

Abstract
Previous work has shown that two molecules of a soluble form of phosphatidylserine, C6PS, bind to human and bovine factor Xa. Activity measurements along with the fluorescence of active-site-labeled human factor Xa showed that two linked sites specifically regulate the active site conformation and proteolytic activity of the human enzyme. These results imply, but cannot demonstrate, a C6PS-induced factor Xa conformational change. The purpose of this paper is to extend these observations to bovine factor Xa and to demonstrate that they do reflect conformational changes. We report that the fluorescence of active-site-labeled bovine factor Xa also varied with C6PS concentration in a sigmoidal manner, whereas amidolytic activity of unlabeled enzyme varied in a simple hyperbolic fashion, also as seen for human factor Xa. C6PS induced a 70-fold increase in bovine factor Xa's autolytic activity, consistent with the 60-fold increase in proteolytic activity reported for human factor Xa. In addition, circular dichroism spectroscopy clearly demonstrated that C6PS binding to bovine factor Xa induces secondary structural changes. In addition, C6PS binding to the tighter of the two sites triggered structural changes that lead to Ca2+-dependent dimer formation, as demonstrated by changes in intrinsic fluorescence and quantitative native gel electrophoresis. Dimerization produced further change in secondary structure, either inter- or intramolecularly. These results, along with results presented previously, support a model in which C6PS binds in a roughly sequential fashion to two linked sites whose occupancy in both human and bovine factor Xa elicits different structural and functional responses.
INTRODUCTION
The activation of prothrombin to thrombin is a crucial step in the blood coagulation process. It is accomplished by the prothrombinase complex, which consists of a serine protease, factor Xa, and a protein cofactor, factor Va, interacting on an appropriate acidic membrane surface (Jackson and Nemerson, 1980; Mann et al., 1990). When compared with factor Xa in the presence of factor Va in solution, the activation of prothrombin to thrombin by the prothrombinase complex on an appropriate phospholipid membrane surface is enhanced ∼1000-fold (Nesheim et al., 1979; Rosing et al., 1980). Activated platelets (Kane et al., 1980; Tracy and Mann, 1986) or other cells of the circulatory system (Tracy, 1988; Tracy et al., 1985) can provide the active surface. Several studies (Comfurius et al., 1994; Jones et al., 1985; Rosing et al., 1988) have shown that PS specifically enhances prothrombin activation. We now know that this is due to specific regulatory sites on factors Xa and Va (Banerjee et al., 2002a; Koppaka et al., 1996; Srivastava et al., 2002; Zhai et al., 2002).
Binding of human factor Xa to a membrane seems to involve two to four molecules of membrane-associated PS (Cutsforth et al., 1989). A soluble form of PS, C6PS, binds human factor Xa in solution (Koppaka et al., 1996). Two C6PS molecules bind to either human or bovine factor Xa, one to a pair of epidermal-growth-factor-like domains (EGFNC) when these are linked to an N-terminal γ-carboxy-glutamic-acid-rich domain, and one to the catalytic domain (Srivastava et al., 2002). The EGFNC site (site 1), located near the membrane surface, alters the catalytic domain at the other end of human factor Xa, including the affinity of the C6PS site in this domain (site 2) (Banerjee et al., 2002a). Binding of C6PS to the catalytic domain is anomalous and appears to involve part of the substrate recognition site (Srivastava et al., 2002). C6PS binding increases human factor Xa's kcat in prothrombin activation by 60-fold (Koppaka et al., 1996), with activity seemingly regulated principally by site 1 (Banerjee et al., 2002a). Site 1 is Ca2+-dependent but not Ca2+-requiring (Srivastava et al., 2002) and minimally binds DAG as a ligand, but requires C6PS to affect activity (Banerjee et al., 2002a). Site 2 requires Ca2+ (Srivastava et al., 2002), and its minimal ligand is GPS (Banerjee et al., 2002a). It can be occupied by acidic lipids other than PS, but not by neutral lipids (Banerjee et al., 2002a; Srivastava et al., 2002).
PS-containing synthetic membranes have been shown to alter the conformation of prothrombinase complex components. The conformation of prothrombin clearly is altered on binding to a PS-containing membrane (Chen and Lentz, 1997). Binding of C6PS alters both the structure and cofactor activity of factor Va (Zhai et al., 2002), such that a fully active prothrombinase is formed in solution when factors Va and Xa are incubated in the presence of C6PS (Majumder et al., 2002). One paper reported a possible effect of PS-containing membranes on factor Xa structure by virtue of a change in fluorescence of an active site label, dansyl-Glu-Gly-Arg-chloromethyl ketone (DEGR) (Husten et al., 1987). We have used the fluorescence of DEGR-Xa to follow binding of different soluble phospholipids, with the inference that different changes in fluorescence reflect different structural changes (Banerjee et al., 2002a). Changes in DEGR-Xa fluorescence in response to C6PS have been interpreted in terms of a model that assumes that occupancy of each of the two sites produces different structural changes (Banerjee et al., 2002a). However, this change in fluorescence of an active-site probe is suggestive of but cannot prove that C6PS induces a conformational change in factor Xa, so we report here additional experiments that demonstrate a C6PS-induced conformational change. Specifically, we show: 1), that PS-specific changes in bovine factor Xa secondary structure accompany soluble phospholipid binding; 2), that occupancy of the two sites seems to produce different structural responses; 3), that conformational changes associated with occupancy of site 1 lead, in a Ca2+-dependent fashion, to factor Xa dimerization in solution; and 4), that changes in the autolytic activity of bovine factor Xa parallel proteolytic activity changes reported for human factor Xa (Banerjee et al., 2002a; Koppaka et al., 1996).
MATERIALS
DEGR-CK was purchased from Calbiochem (La Jolla, CA). C6PC, C6PG, C6PS, DOPG, POPC, and bovine brain PS were purchased from Avanti Polar Lipids (Alabaster, AL). Factor Xa-specific substrate S2765 was purchased from Helena Laboratories (Beaumont, TX). All other chemicals were ACS reagent grade or the best available grade.
METHODS
Short-chain phospholipid preparation
Chloroform was removed from measured amounts of C6PS, C6PG, and C6PC stocks using a stream of nitrogen, and the lipid samples were dissolved in cyclohexane and lyophilized overnight. The lyophilized lipids were then suspended in an appropriate amount of buffer A (50 mM Tris, 0.1 M NaCl, pH 7.5) to reach desired concentrations.
Vesicle preparation
Large unilamellar extrusion vesicles were prepared by the method of Hope et al. (1985). Dried lipid samples were suspended above their phase transition temperatures in an appropriate amount of buffer A to reach a concentration of roughly 15 mM. The vesicles were then extruded seven times, above their phase transition temperature and under pressure (50–60 psi of Ar), through a 0.1-μm polycarbonate filter (Nucleopore, Pleasanton, CA).
Factor X isolation and activation
Bovine factor X was isolated by barium citrate precipitation from freshly collected bovine plasma (Mann, 1976; Tendian and Lentz, 1990). Factor X obtained in this manner was analyzed by SDS-PAGE, concentrated (Centricon-10 concentrator, Amicon Division, W.R. Grace, Danvers, MA) and then stored at −70°C at a concentration ∼1 mg/ml. A final purification step was performed one day before an experiment by dialyzing into low salt buffer, loading onto a Mono Q HR 5/5 ion exchange column (Pharmacia, Norwalk, CT), and eluting by high-performance liquid chromatography on a Perkin-Elmer Isopure LC system using a linear NaCl gradient from 0 to 1M NaCl. The purified factor X was dialyzed into buffer A for activation.
Factor X was activated with a purified fraction of Russel's viper venom factor X activating protein (RVV-X; Hematological Technologies, Essex Junction, VT) that had been covalently linked to Affigel-15 (Bio-Rad, Hercules, CA) agarose beads (Jesty and Nemerson, 1976). The cleavage of a single arginyl-isoleucine peptide bond in the heavy chain of factor X results in the formation of factor Xaα. Factor Xaα is slowly degraded to give rise to factor Xaβ (Fujikawa et al., 1974). The two forms of factor Xa obtained were analyzed by SDS-PAGE on a 10% gel. A high concentration of factor Xaα relative to factor Xaβ was obtained for autolysis experiments by controlling the time of factor X incubation with RVV beads. Sodium citrate was then added to stop the reaction and the RVV beads were removed by centrifugation. We performed a series of experiments to define the optimal reaction time (25 min) to minimize the amounts of factor X and factor Xaβ under our experimental conditions. Under these conditions, the final reaction mixtures contained ∼85–90% factor Xaα, 5–10% factor X, and 2–5% factor Xaβ as averaged from several preparations. The supernatant, containing mainly factor Xaα, was used immediately for autolysis measurements. Factor Xa, which, under normal conditions consists mainly of factor Xaβ, was prepared for structural studies (CD or fluorescence) by incubating RVV beads with factor X in buffer A with 5 mM Ca2+ for at least 90 min at room temperature. Purified factor Xa was obtained by high-performance liquid chromatography using a Mono Q column, as described for factor X. Factor Xa concentration was determined from the rate of S2765 hydrolysis in a plate-reader-based assay (Koppaka et al., 1996), using active-site-titrated factor Xa (Jameson et al., 1973) to construct the standard curve.
DEGR-Xa preparation
DEGR-Xaβ and DEGR-Xaα were prepared by sequential addition of 5 μL aliquots of DEGR-CK (∼1 mg/ml in 0.02 M Tris-HCl, 0.1 M NaCl, pH 7.5 buffer) to 1.0 mL of factor Xaβ or factor Xaα at ∼17 μM. The extent of labeling at the active site was followed by the loss in enzymatic activity, as monitored by the rate of S2765 hydrolysis. DEGR-CK addition continued until no activity remained. DEGR-Xa was then dialyzed in buffer A to remove free reagent (Husten et al., 1987) and analyzed by SDS-PAGE and detected with a Foto/Prep II transilluminator (VWR Scientific, Norwalk, CT) and with Coomassie blue to confirm that the probe was associated with factor Xa.
Kinetic measurements of factor Xa autolysis
Several samples containing 6.5 μM factor Xaα but different concentrations of lipid were followed simultaneously. Each sample was brought to 5 mM in CaCl2 to start the reaction. Aliquots (15 μL) were removed from each sample at different times into preprepared SDS-sample buffer and then immediately heated for ∼2 min at 100°C. A Multichannel Digital Finnpipette (Labsystems, Finland) was used to add calcium to or remove samples from the reaction mixtures to assure that reaction times were carefully coordinated. The relative amounts of factors Xaα and Xaβ were obtained from the relative intensity of the corresponding SDS-PAGE gel bands measured on a scanning densitometer (Mitsubishi Model SLR-2D/1D, Fotodyne, Cyprus, CA).
Fluorescence measurements
The fluorescence intensity of DEGR-Xa was recorded at 22°C with an SLM 48000 fluorescence spectrophotometer (SLM Aminco, Rochester, NY), using an excitation wavelength of 340 nm (band pass 8 nm) and emission wavelength of 550 nm (4-nm band pass). Slits were kept closed except during measurements to avoid photodegradation of the sample. Before each titration, DEGR-Xa was incubated in buffer A with Ca2+ for ∼20 min. Short-chain phospholipids were added in 2 or 3 μL aliquots, with a maximum dilution of 4%. To correct for dilution, we measured the emission intensity of DEGR-Xa in response to titration by buffer A. Fluorescence intensity was recorded ∼2 min after each addition. For each lipid, several parallel measurements were performed and results were averaged.
Data were analyzed according to the binding models described in the Appendix. Nonlinear least squares fitting was accomplished using SigmaPlot (version 4.0 for Windows; Jandel Scientific). We judged the relative appropriateness of different models for describing each data set by comparing the correlation coefficients of the fit (R2) and the sample variances (σ) using an F-test (Bevington, 1969). R2 is the adjusted square of the correlation coefficient; values close to 1 indicate that the model provides a good description of the variation of the dependent variable with the independent variable. σ is the “standard error of the estimate” or “sample variance” (the square root of the quantity, sum of squared residuals divided by the number of degrees of freedom). It gives a measure of the variability of the data about the regression plane. A smaller value means a better fit of the regression equation to the parent data set (Bevington, 1969).
Intrinsic fluorescence of factor Xa was recorded at 22°C while exciting at 285 nm with an 8-nm excitation band pass on the SLM 48000 fluorescence spectrophotometer. Scans were collected from 310 to 390 nm in ratio mode and corrected for instrumental response. Scans of buffer or buffer plus C6PS were recorded and subtracted from scans obtained with factor Xa to correct for background scatter.
Effect of phospholipids on factor Xa activity
The amidolytic activity of factor Xa in the presence of soluble phospholipids and Ca2+ was determined using the synthetic substrate S2765 (0.49 mM) and a microplate reader (SLT 340 ATTC; SLT Instruments, Hillsborough, NC), as described previously (Koppaka et al., 1996). The buffer was a modified form of buffer A containing 0.6 wt % poly(ethylene glycol) to prevent factor Xa from adsorbing to the plate. The effect of C6PS on factor Xa amidolytic activity has been shown previously to be independent of substrate concentration up to 1 mM, allowing us to conclude that the effect of C6PS was not due to C6PS binding to substrate (Koppaka et al., 1996).
Circular dichroism measurements
CD spectra were recorded from 250 to 200 nm on an Aviv Model 620S spectrometer (Aviv Associates, Lake Wood, NJ) in a 1-cm pathlength cell at 25°C with a bandwidth of 1 nm. Samples were prepared in buffer B (1.2 mM Tris, 150 mM NaCl, pH 7.4). Because correlating structural and functional changes was our goal, we adopted this buffer to maintain a similarity to buffer A used in activity and fluorescence measurements. When we collected spectra in a very low-salt, Na+-free buffer down to 185 nm, the effects of Ca2+ and C6PS seen in physiological buffer could not be reproduced (Srivastava and Lentz, unpublished observations). This probably reflects the documented linkage between Na+ and Ca2+ binding to factor Xa (Rezaie and He, 2000; Underwood et al., 2000) and the influence of Ca2+ on C6PS binding (Koppaka et al., 1996). Because a buffer with physiological ionic strength is not very transparent in the deep UV, this precluded complete secondary structure analysis that requires data down to 185 nm, although estimates of α-helix content could be made based on spectra collected to 200 nm (Johnson, 1990). Data points were collected every 0.5 nm with a 5-s average time for each point. The digital data were processed, smoothed, baseline-corrected, and converted to molar ellipticity, [Θ]. For baseline corrections, CD spectra were collected for C6PS, C6PG, and C6PC in buffer with the same concentrations as for sample titration in the presence or absence of Ca2+. All buffer-corrected spectra were flat with roughly zero molar ellipticity from 245 to 250 nm except at high short-chain lipid concentration when this portion of the spectrum dipped dramatically. The lipid concentration range in which this happened for C6PS was shown by quasi-elastic light scattering (QELS) to correspond to the C6PS critical micelle concentration (CMC) (Koppaka et al., 1996). Because the proteolytic activity of factor Xa abruptly increased by 1.7-fold at the C6PS CMC (Koppaka et al., 1996), it is clear that factor Xa binds to C6PS micelles, and we did not attempt to analyze data at or above the CMC of any of the lipids examined.
Native gel electrophoresis to estimate the molecular weight of factor Xa
Polyacrylamide gels were prepared at four different percentages of cross-linking (5%, 6%, 8%, and 10%). Factor Xa and known marker proteins (270 kDa–14.2 kDa; Sigma, St. Louis, MO) were run on these gels in a BioRad Mini-Protean II minigel apparatus (Biorad Corp., Hercules, CA) and stained with colloidal Coomassie blue (Mitra et al., 1994). The Rf of each protein in each gel was measured relative to the tracking dye. For each protein, a plot of log (Rf×100) against the percent gel concentration gave a straight line, the negative slope of which is the retardation coefficient (Ferguson, 1964). A log-log plot of the retardation coefficient against the molecular weights of the marker proteins produces a linear curve from which the molecular weight of any unknown protein may be determined (Bryan, 1977).
RESULTS
Response of bovine DEGR-Xa to C6PS, C6PG, and C6PC parallels response of human DEGR-Xa
One purpose of this paper was to confirm that responses of bovine factor Xa to C6PS are due to structural changes. Because many of our previous studies were performed with human factor Xa, we focus first on demonstrating the essential equivalency of the bovine and human forms of factor Xa in their reactions to short-chain lipid binding. Addition of C6PS to bovine DEGR-Xa, preincubated with 5 mM Ca2+, led to a saturable decrease in emission intensity that paralleled changes observed with human factor Xa (Banerjee et al., 2002a) and indicated a change in the environment of the dansyl moiety at the active site (Fig. 1 A, filled circles). Titration of bovine DEGR-Xa with PS/POPC (1/3) membranes produced a 55% drop in DEGR-Xa fluorescence (data not shown), consistent with but roughly twice that seen with C6PS and consistent with a literature report (Husten et al., 1987).
FIGURE 1.
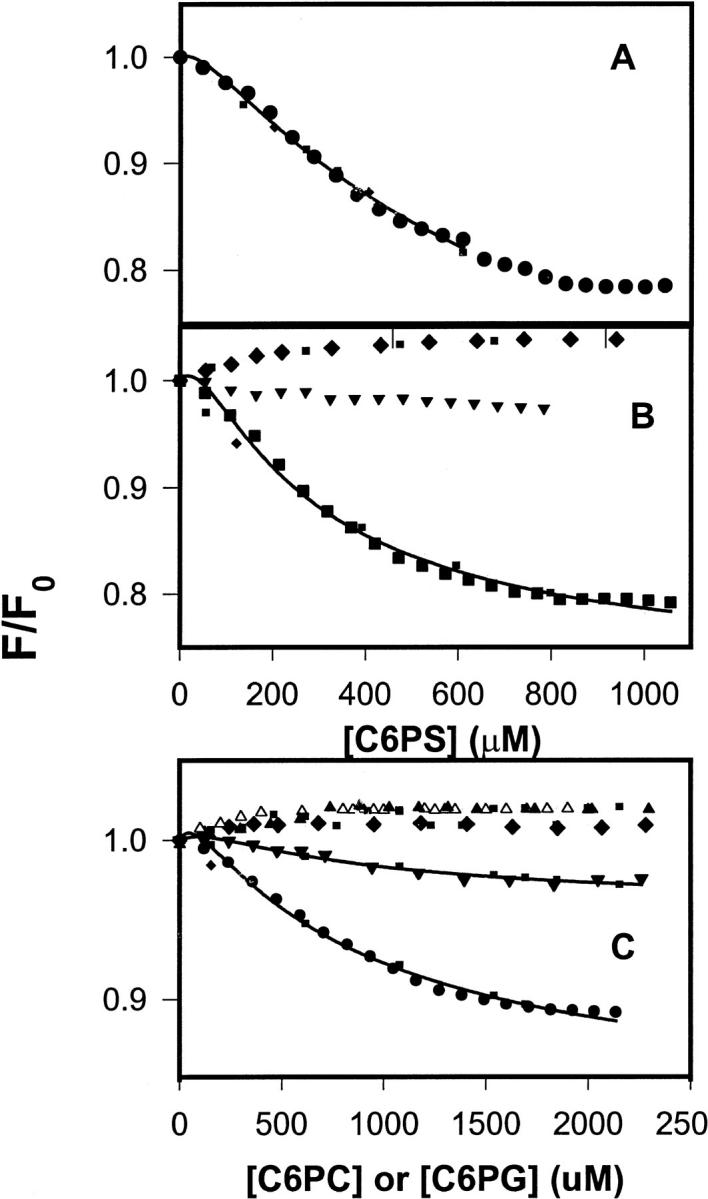
(A) DEGR-Xa (filled circles) at 100 nM in buffer A (50 mM Tris, 0.1 M NaCl, pH 7.5) was titrated at 22°C with C6PS in the presence of 5 mM Ca2+. Fluorescence intensities were measured 2 min after addition of lipid. Fit to the single-site model (Eq. 1) (dotted lines) and the sequential-linked-site model (Eq. 1) (solid line) are shown. Parameters resulting in these fits are given either in the text or in Table 1. (B) DEGR-Xa (100 nM) in buffer A was titrated with C6PS in the presence of 3 mM (squares) and 1 mM (inverted triangles) CaCl2 and in the absence of CaCl2 (diamonds). Fits to the data are as described in A. (C) DEGR-Xa (100 nM) in buffer A was titrated with C6PG (filled circles) and C6PC (filled triangles) in the presence of 5 mM Ca2+; with C6PG in the presence of 1 mM (filled inverted triangles) Ca2+ or no Ca2+ (filled diamonds); with C6PC in the absence of Ca2+ (open triangles). Fits to the data are as described for A.
The discontinuity that occurs in the intensity of bovine DEGR-Xa between 700 and 800 μM C6PS corresponds to the CMC (800 μM) observed for C6PS at 5 mM CaCl2 in the presence of 5 nM human factor Xa (Koppaka et al., 1996). The discontinuity observed at 5 mM Ca2+ was not observed at lower Ca2+ concentrations (Fig. 1 B), because the C6PS CMC increases dramatically with increasing Ca2+ concentrations (Koppaka et al., 1996). Because factor Xa binds to C6PS micelles above the CMC (Koppaka et al., 1996), titration curves were fit to binding models only for lipid concentrations below the CMC.
Because of its S-shape, the C6PS binding isotherm was not well described by a single-site-binding model (R2/σ = 0.98/0.0086; Fig. 1, dotted curve). However, a cooperative model that assumes two linked sites having equivalent structural responses (Klotz and Hunston, 1975) was, as expected, able to reproduce the S-shape of the binding isotherm (fit not shown; stoichiometric dissociation constants Kd1 ∼ 10.7 mM, Kd2 ∼ 14 μM; R2/σ = 0.998/0.0034). Because the C6PS CMC is 1.5 mM in the presence of factor Xa and 3 mM Ca2+ (Koppaka et al., 1996), we were able to obtain data up to higher concentrations of C6PS at 3 mM Ca2+ (Fig. 1 B, squares), making the failure of the single-site-binding model especially clear (dotted curve; R2/σ = 0.97/0.0097). Just as for human factor Xa (Koppaka et al., 1996), C6PS binding was much less tight at Ca2+ concentrations below 3 mM (Fig. 1 B). In the absence of Ca2+, the fluorescence of DEGR-Xa (Fig. 1 B, filled diamonds) increased in response to C6PS, as we have observed for human DEGR-Xa (Banerjee et al., 2002a). This increase was well described by a single site model (Fig. 1 B, dotted curve through filled diamonds) with a kd of 200 ± 17 μM and a ΔRsat of 0.047 ± 0.001.
Although we did not examine all the short-chain lipid analogs that we did for human factor Xa, we did consider responses to C6PG and C6PC. The response of DEGR-Xa fluorescence to titration with C6PG at 5 mM Ca2+ (Fig. 1 C, filled circles) shows that this lipid binds to bovine factor Xa and elicits a sigmoidal response, as it does with human DEGR-Xa (Banerjee et al., 2002a). No discontinuity was detected in the titration of DEGR-Xa with C6PG, inasmuch as the C6PG CMC at 5 mM Ca2+ in the absence of protein is much larger (∼14 mM) than that of C6PS (0.95 mM) (Koppaka et al., 1996). In the absence of Ca2+, C6PG produced a positive change in DEGR-Xa fluorescence (Fig. 1 C, diamonds), although this change was so small (ΔRsat = 0.010 ± 0.001) that the apparent kd for this interaction could not be determined with certainty.
The response of DEGR-Xa to C6PC at 5 mM Ca2+ (Fig. 1 C, closed triangles) was qualitatively different from the response to either C6PS or C6PG in that there was no sigmoidal shape to the titration curve, and C6PC caused a slight increase rather than a decrease in emission intensity. The C6PC CMC under these conditions is ∼9 mM (Koppaka et al., 1996). The dotted curve drawn through these data represents the best fit of the data to a single-site-binding model, which yielded a kd of 436 ± 160 μM and a ΔRsat of 0.025 ± 0.003. The response to C6PC in the absence of Ca2+ also fit a single-site binding model (dotted curve though the open triangles in Fig. 1 C) with similar binding parameters (kd = 150 ± 18 μM; ΔRsat = 0.022 ± 0.004. It is clear from Fig. 1 that the responses of DEGR-Xa to C6PS, C6PG, and C6PC were qualitatively the same in the absence of Ca2+ and similar to the response of C6PC in the presence of Ca2+ (Fig. 1 C, closed triangles), although the asymptotic fluorescence changes associated with C6PS, C6PG, and C6PC binding in the absence of Ca2+ were all different. Human Xa showed the same behavior (Banerjee et al., 2002a).
Effect of short-chain phospholipid on bovine factor Xa amidolytic activity
We reported previously that both soluble C6PS and membrane-associated PS inhibited amidolytic activity of human factor Xa to a comparable extent (Fig. 6 of Koppaka et al., 1996). To assure that the bovine protein had a functional response to C6PS similar to that of the human protein, we report in Fig. 2 the amidolytic activity of bovine factor Xa (relative to the activity in the absence of C6PS) as a function of C6PS concentration in the presence (triangles: 2 mM; circles: 5 mM) and in the absence (squares) of Ca2+. As for human factor Xa (Koppaka et al., 1996), the amidolytic activity of bovine factor Xa was inhibited by C6PS. The site dissociation constant (98 μM) obtained from a least-squares fit of the 5 mM Ca2+ data to a single-site model is somewhat higher than we have reported for C6PS effect on human factor Xa (67 μM) (Koppaka et al., 1996). Binding was weaker at lower Ca2+ concentrations (kd ≈ 190 μM at 2 mM Ca2+), as also seen for the human protein (Koppaka et al., 1996). In the absence of Ca2+, C6PS had no effect on activity of either the bovine or human protein.
FIGURE 6.

Dependence of Xa dimerization on experimental conditions. (A) The variation of the dissociation constant (kddimer) of bovine factor Xa with Ca2+ concentration at 600 μM C6PS. (B) A plot of dimer association constant Kdimer versus C6PS concentration at 5 mM Ca2+. A fit of these values to a hyperbolic curve gave an apparent C6PS kd of 96 μM with Kdsatdimer = 17.2 nM.
FIGURE 2.

Relative rates of S-2765 hydrolysis by bovine factor Xa (5 nM) as a function of C6PS in the presence of 5 mM Ca2+ (filled circles), 2 mM Ca2+ (filled triangles), and in the absence of Ca2+ (filled squares) were determined at 37°C in a buffer containing 50mM Tris, 175 mM NaCl, 0.6% PEG, at pH 7.6. The data are expressed as a fraction of the rate of S2765 hydrolysis by factor Xa in the absence of C6PS. The lines indicate the fits of the data to a model assuming a single C6PS site on factor Xa, yielding dissociation constants of 190 and 98 μM in the presence of 2 and 5 mM Ca2+, respectively.
Effect of soluble phospholipids on the rate of factor Xaα autolytic activity
Bovine factor X can be activated by two alternative pathways (Fujikawa et al., 1974; Jesty et al., 1974, 1975). The first is initiated by the action of factor VIIa or RVV on factor X to cleave an activation peptide from the amino terminus of the heavy chain to produce factor Xaα. Factor Xaα is then converted autocatalytically to factor Xaβ with the loss of a 17-residue glycopeptide from the carboxy terminus of the heavy chain. The conversion of factor Xaα to factor Xaβ is lipid dependent. The alternative pathway is initiated by the action of factor Xa on factor X in the presence of lipid to produce factor Xaβ.
Fig. 3 shows the time course of the appearance of factor Xaβ due to autolysis of factor Xaα, as monitored by SDS-PAGE in the presence of 5 mM CaCl2 and various concentrations of soluble phospholipids or membranes. The fraction of factor Xa in the β (autolyzed) form was estimated from intensities of SDS-PAGE gel bands (gels not shown). Of the three short-chain lipids examined (C6PC, closed circles; C6PG, filled inverted triangles; C6PS, filled triangles), only C6PS produced a significant enhancement (≥70-fold) in the rate of factor Xaα autolysis relative to the rate in the absence of lipid (open diamonds). This rate enhancement occurred below the CMC for C6PS observed in the presence of factor Xa (0.8 mM (Koppaka et al., 1996)). At or above the CMC, C6PS micelles induced an even greater increase (from ≥170- to 230-fold) in the rate of Xaα autolysis (0.8 mM C6PS, filled squares; 1.36 mM, filled diamonds). The rate enhancement of factor Xaα autolysis by C6PS is contrasted in Fig. 3 to the rate enhancement induced by PS/POPC (1/3) membranes (220-fold; open circles). Clearly, the largest part of the autolysis rate enhancement in the presence of a PS-containing membrane is due to the PS molecule, but there is roughly a two- to threefold increased rate due to the presence of an interface, be it a membrane or micelle interface. These data agree with the 60-fold increase in proteolytic activity that we have reported (Koppaka et al., 1996) for the effect of C6PS on the proteolytic activity of human factor Xa against prothrombin and show conclusively that the effect of C6PS is on factor Xa and not on its substrate prothrombin. They also agree with our finding for human factor Xa that the effect of a PS membrane or micelle surface is only two- to threefold greater than the effect of C6PS in solution (Banerjee et al., 2002b). DOPG/POPC membranes (open squares) did enhance the reaction rate by 18-fold, but the rate was much lower than that observed in the presence of soluble or membrane-associated PS. POPC membranes produced only a fivefold increase in the rate of factor Xa autolysis (open triangles).
FIGURE 3.

Fractions of factor Xa present as factor Xaβ were obtained from quantitative densitometry of SDS-PAGE gels (not shown) and are plotted as a function of time of incubation at 22°C with different lipids: 0.43 mM C6PS (filled triangles); 0.8 mM C6PS (filled squares); 1.36 mM C6PS (filled diamonds); 1 mM C6PG (filled inverted triangle); 1 mM C6PC (filled circle); 1 mM DOPG/POPC membranes (open squares); 1 mM POPS/POPC membranes (open circles); 1 mM POPC membranes (open triangles); and no lipid (open diamonds).
In summary, both active-site-labeled bovine DEGR-Xa and unlabeled bovine Xa respond to different soluble lipids in a fashion analogous to the response of the human protein, except that lipid binding to the human protein was in general a bit tighter than was binding to the bovine protein. The response of factor Xa activity to C6PS was hyperbolic whereas that of DEGR-Xa fluorescence was sigmoidal in shape.
C6PS triggers factor Xa dimerization
To test the possibility that the difference in structural and functional responses might be that these experiments were performed at very different factor Xa concentrations (100 nM for fluorescence and 5 nM for activity measurements), we titrated 1 nM DEGR-Xa in the presence of 5 mM Ca2+ with C6PS. The response was no longer sigmoidal (Fig. 4, open triangles). Nor was it simply hyperbolic, inasmuch as a single-site model could not describe the data either, as seen from the dotted curve drawn through the triangles in Fig. 4. The difference in response of DEGR-Xa to C6PS at 1 and 100 nM DEGR-Xa might be an artifact associated with pressing the limits of sensitivity of our fluorometer at 1 nM DEGR-Xa. To test for this possibility, we performed a titration at yet a third DEGR-Xa concentration (150 nM). The resulting response curve (Fig. 4, open circles) showed a clear sigmoid shape but differed in the magnitude of the response from data obtained at 100 nM factor Xa (compare to Fig. 1 A). Clearly, the response to C6PS varied with protein concentration, suggesting that factor Xa might self-associate, at least under the influence of C6PS, and that self-association might influence the observed response to C6PS.
FIGURE 4.

DEGR-Xa at 1 nM (open triangles) and 150 nM (open circles) in buffer A (50 mM Tris, 0.1M NaCl, pH 7.5) was titrated with C6PS in the presence of 5 mM Ca2+, at 22°C, as described for Fig. 1. The response of DEGR-Xa fluorescence to C6PS titration in the absence of Ca2+ is also shown for 1 nM (open diamonds) and 100 nM (closed diamonds) protein. Lines drawn through the data show fits as described in Fig. 1.
To test for possible C6PS-induced factor Xa self-association, we recorded factor Xa intrinsic fluorescence spectra as a function of protein concentration, in the presence and absence of 600 μM C6PS and/or 5 mM Ca2+ (data not shown) and found complex changes in intrinsic fluorescence with increasing factor Xa concentration. In Fig. 5, we present the intensity of the Trp peak of factor Xa intrinsic fluorescence as a function of factor Xa concentration. Not surprisingly, the intensity of factor Xa alone increased linearly with factor Xa concentration in the presence (frame A, closed circles) and absence (frame C, closed squares) of Ca2+. Even in the presence of C6PS, the intrinsic fluorescence increased in a linear fashion in the absence of Ca2+ (Fig. 5 C, closed inverted triangle). However, the fluorescence intensity in the presence of both Ca2+ and C6PS increased in a saturable fashion and then increased linearly with factor Xa concentration (Fig. 5 B, filled triangle). The linear increase of fluorescence represented by the straight line in Fig. 5 B was subtracted from the whole curve in this frame, and the saturable residual curve is shown in the inset to Fig. 5 B. These data were fit to a dimerization model (Eq. 2 in Appendix) to yield a dimer dissociation constant of 20 nM. Similar experiments were performed at several Ca2+ concentrations with the resulting dimer dissociation constants shown as a function Ca2+ concentration in Fig. 6 A. The response of human factor Xa activity to C6PS, which is dominated by binding to site 1 (Banerjee et al., 2002a), shows a very similar Ca2+ dependence (Koppaka et al., 1996). Fig. 6 B shows the variation of dimer association constants with C6PS concentration. A hyperbolic fit to this curve gave an apparent Kd of 96 μM, the same Kd obtained for site 1 (Kd ≈ 98 μM, Fig. 2). Thus, both the Ca2+- and C6PS-dependences of dimerization indicate that occupancy of site 1 site triggers dimerization.
FIGURE 5.

The intrinsic tryptophan fluorescence intensity of factor Xa recorded at 344 nm is plotted as a function of factor Xa concentration in the presence of Ca2+ (A, closed circle) and in the absence of 5 mM Ca2+ in the presence (closed inverted triangle) and absence (closed square) of 600 μM C6PS (C). (B) Factor Xa intrinsic fluorescence intensity is plotted as a function of factor Xa concentration in the presence of 650 μM soluble C6PS (closed triangle). The lower inset to B shows the data in this frame with the limiting linear increase of fluorescence intensity (line through the data at higher Xa concentrations) subtracted. When these data were fit to a simple dimerization model (see text), a dimerization kd of ∼20 nM was obtained.
To obtain a direct measure of factor Xa self-association under a variety of conditions, native polyacrylamide gel electrophoresis was performed. A sample 7% gel is shown as an inset to Fig. 7 and demonstrates that both monomer and dimer were easily resolved. No higher order aggregates were observed at the stacking/running gel interface of any experiment (Fig. 7, top of gel). From these gels, the retardation coefficient of each protein was obtained as described in Methods. Fig. 7 shows a plot of log (retardation coefficient) versus log (molecular weight) (Ferguson, 1964) for four marker proteins (filled circles) and 100 nM factor Xa in the absence of Ca2+(filled triangle; 47 ± 1.5 kDa). The molecular mass of bovine factor Xa is reported by amino acid and carbohydrate analysis to be 45 kDa (Jesty and Nemerson, 1976). In the presence of 400 μM C6PS, the molecular mass of factor Xa was 114 ± 1.7 kDa at 5 mM Ca2+ (Fig. 7, star). From the ratio of these molecular masses (2.4), it seems most likely that factor Xa forms a dimer in the presence of soluble C6PS, with the shape of the dimer causing it to run at slightly greater than twice the molecular mass of the monomer. The fraction of factor Xa involved in dimer under these conditions was 85% (calculated from the dimerization constant recorded in Fig. 6 and obtained as in Fig. 5), whereas the fractions of material in the dimer and monomer bands of lane 4 of Fig. 7 were found from densitometry to be 83 and 17%, respectively. The fact that the distribution of material in the gel reflects the equilibrium distribution in solution suggests that dissociation is slow. It also provides direct validation of our use of intrinsic fluorescence to obtain dimerization constants. A gel run in the presence of 400 μM C6PS but at 1 nM FXa showed that only monomer with a molecular mass of 46 ± 1.6 kDa (data not shown) existed under the conditions of the open triangles in Fig. 4. A control experiment with 100 nM Xa, 2 mM Ca2+, and 100 nM Xa showed a monomer (Fig. 7, diamond) of 47 ± 0.8 kDa, verifying the Ca2+-dependence of factor Xa dimerization shown in Fig. 6 A. Finally, an experiment performed at 5 mM Ca2+ with 400 μM C6PC and 100 nM Xa also showed only monomer Xa (data not shown), demonstrating the specificity of C6PS-induced dimerization. Sedimentation velocity experiments have shown bovine factor Xa to be a 45 kDa monomer in solution in the presence of Ca2+ (Pryzdial and Mann, 1991). Our results agree with this, but clearly show that factor Xa dimerizes in the presence of C6PS.
FIGURE 7.

Factor Xa was analyzed by native polyacrylamide gel electrophoresis in buffer A plus 5 mM CaCl2 as described in Methods. Retardation coefficient for different reference proteins were obtained from plots of log Rf versus percent cross-linking (5%, 6%, 8%, and 10%). A log-log plot of retardation coefficient is shown for these reference proteins (closed circles). Points have been added to this plot for retardation coefficients obtained for 100 nM factor Xa with 5 mM Ca2+ in the absence (closed triangle) or presence (closed star) of 400 μM C6PS and also with 2 mM Ca2+ and 400 μM C6PS (closed diamond). The inset shows lanes from the 7% gel (lane 1—markers, lane 2—100 nM Xa with 2 mM calcium and 400 μM C6PS, lane 3—100 nM Xa with 5 mM Ca2+ without C6PS, lane 4—100 nM Xa with 5 mM Ca2+ and 400 μM C6PS) with bands noted by arrows from the points on the Ferguson plot (Ferguson, 1964).
We conclude that bovine factor Xa forms a dimer under the influence of C6PS binding to site 1 and that the different responses seen for titration of DEGR-Xa fluorescence at 100 and 1 nM factor Xa probably reflect different structural responses elicited by C6PS from the two aggregation states of factor Xa. Thus, binding of C6PS to bovine factor Xa alters its activity, active site environment, and aggregation state in solution. This strongly implies a structural change, the possibility of which we address next with a direct structural measurement.
Effects of short-chain phospholipids on factor Xa secondary structure
CD specta of factor Xa at various concentrations of soluble lipids (Fig. 8) are notable for their lack of a pair of negative peaks at 208 and 222 nm that characterize α-helical secondary structure (Johnson, 1990). Instead, the spectrum is dominated by a single negative band whose peak varies from 203 to 206 nm, depending on the presence of Ca2+ and/or C6PS. This implies that factor Xa in solution consists mainly of β-structures and little α-helix, as confirmed by spectral analysis (Sreerama and Woody, 2000) (11% α-helix, with and without Ca2+). Of course, analysis of these spectra taken only to 200 nm could yield no useful information about β-or disordered structures (Johnson, 1990). However, analysis of spectra carefully collected to 185 nm in salt-free buffer confirmed the presence of only 11% α-helix, with β-structures (sheets and turns) accounting for 60% of the spectral shape. Although the crystal structure of whole bovine factor Xa has not been determined, the crystal structure of the highly homologous human factor IXa has 13% α-helix and 23% β-sheet, with β-turns not tabulated (from the Protein Data Bank based on Brandstetter et al. (1995).
FIGURE 8.

(A) Factor Xa (0.6 μM) in buffer B (1.2 mM Tris, 150 mM NaCl, pH 7.4) was examined at 24°C in the presence of 3 mM CaCl2 (solid line); and in the presence of 0.6 mM C6PS (dot-dash-dot line), C6PG (dotted line), and C6PC (dot-dot-dashed line) with 3 mM CaCl2. (B) Factor Xa (0.6 μM) in buffer (1.2 mM Tris, 150 mM NaCl, pH 7.4) was examined at 24°C (solid line); and in the presence of 0.6 mM C6PS (dot-dash-dot line), C6PG (dotted line), and C6PC (dot-dot-dashed line) in the absence of CaCl2. In the lower insets to A and B, the effects of C6PS (circles), C6PG (squares), and C6PC (triangles) concentrations on Θ222/Θ208 ratios measured from CD spectra of factor Xa in the presence of 3 mM Ca2+ (A) or in the absence of Ca2+ (B). The upper inset to A shows the variation of Θ222/Θ208 ratio with fraction of Xa present as dimer (obtained using kdimer from the upper inset to Fig. 6 B) for factor Xa concentrations of 200, 300, and 600 nM.
Because the major features of the spectra in Fig. 8 are consistent with what information we have about the secondary structure of factor Xa, we may use this approach to monitor changes in factor Xa secondary structure associated with binding of soluble lipids. Because we could not obtain the deep UV spectra needed for a detailed secondary structure analysis (Johnson, 1990), we used the ratio of ellipticity at 222 nm to that at 208 nm (Θ222/Θ208) (Greenfield and Fasman, 1969) as well as estimates of the fraction of α-helix (Sreerama and Woody, 2000) to monitor changes in secondary structure induced by binding of soluble lipids.
Although the CD spectrum of factor Xa was slightly red-shifted on addition of Ca2+ (compare solid lines in Fig. 8 B to Fig. 8 A), there was no significant change in the Θ222/Θ208 ratio (0.52 in both cases) or in our estimate of helical content (11% in both cases). Fig. 8 A also shows that addition of 600 μM C6PC (dot-dot-dashed line), C6PG (dotted line), or C6PS (dot-dashed line) to factor Xa in the presence of 3 mM Ca2+ caused shifts in bovine factor Xa secondary structure. All three lipids changed the shape of the CD spectrum. These changes were clearly detectable both as reproducible changes in the Θ222/Θ208 ratio at increasing concentrations of C6PC (triangles), C6PG (squares), or C6PS (circles) (Fig. 8 A, inset), and as changes in α-helical content (−1, +2.8, and +6%, respectively). These results are consistent with the different changes in DEGR-Xa fluorescence seen for the three different lipid species, in that C6PC- and C6PG-induced different conformational changes in factor Xa, with both of these different from the large changes induced by C6PS. Because the Θ222/Θ208 ratio increased for all three lipids, but the α-helical content decreased for C6PC, it is clear that changes in the Θ222/Θ208 ratio reflected more than just changes in helix content.
Fig. 8 B shows that the CD spectrum of factor Xa also changed upon addition of C6PS, C6PG, and C6PC in the absence of Ca2+, but these soluble lipids induced different conformational changes (inset) than seen in the presence of Ca2+, consistent with our DEGR-Xa titration results (Fig. 1). As for the DEGR-Xa results, the response of factor Xa secondary structure to C6PC was the same in the presence (Fig. 8 A, inset) or absence (Fig. 8 B, inset) of Ca2+.
Inasmuch as C6PS induces dimerization in the presence of Ca2+, the changes observed in the presence of Ca2+ could represent changes in secondary structure associated with intermolecular structures formed upon dimer formation. Even minimal conditions for CD experiments (2mM Ca2+ and 200 nM Xa) will produce a small amount of dimer (∼5–7% of Xa from Fig. 6 B) when C6PS occupies site 1. It was thus impossible to measure directly the effect of C6PS on the CD spectrum of monomeric factor Xa without inducing some dimer formation. Thus, we resorted to taking a series of spectra under conditions that should result in decreasing dimer formation. Spectra taken in the presence of 3 mM Ca2+ and 600 μM C6PS, but at 300 and 200 nM factor Xa, showed decreasing negative peaks at 208 nm (data not shown). A spectrum taken at 5 mM Ca2+, 600 μM C6PS, and 600 nM factor Xa, for which even more Xa should be dimer, had an even larger negative ellipticity minimum (data not shown). From the factor Xa dimerization constant observed at 3 and 5 mM Ca2+ and 600 μM C6PS (Fig. 6 A), we calculated the fraction of Xa involved in dimer formation under these varying conditions and plot the Θ222/Θ208 ratio as a function of this in the upper inset to Fig. 8 A. The intercept of a straight line through these data predicts that factor Xa monomer bound to C6PS in the presence of Ca2+ would give a Θ222/Θ208 ratio of 0.44, nearly the same value seen for monomer factor Xa at saturating C6PS concentration in the absence of Ca2+ (0.45; Fig. 8 B, inset). This implies that the secondary structural change seen in response to C6PS in the presence of Ca2+ results in part from dimer formation and in part from a change in secondary structure associated with C6PS binding to monomer factor Xa.
DISCUSSION
Both human and bovine factor Xa have two soluble-lipid-binding sites, site 1 in the EGFNC domain pair that is Ca2+-dependent and site 2 in the catalytic domain that is Ca2+ requiring and probably corresponds to part of a protein recognition site rather than being a lipid regulatory site (Srivastava et al., 2002). In addition, other studies of DEGR-Xa fluorescence and factor Xa activity have shown that these two sites in human factor Xa are linked, that they have different minimal lipid specificities, and that site 1 is principally involved in regulating factor Xa activity (Banerjee et al., 2002a). While these studies suggest that factor Xa activity is regulated via structural changes induced by C6PS binding, they provided no direct proof of C6PS-induced protein structural changes. To demonstrate C6PS-induced structural changes, we focused in the current paper on bovine factor Xa, which is available more inexpensively in the large quantities needed for structural work. The first part of the results in this paper establish that bovine factor Xa behaves analogously to human factor Xa with respect to C6PS binding, making it reasonable to assign responses observed for one of these very similar forms of factor Xa to the other. The second part of this work leads to three significant conclusions about the effect of PS molecules on bovine factor Xa:
The two sites on bovine factor Xa differ in their response to Ca2+ and to different lipid species. Site 1 is occupied by C6PG and C6PC with similar affinity (kd ∼ 200 μM) in the presence or absence of Ca2+ but with higher affinity by C6PS in the presence (kd ∼ 90–100 μM) than in the absence (kd ∼ 200 μM) of Ca2+. This is the EGFNC site (Srivastava et al., 2002) that recognizes DAG as a minimal ligand in human factor Xa (Banerjee et al., 2002a). Site 2 does not bind C6PC and requires Ca2+ to bind C6PS (kd ∼ 160–250 μM) or C6PG (kd ∼ 700–1100 μM). This site is in the catalytic-domain site (Srivastava et al., 2002) and in human factor Xa recognizes GPS as a minimal ligand (Banerjee et al., 2002a).
Description of C6PS or C6PG binding to these sites minimally requires the sequential-linked-site model. The requirement for something like this model derives from the different response of DEGR-X fluorescence (sigmoidal; Fig. 1) and factor Xa activity titration (hyperbolic; Fig. 2) to C6PS, a situation that also exists for human factor Xa (Banerjee et al., 2002a; Koppaka et al., 1996). There is no way to justify these observations within the context of the traditional linked-site model (Klotz and Hunston, 1975) in which both sites respond with the same structural or functional change. This is because assigning a site dissociation constant (kd) of 90–100 μM (the binding constant obtained from titration of factor Xa activity, Fig. 2), and using published procedures for calculating site constants from stoichiometric constants with one site constant known (Klotz and Hunston, 1975) led to physically unreasonable estimates of the other two independent site dissociation constants for the interaction of C6PS with factor Xa. The basic problem is that the fit provided by the linked-site model requires that tight binding should occur only once a weak site is occupied, and this was not the behavior seen for the response of activity to C6PS (Fig. 2). Because competitive and cooperative binding experiments with different lipid species demonstrate that the two lipid binding sites on human factor Xa are linked (Banerjee et al., 2002a), we have concluded that a more complex linked-site model is needed to describe our data for C6PS binding to factor Xa. The sequential-linked-site model described in the Appendix is the simplest model that we have found accounts for our observations. In fitting our data to this model, we fixed the dissociation constant assigned to site 1 (90 μM), leaving three adjustable parameters, which, for the most part, were well defined by the regression process. This model provided an adequate description of data at both low (1nM) and high (100 or 150 nM) DEGR-Xa concentrations, as documented in Figs. 1 and 4 (solid lines) and Table 1. A likely explanation for the simple hyperbolic response of factor Xa activity (Fig. 2) is that occupancy of only one of the sites on factor Xa (the first or EGFNC site) dominates the activity response but that occupancy of both sites produces an increase in activity, as demonstrated quantitatively for human factor Xa (Banerjee et al., 2002a). Despite the empirical success of this model, the behavior of factor Xa is clearly more complex than it suggests. Thus, the model assumes that the two sites are occupied sequentially to produce different structural responses in the singly bound and doubly bound species. However, we know that GPS can weakly (kd ≈ 0.8 mM) occupy site 2, but that occupancy is much tighter (kd ≈ 0.09 mM) if DAG occupies site 1 (Banerjee et al., 2002a). This means that the “sequential” aspect of our model is an approximation, but a reasonable approximation that reduces the number of unknown parameters from six to four (see Appendix). The sequential-linked-site model is also an approximation in that we show here that C6PS triggers Xa dimer formation (Figs. 6 and 7), but the model does not take into account dimerization. Thus, any response that we see to C6PS includes structural responses of binding to monomer as well as the consequences of dimer formation. Indeed, a possible interpretation of our DEGR-Xa fluorescence data is that C6PS binding to site 1 in monomer Xa might trigger conformational changes that expose the DEGR probe to water (negative DEGR fluorescence change at low DEGR-Xa concentration, Fig. 4 and Table 1), while these same conformational changes might induce dimer formation that then buries the DEGR probe at a dimer interface (positive DEGR fluorescence change, Fig. 1 and Table 1). At higher C6PS concentration, a negative fluorescence change would dominate. To test this interpretation, we formulated the dimer-sequential model (Appendix) that allows for factor Xa bound to a single molecule of C6PS to exist as either a monomer or a dimer. This introduced another unknown parameter (the fluorescence change associated with the C6PS-bound monomer, ΔR1 in Eq. 3). The extra free parameter naturally improved somewhat the fit to our data, but also led to poorly determined saturating fluorescence changes for each presumed species (see Table 1). Nonetheless, the binding parameters that could be determined from regression of this model to the data (kd1, kd12, and ΔRsat,12) were consistent with the corresponding parameter values obtained using the linked-site, sequential model (Table 1). Thus, while it is possible to explain our results with more complex models that rely on dimer formation, our data were insufficient to define the parameters associated with these models. We conclude that, while this model provides a somewhat improved description of our data (an F-test comparing the fit variances of the dimer-sequential and sequential-linked models gave P ≈ 0.025) (P is the probability that the improvement in fit is random rather than significant (Bevington, 1969), the simpler sequential-linked-site model catches the essence of factor Xa's structural and functional responses to C6PS titration without the need to introduce an unreasonable number of adjustable parameters. This is expected to be true especially for [Xa] > 50 nM and [Ca2+] > 3 mM, for which most Xa will be dimer when site 1 is occupied.
Binding of C6PS to the first factor Xa lipid binding site (EGFNC or DAG site) triggers conformational changes that lead to factor Xa dimerization. Because dimer formation accompanies occupancy of site 1, it is appropriate to comment on the evidence for C6PS-induced structural change. First, the fluorescence decrease of 1 nM DEGR-Xa followed the sequential-linked-site model, with decreases in fluorescence accompanying occupancy of both C6PS sites (Table 1). In gel experiments (Fig. 7), we confirmed that 1 nM factor Xa at 5 mM Ca2+ did not form dimers, so that the changes in dansyl fluorescence reflect changes in the active site environment of monomer factor Xa. Second, a secondary structure change accompanied C6PS binding to site 1 in the absence of Ca2+ (Fig. 8 B), when dimer formation is not observed (Fig. 7). Third, C6PC also induced changes in CD spectra in the presence and absence of Ca2+ (Fig. 8) even though C6PC does not promote dimer formation, as we showed by native polyacrylamide gel. Finally, while it is clear that some part of the structural response seen to C6PS binding in the presence of Ca2+ is due to dimerization (Fig. 8 A, upper inset), linear extrapolation of our data to zero percent dimer yielded a Θ222/Θ208 equal to that induced by C6PS in the absence of Ca2+ (see description of Fig. 8 A). These observations provide broad support for our hypothesis that C6PS induces conformational changes in factor Xa monomer that, in the presence of Ca2+, lead to dimerization, in itself an indication of a structural change.
TABLE 1.
Comparison of models describing the effect of soluble lipid on DEGR-factor Xa fluorescence intensity
| Lipid | Model #sites/Eq. | Parameters* | 100 nM DEGR-Xa 3 mM Ca | 100 nM DEGR-Xa 5 mM Ca | 1 nM DEGR-Xa 5 mM Ca | 150 nM DEGR-Xa 5 mM Ca |
|---|---|---|---|---|---|---|
| C6PS | Sequential | kd1 | 90† | 90† | 110 ± 15 | 90† |
| 2/A4 | kd12 | 222 ± 34 | 682 ± 185 | 1420‡ | 255 ± 22 | |
| ΔRsat,1 | 0.06 ± 0.02 | 0.02‡ | −0.19 ± 0.01 | 0.04 ± 0.01 | ||
| ΔRsat,12 | −0.28 ± 0.01 | −0.41 ± 0.04 | −0.060‡ | −0.36 ± 0.01 | ||
| R2/σ | 0.997/0.0042 | 0.997/0.0053 | 0.993/0.0042 | 0.999/0.0036 | ||
| Dimer/Seq. | kd1 | 90† | 90† | ND | 90† | |
| 2/A6 | kd12 | 208 ± 39 | 435 ± 137 | 185 ± 26 | ||
| ΔRsat,1 | −0.14‡ | −0.11‡ | −0.15‡ | |||
| ΔRsat,12 | −0.30 ± 0.01 | −0.37 ± 0.04 | −0.35 ± 0.01 | |||
| ΔRsat,3 | 0.16‡ | 0.13‡ | 0.16‡ | |||
| R2/σ | 0.998/0.0036 | 0.996/0.0044 | 0.999/0.0031 | |||
| C6PG | Sequential | kd1 | 200† | 200† | NA | NA |
| 2/A4 | kd12 | 1130 ± 270 | 750 ± 110 | |||
| ΔRsat,1§ | 0.013‡ | 0.023 ± 0.008 | ||||
| ΔRsat,12§ | −0.18 ± 0.02 | −0.16 ± 0.01 | ||||
| R2/σ | 0.988/0.004 | 0.994/0.0015 |
Units of dissociation constants are μM. ΔRsat values are fluorescence quantum yield differences between the lipid-bound state and the lipid-free state of DEGR-Xa relative to the quantum yield of the lipid-free state.
This value fixed based on titration of activity for C6PS or results ±Ca2+ for C6PG.
A t-test on this parameter produced a P > 0.05%, where P is the probability of being wrong in concluding that the parameter is zero. This indicates that this parameter value is ill determined.
These parameter values were constrained such that ΔRsat,1 > 0, ΔRsat,12 < 0.
ND (not determined).
NA (not applicable) means that these data did not support this analysis.
Are the effects of soluble C6PS due to micelle formation?
Our conclusions depend on our results reflecting the effects of individual C6PS molecules binding to factor Xa. We have previously characterized by both probe fluorescence and QELS the CMCs of C6PS, C6PC, and C6PG as a function of Ca2+ concentration and in the presence of different concentrations of factor Xa and prothrombin with and without Ca2+ and 0.6 wt% poly(ethylene glycol) (Koppaka et al., 1996). As documented throughout the Results section, we were careful in this work never to analyze data taken at concentrations above the CMC of the short-chain phospholipid under study. We were able to detect micelle formation both by QELS and by monitoring sudden changes in an observable for each experiment as we went above the CMC in C6PS concentration (see Results). The C6PS CMC obtained in these different ways always agreed. Thus, the results we report here reflect the binding of factor Xa to soluble lipids, not to lipid micelles.
Aside from the possibility of micelle formation that our controls rule out, Johnson et al. (1981) suggested that short-chain C6PC may exist in an equilibrium mixture of monomers and dimers below the CMC. Because of the net negative charge on C6PS, this probably does not occur for this short-chain lipid. However, if it did, binding of both monomers and dimers of C6PS to a single site on factor Xa might cause the observed sigmoidal binding behavior. This possibility can be precluded for several reasons. First, the binding of C6PC to DEGR-Xa showed no such behavior (Fig. 2 A, filled triangles), although C6PC is the lipid actually suggested to form dimers below its CMC (Johnson et al., 1981). Second, the responses of amidolytic (Fig. 3) and proteolytic activities of factor Xa (Koppaka et al., 1996) to soluble lipids showed simple hyperbolic, not sigmoidal, responses to C6PS. This can be explained easily in terms of single C6PS molecules binding to and eliciting different responses from two different sites, but is hard to explain in terms of C6PS binding as a monomer or dimer to a single site. Finally, direct equilibrium dialysis measurements show that, while two soluble C6PS molecules bind to factor Xa (Banerjee et al., 2002a), they bind to different regions of the factor Xa molecule (Srivastava et al., 2002). These measurements are unequivocal and are inconsistent with our results being due to binding of C6PS dimers to factor Xa.
Relationship of C6PS binding sites to membrane binding
Binding of prothrombin and factor Xa to membranes has been well studied. The Gla domain is required for the Ca2+-dependent interaction with phospholipid membranes (Henriksen and Jackson, 1975), and a comparison of binding affinity does show that the Gla domain plus first Kringle domain (termed fragment 1) of prothrombin account for most, but not all, of the membrane-binding free energy (Pearce et al., 1993). Several models for the mechanism of membrane binding have been proposed: absorption to clusters or domains of 10 or so acidic lipids in the plane of the membrane (Lim et al., 1977; van Dieijen et al., 1981); specific occupancy of a limited number (two–four) of PS binding sites (Cutsforth et al., 1989; Tendian and Lentz, 1990); membrane insertion of a hydrophobic loop involving residues 5 and 8 of the Gla domain (Christiansen et al., 1995; Falls et al., 2001); or formation of a specific lipid-binding pocket involving residues 11, 33, and 34 (McDonald et al., 1997). While there remains controversy about the mechanism of binding, the prevailing opinion is that membrane binding involves a limited number of sites on factor Xa and that the most significant contribution to the free energy of the process derives from the Gla domain. If so, how can a PS site in the EGFNC domain pair be important to membrane binding? The answer is that this site is likely not a significant contributor to the free energy of membrane binding but rather accounts for the effects of PS-containing membranes on factor Xa behavior. This follows from the fact that, for every observable that was altered by C6PS, there was a qualitatively similar (but not identical) change due to PS-containing membranes. There was a 70-fold increase in bovine Xa autolysis rate when C6PS was present and only a two- to threefold additional increase in the presence of PS-membranes (Fig. 3). C6PS decreased factor Xa amidolytic activity by 55% (Fig. 2), while PS-containing membranes decreased this activity by 35% (Koppaka et al., 1996). Adding soluble C6PS produces a saturable 60-fold increase in the rate of prothrombin activation (Banerjee et al., 2002b; Koppaka et al., 1996), while the rate of prothrombin proteolysis by human factor Xa (in the absence of factor Va) increased by 475-fold at an optimal membrane concentration of 50 μM (Banerjee et al., 2002b; Wu et al., 2002). However, a substantial fraction of this increase was due to a membrane-surface effect: channeling of intermediate back to the enzyme for rapid conversion to thrombin (Banerjee et al., 2002b; Wu et al., 2002). Finally, control experiments showed that DEGR-Xa fluorescence decreased by 55% upon addition of PS-containing membranes compared to the 25% decrease seen with C6PS. A decrease in human DEGR-Xa fluorescence upon titration with PS-containing membranes has previously been interpreted in terms of a membrane-induced change in factor Xa structure (Husten et al., 1987), although, as we point out here, a change in DEGR-Xa fluorescence is insufficient to demonstrate a structural change. However, our results do demonstrate C6PS-induced structural changes in factor Xa, and the parallels between our measurements with C6PS and with PS-membranes suggest that the decrease in DEGR-Xa fluorescence seen in the presence of PS-membranes reflects either structural changes or dimerization, or both, induced by membrane binding. Factor Xa dimerization on a PS-membrane would likely have significant implications for how membranes induce the interaction of factors Xa and Va in vivo to form prothrombin-activating complex. This deserves further study.
Acknowledgments
We thank Dr. A. Srivastava for useful discussions and suggestions.
This work was supported by U.S. Public Health Service grant HL45916 to B.R.L.
APPENDIX
Model for equilibrium binding of water-soluble phospholipids to DEGR-Xa
If two ligand-binding sites are linked, and if their occupancy elicits different structural responses, we can write for the net structural response an expression that involves six unknown parameters. A simplification of this model can be made when one site (second site) is unlikely to be occupied without the other being occupied first. Making this assumption eliminates two parameters. This is termed the sequential-linked-site model, and takes the form:
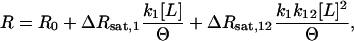 |
(1) |
where 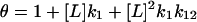 , R is the observed structural response, R0 is the value of R in the absence of ligand, ΔRsat,1 is the structural response to occupancy of the first site, and ΔRsat,12 is the response to occupancy of both sites. When the total protein concentration is much smaller than the ligand concentration, [L] ≅ [L]tot, modeling R as a function of [L] becomes much easier. The quantity Θ is a modified grand partition function describing the distribution of ligands between sites on the protein. The sequential binding model has four parameters: k1, k12, ΔRsat,1, and ΔRsat,12. For a protein with a single binding site, k12 = 0, and this expression reduces to the standard single site binding model, which has only two free parameters, k1 and ΔRsat.
, R is the observed structural response, R0 is the value of R in the absence of ligand, ΔRsat,1 is the structural response to occupancy of the first site, and ΔRsat,12 is the response to occupancy of both sites. When the total protein concentration is much smaller than the ligand concentration, [L] ≅ [L]tot, modeling R as a function of [L] becomes much easier. The quantity Θ is a modified grand partition function describing the distribution of ligands between sites on the protein. The sequential binding model has four parameters: k1, k12, ΔRsat,1, and ΔRsat,12. For a protein with a single binding site, k12 = 0, and this expression reduces to the standard single site binding model, which has only two free parameters, k1 and ΔRsat.
We had to consider one more model when we demonstrated that factor Xa forms dimers in the presence of C6PS. It is easy to show that for a simple dimerization reaction that produces a change in some observable, R, the fraction of protein in a dimer state, f2, is:
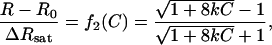 |
(2) |
where C is the total protein concentration, k is the dimer association constant, whose lipid dependence is given in the legend to Fig. 6, and R0 and Rsat are the values of the observable at limiting low and high protein concentrations, respectively. If we presume that PS binding to a single site enables dimer formation, then a fraction (f2) of the total concentration of the species PL1 will be dimer and 1 − f2 will be monomer. We represent the response of the monomer as ΔRsat,1 and that of the dimer as ΔRsat,3 to distinguish it from the response due to occupancy of the second site (ΔRsat,12). We may then write:
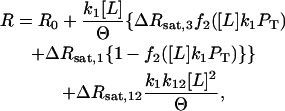 |
(3) |
where we have replaced C in Eq. 2 with the concentration of protein bound to a single PS, k1 × [L] × PT. This makes f2 not only a function of PT, the total protein concentration in an experiment, but also of the phospholipid concentration added during a titration experiment. This is termed the dimer/sequential binding model.
Rinku Majumder and Jianfang Wang contributed equally to this work.
Abbreviations used: PS: phosphatidylserine; C6PC: 1,2-dicaproyl-sn-glycero-3-phosphocholine; C6PG: 1,2-dicaproyl-sn-glycero-3-phospho-rac-1-glycerol; C6PS: 1,2-dicaproyl-sn-glycero-3-phospho-L-serine; POPC: 1,2-palmitoyl-2-oleoyl-3-sn-phosphatidylcholine; DOPG: 1,2-dioleoyl-phosphatidylglycerol; DAG: diacylglycerol; GPS: glycerol phosphorylserine; DEGR-CK: [5-(dimethylamino)-1-naphthalenesulfonyl] glutamylgycylarginyl chloromethyl ketone; DEGR-Xa: factor Xa modified with DEGR-CK; S2765: N-2-benzyloxycarbonyl-d-arginyl-glycyl-Larginine-P-nitroanilide-dihydrochloride; CD: circular dichroism; SDS-PAGE: sodium dodecylsulfate polyacrylamide gel electrophoresis.
References
- Banerjee, M., D. C. Drummond, A. Srivastava, D. Daleke, and B. R. Lentz. 2002a. Specificity of soluble phospholipid binding sites on human factor Xa. Biochemistry. 41:7751–7762. [DOI] [PubMed] [Google Scholar]
- Banerjee, M., R. Majumder, G. Weinreb, J. F. Wang, R. Majumder, and B. R. Lentz. 2002b. Role of procoagulant membranes in human prothrombin activation. 2. Soluble phosphatidylserine upregulates and directs Factor Xa to appropriate peptide bonds in prothrombin. Biochemistry. 41:950–957. [DOI] [PubMed] [Google Scholar]
- Bevington, P. R. 1969. Data Reduction and Error Analysis for the Physical Sciences. McGraw-Hill, New York.
- Brandstetter, H., M. Bauer, R. Huber, P. Lollar, and W. Bode. 1995. X-ray structure of clotting factor IXa: active site and module structure related to Xase activity and hemophilia B. Proc. Natl. Acad. Sci. USA. 92:9796–9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan, J. K. 1977. Molecular weights of protein multimers from polyacrylamide gel electrophoresis. Anal. Biochem. 78:513–519. [DOI] [PubMed] [Google Scholar]
- Chen, Q., and B. R. Lentz. 1997. Fluorescence resonance energy transfer study of shape changes in membrane-bound bovine prothrombin and meizothrombin. Biochemistry. 36:4701–4711. [DOI] [PubMed] [Google Scholar]
- Christiansen, W. T., L. R. Jalbert, R. M. Robertson, A. Jhingan, M. Prorok, and F. J. Castellino. 1995. Hydrophobic amino acid residues of human anticoagulation protein C that contribute to its functional binding to phospholipid vesicles. Biochemistry. 34:10376–10382. [DOI] [PubMed] [Google Scholar]
- Comfurius, P., E. F. Smeets, G. M. Willems, E. M. Bevers, and R. F. Zwaal. 1994. Assembly of the prothrombinase complex on lipid vesicles depends on the stereochemical configuration of the polar headgroup of phosphatidylserine. Biochemistry. 33:10319–10324. [DOI] [PubMed] [Google Scholar]
- Cutsforth, G. A., R. N. Whitaker, J. Hermans, and B. R. Lentz. 1989. A new model to describe extrinsic protein binding to phospholipid membranes of varying composition: application to human coagulation proteins. Biochemistry. 28:7453–7461. [DOI] [PubMed] [Google Scholar]
- Falls, L. A., B. C. Furie, M. Jacobs, B. Furie, and A. C. Rigby. 2001. The omega-loop region of the human prothrombin gamma-carboxyglutamic acid domain penetrates anionic phospholipid membranes. J. Biol. Chem. 276:23895–23902. [DOI] [PubMed] [Google Scholar]
- Ferguson, K. A. 1964. Ferguson plot analysis. Metabolism. 13:985–1002. [DOI] [PubMed] [Google Scholar]
- Fujikawa, K., M. H. Coan, M. E. Legaz, and E. W. Davie. 1974. The mechanism of activation of bovine factor X (Stuart factor) by intrinsic and extrinsic pathways. Biochemistry. 13:5290–5299. [DOI] [PubMed] [Google Scholar]
- Greenfield, N., and G. D. Fasman. 1969. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry. 8:4108–4116. [DOI] [PubMed] [Google Scholar]
- Henriksen, R. A., and C. M. Jackson. 1975. Cooperative calcium binding by the phospholipid binding region of bovine prothrombin: a requirement for intact disulfide bridges. Arch. Biochem. Biophys. 170:149–159. [DOI] [PubMed] [Google Scholar]
- Hope, M. J., M. B. Bally, G. Webb, and P. R. Cullis. 1985. Production of large unilamellar vesicles by a rapid extrusion procedure—characterization of size distribution, trapped volume and ability to maintain a membrane-potential. Biochim. Biophys. Acta. 812:55–65. [DOI] [PubMed] [Google Scholar]
- Husten, E. J., C. T. Esmon, and A. E. Johnson. 1987. The active site of blood coagulation factor Xa. Its distance from the phospholipid surface and its conformational sensitivity to components of the prothrombinase complex. J. Biol. Chem. 262:12953–12961. [PubMed] [Google Scholar]
- Jackson, C. M., and Y. Nemerson. 1980. Blood coagulation. Annu. Rev. Biochem. 49:765–811. [DOI] [PubMed] [Google Scholar]
- Jameson, G. W., D. V. Roberts, R. W. Adams, W. S. Kyle, and D. T. Elmore. 1973. Determination of the operational molarity of solutions of bovine alpha-chymotrypsin, trypsin, thrombin and factor Xa by spectrofluorimetric titration. Biochem. J. 131:107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesty, J., and Y. Nemerson. 1976. The activation of bovine coagulation factor X. Methods Enzymol. 45:95–107. [DOI] [PubMed] [Google Scholar]
- Jesty, J., A. K. Spencer, Y. Nakashima, Y. Nemerson, and W. Konigsberg. 1975. The activation of coagulation factor X. Identity of cleavage sites in the alternative activation pathways and characterization of the COOH-terminal peptide. J. Biol. Chem. 250:4497–4504. [PubMed] [Google Scholar]
- Jesty, J., A. K. Spencer, and Y. Nemerson. 1974. The mechanism of activation of factor X. Kinetic control of alternative pathways leading to the formation of activated factor X. J. Biol. Chem. 249:5614–5622. [PubMed] [Google Scholar]
- Johnson, R. E., M. A. Wells, and J. A. Rupley. 1981. Thermodynamics of dihexanoylphosphatidylcholine aggregation. Biochemistry. 20:4239–4242. [DOI] [PubMed] [Google Scholar]
- Johnson, W. C., Jr. 1990. Protein secondary structure and circular dichroism: a practical guide. Proteins. 7:205–214. [DOI] [PubMed] [Google Scholar]
- Jones, M. E., B. R. Lentz, F. A. Dombrose, and H. Sandberg. 1985. Comparison of the abilities of synthetic and platelet-derived membranes to enhance thrombin formation. Thromb. Res. 39:711–724. [DOI] [PubMed] [Google Scholar]
- Kane, W. H., M. J. Lindhout, C. M. Jackson, and P. W. Majerus. 1980. Factor Va-dependent binding of factor Xa to human platelets. J. Biol. Chem. 255:1170–1174. [PubMed] [Google Scholar]
- Klotz, I. M., and D. L. Hunston. 1975. Protein interactions with small molecules. Relationships between stoichiometric binding constants, site binding constants, and empirical binding parameters. J. Biol. Chem. 250:3001–3009. [PubMed] [Google Scholar]
- Koppaka, V., J. Wang, M. Banerjee, and B. R. Lentz. 1996. Soluble phospholipids enhance factor Xa-catalyzed prothrombin activation in solution. Biochemistry. 35:7482–7491. [DOI] [PubMed] [Google Scholar]
- Lim, T. K., V. A. Bloomfield, and G. L. Nelsestuen. 1977. Structure of the prothrombin- and blood clotting factor X-membrane complexes. Biochemistry. 16:4177–4181. [DOI] [PubMed] [Google Scholar]
- Majumder, R., G. Weinreb, X. Zhai, and B. R. Lentz. 2002. Soluble phosphatidylserine triggers assembly in solution of a prothrombin-activating complex in the absence of a membrane surface. J. Biol. Chem. In press. [DOI] [PubMed]
- Mann, K. G. 1976. Prothrombin. Methods Enzymol. 45:123–156. [DOI] [PubMed] [Google Scholar]
- Mann, K. G., M. E. Nesheim, W. R. Church, P. Haley, and S. Krishnaswamy. 1990. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 76:1–16. [PubMed] [Google Scholar]
- McDonald, J. F., A. M. Shah, R. A. Schwalbe, W. Kisiel, B. Dahlback, and G. L. Nelsestuen. 1997. Comparison of naturally occurring vitamin K-dependent proteins: correlation of amino acid sequences and membrane binding properties suggests a membrane contact site. Biochemistry. 36:5120–5127. [DOI] [PubMed] [Google Scholar]
- Mitra, P., A. K. Pal, D. Basu, and R. N. Hati. 1994. A staining procedure using Coomassie brilliant blue G-250 in phosphoric acid for detection of protein bands with high resolution in polyacrylamide gel and nitrocellulose membrane. Anal. Biochem. 223:327–329. [DOI] [PubMed] [Google Scholar]
- Nesheim, M. E., J. B. Taswell, and K. G. Mann. 1979. The contribution of bovine Factor V and Factor Va to the activity of prothrombinase. J. Biol. Chem. 254:10952–10962. [PubMed] [Google Scholar]
- Pearce, K. H., M. Hof, B. R. Lentz, and N. L. Thompson. 1993. Comparison of the membrane binding kinetics of bovine prothrombin and its fragment 1. J. Biol. Chem. 268:22984–22991. [PubMed] [Google Scholar]
- Pryzdial, E. L., and K. G. Mann. 1991. The association of coagulation factor Xa and factor Va. J. Biol. Chem. 266:8969–8977. [PubMed] [Google Scholar]
- Rezaie, A. R., and X. He. 2000. Sodium binding site of factor Xa: role of sodium in the prothrombinase complex. Biochemistry. 39:1817–1825. [DOI] [PubMed] [Google Scholar]
- Rosing, J., H. Speijer, and R. F. Zwaal. 1988. Prothrombin activation on phospholipid membranes with positive electrostatic potential. Biochemistry. 27:8–11. [DOI] [PubMed] [Google Scholar]
- Rosing, J., G. Tans, J. W. Govers-Riemslag, R. F. Zwaal, and H. C. Hemker. 1980. The role of phospholipids and factor Va in the prothrombinase complex. J. Biol. Chem. 255:274–283. [PubMed] [Google Scholar]
- Sreerama, N., and R. W. Woody. 2000. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287:252–260. [DOI] [PubMed] [Google Scholar]
- Srivastava, A., J. F. Wang, J. Stenflo, A. R. Rezaie, C. T. Esmon, and B. R. Lentz. 2002. Localization of phosphatidylserine binding sites to structural domains of factor Xa. J. Biol. Chem. 277:1855–1863. [DOI] [PubMed] [Google Scholar]
- Tendian, S. W., and B. R. Lentz. 1990. Evaluation of membrane phase behavior as a tool to detect extrinsic protein-induced domain formation: binding of prothrombin to phosphatidylserine/phosphatidylcholine vesicles. Biochemistry. 29:6720–6729. [DOI] [PubMed] [Google Scholar]
- Tracy, P., and K. Mann. 1986. A model for assembly of coagulation factor complexes on cell surfaces: Prothrombin activation on platelets. In Biochemistry of Platelet. D. Phillips and M. Shuman, editors. Academic Press, Toronto. 295–318.
- Tracy, P. B. 1988. Regulation of thrombin generation at cell surfaces. Semin. Thromb. Hemost. 14:227–233. [DOI] [PubMed] [Google Scholar]
- Tracy, P. B., L. L. Eide, and K. G. Mann. 1985. Human prothrombinase complex assembly and function on isolated peripheral blood cell populations. J. Biol. Chem. 260:2119–2124. [PubMed] [Google Scholar]
- Underwood, M. C., D. Zhong, A. Mathur, T. Heyduk, and S. P. Bajaj. 2000. Thermodynamic linkage between the S1 site, the Na+ site, and the Ca2+ site in the protease domain of human coagulation factor Xa. J. Biol. Chem. 275:36876–36884. [DOI] [PubMed] [Google Scholar]
- van Dieijen, G., G. Tans, J. van Rijn, R. F. Zwaal, and J. Rosing. 1981. Simple and rapid method to determine the binding of blood clotting factor X to phospholipid vesicles. Biochemistry. 20:7096–7101. [DOI] [PubMed] [Google Scholar]
- Wu, R. J., C. Zhou, D. D. Powers, R. Majumder, G. Weinreb, and B. R. Lentz. 2002. Role of procoagulant membranes in human prothrombin activation. 1. Prothrombin activation by factor Xa in the absence of factor Va and in the absence and presence of membranes. Biochemistry. 41:935–949. [DOI] [PubMed] [Google Scholar]
- Zhai, X., A. Srivastava, D. C. Drummond, D. Daleke, and B. R. Lentz. 2002. Phosphatidylserine binding alters the conformation and specifically enhances the cofactor activity of bovine factor Va. Biochemistry. 41:5675–5684. [DOI] [PubMed] [Google Scholar]