Friedman. 1996. Ordered water molecules as key allosteric mediators in a cooperative dimeric hemoglobin. Proc. Natl. Acad. Sci. USA. 93:14526–14531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero Neto, J., and M. F. Colombo. 2000. Water regulation of actinomycin-D binding to DNA: the interplay among drug affinity, DNA long-range conformation, and hydration. Biopolymers. 53:46–59. [DOI] [PubMed] [Google Scholar]
- Ruggiero Neto, J., F. Pereira de Souza, and M. F. Colombo. 2001. Hydration effects on DNA double helix stability modulates ligand binding to natural DNA in response to changes in water activity. Cell Mol Biol (Noisy-le-grand). 47:801–814. [PubMed] [Google Scholar]
- Rupley, J. A., E. Gratton, and G. Careri. 1983. Water and globular proteins. Trends Biochem. Sci. 8:18–22. [Google Scholar]
- Seixas, F. A., W. F. de Azevedo, Jr., and M. F. Colombo. 1999. Crystallization and x-ray diffraction data analysis of human deoxyhaemoglobin A(0) fully stripped of any anions. Acta Crystallogr. D Biol. Crystallogr. 55:1914–1916. [DOI] [PubMed] [Google Scholar]
- Shibayama, N., and S. Saigo. 2001. Direct observation of two distinct affinity conformations in the T state human deoxyhemoglobin. FEBS Lett. 492(1–2):50–53. [DOI] [PubMed] [Google Scholar]
- Sidorova, N. Y., and D. C. Rau. 2000. The dissociation rate of the EcoR1-DNA-specific complex is linked to water activity. Biopolymers. 53:363–368. [DOI] [PubMed] [Google Scholar]
- Smith, F. R., E. E. Lattman, and C. W. Carter, Jr. 1991. The mutation beta 99 Asp-Tyr stabilizes Y—a new, composite quaternary state of human hemoglobin. Proteins. 10:81–91. [DOI] [PubMed] [Google Scholar]
- Swaminathan, C. P., N. Surolia, and A. Surolia. 1998. Role of water in the specific binding of mannose and mannooligosaccharides to concanavalin A. J. Am. Chem. Soc. 120:5153–5159. [Google Scholar]
- Szabo, A. 1978. Kinetics of hemoglobin and transition state theory. Proc. Natl. Acad. Sci. USA. 75:2108–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanford, C. 1969. Extension of the theory of linked functions to incorporate the effects of protein hydration. J. Mol. Biol. 39:539–544. [DOI] [PubMed] [Google Scholar]
- Timasheff, S. N. 1998. In disperse solution, “osmotic stress” is a restricted case of preferential interactions. Proc. Natl. Acad. Sci. USA. 95:7363–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossen, K. M., R. Wolz, M. A. Daugherty, and M. G. Fried. 1997. Role of macromolecular hydration in the binding of the Escherichia coli cyclic AMP receptor to DNA. Biochemistry. 36:11640–11647. [DOI] [PubMed] [Google Scholar]
- Xavier, K. A., K. A. Shick, S. J. Smith-Gill, and R. C. Willson. 1997. Involvement of water molecules in the association of monoclonal antibody HyHEL-5 with bobwhite quail lysozyme. Biophys. J. 73:2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey, P. H., M. E. Clark, S. C. Hand, R. D. Bowlus, and G. N. Somero. 1982. Living with water stress: evolution of osmolyte systems. Science. 217:1214–1222. [DOI] [PubMed] [Google Scholar]