

Abstract
A prominent region of the Na+-dependent citrate carrier (CitS) from Klebsiella pneumoniae is the highly conserved loop X-XI, which contains a putative citrate binding site. To monitor potential conformational changes within this region by single-molecule fluorescence spectroscopy, the target cysteines C398 and C414 of the single-Cys mutants (CitS-sC398, CitS-sC414) were selectively labeled with the thiol-reactive fluorophores AlexaFluor 546/568 C5 maleimide (AF546, AF568). While both single-cysteine mutants were catalytically active citrate carriers, labeling with the fluorophore was only tolerated at C398. Upon citrate addition to the functional protein fluorophore conjugate CitS-sC398-AF546, complete fluorescence quenching of the majority of molecules was observed, indicating a citrate-induced conformational change of the fluorophore-containing domain of CitS. This quenching was specific for the physiological substrate citrate and therefore most likely reflecting a conformational change in the citrate transport mechanism. Single-molecule studies with dual-labeled CitS-sC398-AF546/568 and dual-color detection provided strong evidence for a homodimeric association of CitS.
INTRODUCTION
The Na+-dependent citrate carrier (CitS) of Klebsiella pneumoniae is expressed during anaerobic growth on citrate and enables this enterobacterial species to utilize citrate as sole carbon and energy source via the citrate fermentation pathway (Bott et al., 1995; Dimroth and Thomer, 1986). The citS gene encodes a highly hydrophobic integral membrane protein with a postulated topology of 11 transmembrane helices connected by hydrophilic loops (van Geest et al., 1999). CitS catalyzes the highly specific, electroneutral uptake of Hcitrate2- in cotransport with two Na+ ions (Lolkema et al., 1994; Pos and Dimroth, 1996). Citrate counterflow experiments revealed that CitS features two binding sites for citrate, an external high-affinity site and an internal low-affinity site. Active citrate uptake is driven by a chemical ion gradient (ΔpNa+/ΔpH), which is in part generated by the oxaloacetate decarboxylase Na+ pump, a key enzyme of citrate fermentation in K. pneumoniae (Dimroth, 1997). According to multiple-sequence alignments with closely related members of the 2-hydroxy-carboxylate transporter family (TC 2.A.24) (Saier, 2000) the primary structure of CitS shares 26% and 28% identical residues with CitP from Leuconostoc mesenteroides and MleP from Lactococcus lactis, respectively, which function as Hcitrate2-/lactate- and malate2-/lactate- antiporter, respectively (Bandell et al., 1997; Marty-Teysset et al., 1995). In particular, the C-terminal part including loop X-XI of these three transporters is highly conserved. Evidence indicates that this region is involved in substrate binding and hence of particular importance for the function of these transporters (Bandell and Lolkema, 2000a; Bandell and Lolkema, 2000b; Pos et al., 1998).
Single-molecule spectroscopy has developed to a powerful tool to investigate biological systems (Weiss, 1999). This method has distinct advantages over conventional ensemble spectrophotometric studies, e.g., much higher sensitivity, the possibility to discriminate between static and dynamic quenching in a single experiment, or direct observation of binding events by colocalization studies (Schutz et al., 1998). Inherently single-molecule experiments are free from unwanted interactions between proteins due to high-concentration artifacts. Recently, fluorescence quenching has been used to study protein folding (Zhuang et al., 2000) and conformational heterogeneities of membrane proteins (Peleg et al., 2001). We now used fluorescence quenching of single molecules to get further insights into the role of loop X-XI in the citrate translocation mechanism of CitS. Our results show that single-molecule fluorescence studies can be used to examine functional conformational changes in membrane proteins, specifically in transporters. In a second part, single-molecule studies with dual-color labeling allowed us to report on the oligomerization state of CitS.
MATERIALS AND METHODS
Bacterial strains and plasmids
Escherichia coli DH5α (Bethesda Research Laboratories, Frederick, MD) and plasmid pFXa3 (Kaestner et al., 2000) were routinely used for cloning procedures and mutagenesis. E. coli C43(DE3) (Miroux and Walker, 1996) and plasmids derived from pCitSH3 (Pos and Dimroth, 1996) were used for high-level expression of the citS mutants. E. coli BL21(DE3) (Studier and Moffatt, 1986) was used as host for whole-cell transport assays. To test the ability of cells to grow on citrate as sole carbon and energy source Simmons' citrate agar was used (Simmons, 1926).
Construction of single-Cys mutants
The CitS single-Cys mutants were constructed by polymerase chain reaction (PCR) mutagenesis using the two-step method described by Weiner et al. (1994). Four of the five native cysteines (C278, C317, C347, C398, C414) were subsequently replaced with serines resulting in the two CitS single-Cys mutants CitS-sC398 and CitS-sC414. For each mutation, two complementary mutagenic oligonucleotide primers were synthesized containing the desired mutation (indicated by boldface type) and a unique restriction site (underlined and specified in parentheses) to facilitate mutant identification (only the forward primers are shown):
C278S: GGTGCTGTCCACTACAAGCTTCCTGCTGGCC (HindIII)
C317S: CCTCCGGGCTGAGCTCACCGGAAATC (SacI)
C347S: GGGGTCGGCGTCAGCTACACAGATCTACAGGAAATTA-TC (BglII)
C398S: CACTGCAGGCCTGTCCATGGCCAACCGC (NcoI)
C414S: CCTGGAAGTTCTCTCTGCTAGCAACCGTATGAACCTTA-TCTC (NheI)
The mutagenic PCRs for CitS-sC398 (CitS-sC414) were performed in the following order: C278S → C317S → C347S → C414S (C398S). In addition, a Cys-less mutant was constructed by replacing even the last remaining cysteine residue with serine (CitS-C-less). After each PCR the methylated nonmutated parental DNA was digested with DpnI and 2 μl of the reaction were transformed into E. coli DH5α by electroporation (Holo and Nes, 1989). Mutated DNA was identified by the acquisition of the new restriction site and sequenced by using the dideoxy chain termination method (Sanger et al., 1977) to ensure that no other mutations occurred during the amplification process. For expression and purification the mutagenized citS genes in pFXa3 were cloned into pCitSH3 comprising an N-terminally attached 10x polyhistidine tag. Therefore the pFXa3 plasmids were digested with BamHI to produce 1.36 kb fragments carrying the mutagenized citS gene (citS*). Then the BamHI fragment of pCitSH3 was replaced by the mutagenized citS fragments resulting in pCitSH3(sC398), pCitSH3(sC414), and pCitSH3(C-less). Again, the DNA sequence of the ligated fragments was confirmed by sequencing.
Expression and purification of the mutant proteins
For expression and purification of the His-tagged fusion proteins, essentially the same protocol was used as described earlier (Kaestner et al., 2000). For the final Ni2+ affinity chromatography we used a Poros MC20 column (4.6 × 100 mm, PerSeptive Biosystems, Foster City, CA) operated on a BioCAD workstation (PerSeptive Biosystems, Foster City, CA) with ultraviolet (UV) detection at 280 nm. The Triton X-100 extract of the membrane vesicles was applied to the column preequilibrated with 10 column volumes of buffer C (50 mM potassium phosphate, 100 mM potassium chloride, 0.5 mM imidazole, 10% (v/v) glycerol, 0.05% (w/v) n-dodecyl-β-D-maltoside, pH 7.0). The column was washed with a linear gradient of 0.5–180 mM imidazole in 10 column volumes buffer C followed by 10 column volumes of buffer C containing 180 mM imidazole. The fusion proteins were eluted with buffer C containing 500 mM imidazole. The flow rate was 10 ml/min and 1 ml fractions were collected. The fractions containing the purified protein were pooled and concentrated by ultrafiltration using Centricon YM-30 centrifugal filter devices (Amicon, Danvers, MA). To remove imidazole, the samples (2 ml) were dialyzed twice for 6 h at 4°C in 500 ml buffer C using SpectraPor regenerated cellulose membranes (25 kD MWCO, Spectrum Laboratories, Rancho Dominguez, CA).
Reconstitution and transport experiments
The purified proteins were reconstituted using the freeze/thaw/sonication procedure as described previously (Pos and Dimroth, 1996). For transport experiments the proteoliposomes (2 μl) prepared in buffer R (50 mM potassium phosphate, pH 7.5) were diluted into 98 μl of buffer A (50 mM sodium phosphate, pH 6.0) containing 12 μM of [1,5-14C]citrate (180–220 cpm/pmol). The transport assays were terminated according to a time scale (0–120 s) by the addition of 0.9 ml of ice-cold 0.1 M LiCl and rapid filtration through 0.22 μm filters (GSTF, Millipore, Bedford, MA). The filters were washed once with 1 ml of ice-cold 0.1 M LiCl and inserted into wells of a Picoplate (24-well, Canberra Packard). After the addition of 1 ml Microscint-20 (Canberra Packard, Pangbourne, UK), the entrapped [1,5-14C]citrate was determined by liquid scintillation counting on a Microplate scintillation counter (TopCount, Canberra Packard, Pangbourne, UK). Initial rates were determined by the same procedure, but the reaction was immediately stopped after 5 s incubation time.
Labeling with AF546 and AF568
To obtain fluorophore-conjugated CitS, 5 μl of Alexa Fluor 546 C5 maleimide (AF546, Molecular Probes, Leiden, The Netherlands) stock solution (800 μM) in 50 mM potassium phosphate buffer (pH 7.0) were added to 100 μl of purified and concentrated CitS-sC398 or CitS-sC414 (40 μM) and incubated in a thermomixer (Eppendorf, Hamburg, Germany) for 10 min at 25°C in the dark. The reaction was stopped by passage through two centrifuge columns (1 ml, Sephadex G-25 medium) to separate labeled CitS from unreacted fluorophore. The degree of labeling was calculated by measuring the absorption of the fluorophore protein conjugate using a molar extinction coefficient of 92,000 M−1 cm−1 at 575 nm (Molecular Probes, Leiden, The Netherlands) and applying the Beer-Lambert law. For dual-color labeling a stock solution containing equimolar concentrations (800 μM) of Alexa Fluor 568 C5 maleimide (AF568, Molecular Probes, Leiden, The Netherlands) and Alexa Fluor 546 C5 maleimide (AF546) was used. The labeled protein was stored under liquid nitrogen.
SDS-PAGE and BN-PAGE analysis
The fluorophore attachment of fluorophore-protein conjugates was verified by SDS polyacrylamide gel electrophoresis using a 15% separating gel (Laemmli and Faure, 1973). The SDS-PAGE gel was first illuminated at 366 nm and then stained with Coomassie Blue G250.
The Blue Native PAGE analysis of wild-type CitS and CitS-sC398 was performed as described by Schägger and Jagow (1991). The purified samples (25 μg) were applied to a 5–15% gradient polyacrylamide gel with a 4% stacking gel and subjected to electrophoresis at 4°C. Immediately after electrophoresis, the gel was stained with Coomassie Blue G250.
Ni-NTA coating of cover glasses
The glass surface was coated with Ni-NTA using a modified procedure described by Adachi et al. (2000). The cover glasses (24 × 24 mm, Menzel, Braunschweig, Germany) were precleaned by baking at 500°C for 2 h. Successively, the glasses were incubated in sealing solution (2% (v/v) 3-glycidyloxypropyl-trimethoxysilane (Fluka, Buchs, Switzerland), 0.01% (v/v) acetic acid) for 3 h at 90°C; coating solution (2% (w/v) N,N-bis(carboxymethyl)-L-lysine (Fluka, Buchs, Switzerland), 2 mM KHCO3, pH 10.0) for 16 h at 60°C; and Ni2+ solution (10 mM NiSO4, 5 mM glycine, pH 8.0) for at least 2 h at room temperature. After each coating step the glasses were washed with ultrapure water.
Sample preparation
For each series of measurements aliquots of labeled CitS-sC398 were thawed and diluted to a concentration of 30 nM in buffer F (50 mM potassium phosphate, 100 mM potassium chloride, 0.05% n-dodecyl-β-D-maltoside, pH 7.0). A Ni-NTA coated cover glass was attached to the piezo scan stage and covered with 60 μl of buffer F. Upon addition of 2 μl His-tagged CitS or fluorescent derivatives (30 nM), the proteins become immobilized. In the case of fluorescence quenching studies, 5 μl of citrate or Na+ in buffer F in various concentrations was added.
Scanning confocal fluorescence microscopy
The measurements were performed using a home built scanning confocal fluorescence microscope (Prummer et al., 2000). A scheme of the set up is shown in Fig. 1. A pulsed frequency-doubled Nd:YAG laser (Antares; Coherent, Santa Clara, CA) was used to excite the fluorophores. The polarization was controlled by a polarizer, an electrooptical modulator, an analyzer, and a quarter- or a half-wave plate. To achieve a Gaussian beam profile, the light was spatially filtered by an optical fiber. Thereafter, the light was reflected by a dichroic (540DRLP, Omega, Brattleboro, VT) and an ordinary mirror into a high numerical aperture objective lens (Planapo 60×, NA 1.4; Nikon, Tokyo, Japan) resulting in a diffraction limited excitation spot (full-width at half maximum ∼250 nm). A confocal fluorescence image was built up by scanning the sample with the piezo stage pixel-by-pixel through the excitation volume while counting the fluorescence photons at each point.
FIGURE 1.

Schematic drawing of a scanning confocal fluorescence microscope for imaging and spectroscopy of single molecules in aqueous solution. For the immobilization of His-tagged fusion proteins a Ni-NTA coated cover glass was attached to the piezo scan stage and covered with buffer solution.
The fluorescence light emerging from the excitation volume was collected by the objective lens and subsequently filtered by the dichroic mirror and a holographic notch filter (532 nm Notch plus; Kaiser, Ann Arbor, MI). At the polarizing beam splitter the fluorescence light was decomposed in its vertically polarized component, which was detected by a single-photon counting avalanche photo diode (APD 1) (SPCM-AQR-14; Perkin Elmer, Fremont, CA; dark counts less than 100 s−1) and its horizontal component, which was projected onto a dichroic mirror (595DRLP, Omega). The dichroic mirror decomposes the fluorescence light according to its wavelength. Light that had a wavelength shorter than 595 nm was reflected onto APD 2, and light with a longer wavelength than 595 nm passed the dichroic mirror and was detected by APD 3. Artifacts caused by the polarization dependency of the dichroic mirror were avoided by using this polarization resolved detection scheme. The pulses from the three APDs were fed into a time-correlated single-photon counting board (SPC 402, Becker and Hickl, Berlin, Germany), which allowed for the storage of arrival time after pulsed excitation, channel number, and absolute time on a photon-by-photon basis.
From the absolute time when a photon was detected, the intensity was calculated for each channel (I1, I2, I3) with variable integration time. The dichroic ratio
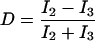 |
depends on the emission spectrum of the fluorophore. It is independent of the emission intensity and was used to identify and distinguish the different fluorophores AF546 and AF568.
In addition, the polarization
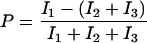 |
was monitored to observe changes in the orientation of the dye labels.
RESULTS
A prominent region of the Na+-dependent citrate carrier CitS and other members of the 2-hydroxycarboxylate (TC 2.A.24) carrier family is the C-terminal part including the hydrophilic loop X-XI and transmembrane segment XI. This region contains numerous conserved residues and has been described to harbor the binding site for the transport substrate. Transporters are generally believed to perform a catalytic cycle, which starts with the binding of the transport substrate(s) from one side of the membrane and succeeds with a conformational change through which the bound substrates gain access to the opposite membrane surface where they dissociate. We reasoned that a conformational change of CitS after citrate binding might be observable by fluorescence quenching of a fluorophore attached to an appropriate site of the molecule.
Single-Cys mutants CitS-sC398 and CitS-sC414
CitS has two endogenous cysteine residues (C398, C414) in the particularly interesting loop X-XI region, which could be used as targets for the binding of the thiol-reactive fluorophores (Fig. 2). However, as five cysteines are present in CitS, single cysteine mutants had first to be created genetically to achieve specific labeling of only a single cysteine residue. With this goal in mind we replaced the five cysteines step-by-step with serine and determined the citrate transport activity of the carrier after each step. Fortunately, all cysteine/serine substitutions were tolerated because the E. coli transformants expressing the various citS mutants (DH5α/pFXa3-citS*) were all able to grow on Simmons' citrate agar as recognized by a color change of the pH indicator bromthymol blue due to alkalinization of the medium. All citrate carriers containing cysteine substitutions were confirmed to be functional by [1,5-14C]citrate uptake into the E. coli cells expressing the CitS mutants. For a more quantitative assessment of the citrate transport activities of particularly interesting mutants, the N-terminally His-tagged fusion proteins CitS-sC398, CitS-sC414, and CitS-C-less were expressed, purified, and reconstituted into proteoliposomes.
FIGURE 2.

Topology model and amino acid sequence alignment of the C-terminal part of CitS from K. pneumoniae (Kp) with homologs of the 2-hydroxy-carboxylate transporter family: CitS from Vibrio cholerae (Vc), CitW from K. pneumoniae (Kp), CitP from Leuconostoc mesenteroides (Lm), and MleP from Lactococcus lactis (Ll). Circles and arrows indicate the position of the target cysteines for thiol-reactive labeling of CitS-sC398 or CitS-sC414. The consensus sequence and the loop X-XI region are indicated by black bars. The boxes represent transmembrane segment X and XI.
Labeling of CitS-sC398 and CitS-sC414 with AF546 and AF568
The thiol-reactive fluorophores AF546 and AF568 were the most suitable probes for our spectroscopic studies with the Nd:YAG excitation laser (532 nm) due to their absorption and emission spectra (Haugland, 2000). For site-specific labeling CitS-sC398 and CitS-sC414 were reacted with AF546 or an equimolar mixture of AF568 and AF546 in case of dual-color labeling. To confirm the labeling reaction, the conjugates CitS-sC398-AF546 (CitS-sC3981) and CitS-sC414-AF546 (CitS-sC4141) were subjected to SDS-PAGE and examined by UV illumination and Coomassie staining (Fig. 4).
FIGURE 4.

(A and B) SDS-PAGE analysis of the protein fluorophore conjugates CitS-sC3981 (left lanes) and CitS-sC4141 (right lanes) and a nonlabeled protein marker (center lane). The gel was examined by UV illumination at 366 nm (A) and Coomassie staining (B). (C) BN-PAGE analysis of purified wild-type CitS (left lane) and CitS-sC398 (right lane) as described under Materials and Methods.
According to our calculations, the degree of labeling was ∼80% for both conjugates. Thus, the region comprising C398 and C414 must be accessible to this rather hydrophilic fluorophore. The effect of the bound fluorescent probe on the activity of the conjugates was again investigated by [1,5-14C]citrate uptake studies with CitS-sC3981 and CitS-sC4141 reconstituted into proteoliposomes (Fig. 3). The initial transport rate of the conjugate CitS-sC3981 (98 nmol mg−1 min−1) was only slightly reduced compared to the nonmodified protein CitS-sC398 (109 nmol mg−1 min−1) and showed almost the same kinetic pattern (Fig. 5). This suggested that the bound fluorophore at position 398 did not affect imperative interactions during citrate translocation or result in partial inactivation. In contrast, with conjugate CitS-sC4141 a complete loss of transport activity was observed. Most likely, the bound probe AF546 at position 414 blocks citrate binding or inhibits mandatory interactions during the transport process. Therefore, we decided to concentrate our studies on the functional conjugate CitS-sC3981.
FIGURE 3.

Citrate uptake into proteoliposomes containing purified wild- type CitS (•), CitS-sC398 (○), CitS-sC414 (▪), CitS-sC398-AF546 (▾), CitS-sC414-AF546 (□), and Cys-less CitS (▿). The transport assay was initiated by diluting 2 μl proteoliposomes into 98 μl buffer A containing 12 μM of [1,5-14C]citrate.
FIGURE 5.
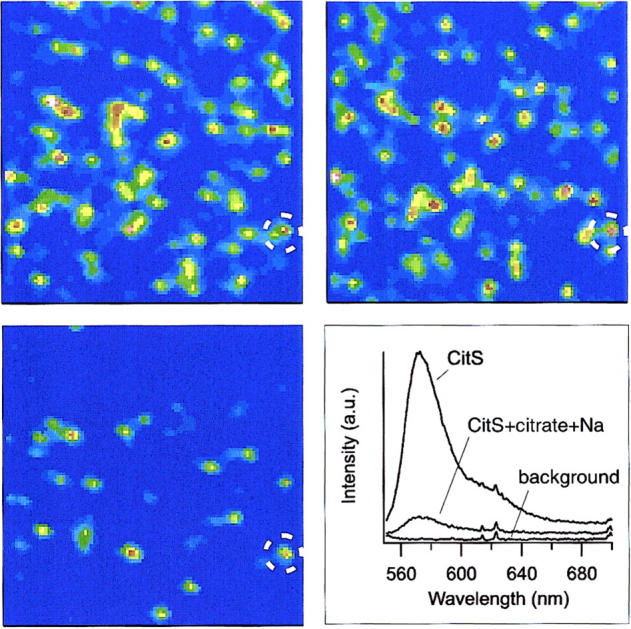
Scanning confocal microscopy images of individual CitS-sC3981 molecules immobilized on Ni-NTA coated cover glass. The first two images (top row) show the same frame (10 μm × 10 μm) recorded between a time interval of 2 min. The third image (bottom left) represents the identical frame upon addition of 250 μM citrate. The dashed circle marks an identical spot in all three images. (Bottom right) Ensemble spectra taken under the same conditions. (From top to bottom) Pure AF546 labeled CitS-sC3981, after addition of citrate and Na+, and background.
Single-molecule fluorescence microscopy with CitS-sC3981
Preliminary experiments with ensemble measurements using the functional conjugate CitS-sC3981 already indicated that the addition of citrate caused fluorescence quenching of the probe bound to C398 in loop X-XI and a slight spectral blue shift (Fig. 5, inset). Because single-molecule fluorescence spectroscopy provides much higher sensitivity and additional spectroscopic information, we decided to use this technique to study the putative conformational changes of the loop X-XI region.
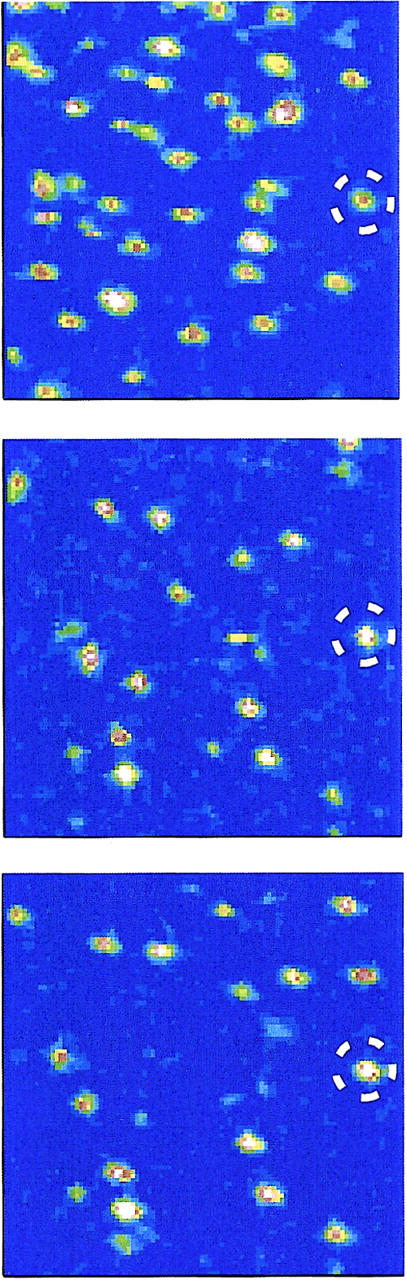
For the single-molecule studies His-tagged CitS-sC3981 was immobilized on a Ni-NTA coated cover glass and subsequently fluorescence images were recorded with an integration time of 1 ms per pixel. The same frame (10 μm × 10 μm) was recorded twice to show the effect of bleaching and additional binding/unbinding of CitS-sC3981 to the glass surface, leading to vanishing and emerging spots, respectively (Fig. 5). Some of the spots moved laterally and indicated partial mobility of conjugates but the majority of bright spots did not change significantly. However, the addition of 250 μM citrate caused strong static quenching of AF546 at position 398, whereas the addition of 250 μM DL-isocitrate, that served as a control, did not alter the fluorescence emission of the bound probe (Fig. 6).
FIGURE 6.
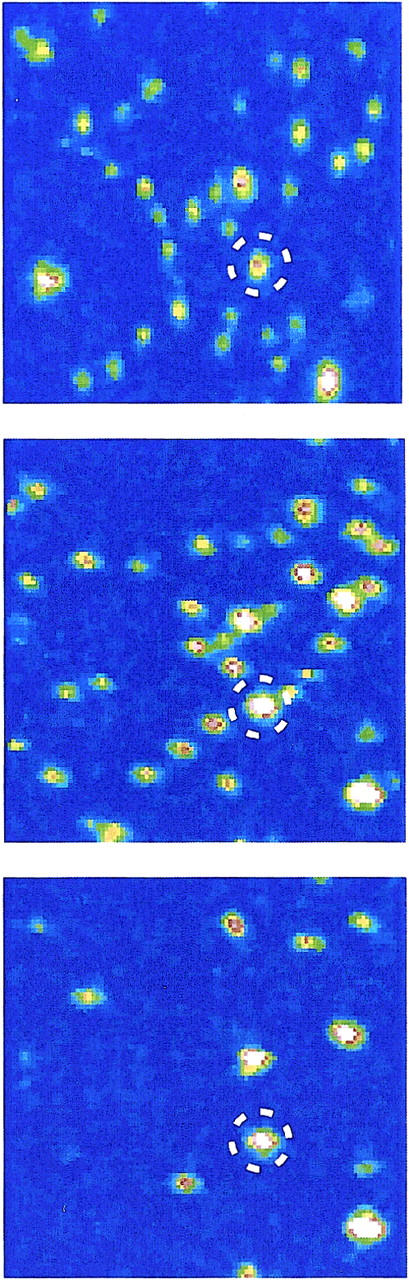
Scanning confocal microscopy images of CitS-sC3981 in the absence of substrate (top), in the presence of 250 μM DL-isocitrate (middle), and upon addition of 250 μM citrate (bottom). All images were recorded consecutively.
Because CitS is known to be the most specific transporter among well-characterized members of the 2-hydroxycarboxylate transport protein family, transporting only citrate in uptake mode as well as in exchange mode (Bandell et al., 1997; Pos and Dimroth, 1996), this finding strongly suggests that the interaction of substrate with CitS induced the quenching, not the mere presence of citrate or other 2-hydroxycarboxylates. Also, a change in the local pH can be excluded as the reason for quenching because the fluorescence properties of the Alexa dyes are only very weakly pH-sensitive as stated by the supplier (Haugland, 2000) and confirmed in this study as shown in the top panel of Fig. 8. To exclude direct interactions between citrate and the bound fluorophore, we conducted an additional control experiment by adding citrate in various concentrations to pure AF546 at pH 7.2, but this time no fluorescence quenching was observed (Fig. 8).
FIGURE 8.
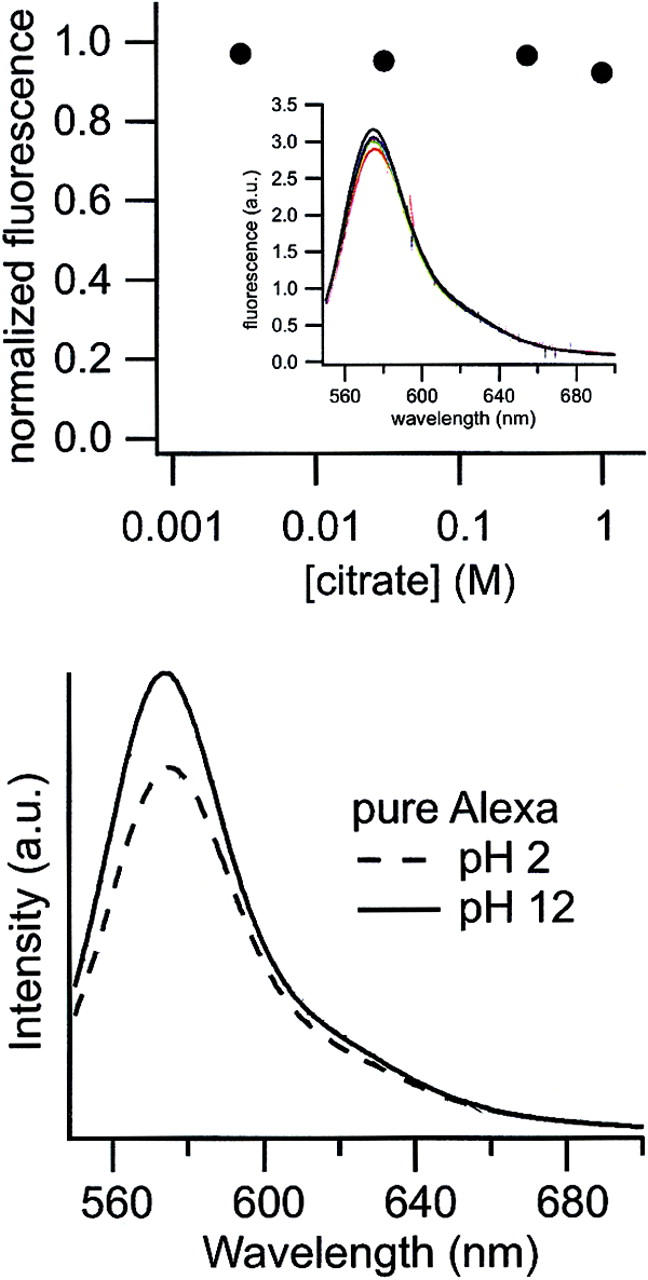
Control experiments. (Bottom) Two emission spectra taken at extreme pH values to prove that a change in pH does not have the observed quenching effect on the fluorophores. (Top) Emission maxima of AF546 taken at increasing citrate concentrations (pH 7.0) to prove that a high level of citrate does not have the observed quenching effect. The values are normalized to the fluorescence intensity without citrate. The respective emission spectra are shown in the inset.
The rather high intensity of the unquenched spots suggests that these spots correspond to inactive protein aggregates or nonspecifically bound protein that might be partly unfolded. We observed an increase of large aggregates when CitS-sC3981 was subjected to room temperature for more than 30 min. Single-molecule experiments offer the unique opportunity to disregard these aggregates from further analysis. Only isolated, diffraction limited spots are taken into account. Within these spots the number of fluorescent molecules can be counted during photobleaching. To test if the presence of the physiological coupling ion Na+ is mandatory for citrate binding interactions with CitS or has any influence on the citrate-induced fluorescence quenching, we first added 250 μM citrate to a Na+-free buffer and subsequently 50 mM Na+ to immobilized CitS-sC3981 and vice versa. To ensure saturating conditions, the used concentration of Na+ was ∼20 times higher than the apparent affinity of wild- type CitS for sodium ions (K0.5 = 2.7 mM) (Kaestner et al., 2000). The images recorded at each step are shown in Fig. 7. Clearly, the addition of citrate already quenched all of the fluorophores bound to functional proteins, whereas the absence or presence of sodium ions had no effect on the observed static quenching of AF546. When first Na+ and then citrate was added, quenching again occurred only at the presence of citrate (data not shown).
FIGURE 7.
Scanning confocal microscopy images of CitS-sC3981 in the absence of substrate (top), in the presence of 250 μM citrate (center), and in the additional presence of 50 mM Na+ (bottom). All images were recorded consecutively.
Dual-color labeling and detection studies with CitS-sC3982
The fluorescence intensity of a number of spots showed strong and irregular variations, usually not seen with single molecules. They could be explained by the existence of CitS oligomers with two or more fluorophores at a distance of 4 nm or less. Fluorescence homotransfer together with some rotational degree of freedom and intersystem crossing could be responsible for the fluctuating intensity. Indeed, there was preliminary evidence for a dimeric arrangement of CitS in detergent micelles and lipid membranes. Such evidences derived from affinity chromatography, where the CitS fusion protein eluted as heterodimer because of a mixed population of a tagged and a nontagged form (purification tag cleaved off) (Pos et al., 1994). Recent blue native PAGE experiments in our lab correspond with these findings because purified wild-type CitS as well as CitS-sC398 showed an electrophoretic mobility of the homodimeric form (∼100 kDa) (Fig. 4 C).
Because single-molecule experiments, in contrast to ensemble measurements by UV/VIS spectrophotometry, allow us to identify heterogeneities or different classes in a population, they can be used to study the state of protein-protein interaction. For this purpose, we performed a dual-color labeling and detection experiment with immobilized CitS-sC398-AF546/568 (CitS-sC3982). Due to our equimolar labeling procedure with the two different Alexa Fluor derivatives AF546 and AF568 and ∼80% degree of labeling, the following dual-labeled dimeric species of CitS would be expected: (CitS-AF546)2, (CitS-sC398-AF546)(CitS-sC398-AF568), and (CitS-sC398-AF568)2. At low surface density of fluorescent spots two-step bleaching and the colocalization of two fluorophores (AF546 and/or AF568) at one spot would prove the existence of the dimeric form. In the case of dual-color labeled CitS-sC3982 a simultaneous change of the dichroic ratio is expected upon stepwise bleaching of the two fluorophores that could serve as additional proof (Schutz et al. 1998).
For each experiment we selected a specific single-molecule spot, collected the fluorescence photons, and computed the intensity and the dichroic ratio as a function of time. By simultaneously monitoring the polarization we could exclude the possibility that the decrease of emission intensity was caused by a reorientation of the fluorophores. Fig. 9 shows a typical time trace of dual-color labeled CitS-sC3982, i.e., (CitS-sC398-AF546)(CitS-sC398-AF568), including consecutive two-step bleaching of AF568 and AF546 (1700 → 800 → ∼100 counts per 0.1 s) and the associated change of the dichroic ratio (D) upon bleaching of the first fluorophore AF568. In summary, 48 spots were selected for evaluation. We observed two-step bleaching for 33 spots of which 17 showed an associated change of the dichroic ratio. Assuming a labeling efficiency of 80%, these numbers indicate a strong evidence of dimer formation of CitS. Intensively fluorescing spots, which exhibited a continuous instead of stepwise bleaching, were also observed. We assigned these spots to nonfunctional aggregates of a large number of molecules and discarded them from further analysis.
FIGURE 9.

Single-molecule fluorescence emission time trace (—) including dichroic ratio (▵) and polarization (○) of dual-color labeled CitS-sC3982. The black arrows indicate the consecutive two-step bleaching of AF568 and AF546 and the associated change of the dichroic ratio upon bleaching of AF568.
DISCUSSION
Single-molecule fluorescence microscopy with immobilized CitS-sC3981 revealed an almost complete fluorescence quenching of the majority of molecules upon citrate addition (Fig. 5). The fluorescence quenching response was highly specific for citrate and was not observed with DL-isocitrate, which is not a substrate of CitS (Fig. 6). Moreover, the addition of citrate to pure AF546 did not cause any significant quenching of the fluorophore, nor did changes of the pH to pH 2 or pH 12 (Fig. 8).
Nontrivial quenching mainly arises from the formation of nonfluorescent products (static quenching), collision with other molecules (dynamic or collisional quenching), and excitation energy transfer (Lakowicz, 1999). In the present study we observed static quenching because only the number of fluorescent molecules decreased, whereas the remaining fluorescent spots showed unchanged photophysical properties. Static quenching can result from the formation of nonfluorescent species, electron or proton transfer, and π-stacking with aromatic systems. Most likely interactions between the fluorophore and proximate aromatic amino acid residues of the protein contribute to the observed effect. Which interaction accounts most for the dramatic quenching of the bound fluorophore AF546 remains to be investigated.
We suppose, therefore, that binding of citrate to CitS elicits a conformational change of the protein concomitant with an alteration of the molecular environment of AF546, and thus with the observed fluorescence quenching. The fluorescent probe is bound to C398 (loop X-XI) within the highly conserved C-terminal region, which probably includes the substrate binding site (Bandell and Lolkema, 2000b) as proposed for the three closely related 2-hydroxy-carboxylate transporters CitS (K. pneumoniae), CitP (L. mesenteroides), and MleP (L. lactis). Hence, the citrate-induced conformational change either involves the loop X-XI region itself or other parts of the molecule interacting with this region. Similar ligand-induced fluorescent changes that were traced back to structural changes associated with substrate binding have been extensively studied for two other secondary active transport proteins using ensemble measurements, the H+-coupled lactose permease (Venkatesan and Kaback, 1998; Wang et al., 1997; Weitzman et al., 1995; Wu et al., 1995) and the H+/Na+-coupled melibiose permease (Cordat et al., 2000; Mus-Veteau and Leblanc, 1996; Mus-Veteau et al., 1995) from E. coli.
CitS has a high-affinity citrate binding site (Ks ∼ 13 μM) proposed to be responsible in vivo for citrate binding from the periplasm and a low-affinity site (Ks ∼ 14 mM) from where citrate is assumed to dissociate into the cytoplasm (Pos and Dimroth, 1996). The concentration of citrate used for the quenching studies (250 μM) was ∼20 times higher than the binding constant for citrate of the high-affinity site to ensure saturating conditions, and 60 times lower than that of the low-affinity binding site. Hence, the observed conformational change is apparently due to the occupation of the high-affinity site with citrate. Sodium ions, which are obligatory required for citrate uptake, had no noticeable effect on the citrate-induced fluorescence quenching, which probably indicates that citrate binding is Na+-independent, but a Na+-dependent step occurs later in the transport cycle.
Interestingly, preliminary data derived from citrate binding studies by isothermal titration calorimetry (Kaestner et al., unpublished; Wiseman et al., 1989) indicated that the overall enthalpy change at the high-affinity site upon citrate binding was slightly endothermic (ΔHhigh = 6 kJ/mol), whereas the observed enthalpy at the low-affinity site was exothermic (ΔHlow = −55 kJ/mol). Similar observations have been made for other proteins with ligand-coupled conformational changes like the cAMP receptor protein (Gorshkova et al., 1995) or the inosine monophosphate dehydrogenase (Bruzzese and Connelly, 1997). There, the exothermic enthalpy of binding (ΔHbind) is overwhelmed by the large endothermic enthalpy of the conformational transition (ΔHconf) that takes place upon binding of the ligand.
For an increasing number of prokaryotic transporters it has been demonstrated that they form dimers. Examples are the tetracycline cation/proton antiporter TetA from Escherichia coli (McMurry and Levy, 1995), the H+-coupled multidrug transporter EmrE from E. coli (Tate et al., 2001), the lactose transporter LacS from Streptococcus thermophilus (Friesen et al., 2000), and the Na+/H+ antiporter NhaA from E. coli (Gerchman et al., 2001). For NhaA even two-dimensional crystals showed tightly packed dimers with well-defined interdimer contacts (Williams et al., 1999). Interestingly, for the phosphate carrier (PIC) from Saccharomyces cerevisiae it was proven that only the dimeric form is completely active, which enables functional cross talk between the two monomers (Schroers et al., 1998). In contrast, the monomeric as well as the dimeric form of lactose permease from E. coli were shown to be functional (Sahin-Toth et al., 1994). Thus, the dimeric arrangement is not necessarily a functional requirement but might be due to protein/lipid interactions and stability reasons in a membrane environment. Moreover, there are theoretical arguments that oligomeric structures are particularly well-suited to integral membrane proteins with transport function (Klingenberg, 1981). Our single-molecule studies with dual-labeled CitS-sC3982 and dual-color detection provided strong evidence for a dimeric association of CitS. The frequent occurrence of two-step bleaching and the simultaneous change of the dichroic ratio upon bleaching of the first fluorophore suggest that functional, detergent-solubilized CitS forms a rather stable homodimer. Together with the biochemical and kinetic indications (Pos et al., 1994; Pos and Dimroth, 1996) these results clearly support our hypothesis that the active form of CitS within the membrane is a homodimer.
In summary, we could demonstrate that fluorescence quenching of single molecules is a powerful technique to follow conformational changes such as that elicited by the binding of citrate within the region of loop X-XI of CitS. Furthermore, with the dual-color labeling and detection technique strong evidence for a dimeric arrangement of CitS was obtained. This technique is generally very useful to determine binding events of single molecules in an unambiguous fashion.
Acknowledgments
We thank Christoph von Ballmoos and Fabienne Henzen for their help in the fluorescence control experiments.
C. N. Kästner, M. Prummer, and B. Sick contributed equally to this work.
References
- Adachi, K., R. Yasuda, H. Noji, H. Itoh, Y. Harada, M. Yoshida, and K. Kinosita, Jr. 2000. Stepping rotation of F1-ATPase visualized through angle-resolved single-fluorophore imaging. Proc. Natl. Acad. Sci. USA. 97:7243–7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandell, M., V. Ansanay, N. Rachidi, S. Dequin, and J. S. Lolkema. 1997. Membrane potential-generating malate (MleP) and citrate (CitP) transporters of lactic acid bacteria are homologous proteins. Substrate specificity of the 2-hydroxycarboxylate transporter family. J. Biol. Chem. 272:18140–18146. [DOI] [PubMed] [Google Scholar]
- Bandell, M., and J. S. Lolkema. 2000a. Arg-425 of the citrate transporter CitP is responsible for high affinity binding of di- and tricarboxylates. J. Biol. Chem. 275:39130–39136. [DOI] [PubMed] [Google Scholar]
- Bandell, M., and J. S. Lolkema. 2000b. The conserved C-terminus of the citrate (CitP) and malate (MleP) transporters of lactic acid bacteria is involved in substrate recognition. Biochemistry. 39:13059–13067. [DOI] [PubMed] [Google Scholar]
- Bott, M., M. Meyer, and P. Dimroth. 1995. Regulation of anaerobic citrate metabolism in Klebsiella pneumoniae. Mol. Microbiol. 18:533–546. [DOI] [PubMed] [Google Scholar]
- Bruzzese, F. J., and P. R. Connelly. 1997. Allosteric properties of inosine monophosphate dehydrogenase revealed through the thermodynamics of binding of inosine 5′-monophosphate and mycophenolic acid. Temperature dependent heat capacity of binding as a signature of ligand-coupled conformational equilibria. Biochemistry. 36:10428–10438. [DOI] [PubMed] [Google Scholar]
- Cordat, E., G. Leblanc, and I. Mus-Veteau. 2000. Evidence for a role of helix IV in connecting cation- and sugar-binding sites of Escherichia coli melibiose permease. Biochemistry. 39:4493–4499. [DOI] [PubMed] [Google Scholar]
- Dimroth, P. 1997. Primary sodium ion translocating enzymes. Biochim. Biophys. Acta. 1318:11–51. [DOI] [PubMed] [Google Scholar]
- Dimroth, P., and A. Thomer. 1986. Citrate transport in Klebsiella pneumoniae. Biol. Chem. Hoppe Seyler. 367:813–823. [DOI] [PubMed] [Google Scholar]
- Friesen, R. H., J. Knol, and B. Poolman. 2000. Quaternary structure of the lactose transport protein of Streptococcus thermophilus in the detergent-solubilized and membrane-reconstituted state. J. Biol. Chem. 275:33527–33535. [DOI] [PubMed] [Google Scholar]
- Gerchman, Y., A. Rimon, M. Venturi, and E. Padan. 2001. Oligomerization of NhaA, the Na+/H+ antiporter of Escherichia coli in the membrane and its functional and structural consequences. Biochemistry. 40:3403–3412. [DOI] [PubMed] [Google Scholar]
- Gorshkova, I., J. L. Moore, K. H. McKenney, and F. P. Schwarz. 1995. Thermodynamics of cyclic nucleotide binding to the cAMP receptor protein and its T127L mutant. J. Biol. Chem. 270:21679–21683. [DOI] [PubMed] [Google Scholar]
- Haugland, R. P. 2000. Handbook of Fluorescent Probes and Research Chemicals. M. T. Spence, editor. Molecular Probes, Inc., Eugene.
- Holo, H., and I. F. Nes. 1989. Appl. Environ. Microbiol. 55:3119–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner, C. N., P. Dimroth, and K. M. Pos. 2000. The Na+-dependent citrate carrier of Klebsiella pneumoniae: high-level expression and site-directed mutagenesis of asparagine-185 and glutamate-194. Arch. Microbiol. 174:67–73. [DOI] [PubMed] [Google Scholar]
- Klingenberg, M. 1981. Membrane protein oligomeric structure and transport function. Nature. 290:449–454. [DOI] [PubMed] [Google Scholar]
- Laemmli, U. K., and K. Faure. 1973. Maturation of the head of bacteriophage T4. J. Mol. Biol. 80:575–599. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Plenum Press, New York.
- Lolkema, J. S., H. Enequist, and M. E. van der Rest. 1994. Transport of citrate catalyzed by the sodium-dependent citrate carrier of Klebsiella pneumoniae is obligatorily coupled to the transport of two sodium ions. Eur. J. Biochem. 220:469–475. [DOI] [PubMed] [Google Scholar]
- Marty-Teysset, C., J. S. Lolkema, P. Schmitt, C. Divies, and W. N. Konings. 1995. Membrane potential-generating transport of citrate and malate catalyzed by CitP of Leuconostoc mesenteroides. J. Biol. Chem. 270:25370–25376. [DOI] [PubMed] [Google Scholar]
- McMurry, L. M., and S. B. Levy. 1995. The NH2-terminal half of the Tn10-specified tetracycline efflux protein TetA contains a dimerization domain. J. Biol. Chem. 270:22752–22757. [DOI] [PubMed] [Google Scholar]
- Miroux, B., and J. E. Walker. 1996. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 260:289–298. [DOI] [PubMed] [Google Scholar]
- Mus-Veteau, I., and G. Leblanc. 1996. Melibiose permease of Escherichia coli: structural organization of cosubstrate binding sites as deduced from tryptophan fluorescence analyses. Biochemistry. 35:12053–12060. [DOI] [PubMed] [Google Scholar]
- Mus-Veteau, I., T. Pourcher, and G. Leblanc. 1995. Melibiose permease of Escherichia coli: substrate-induced conformational changes monitored by tryptophan fluorescence spectroscopy. Biochemistry. 34:6775–6783. [DOI] [PubMed] [Google Scholar]
- Peleg, G., P. Ghanouni, B. K. Kobilka, and R. N. Zare. 2001. Single-molecule spectroscopy of the β2 adrenergic receptor: observation of conformational substrates in a membrane protein. Proc. Natl. Acad. Sci. USA. 98:8469–8474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pos, K. M., M. Bott, and P. Dimroth. 1994. Purification of two active fusion proteins of the Na+-dependent citrate carrier of Klebsiella pneumoniae. FEBS Lett. 347:37–41. [DOI] [PubMed] [Google Scholar]
- Pos, K. M., and P. Dimroth. 1996. Functional properties of the purified Na+-dependent citrate carrier of Klebsiella pneumoniae: evidence for asymmetric orientation of the carrier protein in proteoliposomes. Biochemistry. 35:1018–1026. [DOI] [PubMed] [Google Scholar]
- Pos, K. M., C. N. Kaestner, and P. Dimroth. 1998. Role of conserved residues in the Na+-dependent citrate carrier of Klebsiella pneumoniae. EBEC Short Reports. 10:A–12. [Google Scholar]
- Prummer, M., C. G. Hubner, B. Sick, B. Hecht, A. Renn, and U. P. Wild. 2000. Single-molecule identification by spectrally and time resolved fluorescence detection. Anal. Chem. 72:443–447. [DOI] [PubMed] [Google Scholar]
- Sahin-Toth, M., M. C. Lawrence, and H. R. Kaback. 1994. Properties of permease dimer, a fusion protein containing two lactose permease molecules from Escherichia coli. Proc. Natl. Acad. Sci. USA. 91:5421–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier, M. H., Jr. 2000. A functional-phylogenetic classification system for transmembrane solute transporters. Microbiol. Mol. Biol. Rev. 64:354–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA. 74:5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schägger, H., and G. Jagow. 1991. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199:223–231. [DOI] [PubMed] [Google Scholar]
- Schroers, A., A. Burkovski, H. Wohlrab, and R. Krämer. 1998. The phosphate carrier from yeast mitochondria. Dimerization is a prerequisite for function. J. Biol. Chem. 273:14269–14276. [DOI] [PubMed] [Google Scholar]
- Simmons, J. S. 1926. A culture medium for differentiating organisms of typhiod-colon aerogenes groups and isolation of certain fungi. J. Infect. Dis. 39:209–241. [Google Scholar]
- Studier, F. W., and B. A. Moffatt. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130. [DOI] [PubMed] [Google Scholar]
- Schutz, G. J., W. Trabesinger, and T. Schmidt. 1998. Direct observation of ligand colocalization on individual receptor molecules. Biophys. J. 74:2223–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate, C. G., E. R. Kunji, M. Lebendiker, and S. Schuldiner. 2001. The projection structure of EmrE, a proton-linked multidrug transporter from Escherichia coli, at 7 Å resolution. EMBO J. 20:77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Geest, M., I. Nilsson, G. von Heijne, and J. S. Lolkema. 1999. Insertion of a bacterial secondary transport protein in the endoplasmic reticulum membrane. J. Biol. Chem. 274:2816–2823. [DOI] [PubMed] [Google Scholar]
- Venkatesan, P., and H. R. Kaback. 1998. The substrate-binding site in the lactose permease of Escherichia coli. Proc. Natl. Acad. Sci. USA. 95:9802–9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q., K. Matsushita, B. de Foresta, M. le Maire, and H. R. Kaback. 1997. Ligand-induced movement of helix X in the lactose permease from Escherichia coli: a fluorescence quenching study. Biochemistry. 36:14120–14127. [DOI] [PubMed] [Google Scholar]
- Weiner, M. P., G. L. Costa, W. Schoettlin, J. Cline, E. Mathur, and J. C. Bauer. 1994. Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene. 151:119–123. [DOI] [PubMed] [Google Scholar]
- Weiss, S. 1999. Fluorescence spectroscopy of single biomolecules. Science. 283:1676–1683. [DOI] [PubMed] [Google Scholar]
- Weitzman, C., T. G. Consler, and H. R. Kaback. 1995. Fluorescence of native single-Trp mutants in the lactose permease from Escherichia coli: structural properties and evidence for a substrate-induced conformational change. Protein Sci. 4:2310–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, K. A., U. Geldmacher-Kaufer, E. Padan, S. Schuldiner, and W. Kählbrandt. 1999. Projection structure of NhaA, a secondary transporter from Escherichia coli, at 4.0 Å resolution. EMBO J. 18:3558–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman, T., S. Williston, J. F. Brandts, and L. N. Lin. 1989. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179:131–137. [DOI] [PubMed] [Google Scholar]
- Wu, J., S. Frillingos, and H. R. Kaback. 1995. Dynamics of lactose permease of Escherichia coli determined by site-directed chemical labeling and fluorescence spectroscopy. Biochemistry. 34:8257–8263. [DOI] [PubMed] [Google Scholar]
- Zhuang, X., T. Ha, H. D. Kim, T. Centner, S. Labeit, and S. Chu. 2000. Fluorescence quenching: a tool for single-molecule protein-folding study. Proc. Natl. Acad. Sci. USA. 97:14241–14244. [DOI] [PMC free article] [PubMed] [Google Scholar]