

Abstract
Both the aqueous and the lipid-induced structure of eledoisin, an undecapeptide of mollusk origin, have been studied by two-dimensional proton nuclear magnetic resonance spectroscopy and distance geometry calculations. Unambiguous nuclear magnetic resonance assignments of protons have been made with the aid of correlation spectroscopy experiments and nuclear Overhauser effect spectroscopy experiments. The distance constraints obtained from the nuclear magnetic resonance data have been utilized in a distance geometry algorithm to generate a family of structures, which have been refined using restrained energy minimization and dynamics. These data show that, while in water and dimethyl sulfoxide, eledoisin prefers to be in an extended chain conformation, whereas in the presence of perdeuterated dodecylphosphocholine micelles, a membrane model system, helical conformation is induced in the central core and C-terminal region (K4-M11) of the peptide. N terminus, though less defined, also displays some degree of order and a possible turn structure. The conformation adopted by eledoisin in the presence of dodecylphosphocholine micelles is similar to the structural motif typical of neurokinin-2 selective agonists and with that reported for kassinin in hydrophobic environment.
INTRODUCTION
Eledoisin, an undecapeptide of mollusk origin, with the sequence pGlu-Pro-Ser-Lys-Asp-Ala-Phe-Ile-Gly-Leu-Met-NH2, is a member of the tachykinin family of neuropeptides. It was first isolated from the posterior salivary glands of two mollusk species Eledone muschata and E. aldovandi belonging to the octopod Cephalopoda (De Marco and Gatti, 1975). Other tachykinins from nonmammalian sources include kassinin and physalaemin. These peptides exhibit a wide and complex spectrum of pharmacological and physiological activities such as powerful vasodilation, hypertensive action, and stimulation of extravascular smooth muscle. A recent study (El-Agnaf et al., 1998) has shown similar activity and high level of sequence homology (73%) of eledoisin with β-amyloid protein fragments (Aβ 25–35) and their analogs, which play a major role in the onset and progression of Alzheimer's disease. Hence there is considerable interest in these peptides as potential targets for drug design. The mammalian tachykinins substance P, NKA, and NKB have similar activities to those of the nonmammalian members and have been more widely studied and characterized. All tachykinin peptides share the same consensus C-terminal sequence, that is, Phe-Xxx-Gly-Leu-Met-NH2. The invariant Phe7 residue is probably required for receptor binding. Xxx is either aromatic (phenylalanine, tyrosine) or a branched aliphatic (valine, isoleucine) side chain and is thought to be important in receptor selectivity. This common region, often referred as the message domain, is believed to be responsible for activating the receptor. The divergent N-terminal region or the address domain varies in amino-acid sequence and length and is believed to play a role in determining the receptor subtype specificity (Schwyzer, 1987).
Three distinct receptor subtypes, characterized by different rank order of potencies, have been identified and cloned for tachykinins (Nakanishi, 1991; Masu et al., 1987; Hanley and Jackson, 1987) designated as NK1, NK2, and NK3. These share significant sequence similarity and belong to the super family of G-protein–coupled receptors, whose structure is believed to be similar to the hepta-helical structure of rhodopsin (Palczewski et al., 2000). All tachykinins bind to all the receptor subtypes, with substance P preferring NK1, NKA preferring NK2, and NKB preferring NK3. Physalaemin shows selectivity for NK1 and its affinity is less than that of its mammalian agonist, substance P. By comparison to other tachykinins, the affinity of eledoisin and kassinin for mammalian receptors is weak and their selectivity is much less pronounced. It is thought that the E-type eledoisin receptor (SP-E) which prefers ligands eledoisin, kassinin, and neurokinin B over neurokinin A, physalaemin, and substance P, may be a mixture of NK2 and NK3 binding sites (Schwyzer, 1987). The wide range of physiological activity of tachykinins has been attributed to the lack of selectivity of tachykinins for a particular receptor type (Nakanishi, 1991). The conformational flexibility of the short, linear peptide can also account for the lack of selectivity. The conformational features of tachykinins, which control receptor binding and influence their biological activity, are of significant interest, particularly as the selectivity of these peptides for different receptor sites is not fully understood.
Recently, several studies led to reports on biophysical properties of tachykinin neuropeptides. Various experimental structure studies have been carried out in aqueous and membrane mimetic solutions of SP, its analogs and other tachykinins, NKA, NKB, and physalaemin (Convert et al., 1991; Chassaing et al., 1986a, b; Szollosy et al., 1986; Chassaing et al., 1987; Woolley and Deber, 1987; Convert et al., 1988; Levian-Teitelbaum et al., 1989; Seelig and Macdonald, 1989; Lavielle et al., 1988; Lavielle et al., 1990; Sumner et al., 1990; Williams and Weaver, 1990; Seelig, 1992; Ananthanarayanan and Orlicky, 1992; Whitehead et al., 1998; Young et al., 1994). In general, it has been found that the tachykinins display some elements of secondary structure in appropriate solution environment, though it has been suggested that they undergo rapid conformational exchange (Sumner et al., 1990). There are no discernible trends in the conformation of address segments of these peptides. However, the message domains are similar in each case. In general, the message domain of these peptides undergoes conformational averaging in aqueous environments (Woolley and Deber, 1987; Sumner et al., 1990). In hydrophobic environment, message domain assumes helical conformations (Schwyzer, 1987; Chassaing et al., 1987; Woolley and Deber, 1987; Whitehead et al., 1998; Wu and Yang, 1983; Horne et al., 1993) or exists as a series of turns in dynamic equilibrium (Sumner et al., 1990).
A limited number of studies have been reported on the conformation of tachykinin eledoisin (Wilson et al., 1994) and kassinin (Grace et al., 2001). From circular dichroic (CD) studies eledoisin has been reported to assume a β-structure, resulting perhaps from aggregation, when in contact with phosphatidylcholine membranes (Schwyzer, 1987) and to form an α-helical structure in SDS (Woolley and Deber, 1987). In a study of eledoisin in dimethyl sulfoxide (Yu and Yang, 1991) by NMR and distance geometry technique, no regular conformations were found. However, it has not been possible to get this publication. A detailed study has been reported by Wilson and co-workers on solution conformation on eledoisin, using CD and two-dimensional NMR techniques. In aqueous solution eledoisin was found to be conformationally averaged, but assumed α-helical structure on addition of 50% TFE or SDS (Wilson et al., 1994). Their NMR data also indicated that the helical core of eledoisin was better defined in SDS micelles environment than in TFE.
The role of the lipid phase of the membrane is to facilitate the ligand receptor interactions in at least two sequential steps, binding of the peptide to the membrane, followed by binding of the peptide to the receptor in the membrane (Schwyzer et al., 1986). Although neuropeptides in aqueous solution exist as randomly distributed conformers, the biologically active forms of these neuropeptides are likely to be ordered and stabilized within the lipid bilayers of the cell membrane before binding with their receptors (Schwyzer, 1987; Woolley and Deber, 1987; Schwyzer et al., 1986). Thus, conformational features of eledoisin in lipid medium, which control receptor binding and which govern its activity, are of significant interest.
NMR has been found to be a particularly good method to study micelle-bound peptides. Almost two decades ago, it has been demonstrated that the high-resolution proton NMR spectra can be acquired on peptides bound to micelles of perdeuterated lipid (Brown, 1979). Conditions for determining the conformations of peptides bound to micelles using NMR have been established by several research groups (Lauterwein et al., 1979; Braun et al., 1983; McDonnell and Opella, 1993; Opella, 1997; Kallick et al., 1995; Maurer et al., 1991).
Dodecylphosphocholine is one of the well-characterized model membrane systems in current use, for the study of peptides and proteins which bind to the lipids. It forms a stable micelle which freely rotates in solution, making it an excellent tool to mimic the anisotropic environment of a lipid membrane, while providing motional properties desirable for solution NMR. It has electrostatic and hydrophobic components, which approximate a cell membrane. It has been shown that membrane mimetic systems (Brown, 1979; Lauterwein et al., 1979; Braun et al., 1983; Rizo et al., 1993; Maurer and Rüterjans, 1994; Pellegrini et al., 1996; Cowsik et al., 1997; Grace et al. 2001) are quite capable of inducing structures upon small neuropeptides, which may hold some biological relevance, and here this study has been extended to eledoisin.
In the current study the secondary structure of eledoisin has been investigated in different solvents by CD and NMR spectroscopic techniques. The three-dimensional structure of eledoisin bound to micelles of DPC has been reported for the first time. Also, the conformational properties of eledoisin in dimethyl sulfoxide (DMSO) and perdeuterated DPC micelles have been described as well as compared in the two solvents. Several homonuclear two-dimensional NMR techniques (Wüthrich, 1986), such as TOCSY (Braunschweiler and Ernst, 1983; Davis and Bax, 1985), DQF-COSY (Rance et al., 1983), ROESY (Bothner-By and Noggle, 1979; Bax and Davis, 1985) and NOESY (Macura et al., 1981; Macura and Ernst, 1980; Anil Kumar et al., 1980) have been utilized in deriving the complete proton resonance assignments for eledoisin, in DMSO as well as in lipid. The NOESY crosspeak volumes have further been used to determine the interproton distances in three-dimensional space. An ensemble of model conformations has been generated for eledoisin in lipid using the program DYANA (Dynamic Algorithm for NMR applications; see Güntert and Wüthrich, 1991; Güntert et al., 1991).
EXPERIMENTAL PROCEDURES
Materials and methods
Eledoisin was obtained from Sigma Chemical Company (St. Louis, MO). Perdeuterated DPC (d38) was obtained from Cambridge Isotope Laboratories (Andover, MA). NMR reagents were obtained from Aldrich Chemical Company (Milwaukee, WI).
CD spectropolarimetry
CD spectra have been recorded on a Jasco J-720 spectropolarimeter (JASCO, Tokyo, Japan). The instrument was calibrated using d-10-camphorsulfonic acid (Chen and Yang, 1977). Cells having a path length of 1 mm were employed. The peptide concentration was ∼50 μM. Spectra were the average of four scans recorded with a 1-nm bandwidth, a 0.25-nm step size, and a 0.2-s time constant. After baseline correction, the observed ellipticity was converted to a mean residue ellipticity ([θ] deg.cm2 d mol−1), using the relationship of [θ] = θ/lcN, where θ is the observed ellipticity, l is the path length in millimeters, c is the molar concentration and N is the number of residues in the peptide. The spectra reported have not been smoothened. All the measurements were performed at room temperature. To mimic different biomembrane compartments, different solvents were used. The aqueous environment was mimicked by a 10-mM sodium phosphate buffer at pH 7.2, the charged surface by the anionic detergent SDS and the hydrophobic interior by TFE. CD spectra for all the peptides were recorded in water, Sodium phosphate buffer, in increasing concentrations of TFE, anionic detergent SDS, and Zwitterionic lipid DPC. The spectra recorded in the presence of DPC, SDS micelles, and TFE were corrected by subtracting the spectra of corresponding DPC, SDS, or TFE solutions.
Nuclear magnetic resonance experiments
NMR samples were prepared by dissolving 2 mg of eledoisin in ∼0.4 ml of water (90% H2O, 10% D2O, pH 5.0) and 0.5 ml of DMSO-d6. The experiments in lipid environment were performed with an identical peptide sample to which 25 mg of perdeuterated DPC was added yielding in solution a lipid concentration of 180 mM, which is well above the critical micelle concentration (1 mM) for DPC. The lipid-to-peptide ratio of the NMR sample was 40:1. All NMR spectra in DPC and DMSO were recorded on Bruker DRX 500 and Bruker AMX 400 (Bruker, Zurich, Switzerland) spectrometers, operating at 500- and 400-MHz proton resonance frequency, respectively. The data were processed by the XWINNMR program on a Silicon Graphics Indigo workstation (SGI, Irvine, CA).
All two-dimensional spectra were acquired in the phase-sensitive mode. The homonuclear ROESY, NOESY, and TOCSY spectra were recorded with 64 scans, a relaxation delay of 1.5 s, a spectral width of 5020 Hz in both dimensions, 512 increments in t1, and 2K data points in t2. After zero filling and Sine apodization in t1 and t2 dimensions, the final size of the data matrix was 1K × 1K. The DQF-COSY spectrum was recorded to identify the sequential connectivity among the protons of the same residue. The NOESY spectra were recorded with mixing times of 50, 100, 150, 200, and 300 ms (different mixing times were used to evaluate the linear buildup of NOE and to find the mixing time appropriate to the two-spin approximation). NOESY spectrum recorded with a mixing time of 200 ms was chosen for obtaining the distance constraints.
Structure determination
For the determination of internuclear distances, the NOESY peak volumes on the 200-ms NOESY spectra were classified as strong, medium, and weak corresponding to upper-bound interproton distance restrains of 2.7, 3.5, and 5.0 Å, respectively. The NOEs used for structure calculation have been taken from the NOESY spectrum (200 ms), which lies within the initial buildup of the NOE curve. Hence, two spin approximation has been applied while converting the intensities into distances. Appropriate pseudoatom corrections were applied to nonstereo, specifically assigned methylene and methyl protons. A total of 166 NOE constraints (68 intra residue constraints, 55 constrains of i to i + 1, 11 constraints of i to i + 2, and 28 constraints of i to i + 3) were originally applied to the distance geometry algorithm DYANA (Güntert and Wüthrich, 1991; Güntert et al., 1991). A total of 50 structures were initially generated using DYANA. Dihedral angles (φ), which were derived from the measured 3JNH values, were also used as constraints for the φ values.
RESULTS AND DISCUSSION
Fig. 1 shows the CD spectra of eledoisin in 10-mM phosphate buffer (pH = 7.2), in 75% TFE and SDS micelles (16 mM), and DPC (2 mM), the SDS and DPC concentrations being well above their CMC (8 mM and 1 mM, respectively). The titrations were performed with various concentrations of TFE (10–90%), SDS (4–32 mM), DPPG, and DPC. The spectrum in aqueous solution shows the peptide being primarily unstructured, having a weak maximum ∼220 nm and a strong minimum ∼198 nm (Woody, 1992). The addition of the structure-inducing SDS and DPC micelles induced a shift toward helical conformation (at concentration as low as respective CMC), as all spectra had a minimum at 222 nm (helical ππ* transition) and a second minimum between 203 and 208 nm (overlapping helical and random coil ππ* transition at 208 nm and 200 nm, respectively; see Holzworth and Doty, 1965; Alder et al., 1973; Chang et al., 1978). CD data were also analyzed using two parameters R1 and R2, which are independent of inaccuracies in determined peptide concentration as well as those caused by small shifts in wavelength (Bruch et al., 1991). Here R1 is the ratio of the intensity of the maximum between 190 and 195 nm and the intensity of minimum between 200 and 210 nm and R2 is the ratio of intensity of minimum near 222 nm and the intensity of minimum between 200 and 210 nm. For a random structure R1 is positive, and R2 is close to zero. On the other hand, in a highly helical state, R1 will be close to −2, and R2 will approach 1 (Rizo et al., 1993; Bruch et al., 1991). These parameters calculated for eledoisin in buffer, TFE, DPC, and SDS show an induction of the helical structure on addition of TFE (R1 = −1.8, R2 = 0.73), SDS (R1 = −1.3, R2 = 0.8), and DPC (R1 = −1.2, R2 = 0.5). Thus the CD results indicate that eledoisin associates with SDS and DPC micelles undergoing a conformational transition between a prevalently random coil state (in water) to α-helical state.
FIGURE 1.

CD spectra of eledoisin in buffer (solid line), SDS micelles (dotted line), 75% TFE (dashed line), and DPC (dash/dot line).
Some preliminary one-dimensional and two-dimensional spectra of eledoisin were recorded in aqueous solution at various temperatures. NMR spectra in water indicated presence of aggregation. Addition of DMSO caused sharpening of peaks, and in view of its deaggregating properties, DMSO was used as a medium of two-dimensional NMR study. No further study was carried out in water because of peptide aggregation and also as CD results indicated random structure of eledoisin in water. Eledoisin was also studied in the solvent dimethyl sulfoxide (DMSO-d6) at 400 MHz. Temperature dependent one-dimensional and two-dimensional spectra were recorded and were assigned completely. On the basis of observation of a relatively small number of interresidue crosspeaks in the ROESY spectra, it was concluded that eledoisin adopts essentially a random coil conformation under these conditions. Aliquots of d38-DPC were then added to an aqueous solution of eledoisin and another series of one-dimensional and two-dimensional proton NMR spectra were recorded at 500 MHz. The structural stabilization was apparent in the NMR spectra on addition of 12 mg or more of DPC. All subsequent experiments were performed under these solution conditions (DPC concentration, 180 mM; lipid-to-peptide ratio, 40:1). Some of the residues of eledoisin have a minor conformer in both DMSO as well as in DPC. The peaks from the minor conformer could not be suppressed by temperature or by pH variations. As all the resonances of the minor conformer could not be seen clearly in the spectrum, the structure of the minor conformer could not be obtained.
Spectral assignment
Assignment of the proton spectra of eledoisin in the presence of membrane mimetic solvent (DPC) was accomplished using the technique of sequence-specific resonance assignments developed by Wüthrich (1986). These assignments were made by the interactive interpretation of the two-dimensional DQF-COSY, TOCSY, and NOESY/ROESY spectra. The amide region of the NOESY (200-ms mixing time) spectrum of eledoisin is shown in Fig. 2 b. The assignment of the various resonances in the sequence of the peptide is indicated Fig. 2 a on the spectrum. Complete proton resonance assignments thus obtained are given in Tables 1 and 2 for eledoisin in the presence of membrane mimetic solvent and in DMSO.
FIGURE 2.
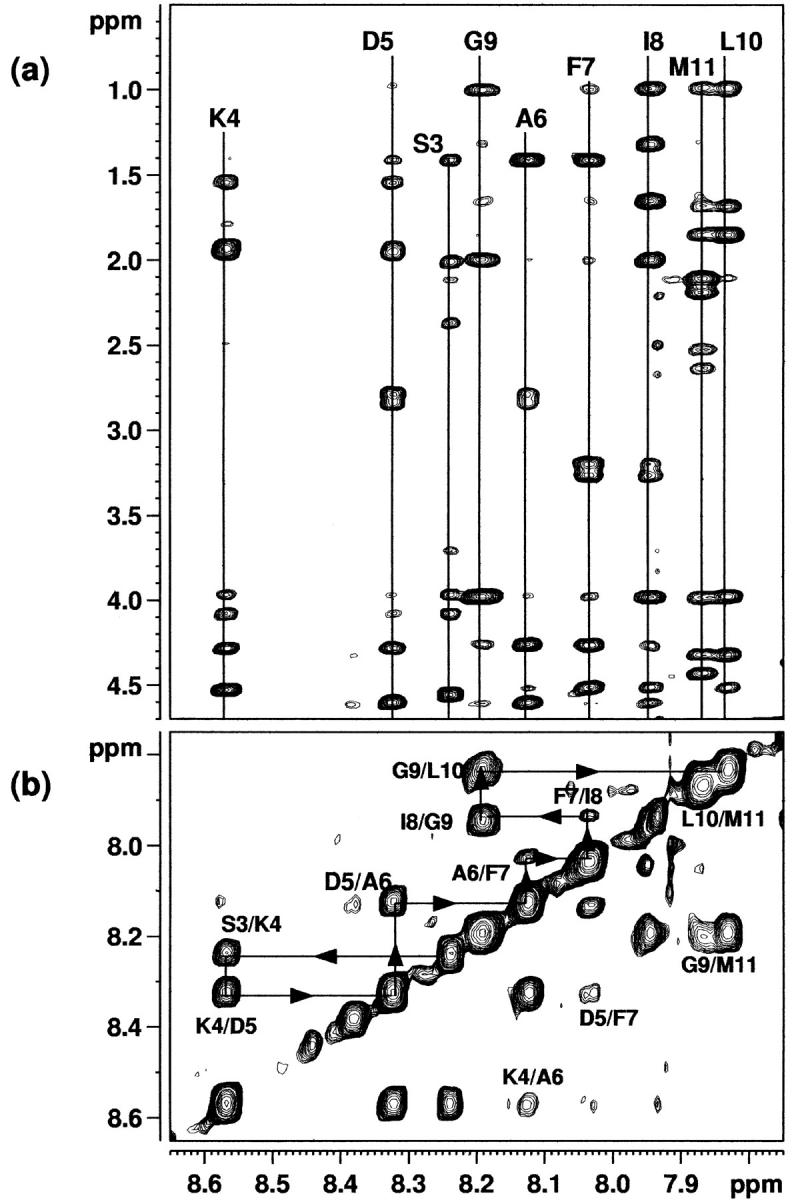
(a) The NH-α,β,γ region of the 500-MHz NOESY spectrum of eledoisin in the presence of DPC, recorded with a mixing time of 200 ms. (b) The NH–NH region of the 500-MHz phase-sensitive NOESY spectrum of eledoisin in the presence of DPC, recorded with a mixing time of 200 ms.
TABLE 1.
Proton NMR assignments of eledoisin in the presence of DPC
| Residue | NH | αH | βH | Others |
|---|---|---|---|---|
| PGlu | 4.31 | 2.68, 2.52 | γCH2: 2.43, 2.06 | |
| Pro | 4.55 | 2.39 | γCH2: 2.13, 2.06 | |
| δCH2: 3.85, 3.73 | ||||
| Ser | 8.25 | 4.53 | 4.08, 3.97 | |
| Lys | 8.57 | 4.27 | 1.95, 1.93 | γCH2: 1.56 |
| δCH2: 1.79 | ||||
| εCH2: 3.06 | ||||
| εNH3: 8.79 | ||||
| Asp | 8.32 | 4.60 | 2.86, 2.79 | |
| Ala | 8.13 | 4.25 | 1.41 | |
| Phe | 8.04 | 4.51 | 3.28, 3.21 | Aro: 7.32 |
| Ile | 7.95 | 3.98 | 2.01 | γCH2: 1.66 |
| γCH3: 1.33 | ||||
| δCH3: 0.98, 1.01 | ||||
| Gly | 8.19 | 3.98 | ||
| Leu | 7.84 | 4.33 | 1.85 | γCH3: 1.69 |
| δCH3: 0.98, 1.01 | ||||
| Met | 7.88 | 4.43 | 2.19 | γCH2: 2.54, 2.65 |
| εCH3: 2.11 | ||||
| NH2 | 7.31, 7.15 |
TABLE 2.
Proton NMR assignments of eledoisin in DMSO-d6
| Residue | NH | αH | βH | Others |
|---|---|---|---|---|
| PGlu | 5.02 | 4.27 | 2.20 | γCH2: 3.49 |
| Pro | 4.21 | 1.96 | γCH2: 1.77 | |
| δCH2: 3.48 | ||||
| Ser | 7.84 | 4.06 | 3.54, 3.43 | |
| Lys | 7.76 | 4.05 | 1.61 | γCH2: 1.21 |
| δCH2: 1.45 | ||||
| εCH2: 2.62 | ||||
| εNH3: 7.16, 6.91 | ||||
| Asp | 7.89 | 4.38 | 2.59, 2.36 | |
| Ala | 7.53 | 4.04 | 1.03 | |
| Phe | 7.77 | 4.39 | 2.93, 2.70 | Aro: 7.06 |
| Ile | 7.63 | 4.05 | 1.62 | γCH2: 1.34 |
| γCH3: 1.00 | ||||
| δCH3: 0.74 | ||||
| Gly | 7.95 | 3.60 | ||
| Leu | 7.76 | 4.16 | 1.50 | γH: 1.37 |
| δCH3: 0.76 | ||||
| Met | 7.78 | 4.10 | 2.33 | γCH2: 1.83 |
| εCH3: 1.72 | ||||
| NH2 | 7.03, 6.88 |
Analysis of chemical shift values
Wishart and co-workers (Wishart et al., 1992) have reported a simple method for predicting the secondary structure of proteins based on changes in their α-H proton chemical shifts. In this method, the observed α-H proton chemical shift of a given residue is compared to the respective random coil chemical shift value for that particular residue. The larger the difference to the random coil value, the more pronounced the secondary structure element should be. A local α-helical structure is identified by a negative secondary shift (resonances shifted to high field relative to the corresponding random coil values). The results for analysis of the α-H proton chemical shifts for eledoisin in DPC show that, for residues 4–8, the difference in the chemical shifts is continually negative, which suggests that in this part of the peptide, a helical secondary structure is favored in the presence of DPC. This is not observed in the case of DMSO, which further confirms the absence of any structure for eledoisin in DMSO as well as in an aqueous medium.
It is very difficult to determine the exact on/off rates for the binding of eledoisin with DPC micelles from the NMR data. However, the percentage of bound conformers to the free conformer may be obtained by determination of the amount of α-helix observed. A semiquantitative estimation of the helical content of eledoisin when going from water to DPC micelles may be obtained from the average upfield shifts of the αH protons. In this procedure, the upfield shifts of the αH protons of the region assumed to be helical are added together and averaged. The averaged upfield shifts are divided by 0.35 (0.35 ppm is assumed to correspond to 100% α-helix) yielding percentage of α-helix (Rizo et al., 1993). The semiquantitative estimate of helical content for residues 4–11 for eledoisin in DPC micelles thus calculated was 31.5%. The prediction of helical content for eledoisin (33%) using the prediction algorithm AGADIR (Muñoz and Serrano, 1994) correlates well with the results from the NMR data.
Analysis of NOE connectivities
Wüthrich and co-workers (Wüthrich, 1986; Wüthrich et al., 1984) have reported that the observation of a group of specific sequential and medium-range NOEs can be used to determine the existence of secondary structural features such as α-helix or β-sheet. The NOEs that are important to characterize the secondary structure of eledoisin in the presence of DPC micelles are summarized in Fig. 3. For eledoisin (S3-M11), intraresidue crosspeaks are more intense, which indicates the presence of helical structure. Similarly, variations in relative intensities of dNN and dαN sequential NOEs support the proposal of a helical structure for eledoisin (S3-M11) as dNN contacts are stronger than dαN contacts (Wüthrich et al., 1984). On the other hand, the ROESY spectrum of eledoisin in DMSO shows only intraresidue and i to i + 1 ROEs among the amide protons and α-protons, confirming the random coil nature of eledoisin in DMSO.
FIGURE 3.

The NOEs that are important to characterize the secondary structure of eledoisin in the presence of DPC micelles. Filled triangles refer to a coupling constant of 4–5 Hz and filled circles to 6 Hz and above.
A dense grouping of NOEs (Fig. 3), the six dαN(i, i + 3), five dαβ(i, i + 3), eight sequential dNN NOEs and three dNN(i, i + 2) NOEs coupled with three dαN(i, i + 2) NOEs support the presence of helical structure in this region of eledoisin (involving residues 3–11). The observation of four dαN(i, i + 2) connectivity (S3–D5, D5–F7, A6–I8, and F7–G9) suggests presence of 310-helical structure. Only one dαN(i, i + 4) connectivity (D5–G9) suggestive of some population of regular α-helical structure was observed. Overlap of some of the resonances under water interfered with the NOE analyses. However, the number of αHi-βHi+3, NOEs observed indicate that eledoisin is substantially folded from residues 3 to 11. Observation of these various types of NOEs simultaneously suggests that some degree of conformational averaging is present around a predominantly helical core. In the N-terminus of the helical segment there are crosspeaks characteristic of a β-turn or 310-helix. Therefore, the possibility of turn conformations must be considered for the region near the N-terminus as seen by the presence of dαN(i, i + 2) NOE between S3 to D5 and D5 to F7. In these structures, distances dαN(i, i + 2) and dαβ(i, i + 2) are very short and the NOE crosspeaks are usually observed (Wüthrich et al., 1984; Dyson et al., 1988). Moreover, side chains of polar residues such as Asp and Ser, when preceding helices, can provide stabilizing C O donors for hydrogen bond interactions in helices. This type of interaction is specially important for stabilizing short-length helix in a small linear peptide.
O donors for hydrogen bond interactions in helices. This type of interaction is specially important for stabilizing short-length helix in a small linear peptide.
It is also possible to acquire information about the φ angles along the peptide chain by measuring 3JNα coupling constants. Helical structures result in coupling constants of 4–5 Hz whereas extended structures have coupling constants in the range of 8–9 Hz (Wüthrich, 1986; Pardi et al., 1984). Wüthrich has suggested that a series of three or more 3JNα coupling constants less than 6 Hz is diagnostic of α-helical structure. All measurable coupling constants for eledoisin are in the range of 4–6 Hz, with the exceptions of S3 (6.4 Hz) and M11 (7 Hz). The apparent 3JNα coupling constant is a weighted average of the population and depends on the distribution of angles over the population (Kessler et al., 1988). Thus the low 3JNα values (<6 Hz) suggests that there is a large population of helical structures in the stretch between residues 4 to 11. M11 also shows a large 3JNα (>6 Hz), which may be due to fraying of helix at the terminus.
From the above NMR results, it is concluded that the residues 3–11 clearly meet the criteria for the existence of helical structure: presence of sequential dNN(i, i + 1) crosspeaks, presence of medium-range dαN(i, i + 3) and dαβ(i, i + 3) crosspeaks, and a series of 3JNα coupling constants of 6 Hz or less. However, the entire peptide is not helical. Due to the lack of medium-range crosspeaks and the large 3JNα coupling constants, we conclude that the first three residues are in turn conformation. No evidence for helix stabilization through salt-bridge formation was observed.
Generation of three-dimensional structure
Given the indication of a helical structure along the central core of eledoisin in DPC, it was of interest to use the observed NOEs to obtain information on the three-dimensional structure of the peptide. This was done using DYANA (Güntert and Wüthrich, 1991; Güntert et al., 1991). Initially, 50 structures were generated by DYANA using simulated annealing protocol, which improves the convergence of the structure calculations by introducing redundant dihedral angle restraints. The 20 conformations with the lowest target function value (i.e., least violations of experimental restraints and van-der-Waals distances) were chosen for further refinement using restrained energy minimization. The resulting structures are shown in Fig. 4, after superimposing the backbone atoms. Pair-wise RMSD calculated for backbone atoms for residues 1–11 for all 20 refined structures ranged from 0.04 to 0.83 Å, with a mean value of 0.39 Å and SD of 0.21 Å.
FIGURE 4.

Stereo view showing the superimposition of the backbone atoms of eledoisin for 20 structures generated by DYANA.
In a stable secondary structure, both Φ and Ψ dihedral angles should have well-defined values. The Ramachandran plots of all 20 refined structures (data not shown) indicate that the backbone dihedral angles consistently lie in the α-region and are solely within the allowed ranges. A helical-type backbone arrangement is indicated for the central region of eledoisin, in particular the stretch from S3 through M11 (Fig. 4), with some dynamic fraying of the helix termini. Measurement of COi, NHi+3 versus COi, NHi+4 distances along the stretch was made and i, i + 3 distances correlated with the eledoisin having a preference for 310 helix over regular helix. For the N-terminus of eledoisin the αHi–αHi+3 distances were measured to be within 7 Å in ensemble of conformations obtained. This distance is the threshold for defining a β-turn. However, it may not be appropriate to interpret this data in terms of a single turn conformation.
NMR spectroscopy is the method of choice for determining the three-dimensional solution structure of peptides. However, a number of factors such as precision in the estimate of NOE values, use of short interproton distances, approximation of the rotational reorientation of the peptide in solution with a single correlation time model, and the internal mobility of the peptide chain complicate the structure determination.
Due to the relatively large size of the DPC micelles, the correlation times in a micellar environment are expected to be much longer than the correlation times observed in an aqueous environment (Rizo et al., 1993). An additional complicating factor that must be recognized is the possibility of different regions of eledoisin experiencing different degrees of association with the micelles due to variations in hydrophobic and electrostatic effects. These factors make accurate quantification of the observed NOEs a major concern. The net result of the longer correlation times is that the observed NOEs may be weighted in favor of conformations that are induced by interactions with the micelles. This could result in an apparently higher percentage of secondary structure than is actually present.
CONCLUSIONS
The NMR studies reported here suggest that, in hydrophobic environment, part of the address domain and the whole of the message domain are folded, whereas in water as well as in DMSO, eledoisin has extended conformation. The likely situation is that eledoisin exists in equilibrium between two states in which there is a possibility of a turn over residues 1–3 followed by a stretch of helical structures for residues 4–11, and another where the helical region extends over residues 3–11. In DPC, the structural equilibrium is biased toward a 310-helix from residues 3 to 11, though small populations of regular α-helix cannot be excluded in the solution ensemble inasmuch as 310-helices are intermediates in the folding/unfolding pathways of regular helices. Also, short linear peptides like eledoisin may be too short to sustain a well-defined regular helix in solution. In support of this, NMR data for other tachykinins like kassinin (Grace et al., 2001), substance P in SDS (Young et al., 1994), DPC (Cowsik et al., 1997; Keire and Fletcher, 1996), and physalaemin in methanol (Chassaing et al., 1986a; Sumner et al., 1990) were also explained in terms of equilibrium between α-helix and 310-helix.
The amidated C-terminus of eledoisin in DPC comprises 310-helix or turn-like elements with some possible fraying of helix terminus. Although dynamic fraying of helical terminus is expected for small linear peptides, this observation is supported by structure activity data reported by Cascieri and co-workers (Cascieri et al., 1986), suggesting that SP-E (NK2/NK3) site has a requirement for a folded conformation of the C-terminal pentapeptide. The “address” segment of eledoisin, while undergoing greater conformational averaging than the message domain, also retains substantial conformational order in DPC. This order may be interpreted as a loosely defined turn or an unstable continuation of 310-helix along the message domain. However, the stability of turns is not known inasmuch as there are few reports on turn occurring in membrane mimetic solvent (Sonnichsen et al., 1992). However, identification of folded conformation in N-terminus under hydrophobic conditions has some significance, as it may represent an essential feature of NK2/NK3 binding. It is significant to note that N terminus of the NK2 selective agonists, Neuropeptide K and NKA, were also found to be folded in TFE (Horne et al., 1993).
Current model of binding of substance P and other agonists at NK1 receptor suggests that the three C-terminal residues (G-L-M-NH2) interact with a transmembrane region of the receptor. Thus, a change in the conformation of the three C-terminal residues will affect receptor binding. In a previous study, our group (Cowsik et al., 1997) determined that the structure adopted by substance P (the primary ligand for NK1 receptor) is comprised of a helical mid-region with an extended C-terminus in the presence of DPC micelles. Such a structure predicted to be biologically active correlates well with that reported for conformationally constrained analogs (Chassaing et al., 1987; Convert et al., 1988) and substance P in the presence of SDS and DPC micelles (Young et al., 1994; Keire and Fletcher, 1996). Further, Whitehead and co-workers have extended this study to NKA and NKB using SDS micelles (Whitehead et al., 1998). Their results also show that a helical structure is the predominant structure adopted by the agonists in the presence of membrane model system. Seelig and co-workers (Seelig and Macdonald, 1989; Seelig et al., 1996) have suggested that a possible reason for helix formation involving F7 and F8 residues of substance P is to provide a hydrophobic face (F7, F8, L10) and a hydrophilic face (Q6, G9) that will position substance P at the receptor binding site in such a fashion as to lead to optimal binding. NKA, NKB, kassinin, and eledoisin lack the Phe-8 residue of substance P and instead have the Val/Ile residue, which extends the helix length to include Gly-8 in case of NKA (Whitehead et al., 1998), both Gly-8 and Leu-9 in case of NKB (Whitehead et al., 1998), Gly-10, Leu-11, and Met-12 in case of kassinin (Grace et al., 2001), and Gly-9, Leu-10, and Met-11 in case of eledoisin (Wilson et al., 1994, and our present investigation). Such a change in helix length alters the positions of the hydrophobic and hydrophilic side chains for the C-terminus, decreasing the ability of eledoisin to bind as effectively to NK1 receptor (Fig. 5). Furthermore, stabilization of helix through an increase in helix length results in a reduction of flexibility of message domain, a situation determined to be unfavorable for NK1 receptor binding (Seelig et al., 1996). Conventional binding assays have shown that substance P is the preferred ligand for NK1 receptor and NKA, NKB, kassinin, and eledoisin bind with orders of magnitude much weaker than substance P. It is interesting to note that increasing helical content in the conformation of C-terminus of eledoisin and prediction of helical content by Seelig et al., (1996) are consistent with the poor binding property of eledoisin to the NK1 receptor. Further, the decreased ability of eledoisin in binding to NK1 can also be attributed to the absence of Proline residue in the position analogous to residue 4 of substance P, which has been indicated by binding studies to be essential for high-affinity binding to NK1 (Cascieri et al., 1992).
FIGURE 5.

A graphic representation of the lipid-bound eledoisin conformation. The peptide backbone is shown as a ribbon tube (blue). Ionic residues are colored red, polar residues are colored purple and the hydrophobic residues are colored yellow. The helical segment is clearly visible.
The binding of tachykinin peptides and their fragments to NK2 receptor sites in hamster urinary bladder membranes was examined and compared with the binding to NK1 receptor sites in rat submandibular glands (Buck and Shatzer, 1988). It was found that NKA and nonmammalian tachykinins eledoisin and kassinin exhibited the highest affinity and selectivity for bladder NK2 receptor binding sites. Structure activity studies indicated that the presence of Aspartate residue in NK2 agonists at the position analogous to residue 5 of substance P was critical for binding to NK2 receptor, suggesting that an ionic interaction may contribute to the binding energy (Cascieri et al., 1992; Buck and Shatzer, 1988). This hypothesis is further strengthened by a study involving probing of the binding domain of the NK2 receptor with fluorescent ligands labeled with environment-sensitive probes (Turcatti et al., 1995). This study indicates that the N-terminal regions of all NK2 agonists bound to the receptor are accessible to the solvent, which points to the extracellular regions of the receptor as the major binding determinant for the N-terminal address region of NK2 agonists (Turcatti et al., 1995). Further, structural comparison of NK2 receptor agonists and antagonists by simulated annealing techniques revealed that there was a close superimposition of the three key residues, Phe, Leu, and Met, which is hypothesized to be the binding site for the NK2 receptor (Giolitti and Maggi 1994). The results from our structural studies agree well with these findings and it can be seen from Fig. 5 that the C-terminus presents a hydrophobic upper half composed of the Phe-7, Ile-8, Leu-10, and Met-11 residues, and the N-terminus a hydrophilic lower half comprised of the Ser-3, Lys-4, and Asp-5 residues. We postulate that the hydrophobic C-terminus interacts with the transmembrane region of the receptor with Phe-7, Leu-10, and Met-11 forming the anchoring points and contributing a major portion of the binding energy. The hydrophilic and solvent accessible N-terminus helps to maintain the peptide conformation and plays an important role in NK2 receptor binding with Lys-4 and Asp-5 as anchoring points. It is interesting to note in this context that the modification of residues Lys-4 and Asp-5 of eledoisin as reported in binding assays (Cascieri et al., 1992; Buck and Shatzer, 1988) modulates its affinity and selectivity for the receptors.
In conclusion, the results obtained in this investigation are consistent with the proposed biologically active conformation of NK2 receptor agonist. Moreover, the NMR results presented here agree well with the theoretical secondary structure prediction for turns and helices in eledoisin using program ALB by Wilson and co-workers (Wilson et al., 1994). Our studies indicate that the helical central core of eledoisin is better defined in DPC micelles than in SDS and TFE. An increase in helical content is observed in presence of lipid micelles with the helix extending from residues 3 to 11, in comparison to SDS wherein the helix extends from 6 to 11 (Wilson et al., 1994). The presence of a loosely defined turn in the N-terminus preceding the helical core in the C-terminus of eledoisin is consistent with that observed in SDS and TFE. The overall conformational features adopted by eledoisin in DPC micelles correlate well with those reported for eledoisin in TFE and SDS micelles (Wilson et al., 1994), and with those of kassinin in DPC (Grace et al., 2001). In bioassays, kassinin has been found to interact with E-type eledoisin receptors in much the same manner as eledoisin (Erspamer et al., 1980; Iversen, 1982). On the basis of this correlation, it is interesting to note that conformation adopted by eledoisin in the presence of DPC micelles provides a biologically relevant structure.
Acknowledgments
The staff of the National 400- and 500-MHz NMR facility at Sophisticated Instruments Facility, Indian Institute of Science in Bangalore, and discussions with Professor Anil Kumar, are gratefully acknowledged. Help from Dr. Mini Thomas for recording CD spectra is gratefully acknowledged.
I.R.C. thanks the Council of Scientific and Industrial Research, India for the Junior Research Fellowship. This work is supported through a grant of University Grants Commission, India.
Abbreviations: DPC, dodecylphosphocholine; DQF-COSY, Double-Quantum Filtered Correlation Spectroscopy; NOESY, two-dimensional Nuclear Overhauser Effect Spectroscopy; ROESY, Rotating frame Overhauser Effect Spectroscopy; TOCSY, Total Correlation Spectroscopy; NK, neurokinin; SP, substance P.
References
- Bax, A., and D. Davis. 1985. MLEV-17 based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65:355–360. [Google Scholar]
- Alder, A. J., N. J. Greenfield, and G. D. Fasman. 1973. Methods Enzymol. 27:675–735. [DOI] [PubMed] [Google Scholar]
- Ananthanarayanan, V. S., and S. Orlicky. 1992. Interaction of substance P and its N- and C-terminal fragments with Ca2+: Implications for hormone action. Biopolymers. 32:1765–1773. [DOI] [PubMed] [Google Scholar]
- Anil Kumar, A. V., R. R. Ernst, and K. Wüthrich. 1980. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 64:2229–2246. [DOI] [PubMed] [Google Scholar]
- Bothner-By, A. A., and J. H. Noggle. 1979. Time development of nuclear Overhauser effects in multispin systems. J. Am. Chem. Soc. 101:5152–5155. [Google Scholar]
- Braun, W., G. Wider, K. H. Lee, and K. Wüthrich. 1983. Conformation of glucagon in a lipid-water interphase by 1H nuclear magnetic resonance. J. Mol. Biol. 169:921–948. [DOI] [PubMed] [Google Scholar]
- Braunschweiler, L., and R. R. Ernst. 1983. Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J. Magn. Reson. 53:521–528. [Google Scholar]
- Brown, L. R. 1979. Location and orientation relative to the micelle surface for glucagon in mixed micelles with dodecylphosphocholine. Biochim. Biophys. Acta. 557:135–148. [DOI] [PubMed] [Google Scholar]
- Bruch, M. D., M. M. Dhingra, and L. M. Gierasch. 1991. Side chain-backbone hydrogen bonding contributes to helix stability in peptides derived from an alpha-helical region of carboxypeptidase A. Proteins Struct. Funct. Genet. 10:130–139. [DOI] [PubMed] [Google Scholar]
- Buck, S. H., and S. A. Shatzer. 1988. Agonists and antagonist binding to tachykinin peptide NK-2 receptors. Life Sci. 42:2701–2708. [DOI] [PubMed] [Google Scholar]
- Cascieri, M. A., G. G. Chicchi, R. M. Freidinger, C. D. Colton, D. S. Perlow, B. Williams, N. R. Curtis, A. T. McKnight, J. J. Maguire, and D. F. Veber. 1986. Conformationally constrained tachykinin analogs which are selective ligands for the eledoisin binding site. Mol. Pharmacol. 29:34–38. [PubMed] [Google Scholar]
- Cascieri, M. A., R. R. Huang, T. M. Fong, A. H. Cheung, S. Sadowski, E. Ber, and C. D. Strader. 1992. Determination of the amino acid residues in substance P conferring selectivity and specificity for the rat neurokinin receptors. Mol. Pharmacol. 41:1096–1099. [PubMed] [Google Scholar]
- Chang, T. C., C. S. Wu, and J. T. Yung. 1978. Circular dichroic analysis of protein conformation: Inclusion of the β-turns. Anal. Biochem. 91:13–31. [DOI] [PubMed] [Google Scholar]
- Chassaing, G., O. Convert, S. Lavielle, and O. Ploux. 1987. In Substance P and Neurokinins. J. L. Henry, R. Couture, A. C. Cuello, G. Pelletier, R. Quirion, and D. Regoli, editors. Springer-Verlag, New York, 63–65.
- Chassaing, G., O. Convert, and S. Lavielle. 1986a. Preferential conformation of substance P in solution. Eur. J. Biochem. 154:77–85. [DOI] [PubMed] [Google Scholar]
- Chassaing, G., O. Convert, and S. Lavielle. 1986b. Conformational analogy between substance P and physalaemin. Biochim. Biophys. Acta. 873:97–104. [DOI] [PubMed] [Google Scholar]
- Chen, G. C., and J. T. Yang. 1977. Anal. Letters. 10:1195–1207. [Google Scholar]
- Convert, O., H. Duplaa, S. Levielle, and G. Chassaing. 1991. Influence of the replacement of amino acid by its D-enantiomer in the sequence of substance P. 2. Conformational analysis by NMR and energy calculations. Neuropeptides. 19:259–270. [DOI] [PubMed] [Google Scholar]
- Convert, O., O. Ploux, S. Lavielle, M. Cotrait, and G. Chassaing. 1988. Analysis of tachykinin-binding site interactions using NMR and energy calculation data of potent cyclic analogues of substance P. Biochim. Biophys. Acta. 954:287–302. [DOI] [PubMed] [Google Scholar]
- Cowsik, S. M., C. Lucke, and H. Rüterjans. 1997. Lipid-induced conformation of substance P. J. Biomol. Struct. Dyn. 15:27–36. [DOI] [PubMed] [Google Scholar]
- Davis, D. G., and A. Bax. 1985. Separation of chemical exchange and cross-relaxation effects in two-dimensional NMR spectroscopy. J. Magn. Reson. 64:533–535. [Google Scholar]
- De Marco, A., and G. Gatti. 1975. 1H- and 13C-NMR spectra of eledoisin and intermediate oligopeptides. Int. J. Pep. Pro. Res. 7:437–444. [DOI] [PubMed] [Google Scholar]
- Dyson, H. J., M. Rance, R. A. Houghten, R. A. Lerner, and P. E. Wright. 1988. Folding of immunogenic peptide fragments of proteins in water solution. I. Sequence requirements for the formation of a reverse turn. J. Mol. Biol. 201:161–200. [DOI] [PubMed] [Google Scholar]
- El-Agnaf, O. M. A., G. B. Irving, G. Fitzpatrick, W. G. Keneeth, and D. J. S. Guthrie. 1998. Comparative studies on peptides representing the so-called tachykinin-like region of the Alzheimer A-beta peptide. Biochem. J. 336:419–427 [Abeta(25–35)]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erspamer, G. F., V. Erspamer, and D. Piccinelli. 1980. Parallel bioassay of physalaemin and kassinin, a tachykinin dodecapeptide from the skin of the African frog Kassina senegalensis. Naunyn Schmiedebergs Arch. Pharmacol. 311:61–65. [DOI] [PubMed] [Google Scholar]
- Giolitti, A., and C. A. Maggi. 1994. Structural comparison of NK2 receptor agonists and antagonists. J. Comput. Aided Mol. Des. 8:341–344. [DOI] [PubMed] [Google Scholar]
- Grace, C. R., A. M. Lynn, and S. M. Cowsik. 2001. Lipid induced conformation of tachykinin peptide kassinin. J. Biomol. Struct. Dyn. 18:611–625. [DOI] [PubMed] [Google Scholar]
- Güntert, P., W. Braun, and K. Wüthrich. 1991. Efficient computation of three-dimensional protein structures in solution from nuclear magnetic resonance data using the program DYANA and the supporting programs CALIBA, HABAS and GLOMSA. J. Mol. Biol. 217:517–530. [DOI] [PubMed] [Google Scholar]
- Güntert, P., and K. Wüthrich. 1991. Improved efficiency of protein structure calculations from NMR data using the program DYANA with redundant dihedral angle constraints. J. Biomol. NMR. 1:447–456. [DOI] [PubMed] [Google Scholar]
- Hanley, M. R., and T. Jackson. 1987. Substance K receptor: Return of the magnificent seven. Nature. 329:766–767. [DOI] [PubMed] [Google Scholar]
- Holzworth, G., and P. Doty. 1965. J. Am. Chem. Soc. 87:218–228. [DOI] [PubMed] [Google Scholar]
- Horne, J., M. Sadek, and D. J. Craik. 1993. Determination of the solution structure of neuropeptide K by high resolution nuclear magnetic resonance spectroscopy. Biochemistry. 32:7406–7412. [DOI] [PubMed] [Google Scholar]
- Iversen, L. L. 1982. Substance P. Br. Med. Bull. 38:277–282. [DOI] [PubMed] [Google Scholar]
- Kallick, D. A., M. R. Tessmer, C. R. Watts, and C. Li. 1995. The use of dodecylphosphocholine micelles in solution NMR. J. Magn. Reson. B. 109:60–65. [DOI] [PubMed] [Google Scholar]
- Keire, D. A., and T. G. Fletcher. 1996. The conformation of substance P in lipid environments. Biophys. J. 70:1716–1721. [DOI] [PMC free article] [PubMed]
- Kessler, H., C. Griesinger, J. Lautz, A. Muller, W. F. Van Gunsteren, and H. J. C. Berendsen. 1988. Conformational dynamics detected by nuclear magnetic resonance NOE values and J-coupling constants. J. Am. Chem. Soc. 110:3393–3396. [Google Scholar]
- Lauterwein, J., L. Bosch, R. Brown, and K. Wüthrich. 1979. Physicochemical studies of the protein-lipid interactions in melittin containing micelles. Biochem. Biophys. Acta. 556:244–264. [DOI] [PubMed] [Google Scholar]
- Lavielle, S., G. Chassaing, O. Ploux, D. Loeuillet, J. Besseyre, S. Julien, A. Marquet, O. Convert, J. C. Beaujouan, Y. Torrens, L. Bergstrom, M. Saffroy, and J. Glowinski. 1988. Analysis of tachykinin binding site interactions using constrained analogues of tachykinins. Biochem. Pharmacol. 37:41–49. [DOI] [PubMed] [Google Scholar]
- Lavielle, S., G. Chassaing, D. Loeuillet, O. Convert, Y. Torrens, J. C. Beaujouan, M. Saffroy, F. Petitet, L. Bergstrom, and J. Glowinski. 1990. Selective agonists of tachykinin binding sites. Fundam. Clin. Pharmacol. 4:257–268. [DOI] [PubMed] [Google Scholar]
- Levian-Teitelbaum, D., N. Kolodny, M. Chorev, Z. Selinger, and C. Gilon. 1989. 1H-NMR studies of receptor-selective substance P analogues reveal distinct predominant conformations in DMSO-d6. Biopolymers. 28:51–64. [DOI] [PubMed] [Google Scholar]
- Macura, S., and R. R. Ernst. 1980. Elucidation of cross-relaxation in liquids by two-dimensional NMR spectroscopy. Mol. Phys. 41:95–117. [Google Scholar]
- Macura, S., Y. Huang, D. Suter, and R. R. Ernst. 1981. Two-dimensional chemical exchange and cross-relaxation spectroscopy of coupled nuclear spins. J. Magn. Reson. 43:259–281. [Google Scholar]
- Masu, Y., K. Nakayama, H. Tamaki, M. Harada, Y. Kuno, and S. Nakanishi. 1987. cDNA cloning of bovine Substance-K receptor through oocyte expression system. Nature. 329:836–838. [DOI] [PubMed] [Google Scholar]
- Maurer, T., and H. Rüterjans. 1994. Solution structure of seminal plasmin in the presence of micelles. Eur. J. Biochem. 220:111–116. [DOI] [PubMed] [Google Scholar]
- Maurer, T., C. Lücke, and H. Rüterjans. 1991. Investigation of the membrane-active peptides melittin and glucagon by photochemically induced dynamic-nuclear-polarization (photo-CIDNP) NMR. Eur. J. Biochem. 196:135–140. [DOI] [PubMed] [Google Scholar]
- McDonnell, P. A., and S. J. Opella. 1993. Effect of detergent concentration on multidimensional solution NMR spectra of membrane proteins in micelles. J. Magn. Reson. B. 102:120–125. [Google Scholar]
- Muñoz, V., and L. Serrano. 1994. Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Biol. 1:399–409. [DOI] [PubMed] [Google Scholar]
- Nakanishi, S. 1991. Mammalian tachykinin receptors. Annu. Rev. Neurosci. 14:123–136. [DOI] [PubMed] [Google Scholar]
- Opella, S. J. 1997. NMR and membrane proteins. Nature Struc. Biol. NMR Supp. 10:845–848. [PubMed] [Google Scholar]
- Palczewski, K., T. Kumasaka, T. Hori, C. A. Behnke, H. Motoshima, B. A. Fox, I. LeTrong, D. C. Teller, T. Okada, R. E. Stenkamp, M. Yamamoto, and M. Miyano. 2000. Crystal structure of rhodopsin: A G-protein-coupled receptor. Science. 289:739–745. [DOI] [PubMed] [Google Scholar]
- Pardi, A., M. Billeter, and K. Wüthrich. 1984. Calibration of the angular dependence of the amide proton-C alpha proton coupling constants, 3JHN alpha, in a globular protein. Use of 3JHN alpha for identification of helical secondary structure. J. Mol. Biol. 180:741–751. [DOI] [PubMed] [Google Scholar]
- Pellegrini, M., M. Royo, M. Chorev, and D. F. Mierke. 1996. Conformational characterization of a peptide mimetic of the third cytoplasmic loop of the G-protein coupled parathyroid hormone/parathyroid hormone related protein receptor. Biopolymers. 40:653–666. [DOI] [PubMed] [Google Scholar]
- Rance, M., O. W. Sorensen, G. Bodenhausen, G. Wagner, R. R. Ernst, and K. Wüthrich. 1983. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 117:479–485. [DOI] [PubMed] [Google Scholar]
- Rizo, J., F. J. Blanco, B. Kobe, M. D. Bruch, and L. M. Gierasch. 1993. Conformational behavior of Escherichia coli OmpA signal peptides in membrane mimetic environments. Biochemistry. 32:4881–4894. [DOI] [PubMed] [Google Scholar]
- Schwyzer, R., D. Erne, and K. Rolka. 1986. Prediction of preferred conformation, orientation and accumulation of substance P on lipid membranes. Helv. Chim. Acta. 69:1789–1797. [Google Scholar]
- Schwyzer, R. 1987. Membrane-assisted molecular mechanism of neurokinin receptor subtype selection. EMBO J. 6:2255–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig, A. 1992. Interaction of a substance P agonist and of substance P antagonists with lipid membranes: A thermodynamic analysis. Biochemistry. 31:2897–2904. [DOI] [PubMed] [Google Scholar]
- Seelig, A., and P. M. Macdonald. 1989. Binding of a neuropeptide, substance P, to neutral and negatively charged lipids. Biochemistry. 28:2490–2496. [DOI] [PubMed] [Google Scholar]
- Seelig, A., T. Alt, S. Lotz, and G. Holzemann. 1996. Binding of substance P agonists to lipid membranes and to the neurokinin-1 receptor. Biochemistry. 35:4365–4374. [DOI] [PubMed] [Google Scholar]
- Sonnichsen, F. D., J. E. Van Eyk, R. S. Hodges, and B. D. Sykes. 1992. Effect of trifluoroethanol on protein secondary structure: an NMR and CD study using a synthetic actin peptide. Biochemistry. 31:8790–8798. [DOI] [PubMed] [Google Scholar]
- Sumner, S. C. J., K. S. Gallagher, D. G. Davis, D. G. Covell, R. L. Jernigan, and J. A. Ferretti. 1990. Conformational analysis of the tachykinins in solution: Substance P and physalaemin. J. Biomol. Struct. Dyn. 8:687–707. [DOI] [PubMed] [Google Scholar]
- Szollosy, A., A. Otter, J. M. Stewart, and G. Kotovych. 1986. An NMR study of the conformations of N-terminal substance P fragments and antagonists. J. Biomol. Str. Dynam. 4:501–519. [DOI] [PubMed] [Google Scholar]
- Turcatti, G., H. Vogel, and A. Chollet. 1995. Probing the binding domain of the NK2 receptor with fluorescent ligands: Evidence that heptapeptide agonists and antagonists bind differently. Biochemistry. 34:3972–3980. [DOI] [PubMed] [Google Scholar]
- Whitehead, T. L., S. D. McNair, C. E. Hadden, J. K. Young, and R. P. Hicks. 1998. Membrane-induced secondary structures of neuropeptides: A comparison of the solution conformations adopted by agonists and antagonists of the mammalian tachykinin NK1 receptor. J. Med. Chem. 41:1497–1506. [DOI] [PubMed] [Google Scholar]
- Williams, R. W., and J. L. Weaver. 1990. Secondary structure of substance P bound to liposomes in organic solvents and in solution from Raman and CD spectroscopy. J. Biol. Chem. 265:2505–2513. [PubMed] [Google Scholar]
- Wilson, J. C., K. J. Nielsen, M. J. Mcleish, and D. J. Craik. 1994. A determination of the solution conformation of the nonmammalian tachykinin eledoisin by NMR and CD spectroscopy. Biochemistry. 33:6802–6811. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., B. D. Sykes, and F. M. Richards. 1992. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 31:1647–1651. [DOI] [PubMed] [Google Scholar]
- Woody, R. W. 1992. Circular dichroism and conformation of unordered polypeptides. Adv. Biophys. Chem. 2:37–79. [Google Scholar]
- Woolley, G. A., and C. M. Deber. 1987. Peptides in membranes: Lipid-induced secondary structure of substance P. Biopolymers. 26:S109–S121. [DOI] [PubMed] [Google Scholar]
- Wu, C., and C. T. Yang. 1983. Instability of terminal amino acid residues in oligopeptides in sodium dodecyl sulfate solution. Biochem. Biophys. Acta. 746:72–80. [Google Scholar]
- Wüthrich, K., M. Billeter, and W. Braun. 1984. Polypeptide secondary structure determination by nuclear magnetic resonance observation of short proton-proton distances. J. Mol. Biol. 180:715–740. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of Proteins and Nucleic Acids. J. Wiley & Sons, New York.
- Young, J. K., C. Anklin, and R. P. Hicks. 1994. NMR and molecular modeling investigations of the neuropeptide substance P in the presence of 15 mM sodium dodecyl sulfate micelles. Biopolymers. 34:1449–1462. [DOI] [PubMed] [Google Scholar]
- Yu, C., and T. H. Yang. 1991. J. Chin. Biochem. Soc. 20:99–105. [Google Scholar]