

Abstract
Fluorescence from a single DNA molecule passing through a laser beam is proportional to the size (contour length) of the molecule, and molecules of different sizes can be counted with equal efficiencies. Single-molecule fluorescence can thus determine the average length of the molecules in a sample and hence the frequency of double-strand breaks induced by various treatments. Ionizing radiation-induced frank double-strand breaks can thus be quantified by single-molecule sizing. Moreover, multiple classes of clustered damages involving damaged bases and abasic sites, alone or in combination with frank single-strand breaks, can be quantified by converting them to double-strand breaks by chemical or enzymatic treatments. For a given size range of DNA molecules, single-molecule sizing is as or more sensitive than gel electrophoresis, and requires several orders-of-magnitude less DNA to determine damage levels.
INTRODUCTION
DNA damages induced by ionizing and ultraviolet radiation and reactive chemical species, both endogenous and exogenous, are believed to be major causes of mutations and cancer induction. Quantifying such damages and the ability of cells to repair them is critical for understanding fundamental processes in cancer biology and developing protocols for limiting exposures for individuals and various human populations. Many types of DNA damages are efficiently repaired by either base excision or nucleotide excision repair pathways, which use the complementary sequence of the opposing strand of duplex DNA to restore a damaged region and the adjoining regions of the damaged strand (Friedberg et al., 1995). Such repair mechanisms may not function properly, however, if both strands of DNA have been damaged. Thus, bistranded clustered damages may be difficult to repair, or difficult to repair accurately, and thus of particular importance in eliciting the long-term effects of carcinogens. Double-strand breaks have long been considered as particularly difficult to repair, and thus critical lesions induced by ionizing radiation (von Sonntag, 1987). A double-strand break can be considered to be a cluster of single-strand breaks with at least one single-strand break on each strand within a short distance, as indicated by Fig. 1 B. The existence of other classes of clustered lesions involving oxidized bases and abasic sites as well as single-strand breaks has been postulated (Goodhead, 1994; Ward, 1994) and, more recently, observed both for DNA irradiated in vivo and isolated from irradiated cells (Sutherland et al., 2000a,b). The induction of damage clusters, including double-strand breaks, is found to be a linear function of radiation dose (Sutherland et al., 2000b), presumably reflecting the fact that a single radiation event, such as an x-ray or a charged particle, is energetically capable of producing multiple reactive species, such as hydroxyl radicals, within a small volume of space. Thus, clustered damages are presumably produced by low doses of ionizing radiation such as those to which human populations may be exposed.
FIGURE 1.

Schematic diagram of the induction of frank double-strand breaks and other bilateral clustered damages in DNA. Ionizing radiation, such as γ-rays, generates hydroxyl radicals and other reactive species that attack undamaged DNA (A), forming frank single-strand breaks (— —) and damaged bases (— ▪ —), as shown in (B). Multiple closely-spaced breaks involving opposing strands result in double-strand breaks (shown within a circle) and spawn two smaller DNA molecules (C). Subsequent treatments with a damage-specific enzyme result in the generation of additional single-strand breaks (D), yielding additional double-strand breaks if the damaged bases are closely spaced or if a damaged base is located near a single-strand break on the opposing strand. These double-strand breaks result in the generation of additional, smaller double-stranded DNA fragments (E). Isolated single-strand breaks or damaged bases, which are actually more numerous than clustered damages, do not generate increased numbers of smaller double-stranded DNA molecules.
Understanding of the biological significance of clustered damages in DNA requires the ability to quantify their formation and repair. Such methods should be accurate, able to quantify low levels of damage, require only small quantities of DNA, and be fast and cost-effective. Recent demonstrations of the existence of damage clusters other than double-strand breaks have been based on the reduction of the average length of the DNA molecules treated with lesion-specific repair enzymes that can convert clusters into double-strand breaks (Sutherland et al., 2000a,b), as indicated by the diagrams on the right side of Fig. 1. Nonenzymatic chemical reactions can also perform this function (Georgakilas et al., 2002). In these studies, average lengths of the DNA molecules in a sample were determined by separating the DNA as a function of molecular size by gel electrophoresis (Freeman et al., 1986; Sutherland et al., 2001b).
Here, we describe an approach for determining the average size of the DNA molecules in a sample in which individual fluorochrome-labeled duplex DNA molecules pass sequentially through a laser beam. The resulting flash of fluorescence is recorded and used to determine the size of each molecule. We confirm that the amplitudes of the laser-induced flashes are a linear function of the size of the DNA and demonstrate that molecules of different sizes can be counted with equal efficiencies. We find that single-molecule sizing is capable of detecting lower levels of damage compared to gel electrophoresis for a given size of DNA in the starting population. Moreover, single-molecule sizing offers huge potential reductions in the quantity of DNA required for a determination of damage clusters.
THEORY
The theory underlying quantitation of DNA damage from the average lengths of heterogeneous DNA populations was originally developed for DNA separation by centrifugation, and later adapted for gel electrophoresis (Freeman et al., 1986; Sutherland et al., 1980, 2001b). For both approaches, the analysis is complicated by the nonlinear relationship between the size of DNA molecules and the speed they move under the influence of a centripetal or electrical force. The linear relationship between fluorescence and molecular size required in single-molecule sizing simplifies the theory. The theory presented below is equally applicable to the measurement of both double- and single-strand breaks in DNA, although the experiments described in this paper deal only with double-strand breaks.
Average length analysis by single-molecule sizing
Consider a population of N duplex DNA molecules in which the ith molecule contains Li basepairs. The average length of the molecules in the population is given by the expression in Eq. 1. Formally, this is called the number average length of the molecules in the population to distinguish it from certain other types of averages that are used to describe the distribution of lengths in a population of polymers (Cantor and Schimmel, 1980). The summation in the numerator of the expression on the right side of Eq. 1 is just the total number of basepairs in the sample. The “0” subscript indicates that this is an initial or untreated sample, in contrast to the sample described below.
 |
(1) |
Suppose that the sample is treated in such a way that M double-strand breaks are introduced into the population of the original N molecules. These breaks could be randomly distributed, or they could be produced at specific sites in the molecules. Double-strand breaks produced by ionizing radiation are, to some approximation, randomly distributed, whereas the strand breaks produced by restriction endonucleases are located at specific sites. Regardless of the distribution, each strand break results in the formation of two (smaller) molecules for each molecule that existed before the break occurred. Thus the treated population consists of N + M molecules, and the average length of the molecules in this population is shown in Eq. 2, where the “+” subscript indicates that this is a treated sample.
 |
(2) |
The number of molecules is increased by the induction of strand breaks, but the number of basepairs is unchanged. Thus, the summation in the numerator of Eq. 2 is equal to the corresponding expression in Eq. 1, and both can be replaced by Nbp, the number of basepairs in the population. Subtracting the reciprocal of the average length of the molecules in the initial or untreated sample from the reciprocal of the average length of the molecules in the treated sample gives Φ, the number of strand breaks divided by the total number of basepairs in the sample, as shown in Eq. 3.
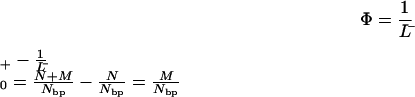 |
(3) |
If the lengths of the molecules in the population are measured in units of basepairs, then Φ is the probability of a strand break per basepair. However, the lengths of the DNA molecules in the population can also be specified in some larger unit, such as kilobasepairs (kbp), megabasepairs (Mbp), or gigabasepairs (Gbp), in which case, Φ is the number of breaks per kbp, Mbp, or Gbp. In this case, Φ can be described as the frequency of breaks induced by the treatment. Thus, we can determine the frequency (breaks/Mbp) or probability per basepair of producing a double-strand break by measuring the average length of the molecules in a population before and after some treatment or combination of treatments that produces strand breaks.
The average length of the molecules in a population can be obtained by measuring the average length of the molecules in a representative sample because both the numerator and denominator of Eq. 1 and Eq. 2 scale as the volume of the sample. That is, we need not examine all of the molecules in a population, but only the molecules in a representative sample derived from that population. It follows that the determination of average lengths is insensitive to the volume of the sample chosen, so small differences in the volume of sample should have no significant effect on the determination of the frequency of breaks.
Determining number average lengths by single-molecule fluorescence
Suppose the DNA molecules in a sample are labeled with a fluorochrome such that the number of labels are proportional to the number of basepairs in the molecule, but independent of base composition (the AT/GC ratio) or base sequence. When such a molecule passes through a beam of light of a wavelength that stimulates the fluorescence of the label, typically generated by a laser, the resulting fluorescence is proportional to the number of basepairs in the molecule. Assuming that the efficiency with which fluorescence is measured is constant in time and independent of the size of the DNA molecule, the arguments of the summations appearing in the above equations can be replaced by the ratio fi/fbp, where fi is a measure of the fluorescence generated by passage of the ith molecule through the laser beam and fbp is the average fluorescence per basepair obtained under identical experimental conditions. Thus, the average length of a population of DNA molecules can be determined from an experiment in which the fluorescence of a population of individual DNA molecules is examined as indicated in Eq. 4, where Ne is the number of molecules examined in a particular experiment. The fluorescence per basepair, fbp, thus serves as a calibration factor.
 |
(4) |
Resolution
The theory presented above presumes a one-to-one correspondence between DNA size and the signal returned by the measuring system. In practice, this is never the case because of the finite resolution of the measuring system. That is, a sample consisting of a homogeneous sample of DNA molecules returns a finite distribution of signals. The effects of the finite width of bands on gels on the determination of average lengths was discussed previously (Sutherland et al., 2001b). The finite widths of the distributions of homogeneous populations in a single-molecule histogram will not affect the determination of the average length of that population so long as the distribution is symmetrical, with larger and smaller lengths being reported with equal frequency. However experimental data will always have some degree of skew in such size distributions. Improving resolution can reduce effects of such asymmetries on determinations of average lengths and also reduce the number of molecules that must be detected to obtain a given degree of statistical accuracy.
MATERIALS AND METHODS
DNA preparation, irradiation, and labeling
DNA from bacteriophage T7 was prepared by phenol extraction as described previously (Sutherland et al., 2000a). T7 DNA (50 μg/ml) in 20 mM NaPO4 buffer (pH 7.4) received doses from 0.1 to 5 Gy of 512 keV γ-rays from a 137Cs source in the Brookhaven Controlled Environment Radiation Facility. All stock solutions were stored at −20°C. BglI (New England Biolabs, Beverly, MA) restriction digests of T7 DNA were prepared according to the manufacturer's instructions; the restriction digestion was terminated by the addition of EDTA to 10 mM and heating to 65°C for 20 min. BglI generates three fragments of T7 DNA with lengths of ∼22.5, 13.5, and 4 kbp. The completeness of digestion was verified by gel electrophoresis.
For experiments in which both double-strand breaks and clustered oxidized purine sites were quantified, bacteriophage T7 DNA in 20 mM potassium phosphate buffer, pH 7.4, was irradiated in plastic tubes, then equilibrated with 70 mM Hepes-KOH, pH 7.6/100 mM KCl/1 mM EDTA. The DNA was subsequently incubated at 37°C for 60 min with a saturating amount of Deinococcus radiodurans Fpg protein. The enzyme was inactivated by adding proteinase K and EDTA to final concentrations of 1.33 mg/ml and 0.1 M respectively, and incubating at 37°C overnight.
YOYO-1 (CAS# 143413-85-8, Molecular Probes, Eugene, OR), an intercalating dye (1 mM solution in DMSO), was stored at −20°C. A 10−6M solution of dye in 40 mM Tris-acetate, 2 mM EDTA, pH 8.0 was filtered through a 0.2 μm filter. DNA samples were labeled with YOYO-1 at a ratio of five DNA basepairs to one dye molecule with concentrations determined spectrophotometrically. These DNA/dye samples were incubated at least 1 h at room temperature in the dark and then were diluted further to a final DNA concentration of 0.3–0.8 pM. These samples were stored at 4°C and used within one week.
Single DNA molecule sizing system
Microfluidic device
Microfabricated devices (chips) for DNA sizing experiments were described previously (Chou et al., 1999; Fu et al., 1999). The channel on the chip is 100 μm wide and 3 μm deep, narrowing to a width of 5 μm near the center. The end of each channel is connected to a cylindrical reservoir 2 mm in diameter. Chips were treated with a solution of diluted HCl (0.02% in water; Chou et al., 1999; Fu et al., 1999), causing the naturally hydrophobic surface of the channel to became hydrophilic. Before and after each experiment the device was cleaned with isopropyl alcohol filtered through a 0.2-μm filter, then dried. One chip can be reused many times, and data for this paper were taken with a single chip. Before the laser sizing experiment, the bottom of the chip was sealed with a coverslip (22 × 22 mm, #0, Erie Scientific, Portsmouth, NH). A small top coverslip covered the entire channel but only partially covered the reservoirs. Before use, all coverslips were cleaned by double-distilled water and dried. The solution of stained DNA molecules entered the channel by capillary action, which determined the flow rate through the channel.
Optical system
Fig. 2 shows a schematic diagram of the optical system. Fluorescence excitation at 488 nm is provided by an air-cooled argon ion laser (model LS300T, American Laser Corporation, Salt Lake City, Utah). Output power was 20 mW, which varied less than 2% over 5 h. At this power, the signal-to-noise ratio was ≈50 for intact T7 molecules and up to this level the amplitude of dye fluorescence was a linear function of laser power (data not shown). A Plan-Apochromat 40 × 0.75 microscope objective (Carl Zeiss, Thornwood, NY) was used to focus the laser beam and collect the emitted fluorescence. The e−1 beam diameter was adjusted to 60 μ with an auxiliary lens. The dimensions across the beam were verified by beam occultation with a razor blade affixed to a precision translation stage. This size of the laser beam was large enough to provide uniform illumination of the 5-μm wide channel and completely cover the biggest molecules in our samples. The contour length of unlabeled DNA molecules is 0.34 L (μm), where L is the size of the molecule in kbp. Intercalation by YOYO-1 at 1 dye/5 bp increases the contour length of T7 DNA from 13.6 to 16.5 μm. The actual spatial extent of the DNA is less than this extreme dimension due to folding of the semi-rigid, rod-like molecule. Measurements of the laser beam diameters were repeated at different distances from the objective near its focal plane and demonstrated that the laser beam was essentially uniform over a depth of 10 μ, thus providing uniform illumination of a 3-μm deep channel.
FIGURE 2.

Schematic diagram of the optical system. The same microscope objective lens focuses the laser beam on the sample and collects the fluorescence light. The laser beam that excites the fluorescence is separated from the fluorescence by a dichroic mirror and filters, as described in the text.
A 488-nm holographic SuperNotch-Plus filter (Kaiser Optical, Ann Arbor, MI), a band-pass filter (model HQ535/50, Chroma Technology Corp., Brattleboro, VT) and a dichroic mirror (model 505DRLP, Omega Optical, Brattleboro, VT) were used to separate fluorescence from the scattered laser light. Fluorescence was focused by a 100-mm focal length lens onto the 400-μm pinhole immediately in front of a thermoelectrically cooled photo-multiplier tube (model C31034, BURLE, Lancaster, PA) operated at a cathode potential of −1400 V. The first-stage preamplifier (model 70710, Oriel Instruments, Stratford, CT) converted photocurrent to voltage and provided a gain of 106 V/A. A second-stage low noise amplifier (model SR560, Stanford Research System, Sunnyvale, CA) provided additional voltage gain of 50 and acted as a band-pass filter with response from 0.3 to 30 Hz. The output voltage from this amplifier was digitized at 500 Hz by a Multifunction PCI I/O Board (model number PCI-MIO-16XE-50, National Instruments, Austin, TX) and recorded under the control of custom software written in Visual Basic (Microsoft Corp., Redmond, WA) running on a computer, with an Intel (Santa Clara, CA) Pentium processor under Microsoft NT-4.0. Fluorescence vs. time data sets were analyzed offline using Origin (Version 6, OriginLab, Northampton, MA) or with a Visual Basic program incorporating similar functionality.
RESULTS
Restriction digests
Bacteriophage T7 contains 39,937 bp (Dunn and Studier 1983). Terminal digestion of T7 DNA with BglI produces three fragments with 22,415, 13,523, and 3999 basepairs, respectively, which should be generated in equal numbers. Fluorescence signals from such a digest of T7 DNA that had been mixed with an approximately equal quantity of intact T7 DNA are shown in Fig. 3. The flashes of fluorescence fall into four distinct groups corresponding to the intact T7 plus the three sizes of fragments which are apparent in the histogram derived from the complete data set (Fig. 4).
FIGURE 3.

Fluorescence signal (in volts) from DNA molecules detected as they pass through the flow cell. The height of the pulse is proportional to the contour length (size) of the DNA molecule. The sample was a BglI digest of bacteriophage T7 DNA to which was added an approximately equal mass of intact T7 DNA. The lower panel shows 8 s of data from an experiment that lasted 15 min. Four classes of pulse amplitudes are resolved, as expected for this sample. The upper panel shows 1 s of the trace on an expanded time scale and displays individual values of the digitized fluorescence. One light flash from each of the four classes of molecules is present. The DNA flow rate and size of the focused laser beam resulted in a transit time of ∼50 ms for each DNA molecule. Fluorescence data are recorded every 2 ms, so a data set lasting 15 min contains 450,000 values.
FIGURE 4.

Histogram of the distribution of the fluorescence flashes of a mixture of intact T7 DNA and a terminal BglI digest of T7 DNA. The binning increment is 120 mV. A data set consisting of a fluorescence-time trace, a portion of which is shown Fig. 3, was analyzed to produce this histogram.
Linearity of response
If Eq. 4 is to be equivalent to Eq. 1 or Eq. 2 there must be a linear relationship between the intensity of the flash of light generated when a fluorochrome-labeled DNA molecule passes through the laser beam and the size (contour length) of the molecule, i.e., fi = fbp Li. Restriction digests of viral DNA are an excellent standard for testing this requirement because the lengths of individual fragments are known from the sequence of the DNA. Fig. 5 shows that the centroids of the four peaks in the histogram are linear with the known sizes of the restriction fragments and the intact T7 DNA, consistent with previous reports (Castro et al., 1993; Chou et al., 1999; Goodwin et al., 1993; Huang et al., 1999). The slope of this line gives fbp. The data fit a straight line (R2 = 0.9996), with fbp = 116 μV/bp. The 197 mV zero-offset of this line represents the level of background signal, which is subtracted from all experimental values. The value of the calibration factor, fbp, is dependent on experimental conditions, including the alignment of the sample cell with respect to the focused laser beam.
FIGURE 5.

Mean position (fluorescence) of each of the four peaks in Fig. 4 plotted vs. known lengths of the BglI restriction fragments of T7 DNA. The points fall very close to a straight line (R2 = 0.9993), the slope of which (116 ± 3 mV/kbp) is the calibration constant, fbp for this data set. The intersection of this line and the fluorescence axis, f0, is 197 ± 0.07 mV, which corresponds to the background level of fluorescence observed in the fluorescence trace shown in Fig. 3. The line shown in the Figure can be written as f(L) = f0 + fbp L.
Detection efficiency independent of DNA size
A second condition necessary for determination of average lengths, but not for the determination of unknown lengths, is that DNA molecules of different sizes are recorded with equal efficiencies. Restriction digests of viral DNAs are also an excellent system to test this requirement because in a terminal restriction digest, the number of molecules of each size is equal. The histograms shown in Fig. 4 and data shown in Table 1 demonstrate that this condition is fulfilled over this size range. The average number of molecules detected for the three size classes is 371. Assuming random variations in these frequencies, we estimate the uncertainty as the square root of this number, which is 19.3, and all three experimental values are within the range 371 ± 19.3.
TABLE 1.
Number of flashes recorded for each size class for the BglI fragments in the histogram in Fig. 4
| DNA size (bp) | 3,999 | 13,523 | 22,415 | ∑ = 39,937 |
| Number of flashes | 375 | 379 | 357 | ∑ = 1111 |
Computation of number average lengths of restriction fragments
The application of Eq. 4 to the set of flashes with amplitudes less than 3.5 volts in the data set shown in part in Fig. 3 and used to generate the histogram used in Fig. 4 returns an average length for the BglI fragments of T7 DNA of 13.8 kbp, in agreement with the value of 13.3 kbp, the result expected for any process that divides each T7 DNA into three pieces. This is an alternate verification that the fluorescence amplitudes are linear with DNA size and that molecules of different sizes are counted with equal efficiencies.
Radiation-induced frank double-strand breaks
Histograms for single molecules of samples of intact and irradiated T7 DNA are shown in Fig. 6. Most of the T7 DNA molecules appear in the major peak for both samples. Those molecules that have suffered a radiation-induced double-strand break appear in the histogram of the irradiated sample as a uniform distribution with amplitudes less than intact T7 DNA, i.e., extending to the left of the intact band. The fluorescence per basepair, fbp, for this data set was determined from the centroid of the peak of the intact T7 DNA. The average length of the molecules in the irradiated sample computed using Eq. 4 is 37.14 kbp. This experiment was repeated for other samples of DNA that were exposed to a range of γ-ray doses from 0 to 5 Gy, with the results shown in Table 2. These average lengths were used to compute the frequency of double-strand breaks using Eq. 3, with the results shown in Table 2 and Fig. 7. Note that the calculation of the average length of the population of DNA molecules proceeds directly from the fluorescence data (Fig. 3), without generating histograms as an intermediary step. The average length of the unirradiated sample was determined by single-molecule sizing to be 39.895 kbp, less than the value of 39.937 determined from sequencing. This small (0.1%) difference is within the range of experimental uncertainty.
FIGURE 6.

Histograms of the distribution of single-molecule fluorescence of T7 DNA that was not irradiated (lower panel) or irradiated with 1 Gy of γ-rays (upper panel), before single-molecule sizing analysis. Because only a small fraction of the initial population of T7 DNA molecules were broken in the irradiated sample, the distribution of smaller molecules (to the left of the main peak) is approximately uniform as a function of their size. Values of peak fluorescence less than the level of background noise are not plotted.
TABLE 2.
Average size of DNA populations and double-strand break frequencies after γ-irradiation
| Dose (Gy) | Average Size (kbp) | DSB/Mbp |
|---|---|---|
| 0 | 39.895 | — |
| 0.1 | 39.365 | 0.337 |
| 0.2 | 37.75 | 1.424 |
| 0.5 | 37.95 | 1.285 |
| 1 | 37.14 | 1.859 |
| 1.5 | 34.82 | 3.653 |
| 2 | 34.73 | 3.728 |
| 2.5 | 33.567 | 4.725 |
| 5 | 27.24 | 11.645 |
FIGURE 7.

Dose response curve for frank double-strand break production in T7 DNA by γ-rays from a 137Cs source. In the upper panel, the frequency of breaks determined by single-molecule sizing (▴) and by gel electrophoresis (□, Chen and Sutherland, 1989 and ⋄, Sutherland et al., 2000b) are plotted vs. dose, with linear scales used for each axis. The straight line was fit to the single-molecule data. In the lower panel, the single-molecule data and gel-electrophoresis data are plotted on double logarithmic axes so that the electrophoresis data obtained at higher doses can be included. The solid line was fit to electrophoresis data (Sutherland et al., 2000b) over the dose range of 1–50 Gy and extrapolated into the range covered by the single-molecule data.
Other clustered DNA damages
Double-strand breaks are known to be an important form of DNA damage because they are difficult to repair or to repair accurately. Recently, however, other classes of damage clusters have been postulated (Goodhead, 1994; Ward, 1994) and measured experimentally (Sutherland et al., 2000a,b). Such clustered damages are presumed to have biological effectiveness comparable to double-strand breaks and their biological activities are a matter of intense interest (Georgakilas et al., 2002; Sutherland et al., 2001a; Sutherland et al., 2002a,b). Thus, any assay to be used for studying radiation-induced DNA damage should be capable of quantifying both double-strand breaks and the many other classes of clustered damages. The detection of multiply-damaged sites in addition to double-strand breaks is demonstrated in Fig. 8. The induction of frank double-strand breaks by the gamma rays reduces the fraction of the events in the main T7 peak and produces a continuum of smaller molecules, which are not present in the unirradiated DNA. The induction of additional double-strand breaks by treatment of the irradiated DNA with Fpg protein, which makes single-strand breaks at certain oxidized purines (Senturker et al., 1999), further reduces the fraction of events corresponding to intact T7 DNA molecules and increases the fraction of the DNA in the region of smaller molecules. The frequency of frank, radiation-induced double-strand breaks is computed by comparing the differences in the average lengths of irradiated and unirradiated samples using Eq. 3. The frequencies of the various classes of other clustered damages are obtained by comparing the average lengths of a sample that has been treated with an enzyme or other agent that makes strand breaks at a specific class of damage, with the average length of the DNA that received the same dose of radiation but has not been further treated, minus the frequency of breaks introduced by a similar treatment of the unirradiated DNA (if any). The data shown in Fig. 8 include only one such agent, but many others with different lesion specificities are available (Georgakilas et al., 2002; Sutherland et al., 2000b; Wallace 1998) while still others may appear in the future.
FIGURE 8.

Histograms for DNA from bacteriophage T7 that has not been irradiated (⋄), irradiated with a dose of 1 Gy of γ-rays from a 137Cs source (▵) and irradiated and then treated with D. radiodurans Fpg protein that makes single-strand breaks at certain oxidized purines (□). To facilitate comparisons, the histograms are plotted as a function of molecular length and have been normalized by dividing by the total number of peaks recorded for each histogram so that they have the same area and the vertical axis becomes the fraction of events recorded per binning interval for each sample. The right scale represents the events for the main plot, while the left scale is expanded by a factor of five and offset by 20% to facilitate comparisons of the low number of events recorded for smaller molecules. The data show that radiation alone decreases the fraction of the events corresponding to intact T7 DNA molecules and increases the events corresponding to fragments of T7 DNA. Subsequent treatment with Fpg protein produces a further drop in the fraction of intact molecules and an increase in the fragments. In the case of the untreated DNA and the irradiated-only sample, the distribution of samples is, within experimental uncertainty, independent of fragment length, whereas for enzyme-treated sample, the smaller fragments may occur at a slightly higher frequency, reflecting an observable rate of multiple breaks in some of the intact molecules.
DISCUSSION
Our objectives are to demonstrate that single-molecule sizing can measure the average length of populations of DNA molecules, and hence the induction of double-strand breaks by agents such as ionizing radiation, and to determine basic performance characteristics of this approach to quantifying DNA damage. Restriction digests are useful in demonstrating linearity of response with DNA size and detection efficiency independent of DNA size. The theory of average length analysis is equally applicable to breaks distributed randomly along the length of DNA molecules and to scissions introduced by restriction endonucleases, which are formed at the same place in each molecule. We also investigated the use of single-molecule sizing for determining the average lengths of populations subjected to various levels of γ-rays that generate double-strand breaks throughout the length of the DNA, and evaluated the advantages and limitations of single-molecule sizing compared to alternate approaches such as gel electrophoresis.
Calibration of fluorescence intensity
The calibration factor, fbp, introduced in Eq. 4 is sensitive to many experimental details and hence must be determined routinely for experimental samples. For that reason, we used samples that are inherently self-calibrating, in that they all contain at least one DNA species of known length. Although not required for demonstrating the feasibility of the approach to quantifying DNA damage, the ability to calibrate the fluorescence sensitivity independently of the experimental sample is desirable. One approach is to mix calibrating molecules of known length with the experimental molecules containing strand breaks. The data in Fig. 4 demonstrate this approach; intact T7 DNA was added to the restriction digest and did not to interfere with the determination of their average molecular lengths.
The linear fit to the data relating fluorescence amplitudes and DNA size shown in Fig. 5 results in a zero length intercept of ≈200 mV. When only one size standard is available, as is the case for the data in Fig. 6 and Fig. 8, the zero length intercept can be determined from the baseline level of fluorescence, which in Fig. 3 is ≈200 mV.
Radiation-induced double-strand breaks
Each absorbed γ-ray deposits sufficient energy in a small volume to generate numerous hydroxyl radicals and other reactive species. These reactive species primarily produce isolated lesions in DNA that may lead to single-strand breaks. Sometimes, however, multiple damages result within a short segment of DNA. When the reactive species responsible for the generation of a double-strand break or other damage cluster result from the same radiation event, the dose response function should be a linear function of dose. However, if the reactive species that generate a multiply-damaged site arise from two radiation events, the response should scale as the square of the dose.
The data presented in Fig. 7 are fit by a linear function with a slope of 2.19 DSB Mbp−1 Gy−1. Data from two previous studies of double-strand break induction in T7 DNA under similar solution conditions, but using gel electrophoresis give similar results (Chen and Sutherland, 1989; Sutherland et al., 2000b). One found a linear response with a 13% greater slope (2.48 vs. 2.19; Sutherland et al., 2000b) whereas the other reported a slightly lower linear coefficient (2.12 DSB Mbp−1 Gy−1), but with a small quadratic component (0.128 DSB Mbp−1 Gy−2). The straight line shown in the lower part of Fig. 7 was obtained by fitting a straight line to the log–log data set for the recent electrophoretic data (Sutherland et al., 2000b), which is equivalent to fitting an equation of the form Φ = αDn to the linear data. The exponent, n, is found to be 1.00 and the intercept is 0.38, which corresponds to α = 2.39 DSB Mbp−1 Gy−1, in good agreement with the slopes obtained from the linear fittings. All of the data presented in Fig. 7 are close to this simple linear function with a slope in the range of 2–2.5 DSB Mbp−1 Gy−1 for doses from 0.1 to 50 Gy. Presumably there is a quadratic component at higher doses, as suggested by the early electrophoresis data (Chen and Sutherland, 1989), but it will have essentially no impact on the lower end of the dose range.
The formation of damage clusters by single radiation events means that such damages will occur even at the low radiation doses encountered in the natural environment and various occupational situations, and emphasizes the importance of quantifying such damages at these low doses. Comparison of our single-molecule sizing results with gel electrophoresis data indicate that for a given size of undamaged DNA molecules, single-molecule sizing is able to quantify damage levels induced by lower doses of radiation. For example, in the case of T7 DNA (40 kbp), the lowest dose reported for the gel electrophoresis measurements was 1 Gy (Sutherland et al., 2000b). In contrast, single-molecule sizing damage levels can extend to 0.1 Gy, as shown in Table 2 and Fig. 7. This corresponds to detecting the breakage of ∼1% of the intact molecules in the population. At the present level of development, gel electrophoresis can quantify lower levels of DNA damage than can single-molecule sizing because much larger molecules can be analyzed using pulsed-field methods. However, the results presented here indicate that for equal sizes of the starting material, single-molecule sizing is more sensitive.
Quantity of DNA required
The greatest potential advantage in using single-molecule sizing to quantify DNA damage is a huge reduction in the quantity of DNA required to determine the average lengths of a representative population. The minimum number of molecules that must be observed will depend on the resolution of the sizing system and the statistical accuracy required for a particular application. An estimate of the potential for reducing DNA quantity can be obtained from the data presented in Table 1, which demonstrates that a reasonable estimate of the average length of the molecules in a population can be measured by observing as few as a thousand DNA molecules. Assuming an average DNA size of 105 bp per molecule, this corresponds to ∼100 femtograms of DNA, compared to ∼10 ng of DNA required for a determination of the average length of a DNA sample by gel electrophoresis. This approximately 100,000-fold reduction (105) in the quantity of DNA required for an assay suggests that it may eventually be possible to determine damage levels in the genomes of single cells. However, realization of this objective must await advances in sample handling, as the total quantity of DNA loaded into the sample reservoirs presently far exceeds the quantity required to pass through the laser beam.
Double-strand breaks with staggered ends
The theory presented above assumes that all of the double-strand breaks induced by either restriction enzymes or resulting directly or indirectly from radiation damage are blunt-ended. In reality, all classes of double-strand breaks may have staggered ends, resulting from two single-strand breaks that are not exactly opposite one another. The unpaired bases in such staggered ends may not bind fluorescent dyes as efficiently as normal basepairs, hence leading to an underestimate of the total bases in the sample, i.e., the numerator of Eq. 4, and, thus, of the average length of the populations. This effect will tend to overestimate the frequency of DNA damages. The maximum length of the unpaired single-stranded DNA at a break is on the order of one helical turn of double-stranded DNA, i.e., 10 bases. The stability of noncovalently linked multimers of phage λ DNA suggests that if the two single-strand breaks that make up a double-strand break are separated by much more than 10 basepairs, the DNA will not separate into two molecules, but the presence of damage sites, dye molecules, and the sequence of the intervening bases could modify this value. However, the minimum size of DNA that can be detected at present is about 1,000 basepairs. Thus, for the smallest observable molecules in a DNA population, the error in the determination of average lengths due to staggered ends of double-strand breaks is only about 1% and this decreases as one moves across a DNA distribution in the direction of increasing molecular size, reaching only 0.01% for molecules 105-basepairs long. The effect also becomes smaller at low doses. Therefore, the magnitude of this effect makes it insignificant. A more important problem is the limited ability to resolve small DNA molecules, which is discussed below.
Minimum resolvable DNA size and radiation dose limits
Random introduction of strand breaks in a population of DNA molecules of a single size will generate a new population in which a certain fraction of the population is undamaged, and the remainder is converted to smaller sizes. The distribution of sizes can be predicted by Poisson statistics. Results derived from Poisson analysis also can be applied to populations that initially are heterogeneous in length because such populations can be considered a linear sum of homogeneous populations (Sutherland, 1999). For sufficiently low doses, most of the molecules in the original population will remain intact, and all daughter molecules will be derived from a parent that has been broken exactly once. That is, all Poisson coefficients greater than 1 can be ignored. In this situation, the daughter fragments will be uniformly distributed across all lengths less than that of the unbroken parents. This is the pattern present in the histogram shown in the upper panel in Fig. 6 and in Fig. 8.
Below a certain critical molecular size the flash produced by a DNA molecule passing through the laser beam cannot be distinguished from noise in the detection system (see Fig. 3) and hence cannot be detected by single-molecule fluorescence. (Note that the limit of detection is determined by the level of random variations in the baseline level and not by the average value of the displacement from zero. These variations are due to electronic noise in the detection circuitry plus shot noise in the signal due to a low level of scattered incident light reaching the detector.) In the low-dose limit described above, we could correct for the these “missing” molecules by assuming the number of molecules-per-unit flash size is constant down to zero size. This correction, which was not applied to the data presented in Table 2 and Fig. 7, will increase the number of lesions calculated from Eq. 3 by ∼17%. For higher doses in which a significant number of parent molecules are broken more than once, this simple correction is no longer adequate because the probability of unresolved small molecules exceeds the number of larger daughter fragments. Thus, determination of DNA damage by single-molecule sizing is optimally suited to situations in which the fraction of damaged molecules is low. As the size of the undamaged population of DNA increases, the flashes will become larger, and hence a smaller proportion of damaged molecules will be unresolvable from noise, so the situation discussed here should become less important. Note that determination of average length by gel electrophoresis is also limited in resolving small molecules (Sutherland et al., 2001b).
Maximum DNA concentrations
Two DNA molecules passing through the laser beam at the same time will generate a compound flash that may be incorrectly scored, hence tending to overestimate the average length of the population. Such errors can be controlled by monitoring the molecular concentration of the sample being studied. Assuming the passage of a DNA molecule through the laser beam is a random event, the probability that the flash generated by molecule n + 1 will overlap in time with the flashes of the other n molecules in the sample is given by Eq. 5, where Δt is the transit time of a DNA molecule through the laser beam, t is the duration of data collection, and PP is the Poisson probability that these will be simultaneous events. For the data set from which Fig. 5 was obtained, n = 1112, t = 900 s, and Δt = 50 ms FWHM, we predict a coincidence frequency of 6%, which is at the high end of the acceptable range. An alternate approach is to use computationally intensive pulse-shape fitting software that can resolve partially overlapping flashes (Petty et al., 1995), and should also improve resolution:
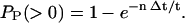 |
(5) |
Laser polarization
Recent reports indicates that the signal obtained in a single-molecule sizing experiment may depend on the polarization of the incident laser beam and that polarization effects may result in nonlinearity of fluorescence due to a greater degree of orientation of longer DNA molecules (Agronskaia et al., 1999; Werner et al., 2000). We have observed that switching the polarization of our laser beam from perpendicular to parallel with respect to the direction of flow decreases the signal response (fbp). However, the linearity of response demonstrated in Fig. 5 indicates that polarization effects are not significant for our experimental arrangement over the range of size of DNA molecules that we studied. Similar linearity of response has been demonstrated for single-molecule sizing using sheath-flow DNA transport for molecules up to 350 kbp (Huang et al., 1999). Polarization of the excitation beam perpendicular to the direction of flow, which is the configuration we use, seems to produce a linear response, whereas polarization parallel to the flow can result in nonlinearities (Werner et al., 2000). However, the use of magic-angle polarization can presumably eliminate such polarization artifacts (Agronskaia et al., 1999).
Future directions
We have demonstrated that single-molecule sizing by laser-excited fluorescence can be used to determine the average lengths of heterogeneous populations of DNA molecules as they flow through narrow channels, and thus can be used to quantify multiply-damaged sites in DNA inflicted by ionizing radiation, and presumably other carcinogens. Our results indicate that this approach can detect lower levels of damage than can average length analysis based on gel electrophoresis when similar sizes of DNA are analyzed by both methods. To reach or exceed the absolute limits of sensitivity achievable with electrophoresis, it will be necessary to use much larger DNA molecules. Our results also indicate that single-molecule sizing should be able to record damage levels using vastly smaller quantities of DNA than is possible with gel electrophoresis. Realizing this potential will require significant improvements in DNA sample handling so that the number of molecules in the total sample are reduced to the levels that are actually required to determine the average length of the population. The widths of the peaks shown in Fig. 4 increase as the size of the molecules increase, but at a less rapid rate. Thus, resolution actually improves with increasing DNA size. Factors responsible for the widths of these peaks need to be analyzed and experimental protocols modified to improve resolution over the entire size range.
Acknowledgments
We thank Dr. Megumi Nada for samples of irradiated T7 DNA treated with the D. radiodurans Fpg protein, Dr. J. Laval (Centre National de la Recherche Scientifique, Institute Gustave Roussy) for providing this protein, and Ms. Paula Bennett for assistance with the samples of DNA used in this report.
This work was supported by grant CA88335 from the Innovative Technologies for the Molecular Analysis of Cancer program of the National Cancer Institute, National Institutes of Health, to J.C.S., and grants from the Low Dose Radiation Research Program of the Office of Biological and Environmental Research, United States Department of Energy and the National Cancer Institute (CA 86897), to B.M.S. Brookhaven National Laboratory is operated under contract with the United States Department of Energy.
References
- Agronskaia, A., J. M. Schins, B. G. d. Grooth, and J. Greve. 1999. Polarization effects in flow cytometric DNA sizing. Applied Optics. 38:714–719. [DOI] [PubMed] [Google Scholar]
- Cantor, C. R., and P. R. Schimmel. 1980. Biophysical Chemistry. W. H.Freeman & Company, New York.
- Castro, A., F. R. Fairfield, and E. B. Shera. 1993. Fluorescence detection and size measurement of single DNA molecules. Anal. Chem. 65:849–852. [Google Scholar]
- Chen, C.-Z., and J. C. Sutherland. 1989. Gel electrophoresis method for quantitation of gamma ray induced single- and double-strand breaks in DNA irradiated in vitro. Electrophoresis. 10:318–326. [DOI] [PubMed] [Google Scholar]
- Chou, H.-P., C. Spence, A. Scherer, and S. Quake. 1999. A microfabricated device for sizing and sorting DNA molecules. Proc. Natl. Acad. Sci. USA. 96:11–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, J. J., and F. W. Studier. 1983. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 166:477–535. [DOI] [PubMed] [Google Scholar]
- Freeman, S. E., A. D. Blackett, D. C. Monteleone, R. B. Setlow, B. M. Sutherland, and J. C. Sutherland. 1986. Quantitation of radiation-, chemical-, or enzyme-induced single strand breaks in nonradioactive DNA by alkaline gel electrophoresis: application to pyrimidine dimers. Anal. Biochem. 158:119–129. [DOI] [PubMed] [Google Scholar]
- Friedberg, E. C., G. C. Walker, and W. Siede. 1995. DNA Repair and Mutagenesis. American Society for Microbiology, Washington, DC. 698.
- Fu, A. Y., C. Spence, A. Scherer, F. Arnold, and S. R. Quake. 1999. A microfabricated fluorescence-activated cell sorter. Nature Biotech. 17:1109–1111. [DOI] [PubMed] [Google Scholar]
- Georgakilas, A. G., P. V. Bennett, and B. M. Sutherland. 2002. High efficiency detection of bistranded abasic clusters in γ-irradiated DNA by putrescine. Nucleic Acids Res. 30:2800–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodhead, D. T. 1994. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int. J. Radiat. Biol. 65:7–17. [DOI] [PubMed] [Google Scholar]
- Goodwin, P. M., M. E. Johnson, J. C. Martin, W. P. Ambrose, B. L. Morrone, J. H. Jett, and R. A. Keller. 1993. Rapid sizing of individual fluorescently stained DNA fragments by flow cytometry. Nucleic Acids Res. 21:803–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Z., J. H. Jett, and R. A. Keller. 1999. Bacteria genome fingerprinting by flow cytometry. Cytometry. 35:169–175. [DOI] [PubMed] [Google Scholar]
- Petty, J. T., M. E. Johnson, P. M. Goodwin, J. C. Martin, J. H. Jett, and R. A. Keller. 1995. Characterization of DNA size determination of small fragments by flow cytometry. Anal. Chem. 67:1755–1761. [Google Scholar]
- Senturker, S., C. Bauche, J. Laval, and M. Dizdaroglu. 1999. Substrate specificity of Deinococcus radiodurans Fpg protein. Biochemistry. 38:9435–9439. [DOI] [PubMed] [Google Scholar]
- Sutherland, B. M., P. Bennett, O. Sidorkina, and J. Laval. 2000a. Clustered damages and total lesions induced in DNA by ionizing radiation: oxidized bases and strand breaks. Biochemistry. 39:8026–8031. [DOI] [PubMed] [Google Scholar]
- Sutherland, B. M., P. V. Bennett, H. Schenk, O. Sidorkina., J. Laval, J. Trunk, D. Monteleone, and J. C. Sutherland. 2001a. Clustered DNA damages induced by high and low LET radiation, including heavy ions. Physica Medica. 17:204–206. [PubMed] [Google Scholar]
- Sutherland, B. M., P. V. Bennett, O. Sidorkina, and J. Laval. 2000b. Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc. Natl. Acad. Sci. USA. 97:103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland, B. M., P. V. Bennett, J. C. Sutherland, and J. Laval. 2002a. Clustered DNA damages induced by X-rays in human cells. Radiat. Res. 156:611–616. [DOI] [PubMed] [Google Scholar]
- Sutherland, B. M., N. Cintron-Torres, P. V. Bennett, P. Guida, M. Hada, H. Schenk, J. G. Trunk, D. C. Monteleone, J. C. Sutherland, and J. Laval. 2002b. Clustered DNA damages induced in human hematopoietic cells by low doses of ionizing radiation. Jpn. J. Radiat. Res. In press. [DOI] [PubMed]
- Sutherland, B. M., L. C. Harber, and I. E. Kochevar. 1980. Pyrimidine dimer formation and repair in human skin. Cancer Res. 40:3181–3185. [PubMed] [Google Scholar]
- Sutherland, J. C. 1999. Quantitation of DNA damage by gel electrophoresis and the method-of-moments: evaluation of accuracy using calculated lane profiles. Photochem. Photobiol. 69S:86. [Google Scholar]
- Sutherland, J. C., D. C. Monteleone, J. G. Trunk, P. V. Bennett, and B. M. Sutherland. 2001b. Quantifying DNA damage by gel electrophoresis, electronic imaging and number-average length analysis. Electrophoresis. 22:843–854. [DOI] [PubMed] [Google Scholar]
- von Sonntag, C. 1987. The Chemical Basis of Radiation Biology. Taylor and Francis, London, UK.
- Wallace, S. S. 1998. Enzymatic processing of radiation-induced free radical damage in DNA. Radiat. Res. 150:S60–S79 (Suppl.). [PubMed] [Google Scholar]
- Ward, J. F. 1994. The complexity of DNA damage: relevance to biological consequences. Int. J. Radiat. Biol. 66:427–432. [DOI] [PubMed] [Google Scholar]
- Werner, J. H., E. J. Larson, P. M. Goodwin, W. P. Ambrose, and R. A. Keller. 2000. Effects of fluorescence excitation geometry on the accuracy of DNA fragment sizing by flow cytometry. Applied Optics. 39:2831–2839. [DOI] [PubMed] [Google Scholar]