

Abstract
The “Phot” protein family comprises blue-light photoreceptors that consist of two flavin mononucleotide (FMN)-binding LOV (light, oxygen, and voltage) domains and a serine/threonine kinase domain. We have investigated the LOV1 domain of Phot1 from Chlamydomonas reinhardtii by time-resolved absorption spectroscopy. Photoexcitation of the dark form, LOV1-447, causes transient bleaching and formation of two spectrally similar red-shifted intermediates that are both assigned to triplet states of the FMN. The triplet states decay with time constants of 800 ns and 4 μs with an efficiency of >90% into a blue-shifted intermediate, LOV1-390, that is attributed to a thiol adduct of cysteine 57 to FMN C(4a). LOV1-390 reverts to the dark form in hundreds of seconds, the time constant being dependent on pH and salt concentration. In the mutant C57S, where the thiol adduct cannot be formed, the triplet state displays an oxygen-dependent decay directly to the dark form. We present here a spectroscopic characterization of an algal sensory photoreceptor in general and of a LOV1 domain photocycle in particular. The results are discussed with respect to the behavior of the homologous LOV2 domain from oat.
INTRODUCTION
Phototropins and homologs, recently renamed Phot proteins (Briggs et al., 2001), comprise a class of blue-light receptors that are involved in phototropic plant movement (Christie et al., 1998, 1999; Huala et al., 1997), chloroplast relocation (Jarillo et al., 2001; Kagawa et al., 2001), and stomatal opening in guard cells (Kinoshita et al., 2001). All Phot proteins contain two LOV (light-, oxygen-, voltage-sensitive) domains and a downstream serine/threonine kinase. Heterologous expression studies have shown that one flavin mononucleotide (FMN) is bound to each LOV domain (Christie et al., 1999).
Absorption spectra have been published for LOV1 and LOV2 (Christie et al., 1999), and it has been shown that both LOV domains are bleached under continuous illumination (Salomon et al., 2000), converting the LOV domain into a blue-shifted species (λmax = 390 nm) that only slowly recovers within many seconds in darkness. Before time-resolved spectroscopy was applied, Salomon et al. (2000) proposed that a flavin C(4a) thiol adduct is formed between FMN and a cysteine residue. This statement has been supported by two experimental approaches. First, replacing the cysteine of the highly conserved amino acid segment NCRFLQG by alanine led to an unbleachable LOV domain (Salomon et al., 2000). Second, the LOV2 domain of Avena sativa (oat) Phot1 was reconstituted with various 13C/15N-labeled isotopomers of FMN and investigated by NMR spectroscopy. The light-driven formation of the flavin C(4a) thiol adduct caused a large upfield shift of the C(4a) signal (Salomon et al., 2001).
Time-resolved spectroscopy in the time range of 30 ns–200 s, which allows conclusions about the photocycle, has only been carried out with a LOV2 domain of Phot1 from A. sativa (Swartz et al., 2001). The authors identified a triplet-like species that is formed within <30 ns after light excitation and decays with a time constant of 4 μs. The triplet converts into a 390-nm absorbing metastable form that decays with τ = 50 s. The LOV-390 was likely to resemble the FMN-cysteine adduct. Based on pH titration of the chromophore fluorescence, the authors suggested that the reactive cysteine species is a thiolate.
Recently, the first Phot gene in a green alga was discovered in Chlamydomonas reinhardtii. The gene encodes a 75-kDa protein that is smaller than its relatives from higher plants (120 kDa). Expression analysis revealed rather constant levels of Phot protein in the darkness during the cell cycle, whereas expression was reduced in vegetative cells in the light (Huang et al., 2002). When Phot expression was down-regulated by using an antisense approach, the blue-light dependent formation of sexually competent gametes under nitrogen starvation was inhibited (Huang and Beck, 2002). Thus the regulation of gametogenesis under low nutrient conditions is mediated by Phot1. The LOV1 domain from the algal Phot1 receptor was expressed in Escherichia coli. Not only the sequence but also the basic absorption and fluorescence properties are similar to LOV domains from higher plant Phot1 receptors (Holzer et al., 2002). A preliminary study of the kinetic properties of C. reinhardtii LOV1 showed an exceptionally slow dark decay rate after blue light irradiation (Kasahara et al., 2002). The LOV1 domain from C. reinhardtii was crystallized and the structure in the dark and illuminated state was determined to 1.9 Å and 2.8 Å resolution, respectively (Fedorov et al., 2003). Analysis of the crystal structure revealed that during illumination the Cys-57 sulfur atom interacts covalently with the FMN C(4a) atom at a distance of 1.9 Å. The overall structure is similar to the LOV2 structure of the phy3-receptor from the fern Adiantum capillus-veneris (Crosson and Moffat, 2001, 2002).
Here, we present the photocycle of the LOV1 domain from the C. reinhardtii Phot1 protein. The analysis of dynamic spectroscopic changes that follow light excitation together with the structural information led to the development of a light-triggered molecular reaction pathway. The results are compared with published data on LOV2 from higher plant Phot1.
MATERIALS AND METHODS
LOV1 expression and purification
The full length cDNA-clone (Genebank accession No. AJ 416557) (Huang et al., 2002) of the C. reinhardtii phot1 (formerly named nph1 for nonphototropic hypocotyl 1) was received from C. F. Beck and K. Huang, Freiburg, Germany. The gene fragment encoding the FMN-binding LOV1 domain (amino acids 16–133) was amplified by PCR and inserted into the XhoI and BamHI sites of the E. coli expression vector pET16 (Novagen, Bad Soden, Germany) in such a way that the protein carries 1 Gly, 10 His, and a protease cleavage site (factor Xa) derived from the vector sequences at the N-terminal end. The protein was expressed in E. coli strain BL21 and purified via a Ni-NTA column (Qiagen, Hilden, Germany). The C57S mutant was generated by site-directed mutagenesis, and expressed and purified like the wild-type LOV1.
In a second approach, the gene fragment encoding LOV1 was amplified by PCR using oligonucleotide primers that contain EcoRI and HindIII restriction sites. The PCR product was digested with EcoRI and HindIII and cloned into the pMALC2-vector (New England Biolabs, Frankfurt, Germany). A short DNA-sequence encoding 9 His was inserted into the EcoRI site. The fusion protein MBP-LOV1 was expressed in BL21 and purified via amylose resin (New England Biolabs) according to the instructions of the manufacturer.
The chromophore in LOV1-WT and LOV1-C57S is flavin mononucleotide (FMN, riboflavin-5′-phosphate) as determined by chromophore extraction in 1% TCA, protein precipitation, and electrospray mass spectroscopy (SSQ 7000, Thermo Finnigan, San Jose, CA, USA). For injection, the samples were diluted in 0.5% acetic acid, 50% acetonitrile. The determined molar mass of the apoprotein is 15,609 g mol−1, which is 130 g mol−1 below the expected value (15,739 g mol−1), indicating that the N-terminal methionine was cleaved off. The molar mass of 457 g mol−1 for the chromophore corresponds to that of FMN. For spectroscopic experiments, LOV1-WT and LOV1-C57S are diluted in 10 mM phosphate buffer, pH 8, containing 10 mM NaCl.
UV/Vis spectra and dependence of slow kinetics on pH and salt concentration
Absorption spectra were measured with a Lambda 9 spectrophotometer (PerkinElmer, Frankfurt, Germany). The time sequence of absorption spectra was recorded with a Specord S100B diode array spectrometer (Analytik Jena, Jena, Germany). Samples at different pH values and different salt concentrations were obtained by adding 1 M phosphate buffer and 1 M sodium chloride solutions to the standard protein samples. The samples were kept at 20°C and irradiated for 30 s with a 50-W tungsten lamp (Osram, München, Germany) through a 435-nm cutoff filter (GG435, Schott, Germany). Subsequently, time traces at 475 nm were recorded in the dark.
Time-resolved UV/Vis experiments at single wavelengths
The sample was excited at the long side of a rectangular (10 × 2 mm) fluorescence cuvette by a light pulse of 446-nm wavelength, 15-ns duration, 5 × 10 mm cross section, and 1 mJ energy from a Scanmate 2E dye laser (Lambda Physik, Göttingen, Germany) pumped by the third harmonic of an SL-803 Nd:YAG (Spectron Laser Systems, Rugby, England). The transient absorption was measured along the 10-mm path perpendicular to the excitation by a pulsed 150-W Xe lamp (MSP-05, Müller, Moosinning, Germany). Two monochromators, one before and one after the sample, served to select the wavelength with a resolution of 3 nm and eliminate fluorescence and stray light. The transmitted signal was detected by a R446 photomultiplier (Hamamatsu Photonics, Herrsching, Germany) and recorded with a 500-MHz digital storage oscilloscope (TDS 744A, Tektronix, Beaverton, OR, USA) triggered by the excitation pulse via a photodiode. A homemade trigger generator regulated the selection of single laser shots from the 20-Hz pulse train of the laser and the synchronization of the experiment. Each data set was generated by taking four averaged traces in succession: the signal trace, its reference trace, a fluorescence signal without measuring pulse, and a baseline without both light sources. In the case of the wild-type LOV1, a sampling rate of 0.03 Hz was chosen to minimize accumulation of the long-lived intermediate. The temperature was maintained at 20°C. Measurements under pure nitrogen and xenon atmosphere were performed by bubbling the gas through the solution for 20 min.
Time-resolved UV/Vis experiments with CCD camera detection
A CCD camera detection setup was used to acquire time-resolved difference spectra. A tunable optical parametric oscillator (GWU Lasertechnik, Erftstadt, Germany) pumped by the third harmonic of a Quanta Ray GRC 12S Nd:YAG laser (Spectra-Physics, Mountain View, CA, USA) was used for excitation. Pulses of 5-mJ energy and 5-ns duration at 450 nm were guided to the sample by a quartz fiber bundle to achieve homogenous illumination. The repetition rate of the laser was adjusted to the photocycling time of the enzyme, i.e., as low as 0.01 Hz for wild-type LOV1 and as high as 10 Hz for the C57S mutant. The probe light from a continuous Xe-lamp (LXH100, Müller, Moosinning, Germany) was guided through a fiber bundle, attenuated to an intensity of 100 μW/cm2 by a neutral gray filter, collimated by a lens, and passed through the cuvette with the sample. The transmitted light was imaged onto a second fiber bundle connected to the entrance slit of a SpectraPro-150 spectrograph (Acton Research, Acton, MA, USA) equipped with an intensified CCD camera (CCD-576G, Princeton Instruments, Monmouth Junction, NJ, USA). The time resolution was determined by the minimal gating time of 7 ns of the image intensifier (micro channel plate). The dispersion of the grating allowed to cover a wavelength range of 350 nm in a single experiment. Spectra with different midpoints of the grating were appended to cover the visible wavelength range from 300 to 850 nm. A delay generator triggered the pulsed laser and synchronized it with the gate of the CCD camera. Gating also effectively suppressed interfering fluorescence and laser stray light.
Data analysis
The data sets from the transient spectral measurements were reduced for global analysis by singular value decomposition (SVD). This method separates the matrix of the wavelength- and time-resolved data into time-dependent and spectral components of descending significance. In the case of the CCD camera data, the MATLAB software (The MathWorks, Natick, MA, USA) was used. The data sets obtained with the diode array spectrometer were analyzed by SVD and target factor analysis with homemade software.
RESULTS
The dark form (LOV1-447)
The LOV1 domain of the algal phot1 was expressed in E. coli and purified via affinity chromatography. The amino acid sequence and a crystal structure with 1.9 Å resolution is presented by Fedorov et al., 2003). The basic absorption properties of LOV1 have been reported before (Holzer et al., 2002). A representative spectrum is shown in the upper part of Fig. 1 (solid line): The absorption bands centered at 447 and 360 nm represent the S0 → S1 and S0 → S2 transitions of the FMN, respectively (Heelis, 1982). They exhibit vibronic fine structure that is smeared out when the chromophore is released from the apoprotein in aqueous solution. This smearing could be due to solvent-induced inhomogeneous broadening or due to the loss of motional constraints imposed by the protein environment.
FIGURE 1.

(Top) Absorption spectrum of wild-type LOV1 (solid line) and the long-lived intermediate LOV1-390 (dotted line) extracted by target factor analysis from the data shown below. (Bottom) Sequence of absorption spectra showing the relaxation of wild-type LOV1 from the photostationary state to the dark form after white-light irradiation. The inset shows the temporal behavior of the absorbance at 475 nm.
The long-lived intermediate
Irradiation of wild-type LOV1 with blue light (λ < 480 nm) leads to partial bleaching of the absorption that recovers in the dark on a timescale of several minutes. The UV/Vis absorption spectrum of this long-lived intermediate was obtained in the following way. The sample was irradiated at pH 8 by a xenon flash. In 20-s time steps, we recorded a sequence of absorption spectra with the diode array spectrometer. The sequence is shown in the lower part of Fig. 1. The inset in this figure shows the temporal behavior of the absorbance at 475 nm. The data were analyzed with target factor analysis (Hendler and Shrager, 1994). This method allows the extraction of the spectrum of the intermediate even in the case when it completely overlaps with that of the dark form. The result is unique if we impose the restrictions that the spectrum must be positive at all wavelengths and should not contain the absorption maximum at 475 nm of the dark form. The spectrum obtained in this way shows a broad absorption band at 390 nm without vibrational structure. It is shown as the dotted line in the upper part of Fig. 1. This intermediate spectrum (LOV1-390) is the first for a LOV1 domain. Salomon et al. (2000) obtained under continuous illumination a similar spectrum in oat LOV2. The oat LOV1 domain was bleached to a photostationary state, suggesting that it was only partially converted to a thiol adduct (Salomon et al., 2000). Our intermediate spectrum can accordingly be assigned to the covalent adduct of flavin C(4a) and the cysteine 57 thiol group. Other cysteine residues (Cys-32 and Cys-83) are too far away from the chromophore to be able to react with the FMN (Fedorov et al., 2003).
The decay kinetics of LOV1-390 was studied by measuring time traces of the recovery of absorbance at 475 nm after illumination. A monoexponential fit to the recovery yielded a time constant of τ = 200 s at 20°C and pH 8 that is close to the value of 168 s determined by Kasahara et al. (2002). A variation of the pH of the sample revealed that the reaction is considerably slower under acidic than under basic conditions (see Fig. 2). Additionally, below pH 6 the time constant is dependent on sodium chloride concentration at a constant concentration of the phosphate buffer (100 mM). Thus it seems that the decay is facilitated by a base located at the surface of the protein. The pH dependence of the single exponential decay time can be understood with the following simple model: The system contains a basic center that is involved in the back reaction. In the case that this base is in the protonated form BH, the back reaction proceeds with a rate constant k1. If the base is in the deprotonated form B−, the rate constant of the process is k2. When the equilibrium between both forms is established much faster than both k1 and k2, a monoexponential decay is observed with the apparent rate constant
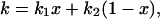 |
(1) |
where x is the relative population of the protonated form. The latter can be related to the concentration ratio by:
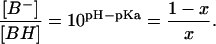 |
(2) |
This leads to the following dependence of the time constant on the pH:
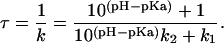 |
(3) |
The fit of Eq. 3 to the experimental data is shown in Fig. 2. The data correspond well to the simple model with only three parameters, using a pKa of the acid-base pair of 5.3 and 5.6 for 10 mM and 90 mM NaCl concentration, respectively. The limiting time constants (τ = 1/k) are 250 s and 950 s at low salt and 180 s and 1900 s at high salt concentration.
FIGURE 2.

pH dependence of the decay time of the wild-type long-lived intermediate LOV1-390 at different sodium chloride concentrations (100 mM phosphate buffer). Time constants were derived from the recovery of absorbance at 475 nm with a maximum deviation of 3% from the average value. The lines are fitted curves corresponding to a general mechanistic model, which yields a pKa between 5 and 6 of the involved acid-base pair.
Inasmuch as the spontaneous back reaction from LOV1-390 to LOV1-447 is slow, moderate blue-light irradiation should lead to a complete conversion into LOV1-390. To investigate this, a sample was illuminated with a 100-W tungsten lamp through a 435-nm cutoff filter until the photostationary state was reached. By varying the intensity with neutral density filters, relative changes in absorbance ΔA/A0 at 475 nm were observed (see Fig. 3). The experimental values could be fitted by an equation of the form:
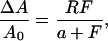 |
(4) |
where F is the light intensity, R is the saturated value at infinite intensity, and a is the intensity at which half of the saturation is achieved. This approach enabled an extrapolation to infinite light intensity that yielded a value of R = 0.84 at 475 nm, i.e., the absorption cannot be bleached below 16% of the initial absorption. A model that yields this functional form in terms of the quantum efficiencies for both reactions and the absorption coefficients of both species at the probe wavelength is presented in the Appendix. The conclusion of our calculations is the following: Under the assumption that the molar extinction coefficient of LOV1-390 at 475 nm is less than 6% of that of LOV1-447, as derived from the spectrum of the intermediate shown in Fig. 1, there must be a photoinduced reaction from LOV1-390 back to LOV1-447.
FIGURE 3.

Relative changes in absorbance of wild-type LOV1 at 475 nm dependent on the relative intensity of irradiation. The solid line is a fitted curve according to Eq. 4 reaching the limiting value of 0.84 at infinite light intensity. The dashed line illustrates the limit of 0.94 expected in the absence of a photoinduced back reaction from LOV1-390 to the dark form. This value results from the absorption of LOV1-390 at 475 nm according to Fig. 1.
The short-lived intermediate
The kinetics of the photoreaction of wild-type LOV1 has been followed after nanosecond excitation with a blue laser pulse. Fig. 4 shows difference spectra in the visible range at indicated times after the pulse. The negative absorbance changes at 447 nm and 475 nm correspond to the depletion of LOV1-447. A red-shifted absorption band at 510 nm and a deep red feature around 700 nm are discernible in the 1 μs spectrum that decay during the transition to the subsequent spectra. A band at 390 nm is present 1 μs after the flash. This band decays only partially and then remains constant in the millisecond range. This residual positive absorption at 390 nm is readily assigned to formation of the thiol adduct of FMN based on the spectrum of the long-lived intermediate shown in Fig. 1. The decay occurs on the timescale of minutes.
FIGURE 4.

Absorption difference spectra of wild-type LOV1 at indicated delay times after excitation with a 450-nm laser pulse. The 1-μs spectrum shows the formation of the intermediate LOV1-715.
The study of the early photointermediates is hampered by the long cycling time of the enzyme. To reduce this, we have employed the C57S mutant where the reactive thiol group is replaced by a hydroxyl group and a flavin-cysteine adduct cannot be formed. This allows a high repetition rate and permits extensive averaging to improve the signal-to-noise ratio. The dark form spectrum of the C57S mutant is shown in Fig. 5 (top). It is very similar to the wild-type spectrum except that all peaks are ∼5 nm blue-shifted. The high similarity is a strong indication that the mutation influences the properties of the chromophore only to a minor extent. In contrast to oat LOV2 (Swartz et al., 2001), the C57S mutant shows the same fine structure as the wild type in the 350-nm band. All absorbance difference bands of the C57S mutant decay within 100 μs after blue-light excitation (see Fig. 5). The kinetics of this decay have been analyzed by SVD. As can be seen in Fig. 6 A, all of the spectral information is included in only one component, whereas all other components contain only noise. The negative bands at 442 nm and 470 nm correspond to the dark form of the LOV1-C57S mutant that is depleted by the laser pulse. Two strong absorption bands appear with maxima at 648 nm and 715 nm. Accordingly, the intermediate is referred to as LOV1-715. It can be attributed to the absorption of a triplet state of FMN along with its vibronic side band in agreement with the assignment of Swartz et al. (2001) for oat LOV2, for which a similar spectrum has been recorded up to 700 nm. Additionally, two bands at 379 nm and around 500 nm appear that might be caused by triplet absorptions with higher energy as inferred from literature data (Sakai and Takahashi, 1996; Bernt et al., 1999). Because the chromophore is left unchanged by the C57S mutation, the triplet excited state must be observed in the wild-type as well. Indeed, all four absorption bands of the C57S difference spectrum also occur in wild-type LOV1 and evidently at the same wavelengths (see Fig. 4). The band at 379 nm overlaps in the wild-type with the rise of LOV1-390 absorption. The relative intensity of the band at 500 nm is higher in the wild-type and C57S mutant spectra than in spectra of the FMN triplet state in aqueous solution (Sakai and Takahashi, 1996). Therefore, a further intermediate might be present such as a radical species absorbing in this spectral region. The time trace of the LOV1-715 decay in the C57S mutant is displayed in Fig. 6 B (filled symbols) along with a biexponential fit. The corresponding time constants are 2 μs (with a relative amplitude of 20%) and 23 μs (80%). A third exponential improves the fit only marginally. Hence at least two species with different kinetics are involved that cannot, however, be distinguished on the basis of their absorption spectra.
FIGURE 5.

(Top) Absorption spectrum of the C57S mutant of LOV1. (Bottom) Absorption difference spectra of the mutant at various delay times after the laser flash. All difference bands decay in the mutant within 100 μs and are assigned to the LOV1-715 intermediate.
FIGURE 6.

Singular value decomposition and exponential fitting of the time-resolved absorbance changes of the LOV1-C57S mutant. Panel A depicts the three most significant spectral components. The time trace of the spectral component LOV1-715 is shown in panel B along with the fitted sum of two exponentials. The resulting time constants are indicated by dashed vertical lines (τ1 = 2 μs and τ2 = 23 μs, with relative amplitudes of 20% and 80% respectively).
Because of the extremely long cycling time, it is difficult to measure the fast kinetics of wild-type LOV1 with the multichannel approach. Therefore, the decay kinetics of wild-type and mutant LOV1 were also measured with the monochromatic setup. As already mentioned, in wild-type LOV1, the short-lived transient LOV1-715 decays into the LOV1-390 intermediate. A representative trace of this decay at 710 nm is depicted in Fig. 7 (top). A biexponential fit to the trace yields time constants of 800 ns (80%) and 4 μs (20%). This fast kinetics explains why in the multichannel approach in Fig. 4 the absorption of LOV1-715 is not detectable after 10 μs.
FIGURE 7.
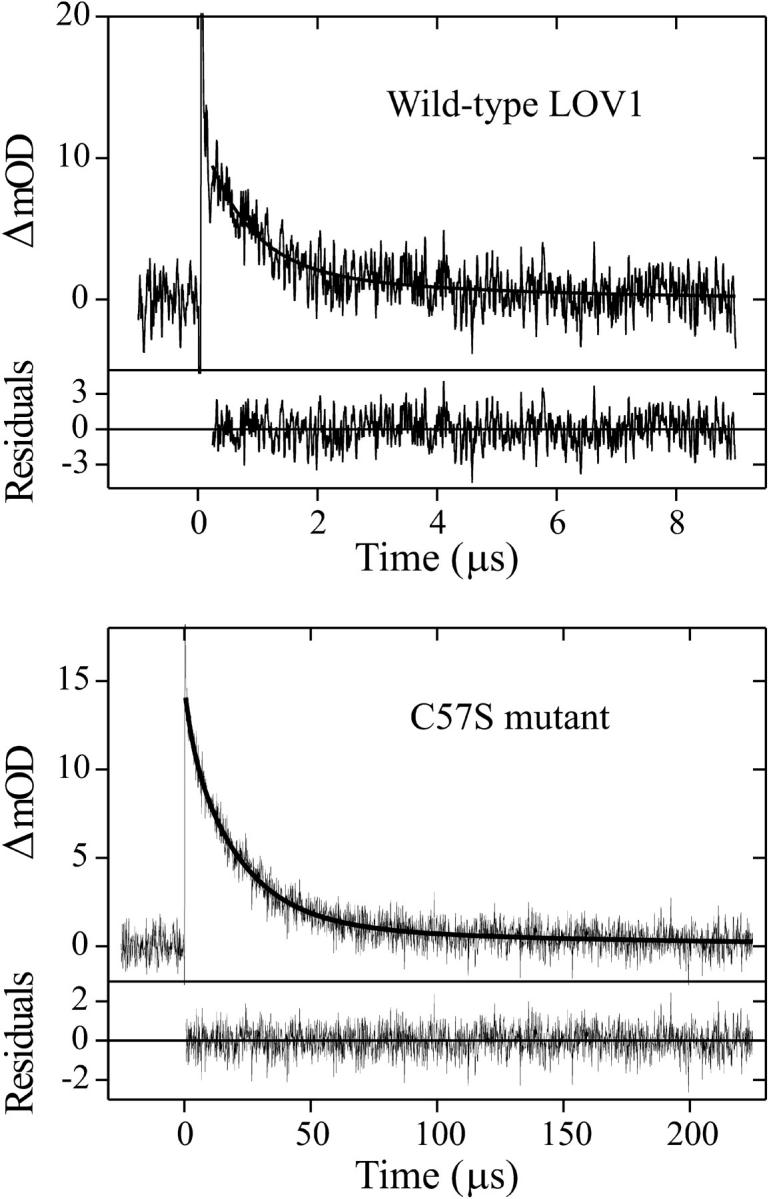
Decay kinetics of the intermediate LOV1-715 obtained by monitoring single wavelengths after excitation with a 446-nm laser pulse. Solid lines indicate a biexponential fit. (Top) Wild-type LOV1-715 decay observed at 710 nm yielding time constants of τ1 = 800 ns (80%) and τ2 = 4 μs (20%). (Bottom) C57S mutant observed at 650 nm with decay time constants of τ1 = 3 μs (25%) and τ2 = 27 μs (75%).
For the C57S mutant, the time trace of the absorbance change at 650 nm is shown in Fig. 7 (bottom). The fitted curve is a sum of two exponentials with time constants of 3 and 27 μs and relative amplitudes of 25 and 75%, respectively. This result agrees well with the analysis of the kinetic data obtained with the multichannel approach (see Fig. 6 B).
Because the decay of LOV1-715 in the C57S mutant is biexponential but only one spectral component is obtained from SVD analysis, we have to postulate two species with identical absorption spectra. The simplest model explaining this kinetics involves separate decays due to the presence of two different quenching channels and excludes any interchange reactions. As the intermediate LOV1-715 has been assigned to the triplet state of FMN, we deduce that molecular oxygen could be a quencher when present close to the chromophore in a certain fraction of the protein sample. The samples had not been prepared under anaerobic conditions. Therefore, this hypothesis was tested by repeating the measurement in solutions saturated with pure nitrogen. The decay of LOV1-715 could then be fitted by a monoexponential function with τ = 27 μs (data not shown). The fast (3 μs) component of aerated C57S mutant solution had disappeared. A correlation between the amount of oxygen dissolved and the relative amplitudes of the two exponential components could not be investigated because measurements in pure oxygen lead to a fast degeneration of the sample. The finding that oxygen has a strong effect on the decay kinetics of the short-lived intermediate LOV1-715 is further support for the assignment of this species to the triplet excited state of the chromophore. In contrast, the biexponential decay of wild-type LOV1-715 was not influenced by oxygen. Experiments in nitrogen-saturated solution did not show a significant change in the time constants of 800 ns and 4 μs, nor in their amplitudes.
Triplet states might also be affected by external heavy atoms. Kinetic measurements in a xenon-saturated solution of the C57S mutant did, however, show a monoexponential decay of LOV1-715 with the same time constant of τ = 27 μs derived from the exclusion of oxygen. A significant heavy atom effect of xenon on the decay was not observed.
DISCUSSION
We have presented the first detailed in vitro characterization of a chromophore binding domain from an algal sensory photoreceptor. The availability of crystal structure data on this particular LOV1 domain facilitates the mechanistic interpretation of the spectral data obtained (Fedorov et al., 2003). The comparison with LOV2 domains shows similarities but is limited by the fact that all previous work has been performed on higher plants. The photocycle has been studied for a LOV2 domain from oat Phot1 (Swartz et al., 2001) and a structure is available for LOV2 from fern phy3 (Crosson and Moffat, 2001, 2002).
The dark form LOV1-447 and the cysteine 57 protonation state
As already mentioned by Holzer et al. (2002), the absorption spectrum of the dark form LOV1-447 and the fluorescence properties are readily explained under the assumption that the Cys-57 is protonated and interacts only weakly with the FMN. This is in contrast to Swartz et al. (2001), who argued in favor of a thiolate in LOV2 from oat Phot1. Our arguments are the following:
The vibrational fine structure in the absorption spectra of flavoproteins is an indication for a hydrophobic environment of the FMN chromophore (Heelis, 1982). Miller et al. (1990) generated a triple mutant of the mercuric ion reductase where three of the four essential cysteines were replaced by alanine. Only the cysteine in the C(4a) region of the FMN (the one forming the thiol adduct) was retained. This mutant shows a fine structured absorption at 470 nm only when the cysteine is protonated. The position of the relevant cysteine thiol group with respect to the FMN is not identical in mercuric ion reductase and Phot1 LOV1 (Fedorov et al., 2003), but the protonation state should at least have some influence on the LOV domain spectra.
In most FMN-containing proteins, a negative charge in form of a thiolate in the environment of the FMN C(4a) induces charge transfer properties as seen from the additional absorption in the range between 500 and 600 nm (Miller et al., 1990; Williams, 1992). Such an absorption has neither been seen in the LOV1 spectrum of the C. reinhardtii Phot1 (see Fig. 1) nor in any other spectrum of a Phot-LOV domain.
Swartz et al. (2001) argued for a thiolate mainly on the basis of the increased fluorescence of the C39S and C39A mutants relative to wild-type LOV2 (factor 1.5–1.8). The observed small increase in fluorescence between pH 2.5 and 3 was interpreted as a protonation of the reactive Cys-39 at this acidic pH. In our understanding, a lower fluorescence quantum efficiency in the wild-type is due to a promotion of intersystem crossing by the sulfur and not an effect of the negative charge (Holzer et al., 2002; Song, 1971). In the above-mentioned mercuric ion reductase mutant, deprotonation of the retained Cys-140, which directly interacts with FMN C(4a), leads to a complete loss of fluorescence (Miller et al., 1990).
FT-IR experiments on this LOV1 domain provide direct evidence for a protonated Cys-57 (Ataka et al., 2003). In the light of these arguments, we do not consider that Cys-57 in the algal LOV1 is deprotonated in the dark form LOV1-447. This conclusion is of importance for the subsequent discussion of the reaction pathway after light excitation.
The photocycle of LOV1
The time-resolved observations collected so far for this algal LOV1 domain can be understood within the framework of the reaction scheme presented in Fig. 8.
FIGURE 8.

Photocycle of wild-type LOV1 with the assigned reaction mechanism. LOV1-447 corresponds to the dark form, LOV1-715 to the excited triplet state of FMN, and LOV1-390 to a flavin-cysteine adduct.
Photoexcitation of the dark form LOV1-447 produces the excited singlet state of the FMN. Measurements of the fluorescence lifetime yield a decay time of the excited state τf = 2.9 ns (Holzer et al., 2002). It should be noted that the observed decay rate of the fluorescence represents a sum of rates of all the contributing decay processes. The true decay time of the fluorescence, i.e., the radiative decay time, can be calculated to τr = 17 ns, taking into account a fluorescence quantum yield of Φf = 0.17 (Holzer et al., 2002). The excited singlet state can alternatively decay via intersystem crossing (ISC) into the triplet state LOV1-715. In the case that internal conversion is negligible, a quantum yield for intersystem crossing Φisc = 0.83 leads to the decay time τisc = 3.5 ns.
In the C57S mutant, the triplet state LOV1-715 cannot proceed to the LOV1-390 intermediate because it lacks the reactive thiol group. Instead, it returns to the dark form with time constants of 3 μs and 27 μs, indicating the presence of two subspecies with identical spectra. The disappearance of the 3 μs subspecies after removal of oxygen may be attributed to a situation where only in a certain fraction of the protein sample oxygen is present as quencher in the vicinity of the chromophore. This fact and the similarity of the transient spectrum to that of triplet FMN support our conclusion that LOV1-715 resembles the triplet state of FMN in LOV1. In the absence of oxygen, the intrinsic time constant of 27 μs for the lifetime of the triplet state is observed.
In wild-type LOV1, the decay of LOV1-715 is faster and again biexponential but oxygen-independent (τa = 800 ns, 80%; τb = 4 μs, 20%). Therefore, two additional decay channels in the wild-type form must exist, neither of which leads back to the dark form. This can be seen in Fig. 4, where no recovery of LOV1-447 is observed within the first 10 μs after excitation. In support of this statement, an overall quantum yield of 0.82 for the LOV1-390 formation has been determined at 20°C for this LOV1-domain (A. Losi, Max-Planck-Institut fuer Strahlenchemie, personal communication, 2002). Taking into account a fluorescence quantum yield of Φf = 0.17 (Holzer et al., 2002), the conclusion can be drawn that LOV1-715 almost quantitatively reacts to LOV1-390. This means that the competing quenching mechanisms leading to LOV1-447 observed in the C57S mutant do not significantly contribute to the wild-type reaction. Correspondingly, an increase of the wild-type rate constants by an external heavy atom effect is not observed.
The two time constants of wild-type LOV1-715 decay indicate the presence of two subspecies, LOV1-715a and LOV1-715b. The simplest interpretation would be a consecutive formation of both species. In that case, the fast time constant of 800 ns would have to be assigned to the reaction from the triplet state LOV1-715a to LOV1-715b and the time constant of 4 μs is the subsequent reaction to LOV1-390. However, a triplet species is still observed 1 μs after excitation (see Fig. 4). That observation is inconsistent with a fast decay of LOV1-715a. Moreover, no conceivable ground state intermediate would show significant absorption at the monitored wavelength of 710 nm (Massey and Palmer, 1966; Nanni et al., 1981). Therefore, the possibility of a consecutive formation can be excluded. Instead, both subspecies must be formed in a parallel reaction and can be assigned to triplet species with similar spectra. According to this scheme, LOV1-715a decays with the time constant of 800 ns into LOV1-390. LOV1-715b decays with the time constant of 4 μs either into LOV1-715a or directly into LOV1-390 (see Fig. 8). These two reaction pathways of LOV1-715b cannot be distinguished kinetically. Both subspecies result in LOV1-390 formation.
Inasmuch as both subspecies are triplet states of the FMN chromophore, the different reactivity may be caused by a different interaction of the FMN with the protein environment. Indeed, crystal structure analysis provides evidence for two conformations of the dark form (Fedorov et al., unpublished). The conformations differ from each other in the distance from the reactive cysteine to the C(4a) position of the FMN. This distance is likely to have an influence on the time constant of the adduct formation.
According to the proposed reaction mechanism, quantum efficiencies may be calculated from the competition between the two observed time constants τa = 800 ns and τb = 4 μs, and the 27 μs time constant τint for the intrinsic lifetime of the triplet state with the formalism:
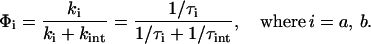 |
(5) |
This leads to Φa = 0.97 for the decay of LOV1-715a and Φb = 0.87 for the decay of LOV1-715b. Taking into account the ratio of LOV1-715a to LOV1-715b, an averaged quantum efficiency of 0.95 is calculated for the reaction from LOV1-715 to LOV1-390. This value is in agreement with the earlier conclusion that the triplet state almost quantitatively reacts to LOV1-390. The overall quantum efficiency for LOV-390 formation in C. reinhardtii LOV1 (0.82) is significantly higher than those determined in LOV1 (0.05) and LOV2 (0.44) from A. sativa (Salomon et al., 2000).
Swartz et al. (2001) suggested for oat LOV2 that the primary mechanistic step in LOV-390 formation is the protonation of the flavin N(5) position through a proton-donating group in the protein. The resulting C(4a) carbocation is attacked by the cysteine thiolate. Such a carbocation should have a red-shifted absorption (Nanni et al., 1981), which has not been observed either for LOV1 or for LOV2. More important, a proton-donating amino acid residue in the vicinity of the chromophore is neither present in C. reinhardtii LOV1 (Fedorov et al., 2003) nor in A. capillus-veneris LOV2 (Crosson and Moffat, 2001). Therefore, we favor a concerted mechanism similar to the one hypothesized for the ground state reaction of mercuric ion reductase ACAA mutant (Miller et al., 1990). Alternatively, a transfer of a hydrogen atom from the thiol group to the triplet state N(5) position and a fast recombination of the resulting radicals is conceivable (Neiß and Saalfrank, 2003). As a result of the reaction, the planar conformation of the FMN is changed to a sp3 hybridization of C(4a) in LOV1-390. This geometry of the chromophore is likely to be constrained and therefore facilitates the thermal back reaction to LOV1-447.
With the decay of LOV1-390, the adduct bonds (C(4a)-S and N(5)-H) are cleaved and the dark form is recovered in a pH-dependent reaction (see Fig. 2). This observation may be attributed to base catalysis involving an acid-base pair with a pKa between 5 and 6. However, there are no protonatable amino acid side chains in the chromophore binding pocket (Fedorov et al., 2003). As has been proposed by Swartz et al. (2001), base catalysis may proceed via a hydrogen bonding network between base, chromophore, and intraproteineous water molecules. His-20 and His-55 of the LOV1 domain are possible candidates with a distance of 14 Å to the FMN. Taking into consideration the crystal structure data, a second interpretation of the kinetics is conceivable (Fedorov et al., 2003). The chromophore FMN itself may be protonated at the phosphate group that exhibits a pKa of 6.2 in aqueous solution (Bidwell et al., 1986). The phosphate group is in direct contact via hydrogen bonding with Arg-58 and Arg-74, which might cause a shift of the pKa value. Moreover, the influence of the sodium chloride concentration on the time constants at an acidic pH suggests that the titratable group is located at the surface of the protein (see Fig. 2). If the phosphate is protonated, the interactions with Arg-58 become unfavorable and Arg-58 will stay closer to the flavin ring and may thereby stabilize the bond between Cys-57 and C(4a) (Fedorov et al., 2003). In the LOV1-390 intermediate, this may lead to different time constants of the flavin-cysteine bond cleavage.
The presence of two LOV domains in Phot blue-light receptors raises the question of the difference in property and function of LOV1 and LOV2. The photocycle of LOV1 domains has not been investigated up to now, because reported low quantum yields in comparison to LOV2 in A. sativa implicated only a minor contribution to the overall signal reception (Salomon et al., 2000). In contrast, we have shown that the LOV1 domain is a highly efficient photochemical transducer and therefore can play, at least in algal Phot1 receptors, an important role in the early steps of signaling. Nevertheless, the interaction between the two LOV domains and the downstream processes have yet to be resolved by concomitant spectroscopic and structural studies of larger segments of the Phot receptor proteins.
Acknowledgments
We thank Markus Fuhrmann, Uwe Kensy, and Christian Neiß for advice and support, Tina Schireis for excellent technical assistance, and Eduard Hochmuth for taking the MS spectra. The work was supported by the Deutsche Forschungsgemeinschaft (GK 640 and SFB 521). B.D. and P.H. acknowledge the support by the “Fonds der Chemischen Industrie”. D.H. and J.H. are grateful to Georg Büldt (Forschungszentrum Jülich) for generous support.
APPENDIX: MODEL FOR A THERMALLY AND PHOTOCHEMICALLY REVERSIBLE PHOTOREACTION
We consider a reaction system in which a species 1 (the dark form) is photochemically converted into an intermediate species 2. This intermediate can return to the dark form either thermally or photochemically. This can be modeled by the following differential equation for the relative population x = c/c0, where c is the concentration of intermediate and c0 is the initial concentration of the dark form:
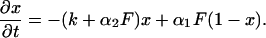 |
In this expression, F is the photon flux and k the rate constant of the spontaneous (thermal) back reaction. The coefficient α1 is related to the extinction coefficient ɛ1(λ) of species 1, the quantum yield of decay to the intermediate Φ1→2 and the normalized quantum emission spectrum of the light source ρ(λ) by
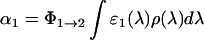 |
and α2 is defined accordingly with the extinction coefficient ɛ2(λ) of species 2 and the quantum yield Φ2→1 of the reverse reaction. The relative population xs of the photostationary state is given by
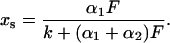 |
The absorbance AF′ of the sample in the photostationary state at a probe wavelength λ′ can be expressed in terms of this relative population by
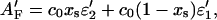 |
with ɛ2′ = ɛ2(λ′) and ɛ1′ = ɛ1(λ′). This can be rearranged to
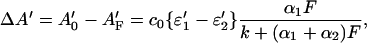 |
where A0′ is the absorbance of the sample without photolytic irradiation. The relative change in absorbance at a probe wavelength λ′ is therefore
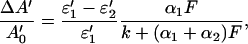 |
defining a of Eq. 4 as
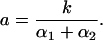 |
Extrapolation to infinite photolytic light intensity yields the relation
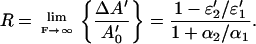 |
Because ɛi′ and αi are both positive quantities, the value of R is bound by R ≤ 1. The upper limit R = 1 is obtained if both conditions ɛ2′ = 0 (i.e., no absorption of the photoproduct) and α2 = 0 (i.e., no photochemical back reaction) are fulfilled. A value of R < 1, on the other hand, provides limits for ɛ2′ or α2, namely
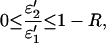 |
which reaches the upper limit for α2 = 0, and
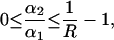 |
where the upper limit corresponds to the case ɛ2′ = 0. The corresponding experiment has been described in the results section. Extrapolation to infinite light intensities reduced the initial absorbance of A0(475 nm) = 0.37 to a limit of AF(475 nm) = 0.06, i.e., R = 0.84. This leads to the following ranges for the ratios of the coefficients ɛi′ and αi:
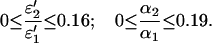 |
Hence, if no photochemical back reaction occurs, the observed photostationary absorption requires ɛ2′ = 0.16 ɛ1′ at 475 nm. The spectrum of the intermediate presented in Fig. 1 shows, however, a much lower extinction coefficient ɛ2′ ≤ 0.06 ɛ1′. (It is noted that there is some arbitrariness in the SVD reconstruction of this spectrum.) If we accept this lower limit for ɛ2′, the range of α2/α1 is reduced to
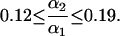 |
Hence there is evidence for a photoinduced back reaction of the long-lived intermediate to the dark form.
References
- Ataka, K., P. Hegemann, and J. Heberle. 2003. Vibrational spectroscopy of an algal Phot-LOV1 domain probes the molecular changes associated with blue-light reception. Biophys. J. 84:466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt, M., I. Adriana, G. W. Haggquist, and N. Razi. 1999. Hydrogen abstraction by triplet flavins. I: time-resolved multi-channel absorption spectra of flash-irradiated riboflavin solutions in water. Spectrochim. Acta. Part A: Mol. Biomol. Spectrosc. 55:2299–2307. [Google Scholar]
- Bidwell, J., J. Thomas, and J. Stuehr. 1986. Thermodynamic and kinetic study of the interactions of Ni(II) with FMN and FAD. J. Am. Chem. Soc. 108:820–825. [Google Scholar]
- Briggs, W. R., C. F. Beck, A. R. Cashmore, J. M. Christie, J. Hughes, J. A. Jarillo, T. Kagawa, H. Kanegae, E. Liscum, A. Nagatanni, K. Okada, M. Salomon, R. Rüdiger, T. Sakai, M. Zakano, M. Wada, and J. C. Watson. 2001. The phototropin family of photoreceptors. Plant Cell. 13:993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie, J. M., P. Reymond, G. K. Powell, P. Bernasconi, A. A. Raibekas, E. Liscum, and W. R. Briggs. 1998. Arabidopsis NPH1: a flavoprotein with the properties of a photoreceptor for phototropism. Science. 282:1698–1701. [DOI] [PubMed] [Google Scholar]
- Christie, J. M., M. Salomon, K. Nozue, M. Wada, and W. R. Briggs. 1999. LOV (light, oxygen, or voltage) domains of the blue-light photoreceptor phototropin (nph1): binding sites for the chromophore flavin mononucleotide. Proc. Natl. Acad. Sci. USA. 96:8779–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosson, S., and K. Moffat. 2001. Structure of a flavin-binding plant photoreceptor domain: insights into light-mediated signal transduction. Proc. Natl. Acad. Sci. USA. 98:2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosson, S., and K. Moffat. 2002. Photoexcited structure of a plant photoreceptor domain reveals a light-driven molecular switch. Plant Cell. 14:1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov, R., I. Schlichting, E. Hartmann, T. Domratcheva, M. Fuhrmann, and P. Hegemann. 2003. Crystal structures and molecular mechanism of a light-induced signaling switch: the Phot-LOV1 domain from Chlamydomonas reinhardtii. Biophys. J. In press. [DOI] [PMC free article] [PubMed]
- Heelis, P. F. 1982. The photophysical and photochemical properties of flavins (isoalloxazines). Chem. Soc. Rev. 11:15–39. [Google Scholar]
- Hendler, R. W., and R. I. Shrager. 1994. Deconvolutions based on singular value decomposition and the pseudoinverse: a guide for beginners. J. Biochem. Biophys. Methods. 28:1–33. [DOI] [PubMed] [Google Scholar]
- Holzer, W., A. Penzkofer, M. Fuhrmann, and P. Hegemann. 2002. Spectroscopic characterization of flavin mononucleotide bound to the LOV1 domain of Phot1 from Chlamydomonas reinhardtii. Photochem. Photobiol. 75:479–487. [DOI] [PubMed] [Google Scholar]
- Huala, E., P. W. Oeller, E. Liscum, E. Han, I. S. Larsen, and W. R. Briggs. 1997. Arabidopsis NPH1: a protein kinase with a putative redox-sensing domain. Science. 278:2120–2123. [DOI] [PubMed] [Google Scholar]
- Huang, K., and C. F. Beck. 2002. Isolation and characterization of a Chlamydomonas gene that encodes a blue-light photoreceptor of the phototropin family. Tenth International Conference on the Cell and Molecular Biology of Chlamydomonas. University of Vancouver, British Columbia. 57a. (Abstr.)
- Huang, K., T. Merkle, and C. F. Beck. 2002. Isolation and characterization of a Chlamydomonas gene that encodes a putative blue-light photoreceptor of the phototropin family. Physiol. Plant. 115:613–622. [DOI] [PubMed] [Google Scholar]
- Jarillo, J. A., H. Gabrys, J. Capel, J. M. Alonso, J. R. Ecker, and A. R. Cashmore. 2001. Phototropin-related NPL1 controls chloroplast relocation induced by blue light. Nature (Lond.). 410:952–954. [DOI] [PubMed] [Google Scholar]
- Kagawa, T., T. Sakai, N. Suetsugu, K. Oikawa, S. Ishiguro, T. Kato, S. Tabata, K. Okada, and M. Wada. 2001. Arabidopsis NPL1: a phototropin homolog controlling the chloroplast high-light avoidance response. Science. 291:2138–2141. [DOI] [PubMed] [Google Scholar]
- Kasahara, M., T. E. Swartz, M. A. Olney, A. Onodera, N. Mochizuki, H. Fukuzawa, E. Asamizu, S. Tabata, H. Kanegae, M. Takano, J. M. Christie, A. Nagatani, and W. R. Briggs. 2002. Photochemical properties of the flavin mononucleotide-binding domains of the phototropins from Arabidopsis, rice, and Chlamydomonas reinhardtii. Plant Physiol. 129:762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, T., M. Doi, N. Suetsugu, T. Kagawa, M. Wada, and K. I. Shimazaki. 2001. Phot1 and phot2 mediate blue light regulation of stomatal opening. Nature (Lond.). 414:656–660. [DOI] [PubMed] [Google Scholar]
- Massey, V., and G. Palmer. 1966. On the existence of spectrally distinct classes of flavoprotein semiquinones. A new method for the quantitative production of flavoprotein semiquinones. Biochemistry. 5:3181–3189. [DOI] [PubMed] [Google Scholar]
- Miller, S. M., V. Massey, D. Ballou, C. H. Williams, Jr., M. D. Distefano, M. J. Moore, and C. T. Walsh. 1990. Use of a site-directed mutant to trap intermediates: demonstration that the flavin C(4a)-thiol adduct and reduced flavin are kinetically competent intermediates in mercuric ion reductase. Biochemistry. 29:2831–2841. [DOI] [PubMed] [Google Scholar]
- Nanni, E. J., Jr., D. T. Sawyer, S. S. Ball, and T. C. Bruice. 1981. Redox chemistry of N5-ethyl-3-methyllumiflavinium cation and N5-ethyl-4a-hydroperoxy-3-methyllumiflavin in dimethylformamide. Evidence for the formation of the N5-ethyl-4a-hydroperoxy-3-methyllumiflavin anion via radical-radical coupling with superoxide ion. J. Am. Chem. Soc. 103:2797–2802. [Google Scholar]
- Neiß, C., and P. Saalfrank. 2003. Ab initio quantum chemical investigation of the first steps of the photocycle of phototropin: a model study. Photochem. Photobiol. In press. [DOI] [PubMed]
- Sakai, M., and H. Takahashi. 1996. One-electron photoreduction of flavin mononucleotide: time-resolved resonance Raman and absorption study. J. Mol. Struct. 379:9–18. [Google Scholar]
- Salomon, M., J. M. Christie, E. Knieb, U. Lempert, and W. R. Briggs. 2000. Photochemical and mutational analysis of the FMN-binding domains of the plant blue light receptor, phototropin. Biochemistry. 39:9401–9410. [DOI] [PubMed] [Google Scholar]
- Salomon, M., W. Eisenreich, H. Dürr, E. Schleicher, E. Knieb, V. Massey, W. Rüdiger, F. Müller, A. Bacher, and G. Richter. 2001. An optomechanical transducer in the blue light receptor phototropin from Avena sativa. Proc. Natl. Acad. Sci. USA. 98:12357–12361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, P. S. 1968. On the basicity of the excited state of flavins. Photochem. Photobiol. 7:311–313. [DOI] [PubMed] [Google Scholar]
- Song, P. S. 1971. Chemistry of flavins in their excited states. In Flavins and Flavoproteins. H. Kamin, editor. University Park Press, Baltimore. 37–60.
- Swartz, T. E., S. B. Corchnoy, J. M. Christie, J. W. Lewis, I. Szundi, W. R. Briggs, and R. A. Bogomolni. 2001. The photocycle of a flavin-binding domain of the blue light photoreceptor phototropin. J. Biol. Chem. 276:36493–36500. [DOI] [PubMed] [Google Scholar]
- Williams, C. H., Jr. 1992. Lipoamide dehydrogenase, glutathione reductase, thioredoxin reductase, and mercuric ion reductase—a family of flavoenzyme transhydrogenases. In Chemistry and Biochemistry of Flavoenzymes, Vol. 3. F. Mueller, editor. CRC Press, Boca Raton, FL. 121–211.