

Abstract
The release of surfactant from alveolar type II cells is essential to lower the surface tension in the lung and to facilitate inspiration. However, the factors controlling dispersal and diffusion of this hydrophobic material are still poorly understood. Here we report that release of surfactant from the fused vesicle, termed lamellar body (LB), resisted mechanical forces applied by optical tweezers: At constant trapping force, the probability to expand LB contents, i.e., to “pull” surfactant into the extracellular fluid, increased with time after LB fusion with the plasma membrane, consistent with slow fusion pore expansion in these cells. Elevations of the cytoplasmic Ca2+ concentration ([Ca2+]c) had a similar effect. Inasmuch as surfactant did not disintegrate in the extracellular space, this method permitted for the first time the determination of elastic and recoil properties of the macromolecular complex, yielding a spring constant of ∼12.5 pN/μm. This is the first functional evidence that release of hydrophobic material is mechanically impeded and occurs in an “all-or-none” fashion. This mode of release is most probably the result of cohesive forces of surfactant, combined with adhesive forces and/or retaining forces exerted by a constrictive fusion pore acting as a regulated mechanical barrier, withstanding forces up to 160 pN. In independent experiments equiaxial strain was exerted on cells without optical tweezers. Strain facilitated surfactant release from preexisting fused vesicles, consistent with the view of mechanical impediments during the release process, which can be overcome by cell strain.
INTRODUCTION
In addition to factors regulating the fusion of exocytotic vesicles with the plasma membrane, postfusion events have attracted broad scientific interest. The combination of innovative electrophysiological techniques such as patch clamp and amperometry unraveled early stages of fusion pore dynamics, leading to the awareness that release and dispersal of hydrophilic vesicle contents may be complete or partial, and that the fusion pore is a regulated structure which may play an important role during these early stages of release (de Alvarez et al., 1993; Fisher et al., 2001; Albillos et al., 1997; Breckenridge and Almers, 1987; Ales et al., 1999; Curran et al., 1993).
In contrast to these groundbreaking studies on the rapid release of hydrophilic, avidly dispersing vesicle contents, far less is known about the “fate” of hydrophobic materials such as surfactant, which is—upon formation of the fusion pore—exposed to an aqueous environment. In addition, little is known about cellular structures following initial, channel-like fusion pores, and their role for release at later times (i.e., at times exceeding the “flickering stage” of fusion pore transition).
Surfactant is a lipid-rich, lipoprotein-like material, which is stored as densely packed, circular arrangements of lipid membranes in large vesicles (1–3 μm) termed lamellar bodies (LBs). [Please note that, in this article, we shall use the term “LB” irrespective of whether its limiting membrane has been fused with the plasma membrane or not; i.e., by our definition, LBs may be in a pre- or postfusion state, and may represent both the vesicle contents or the whole vesicle.] The main function of surfactant is to lower the surface tension at the air–fluid interface and to facilitate inspiration. It is secreted from type II cells in a very slow and regulated exocytotic process (Dietl et al., 2001; Frick et al., 2001; Haller et al., 2001a). An elevation of [Ca2+]c above ≈320 nmol/l is an effective trigger for secretion (Haller et al., 1999). Previous studies in isolated type II cell preparations revealed that, depending on the mode of stimulation, the prefusion phase (i.e., the delay between stimulus and LB fusion with the plasma membrane) can last for almost 30 min, although with considerable variations (Haller et al., 1998; Frick et al., 2001). The postfusion phase (i.e., the release of surfactant into the extracellular space through the fusion pore) can even take hours (Haller et al., 2001a), which is also subject to considerable variation between individual LBs. The long postfusion phase may be related to the exocytotic machinery of the type II cell, the above-mentioned physicochemical properties of surfactant, and the composition of the extracellular fluid. It is probably the hydrophobic nature of these surfactant particles which impedes their rapid dissolution and dispersal in the bath solution; therefore, they may remain as distinct spheres for periods up to hours (see Discussion). On the basis of a modified FRAP (fluorescence recovery after photobleaching) method enabling monitoring of single fusion pore dynamics in living cells, we reported recently that fusion pores in type II cells expand slowly and discontinuously within time scales up to hours, greatly varying between individual pores (Haller et al., 2001a). Similar to other cell types (Scepek et al., 1998; Hartmann and Lindau, 1995), fusion pore expansion in type II cells is accelerated by an elevation of [Ca2+]c (Haller et al., 2001a).
We have used an experimental setup enabling the trapping of fused LBs in a laser beam, and combined this with fluorescence methods to distinguish fused from nonfused LBs (see Methods). In addition, we used a mechanical strain device which allows us to observe the process of LB release with high magnification during or after equiaxial strain of the entire cell. These techniques allow for the first time the investigation of the biophysical properties of surfactant during the postfusion phase of exocytosis, even before its release into the extracellular space. In addition, by application of mechanical forces on fused vesicle contents, mechanical barriers within the release process can be identified. Our observations indicate that the cell surface at the site of vesicle fusion does not rapidly flatten out as would be expected for a purely passive structure driven by membrane tension. Instead, it remains an active, controlled membrane infolding. Cohesive forces of surfactant in conjunction with adhesive and/or retaining forces by the fusion pore prevent its partial release for a considerable time but cause fusion-delayed release in an all-or-none fashion. This type of release is facilitated by cell strain.
METHODS
Cell preparation and storage
Type II cells were isolated from male Sprague Dawley rats (∼200 g) according to the procedure of Dobbs et al., (1986) and seeded on glass coverslips at low density (40 cells per mm2). Cells were incubated in DMEM supplemented with 24 mM NaHCO3 in a humidified 5% CO2 atmosphere at 37°C until use. During the experiments, the cells were kept in a perfusion chamber on the stage of an inverted Zeiss 135 TV Axiovert microscope at constant room temperature (21°C). During the experiments, the bath solution contained (in mM): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 5 glucose, 10 HEPES, pH 7.4.
Fluorescence imaging of exocytosis
Our self-assembled combined fluorescence imaging and optical tweezers setup is schematically illustrated in Fig. 1 A. Visualization of vesicle fusion and surfactant release was described in detail in (Haller et al., 1998). In short, fused LBs were identified by the increase in fluorescence of LBs due to partitioning of FM 1-43 (1 μM), an amphiphilic dye (Smith and Betz, 1996), after it passes, from the external solution, through the fusion pore into the lipid layers of surfactant. Importantly, fluorescence of fused vesicles is manyfold brighter than the staining of the thin plasma membrane. In this study, FM 1-43 fluorescence served two purposes: first, to identify fused LBs and second, to record possible alterations in shape and location during traction by the laser tweezers. Furthermore, FM 1-43 fluorescence was combined with transmission imaging by continuously illuminating the cells under study (FM 1-43 fluorescence was excited at 490 nm for 20 ms at a rate of 20 Hz). Images were captured with a 530-nm dichroic mirror by a Peltier cooled slow scan camera of the imaging system (TILL Photonics, Germany).
FIGURE 1.
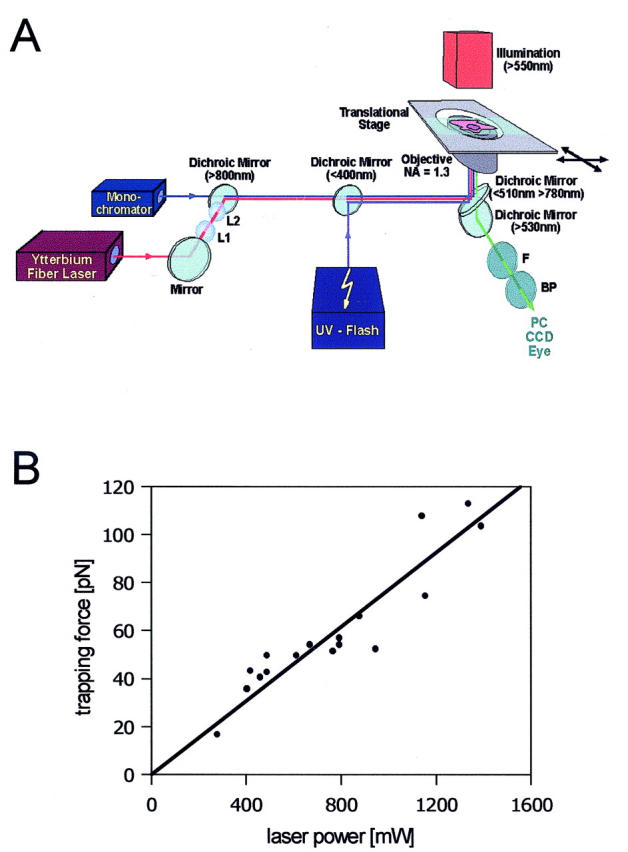
(A) Schematic setup overview. Laser beam, UV-flash, and fluorescence excitation light are guided through a 100× PlanNeofluar objective (NA = 1.3) of an inverted microscope. Cell position is controlled by an electrically driven translation stage. Transmission images, as well as fluorescence images, are captured by a high-resolution slow-scan camera. In the experiments, exocytosed surfactant is pulled into the laser beam. The trapping force can be adjusted by varying the laser power. The trapped surfactant is held in position, whereas the cells can be moved in any horizontal direction by the translation stage. BP, exchangeable band pass filter for emission light (520 ± 7 nm or 650 ± 7 nm); F, shortpass filter (<800 nm); L1 and L2, lenses to adjust beam diameter; and dichroic mirrors are characterized by their cutoff wavelengths. (B) Correlation between laser power and trapping force of free-floating LBs. The “laser power—trapping force correlation” was determined as described in Methods.
Flash photolysis of caged Ca2+
Cells were incubated with NP-EGTA/AM (1–10 μM) for up to 1 h. Uncaging was performed by a pulsed xenon arc lamp (pulse length 0.5 ms, wavelength 320–390 nm).
Optical tweezers
As illustrated in Fig. 1 A, light from an ytterbium fiber laser (LOT-Oriel, Germany) was focused through a microscope objective (Plan Neofluar 100×, NA = 1.3, Zeiss). The fiber laser emits a pure TEM00 mode at 1.064 μm with maximal emission power of 5 W, but typically numbers well below this have been used. This wavelength was chosen to minimize the absorption by water, preventing possible cell damage. For precise control of the displacement of the trapped vesicle with respect to the cell, an electrically driven translation stage (LEP, Germany) was used. The laser power values given in the Results section correspond to the respective emission power of the fiber laser; knowing the transmission factors of the optical components (objective, dichroic mirrors, and lenses) in the laser path (Liu et al., 1995; Svoboda and Block, 1994), we determined the actual laser power at the beam focus to be ∼34% of the laser emission power. A local rise in temperature cannot be excluded but was determined to be well below nonphysiological values (Liu et al., 1995).
Correlation between laser power and force exerted on a fused LB
Optical forces, which arise from transfer of the momentum the light carries itself, have been successfully applied in a variety of biological applications (Svoboda and Block, 1994; Ashkin, 1997; Ashkin et al., 1990; Quake et al., 1997; Sheetz, 2001). The force exerted on a trapped particle depends on its size, geometry, and the difference in refractive index between the particle and its environment, and scales linearly with the incident laser power (Svoboda and Block, 1994; Simmons et al., 1996; Singer et al., 2000; Wright et al., 1994). However, as the refractive index of the fused LB is not precisely known (but >nH2O), and the size of the particle and the focus of the trapping laser are of the same order of magnitude, an experimental force calibration was required for our experiments. A widely used method consists in subjecting the trapped particle to a counterflow of known velocity. This was performed by translational displacement of the object chamber, and therefore the surrounding bath solution, with respect to the trapped particle. Knowing the velocity v, when the particle unsnaps from the trap, the maximum trapping force, which equals the opposing viscous drag force F = 6πηrv, can be calculated according to (Svoboda and Block, 1994; Singer et al., 2000) with r denoting the particle radius and η the viscosity of the surrounding liquid.
As it was the aim of this study to investigate mechanical forces on fused LBs and determine elastic recoil properties of surfactant, a “laser power–trapping force” relationship was first established using single free-floating, secreted LBs. As noted above, secreted LBs, which are always present in the bath of a stimulated cell monolayer, retain their round shape for long periods of time. Based on our calibration method, we obtained this power/force relationship shown in Fig. 1 B. Although we cannot exclude that fully secreted LBs have slightly different hydration states and thus different refractive indices compared to LBs immediately after fusion, this calibration should represent a reasonable estimate of the actual forces exerted on fused LBs on the cell.
Equiaxial cell strain
Cells were grown on elastic, optically clear, silicone membranes (advancedLab, Austria). The strain device (advancedLab, Austria) was mounted on the stage of an inverted microscope. It enables us to exert equiaxial strain of variable strength and frequency to the cells by mechanical deflection of the silicone membrane. An optical positioning system allows to align the center of stretch in the experimental chamber with the optical path of the microscope, thereby minimizing lateral shift of the cells under study. Thus, continuous observation of single cells is possible while inducing equibiaxial strain. Under observation, cells can also be perfused while mechanical stimulation occurs.
RESULTS
Fused LBs exhibit resistance to applied force
For the study of postfusion mechanisms of secretion, we aimed at applying a force on fused LBs before full release. These LBs were identified by their FM 1-43 fluorescence, their reduced Brownian motion compared to freely floating LBs and their apparent “intracellular” location as judged by light transmission microscopy. A force was exerted on these constitutively fused LBs by performing a controlled motion of the translation stage (and thereby the cell under study) in a horizontal direction while keeping the fused LB trapped in the laser beam. LBs were always pulled radially outward from the cell center. We observed three types of response to the applied force: 1) The LBs could not be moved at all. Although they had fused, they behaved just as nonfused, intracellular LBs. Nonfused LBs apparently adhere so firmly to intracellular structures (presumably cytoskeletal elements), that they could not be affected even with maximum trapping force. 2) On very rare occasions, the LB could be readily and completely removed from the cell without a change in shape. 3) The LB could be moved, but with a dramatic change of shape, with one end of the expanded structure remaining firmly attached on the cell. These FM 1-43-stained “stretched LBs” frequently revealed the shape of a “drumstick,” with expanded, spherical endings at the site of origin (cell), or at the site of the pulling laser trap, or both (Fig. 2 A). When the laser power was turned off during stretch, this structure collapsed and regained its original round form, though not completely (Fig. 2 A). To gain further information about this apparent elasticity we investigated the relationship between the length of a stretched LB and the laser power by measuring the moved distance of the stage at which the pulled LB unsnapped from the trap at a constant light power (Fig. 2 B). This expanding distance/laser power relationship is shown in Fig. 2 B and will be discussed below. By confocal laser scanning microscopy, scanning, and transmission electron microscopy (Haller et al., 2001a) we know that some, but not all, LBs are subject to a spontaneous conformational change after fusion, during which parts of vesicle contents protrude through the fusion pore into the extracellular space (Haller et al., 2001a). By conventional microscopy as used here, such spontaneous transitions of surfactant after applying force are difficult to observe, and therefore it may be possible that some transformation of surfactant is necessary for attaining an expandable state. We can be sure, however, that we selectively pulled surfactant from fused vesicles for the following reasons:
Nonfused (“docked”) LBs do not stain with FM1-43 (Haller et al., 1998) and fused LBs are the only organelles in type II cells which brightly fluoresce in the presence of this dye (Haller et al., 1998).
To further exclude that surfactant from nonfused LBs or the entire fused LB along with plasma membrane and cytoplasmic components was pulled, a differential staining protocol for cytoplasm and surfactant from fused vesicles was applied using band pass filters (±7 nm) in the emission spectrum of the cytoplasmic indicator calcein (520 nm) and of FM 1-43 (650 nm), respectively. Fig. 3 shows the calcein (left) and the FM 1-43 staining (middle), and their overlay (right) before and after pulling the fused vesicle. Note that with calcein staining, nonfused LBs appear as dark inclusions. These experiments essentially exclude that the stretched structure contains cytoplasmic components and confirm that surfactant was selectively pulled from the fused vesicle.
Any long-scale movement of intracellular components by the laser trap in such dramatic dimensions (up to >100 μm, i.e., several times the diameter of a type II cell) would inevitably lead to a considerable stretch and hence leakiness of the plasma membrane, in particular because type II cells grown on glass have few and small microvilli and hence little membrane reserve (see scanning electron microscopy in Fig. 8, Haller et al., 2001a). The fact that we did not detect any measurable loss of calcein fluorescence intensity during stretch of fused LBs argues against a significant stretch of the plasma membrane. Likewise, we did not observe FM 1-43 uptake by the cell (from the bath), a very sensitive indicator for the loss of membrane integrity.
FIGURE 2.

(A) Expansion of exocytosed surfactant. Transmission microscopy images demonstrating extension of a single FM 1-43-labeled fused LB. The fused LB was trapped in the laser beam (arrows indicate the position of the laser trap). (Left), Trapped LB before movement of the translational stage (i.e., before cell movement). (Middle), Trapped LB after movement of the stage. Note the extension of surfactant. (Right), Untrapped LB (laser power turned off) following extension. Note the “recoiling” of surfactant. (B) Correlation between trapping force and expansion length of fused LBs. Dots indicate mean ± SE of the mean (n = 34).
FIGURE 3.
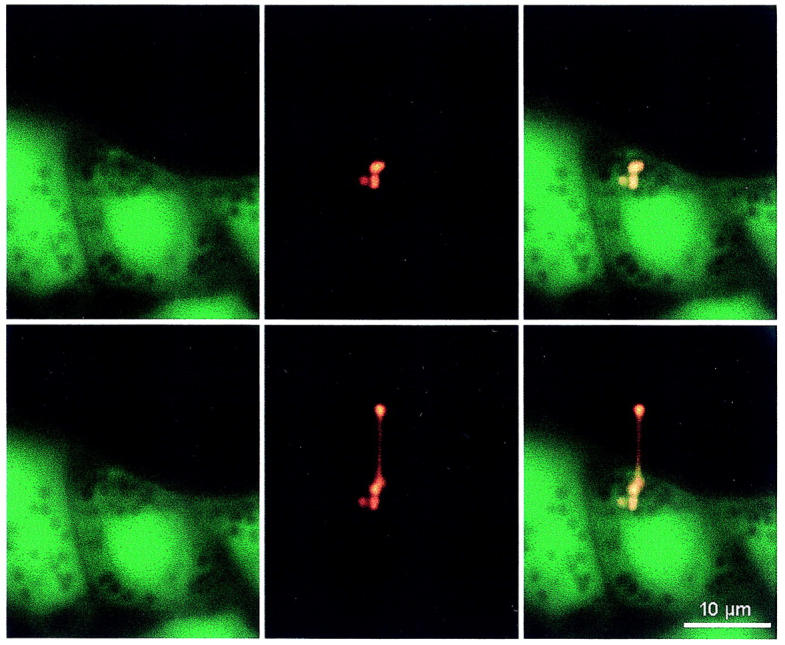
Differential staining of the cytoplasm (Calcein, left images) and of surfactant from fused vesicles (FM 1-43, middle images), in a cell before stretch (upper images), and during stretch (lower images) of a fused LB. The right images represent the overlay of the Calcein and FM 1-43 stainings. Before the experiment, cells had been preincubated for 30 min in 1 μM Calcein. Note that three neighboring vesicles stain with FM 1-43, suggesting compound exocytosis at this site. Only one of these three fused vesicles could be pulled by the laser trap.
Although it was easily possible to expand surfactant material by laser tweezers, it was not possible to completely remove stretched surfactant from the cell, even with high laser power. Instead, rupture of the stretched portion occurred when the laser power was increased. This indicates structural components which keeps fused LBs from being mechanically removed. In previous experiments using laser scanning as well as transmission and scanning electron microscopy (Haller et al., 2001a) we identified the fusion pore as a slowly expanding structure which most likely contributes to this phenomenon. The most convincing evidence for this idea is the fact that fusion pore diameters—even long periods after fusion—are smaller than corresponding LBs (reviewed in Haller et al., 2001b). We assume that in those fused LBs which we could not move, the fusion pore geometry was too restrictive for the surfactant complex to be squeezed through. Only after some degree of fusion pore expansion, surfactant should be able to permeate, as also suggested by the following experiments.
Expansion of fused vesicle contents depends on the “age” of fusion pores and the mode of stimulation
In accordance with the concept of fusion pores as structures impeding release, there should be a temporal transition of resistance to applied force from high to low, and the time course of this transition as well should depend on the mode of stimulation. If indeed this is the case, the percentage of vesicle contents that cannot be moved through the pore should be higher in freshly fused vesicles than in preexisting ones (i.e., constitutive fusions already present at the start of the experiments). In a first set of paired experiments, we tried to expand vesicle contents from such preexisting fusions and compared them with vesicles freshly fused following either flash photolysis of caged Ca2+ or extracellular application of ATP (0–5 min thereafter). The results are shown in Fig. 4 A and confirm that the time lapse between fusion and experiment determines the probability to expand surfactant, according with a slow expansion process of fusion pores. In a second set of experiments, we established a time course of pore transitions in fused vesicles after different types of stimulation. It is evident (Fig. 4 B) that in all treatments, the percentage of vesicles that could be expanded through the pore was considerably higher at 20 min than at 5 min after fusion. In addition, the most impressive transition from immobile to expandable was observed by a combined stimulation of the cells with caged Ca2+ and ATP (Fig. 4 B), consistent with the known synergistic action of Ca2+ and protein kinase C in potentiating surfactant secretion.
FIGURE 4.

(A) Force-induced expansion of fused vesicles is a function of time. Fused LBs were trapped and subject to a constant expanding force (2W of laser emission power) by translational movement of the stage (see Figs. 2 and 3). Bars indicate the percentage of expandable LBs (i.e., LBs which could be “pulled” as shown in Figs. 2 and 3). The remaining fused LBs could not be moved at all. The contents of preexisting fused vesicles (time of fusion min to h before the experiment, n = 65) were in average less strongly attached to the cells than LBs 5 min after stimulated fusion, following either release of caged calcium (n = 10) or addition of 10 μM ATP (n = 15). (B) The time course of force-induced vesicle expansion depends on the mode of stimulation. Again, LBs were expanded by applying a constant trapping force (2W of laser emission power). Light bars indicate the percentage of expandable LBs 5 min after stimulated fusion following either release of caged calcium (n = 10) or addition of 10 μM ATP (n = 15), whereas dark bars indicate the percentage of expandable LBs 20 min thereafter. ATP and flash photolysis of caged Ca2+ have a strong synergistic effect on the expandability of fused vesicles (hatched bar, n = 10).
Cell strain facilitates the release of fused LBs into the extracellular space
The above experiments indicate that mechanical factors govern surfactant release, but they do not suggest a mechanism which might control the barrier for LB release under physiological conditions and how surfactant release may be mechanically modulated without exerting an active force by optical tweezers. The following experiments were thus designed to test the hypothesis that cell strain, which occurs during inspiration in vivo (Tschumperlin and Margulies, 1999) and which increases surfactant secretion in vitro (Wirtz and Dobbs, 1990), is a mechanical factor for release, presumably by a change of the clearance of the fusion pore. The experimental protocol was the following (schematically shown as inset in Fig. 5: first, cells were stimulated with 10 μM ATP to obtain a maximum fusion response, i.e., to have as many LBs as possible in a fused state. 30 min later (when the fusion response was complete), the number of fused LBs was defined as 100%. Then, cells were subject to a gentle superfusion (0.5 ml/s for 3 s) of the extracellular fluid as described in Haller et al. (1998), which is sufficient to “wash out” fused LBs that are not firmly attached to the cell surface. The remaining fused LBs were evidently those which were still strongly kept in place by mechanical barriers preventing washout. A second LB washout protocol was performed 5 min later. In the control group, this was done without strain of the cells. In the experimental group, the second LB washout maneuver was accompanied by a single short-lasting (≈3 s) strain. The results are shown in Fig. 5. Before strain (after the first superfusion), ∼50% of fused LBs remained on cell (i.e., could not be “washed away”). After the second washout period, less than 10% of the fused LBs remained on cell in the strained cells, whereas in the nonstrained cells, the second superfusion had no further effect. These data indicate that cell strain facilitates the release of fused LBs.
FIGURE 5.

Cell strain facilitates the release of fused LBs. LB release was assessed using a “washout” protocol (see Results for details) as schematically shown in the above drawing. The arrowhead indicates a “remaining fused LB” following washout. LB fusion with the plasma membrane was stimulated by 10 μM ATP, and the number of fused LBs was defined as 100% at the time immediately before the first LB washout. Bars represent remaining fused LBs after washout (mean ± SE), which was performed twice in each experiment, the first at 30 min after stimulation with ATP (i.e., at the time when the ATP-induced fusion response was complete) and the second at 5 min later. In the control experiments (light bars), no strain was exerted. In the experimental group (dark bars), the second LB washout was accompanied by a single short-lasting strain of the cells (∼3 s, 20% increase in cell surface area). * indicates p < 0.01.
DISCUSSION
As noted, the release of surfactant from the fused LB into the extracellular space occurred in an apparent “all-or-none” fashion, i.e., surfactant was either fully released or remained, with one end, encapsulated within a fused LB. Only in a few occasions of extensive stretching (>100 μm), we were able to tear the stretched LB into two fractions. This is entirely consistent with our previous observations that surfactant protrusions (i.e., surfactant attached to the cell but extending into the extracellular space) never separate spontaneously and are apparently released as a single unit. Our observations with laser tweezers provide the first direct evidence that surfactant from fused vesicles exhibits viscoelastic properties by strong cohesive forces, which actually prevents a rapid dispersal of the secreted material. The nonlinear laser power/expanding distance relationship in Fig. 2 B is consistent with an elastic material, in which the decreased diameter with continuing stretch diminishes the force which is required for further expansion. Inasmuch as the expanding distance/power ratio is almost constant in the range between 80 and 160 pN (Fig. 2 B), we have determined a spring constant for surfactant in the range of ∼12.5 pN/μm (a spring constant for forces of <80 pN was not calculated to avoid possible errors resulting from some potential interaction between the laser beam and cellular components other than LBs within very small expanding distances). For strongly stretched surfactant, the elastic recoil was never complete (data not shown), indicating some plastic deformation as a result of partial disintegration of the macromolecular complex.
The elastic properties of surfactant immediately after its exocytotic release, as shown here for the first time, are consistent with the ultrastructural finding of nondispersed surfactant complexes in the hypophase of the alveolus (Schurch et al., 1998) and with floating, FM1-43-labeled particles found long after cell stimulation in our experiments. These results raise again the interesting question about how this material is actually processed in the lung, until final dispersal at the air–liquid–interface. Our first assumption that the spreading of pulled surfactant happens spontaneously as soon as the fused LB comes into contact with a gas–liquid–surface was not supported by preliminary experiments, in which we touched the pulled LB with small bubbles of gas (created at the tip of a microwire by electrolysis of the surrounding liquid). Future studies will have to identify the conditions (such as the composition of the extracellular fluid) which determine the elastic/spreading properties of surfactant, which may be an important basis for the treatment of various respiratory diseases. It should be noted here that the dispersal characteristics of surfactant in the hypophase of the lung alveolus might be different than in a standard modified Ringer solution, but this issue remains purely speculative as long as the exact composition of the hypophase is still a “black box.” Interestingly, spontaneous disintegration and dispersal of secreted surfactant clots in extracellular fluid is not appreciably altered by changing the temperature within the range of ∼20°C to 37°C: we determined the mass distribution of free-floating LBs in the bath solution and found no significant dependence on the temperature in this range (Thomas Haller, unpublished observation).
The fusion pore
In addition to cohesive forces of surfactant, the delayed “all-or-none release” appears to depend on forces interacting between the macromolecular surfactant aggregate and the cell surface, i.e., adhesive forces and/or retaining forces by constriction/insufficient relaxation of the fusion pore. As noted above, it is well documented that the fusion pore can act as a barrier for release during early stages of fusion pore expansion, resulting in partial release of secretory products in cell types with hydrophilic vesicle contents. In that case, fusion pores are fluctuating structures, which either fully expand (full fusion) or close again (transient fusion). With regard to the present study, fusion pores rather have to be considered as stable, long-lasting, purse-string-like constrictions at the site of LB fusion. Combining the data of this study with those of a detailed previous investigation (Haller et al., 2001a), strong evidence suggests that this fusion pore does in fact act as mechanical barriers for release:
A multitude of morphological investigations revealed that the aperture of fused LBs is always smaller in diameter than the corresponding LB (reviewed in Haller et al., 2001b).
Accordingly, the time course of phospholipid accumulation in cell supernatants is considerably slower than that of LB fusion (reviewed in Dietl et al., 2001).
These structures expand slowly and by a Ca2+-regulated mechanism (Haller et al., 2001a).
In accordance with point 3, the feasibility to expand fused LBs is a function of time and Ca2+(agonist)-dependent (shown here).
Narrow, apparently constricted rings at the site of LB fusion with the plasma membrane can be well observed in confocal laser scanning microscopy (see Figs. 7 and 8, Haller et al., 2001a).
The concept of fusion pores as long-lasting mechanical barriers imply structures in addition to the mere “fat/meat composition” of initial fusion pores (recently reviewed in Zimmerberg, 2001), which would not be designed to resist force. Our finding that an elevation of [Ca2+]c facilitates release would be consistent with the role of actin as a regulatory component of secretion at a postfusion stage, in addition to its role in subplasmalemmal vesicle transport (Lang et al., 2000). It has been shown that actin filaments are associated with LB (Tsilibary and Williams, 1983) and that actin depolymerization augments surfactant release (Rose et al., 1999).
Adhesive forces
In theory, the hindrance of LB release could also be due—at least in part—to “tethering” of LB contents to the limiting LB membrane. This possibility cannot be entirely excluded because electron micrographs revealed that bell-shaped lamellae of LBs are organized around a cylindrical core, which may include a protrusion of the limiting membrane (Gil, 1985). Given the all-or-none release in combination with the low dispersal characteristics of a fused LB, however, the question remains why this tether should give way to release some time after fusion, and how this should be regulated by [Ca2+]c.
Whether release from a fused vesicle is mainly restricted by the adhesive/cohesive forces of vesicle contents or the constriction of the fusion pore may—in general—depend on the stage of fusion pore expansion and the material to be released. Catecholamine release, for instance, is a fast process, and the amperometric findings of “foot currents” during the flickering stage of the fusion pore was considered as evidence that small, early fusion pores are rate-limiting for this release (Chow et al., 1992; Neher, 1993). On the other hand, the release of many small hydrophilic compounds (serotonin, epinephrine, histamine, etc.) through large fusion pores is limited by ion exchange through the granule matrix, and not by the fusion pore (Marszalek et al., 1996, 1997). Granule matrices (charged gels such as proteins, proteoglycans, or sugars), in turn, may behave similarly in the way they are released as, for example, surfactant or mucins: they are insoluble in water and exhibit elastic properties (Parpura and Fernandez, 1996). It is yet unclear to what extent the release of these materials is restricted by the constriction of the granule neck, as proposed here for surfactant release. The ratio between (long-lasting) fusion pore and granule diameters in an individual cell type may be a hint to this question. A recent atomic force microscopy described persistent structures (“depressions”) consistent with fusion pores sized between 150 and 200 nm in pancreatic acinar cells (Schneider et al., 1997). Inasmuch as zymogen granules are almost as large as LBs, it is therefore well conceivable that the granule neck is in fact also a hindrance for release in cells of the pancreatic acinus. In this context, it was recently shown that Ω-shaped structures in pancreatic acini maintain their profile for up to 8 min, and sequential secretion was suggested as a result of granule–granule fusion (Nemoto et al., 2001). This compound exocytosis also occurs in type II cells, possibly resulting in sequential secretion at a single site (Mair et al., 1999). Preliminary observations (Thomas Haller) suggest that sequential secretion at one site may occur hours following the LB-plasma-membrane-fusion event. Another cell type with granules almost as large as LBs is the mast cell. Freezing electron microscopy techniques revealed dimples, i.e., structures preceding fusion pores, of similar size than in pancreatic cells (∅ ≈ 100 nm), suggesting that this cytoskeleton-associated, filamentous structure might also be a mechanical barrier (Chandler and Heuser, 1980).
Our data indicate that cell strain facilitates the release of LBs into the extracellular space. In this set of experiments, “releasability” was assessed by a bath superfusion protocol instead of optical tweezers. Although it would have been desirable to measure “releasability” during strain also by use of optical tweezers, this type of experiment is limited by the problem that any amount of strain alters the thickness of the Silastic membrane, the optical path (silicone has a different refractive index than glass and immersion oil), and, thereby, the applied force. As each strain was accompanied by a Ca2+ signal (data not shown), consistent with previous observations (Wirtz and Dobbs, 1990), the effect of strain could be mediated by Ca2+-activated fusion pore expansion (Haller et al., 2001a). Alternatively, cell strain could exert a direct mechanical effect on fused vesicles, for example, by an interaction between cytoskeletal elements and the plasma membrane (Sheetz, 2001). In the lung, where the most important physiological stimulus for surfactant secretion is probably cell strain during a deep inspiration (Wirtz and Dobbs, 1990), strain-induced fusion pore regulation may actually determine the supply of surfactant to the air-liquid interface.
In summary, LB release is governed by mechanical forces rather than by the laws of diffusion. In this process, the number of vesicles fusing with the plasma membrane determines the amount of secretion, whereas the time course of release is determined by the fusion pore and/or other structures. In contrast to the classical definition of exocytosis, amount of secretion and time course of release are dissociated and subject to different modes of regulation.
Acknowledgments
We thank Drs. Kristian Pfaller and Michael Hess for valuable ultrastructural information about type II cell morphology, and Drs. Günther Putz and Leland Dobbs for helpful discussions. The technical assistance of Irina Öttl and Gerlinde Siber is gratefully acknowledged.
This work was supported by grants P15742 and P14263 from the Austrian Science Foundation.
Wolfgang Singer and Manfred Frick contributed equally to this work.
References
- Albillos, A., G. Dernick, H. Horstmann, W. Almers, T. de Alvarez, and M. Lindau. 1997. The exocytotic event in chromaffin cells revealed by patch amperometry. Nature. 389:509–512. [DOI] [PubMed] [Google Scholar]
- Ales, E., L. Tabares, J. M. Poyato, V. Valero, M. Lindau, and T. de Alvarez. 1999. High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nat. Cell Biol. 1:40–44. [DOI] [PubMed] [Google Scholar]
- Ashkin, A. 1997. Optical trapping and manipulation of neutral particles using lasers. Proc. Natl. Acad. Sci. USA. 94:4853–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkin, A., K. Schutze, J. M. Dziedzic, U. Euteneuer, and M. Schliwa. 1990. Force generation of organelle transport measured in vivo by an infrared laser trap. Nature. 348:346–348. [DOI] [PubMed] [Google Scholar]
- Breckenridge, L. J., and W. Almers. 1987. Final steps in exocytosis observed in a cell with giant secretory granules. Proc. Natl. Acad. Sci. USA. 84:1945–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler, D. E., and J. E. Heuser. 1980. Arrest of membrane fusion events in mast cells by quick-freezing. J. Cell Biol. 86:666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow, R. H., L. von Ruden, and E. Neher. 1992. Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature. 356:60–63. [DOI] [PubMed] [Google Scholar]
- Curran, M. J., F. S. Cohen, D. E. Chandler, P. J. Munson, and J. Zimmerberg. 1993. Exocytotic fusion pores exhibit semi-stable states. J. Membr. Biol. 133:61–75. [DOI] [PubMed] [Google Scholar]
- de Alvarez, T. G., C. R. Fernandez, and J. M. Fernandez. 1993. Release of secretory products during transient vesicle fusion. Nature. 363:554–558. [DOI] [PubMed] [Google Scholar]
- Dietl, P., T. Haller, N. Mair, and M. Frick. 2001. Mechanisms of surfactant exocytosis in alveolar type II cells in vitro and in vivo. News Physiol. Sci. 16:239–243. [DOI] [PubMed] [Google Scholar]
- Dobbs, L. G., R. Gonzalez, and M. C. Williams. 1986. An improved method for isolating type II cells in high yield and purity. Am. Rev. Respir. Dis. 134:141–145. [DOI] [PubMed] [Google Scholar]
- Fisher, R. J., J. Pevsner, and R. D. Burgoyne. 2001. Control of fusion pore dynamics during exocytosis by Munc18. Science. 291:875–878. [DOI] [PubMed] [Google Scholar]
- Frick, M., S. Eschertzhuber, T. Haller, N. Mair, and P. Dietl. 2001. Secretion in alveolar type II cells at the interface of constitutive and regulated exocytosis. Am. J. Respir. Cell Mol. Biol. 25:306–315. [DOI] [PubMed] [Google Scholar]
- Gil, J. 1985. Histological preservation and ultrastructure of alveolar surfactant. Annu. Rev. Physiol. 47:753–763. [DOI] [PubMed] [Google Scholar]
- Haller, T., K. Auktor, M. Frick, N. Mair, and P. Dietl. 1999. Threshold calcium levels for lamellar body exocytosis in type II pneumocytes. Am. J. Physiol. 277:L893–L900. [DOI] [PubMed] [Google Scholar]
- Haller, T., P. Dietl, K. Pfaller, M. Frick, N. Mair, M. Paulmichl, M. W. Hess, J. Furst, and K. Maly. 2001a. Fusion pore expansion is a slow, discontinuous, and Ca2+-dependent process regulating secretion from alveolar type II cells. J. Cell Biol. 155:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller, T., J. Ortmayr, F. Friedrich, H. Volkl, and P. Dietl. 1998. Dynamics of surfactant release in alveolar type II cells. Proc. Natl. Acad. Sci. USA. 95:1579–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller, T., K. Pfaller, and P. Dietl. 2001b. The conception of fusion pores as rate-limiting structures for surfactant secretion. J. Comp. Biochem. Physiol. 129:227–231. [DOI] [PubMed] [Google Scholar]
- Hartmann, J., and M. Lindau. 1995. A novel Ca2+-dependent step in exocytosis subsequent to vesicle fusion. FEBS Lett. 363:217–220. [DOI] [PubMed] [Google Scholar]
- Lang, T., I. Wacker, I. Wunderlich, A. Rohrbach, G. Giese, T. Soldati, and W. Almers. 2000. Role of actin cortex in the subplasmalemmal transport of secretory granules in PC-12 cells. Biophys. J. 78:2863–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., D. K. Cheng, G. J. Sonek, M. W. Berns, C. F. Chapman, and B. J. Tromberg. 1995. Evidence for localized cell heating induced by infrared optical tweezers. Biophys. J. 68:2137–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair, N., T. Haller, and P. Dietl. 1999. Exocytosis in alveolar type II cells revealed by cell capacitance and fluorescence measurements. Am. J. Physiol. 276:L376–L382. [DOI] [PubMed] [Google Scholar]
- Marszalek, P., B. Farrell, and J. M. Fernandez. 1996. Ion-exchange gel regulates neurotransmitter release through the exocytotic fusion pore. Soc. Gen. Physiol. Ser. 51:211–222. [PubMed] [Google Scholar]
- Marszalek, P. E., B. Farrell, P. Verdugo, and J. M. Fernandez. 1997. Kinetics of release of serotonin from isolated secretory granules. II. Ion exchange determines the diffusivity of serotonin. Biophys. J. 73:1169–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher, E. 1993. Cell physiology. Secretion without full fusion. Nature. 363:497–498. [DOI] [PubMed] [Google Scholar]
- Nemoto, T., R. Kimura, K. Ito, A. Tachikawa, Y. Miyashita, M. Iino, and H. Kasai. 2001. Sequential-replenishment mechanism of exocytosis in pancreatic acini. Nat. Cell Biol. 3:253–258. [DOI] [PubMed] [Google Scholar]
- Parpura, V., and J. M. Fernandez. 1996. Atomic force microscopy study of the secretory granule lumen. Biophys. J. 71:2356–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quake, S. R., H. Babcock, and S. Chu. 1997. The dynamics of partially extended single molecules of DNA. Nature. 388:151–154. [DOI] [PubMed] [Google Scholar]
- Rose, F., C. Kurth-Landwehr, U. Sibelius, K. H. Reuner, K. Aktories, W. Seeger, and F. Grimminger. 1999. Role of actin depolymerization in the surfactant secretory response of alveolar epithelial type II cells. Am. J. Respir. Crit. Care Med. 159:206–212. [DOI] [PubMed] [Google Scholar]
- Scepek, S., J. R. Coorssen, and M. Lindau. 1998. Fusion pore expansion in horse eosinophils is modulated by Ca2+ and protein kinase C via distinct mechanisms. EMBO J. 17:4340–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, S. W., K. C. Sritharan, J. P. Geibel, H. Oberleithner, and B. P. Jena. 1997. Surface dynamics in living acinar cells imaged by atomic force microscopy: identification of plasma membrane structures involved in exocytosis. Proc. Natl. Acad. Sci. USA. 94:316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurch, S., F. H. Green, and H. Bachofen. 1998. Formation and structure of surface films: captive bubble surfactometry. Biochim. Biophys. Acta. 1408:180–202. [DOI] [PubMed] [Google Scholar]
- Sheetz, M. P. 2001. Cell control by membrane-cytoskeleton adhesion. Nat. Rev. Mol. Cell Biol. 2:392–396. [DOI] [PubMed] [Google Scholar]
- Simmons, R. M., J. T. Finer, S. Chu, and J. A. Spudich. 1996. Quantitative measurements of force and displacement using an optical trap. Biophys. J. 70:1813–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, W., S. Bernet, N. Hecker, and M. Ritsch-Marte. 2000. Three-dimensional force calibration of optical tweezers. J. Modern Optics. 45:2921–2931. [Google Scholar]
- Smith, C. B., and W. J. Betz. 1996. Simultaneous independent measurement of endocytosis and exocytosis. Nature. 380:531–534. [DOI] [PubMed] [Google Scholar]
- Svoboda, K., and S. Block. 1994. Biological applications of optical forces. Annu. Rev. Biophys. Biomol. Struct. 23:247–285. [DOI] [PubMed] [Google Scholar]
- Tschumperlin, D. J., and S. S. Margulies. 1999. Alveolar epithelial surface area-volume relationship in isolated rat lungs. J. Appl. Physiol. 86:2026–2033. [DOI] [PubMed] [Google Scholar]
- Tsilibary, E. C., and M. C. Williams. 1983. Actin and secretion of surfactant. J. Histochem. Cytochem. 31:1298–1304. [DOI] [PubMed] [Google Scholar]
- Wirtz, H. R., and L. G. Dobbs. 1990. Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science. 250:1266–1269. [DOI] [PubMed] [Google Scholar]
- Wright, W. H., G. J. Sonek, and M. W. Berns. 1994. Parametric study of the forces on microspheres held by optical tweezers. Appl. Optics. 33:1735–1748. [DOI] [PubMed] [Google Scholar]
- Zimmerberg, J. 2001. How can proteolipids be central players in membrane fusion? Trends Cell Biol. 11:233–235. [DOI] [PubMed] [Google Scholar]