

Abstract
Fast Ca2+ release kinetics were measured in cardiac sarcoplasmic reticulum vesicles actively loaded with Ca2+. Release was induced in solutions containing 1.2 mM free ATP and variable free [Ca2+] and [Mg2+]. Release rate constants (k) were 10-fold higher at pCa 6 than at pCa 5 whereas Ryanodine binding was highest at pCa ≤5. These results suggest that channels respond differently when exposed to sudden [Ca2+] changes than when exposed to Ca2+ for longer periods. Vesicles with severalfold different luminal calcium contents exhibited double exponential release kinetics at pCa 6, suggesting that channels undergo time-dependent activity changes. Addition of Mg2+ produced a marked inhibition of release kinetics at pCa 6 (K0.5 = 63 μM) but not at pCa 5. Coexistence of calcium activation and inhibition sites with equally fast binding kinetics is proposed to explain this behavior. Thimerosal activated release kinetics at pCa 5 at all [Mg2+] tested and increased at pCa 6 the K0.5 for Mg2+ inhibition, from 63 μM to 136 μM. We discuss the possible relevance of these results, which suggest release through RyR2 channels is subject to fast regulation by Ca2+ and Mg2+ followed by time-dependent regulation, to the physiological mechanisms of cardiac channel opening and closing.
INTRODUCTION
During each cardiac action potential, a small Ca2+ influx through plasma membranes L-type channels initiates the intracellular free [Ca2+] increase responsible for the cyclic contractions of mammalian cardiac muscle. The resulting local increase in free [Ca2+] activates subplasmalemmal sarcoplasmic reticulum (SR) Ca2+-release channels, which, through Ca2+-induced Ca2+ release (CICR), increase cytoplasmic free [Ca2+] to the levels required for contraction (Bers, 2001, 2002). The cytosolic Ca2+ transient reflects the concerted activation of many cardiac specific isoforms of the Ryanodine receptor (RyR) release channels (Otsu et al., 1990; Witcher et al., 1991). Due to their central role in cardiac excitation contraction coupling, the regulation of cardiac release channels (RyR2 channels) has been a matter of extensive investigation (reviewed in Coronado et al., 1994; Zucchi and Ronca-Testoni, 1997). Yet, important questions regarding the regulation of RyR2 channels, such as the physiological mechanisms responsible for channel closing during each cardiac contraction/relaxation cycle, remain unresolved.
By measuring intracellular Ca2+ transients, it has been possible to analyze the response of groups of RyR2 channels in whole cardiac cells (Beuckelmann and Wier, 1988; Blatter et al., 1997). However, cellular regulatory mechanisms do not permit induction of well-defined changes in ionic concentrations (Ca2+, Mg2+), pH, or channel redox status that modify channel activity in vitro. Studies of fast (milliseconds) Ca2+ release fluxes using stopped flow optical techniques has advanced current understanding of the regulation of the skeletal RyR1 channel isoforms. These studies have provided information on how transverse tubule depolarization, Ca2+, Mg2+, and redox state affect channel function (Ikemoto et al., 1994; Yamaguchi et al., 1997; Donoso et al., 2000). In contrast, there are few reports of RyR2-mediated fast Ca2+ release fluxes determined with stopped flow optical techniques (Yano et al., 1998; Ono et al., 2000). The properties of RyR2 channels have been analyzed in vitro using methods that lack the ms time resolution of optical techniques. These methods include assessing [3H]-Ryanodine binding (Xu et al., 1996; Liu et al., 1998; Fruen et al., 2000), characterizing single channel activity in bilayers (Laver et al., 1995, 1997; Du et al, 2001), or measuring Ca2+ release fluxes with other procedures (Meissner et al., 1986; Rousseau et al., 1986; Xu et al., 1996; Fruen et al., 2000).
Measurements with ms temporal resolution are likely to better reflect the physiological response of RyR2 channels to the sudden [Ca2+] changes that occur during cardiac channel activation (Bers, 2001, 2002). Stopped flow systems allow mixing vesicles with releasing solutions in less than 2 ms, and yield data with high temporal resolution because records are collected with several hundred data points. Determinations of fast Ca2+ release fluxes have the additional advantage that the rate constant of Ca2+ release correlates with average open probability and conductance of the releasing channels (Miller, 1984; Donoso et al., 1995; Mészáros et al., 1998). For these reasons, in this study we measured fast Ca2+ release fluxes to characterize how the RyR2 channel population present in cardiac SR vesicles responds in different conditions to sudden [Ca2+] changes. Our results show maximal activation of release kinetics at pCa 6 in the absence of free [Mg2+], with release rate constants as high as 110 s−1, whereas maximal activation of [3H]-Ryanodine binding required pCa ≤5. Furthermore, at pCa 6, Ca2+ release exhibited double exponential kinetics, regardless of variations in luminal calcium content from <10 nmol/mg protein to 80 nmol/mg protein. These results suggest that the double exponential behavior is not due to a decrease in luminal calcium content but probably reflects an intrinsic time-dependent decrease in cardiac release channel activity after activation. Addition of up to 0.8 mM free [Mg2+] did not modify release kinetics at pCa 5 but markedly inhibited release rate constants at pCa 6, with K0.5 = 63 μM. Caffeine (6 mM) reversed the inhibition exerted by 0.3 mM free [Mg2+] at pCa 6. Addition of thimerosal activated release kinetics reversibly at pCa 5, but had a modest effect on channel inhibition by Mg2+ at pCa 6.
We discuss the possible relevance of these results, including modulation of channel activity by redox modification, to the physiological mechanisms that underlie RyR2 channel opening and closing.
MATERIALS AND METHODS
Isolation of SR vesicles
Sarcoplasmic reticulum vesicles were isolated from canine hearts following a modification of previous methods (Inui et al., 1988). All procedures were done according to the “Position of the American Heart Association on Research and Animal Use” and the guidelines of the Animal Care Committee of the Faculty of Medicine, University of Chile. Briefly, dogs of either sex weighing 12–16 kg were anaesthetized with pentobarbital. The hearts were quickly removed and perfused through the aorta with a cold cardioplegic solution (in mM): 2 CaCl2, 16 MgCl2, 102 NaCl, 20 KCl, 2 EGTA, 20 MOPS adjusted with Tris base to pH 6.8. All the following procedures were carried out at 4°C. The left ventricle was finely minced and homogenized in four volumes of ice-cold homogenization buffer. This solution contained (in mM): 300 sucrose, 20 MOPS adjusted with Tris base to pH 6.8, and a combination of protease inhibitors (4 μg/ml Leupeptin, 4 μg/ml Pepstatin A, 1 mM Benzamidine, 1 mM phenylmethylsulfonyl fluoride). Tissues were homogenized in a Heidolph Diax 600 homogenizer using three 15 s bursts at 24,000 rpm followed each time by 5 s rest intervals. The homogenate was sedimented at 3800 × g for 15 min and the resulting pellet was homogenized in three volumes of homogenization buffer as above, and sedimented at 3800 × g for 15 min. The combined supernatants were filtered through cheesecloth and were sedimented at 28,000 × g for 15 min. Solid KCl was added to the supernatant to a final concentration of 0.65 M before centrifugation at 120,000 × g for 1 h. The pellet, enriched in SR vesicles, was resuspended in homogenization buffer to a final protein concentration of 10 mg per ml, fractionated in small aliquots and snap-frozen in liquid N2. Vesicles were stored at −80°C for up to 2–3 weeks.
Active loading of cardiac SR vesicles with calcium
SR vesicles (1 mg/ml) were added to a solution containing (in mM): 0.05 CaCl2, 100 KCl, 10 phosphocreatine, 15 U/ml creatine kinase, 20 imidazole-MOPS, pH 7.2, and 0.1 μM Calcium Green 2 or Calcium Green 5N. Calcium uptake was determined at 25°C by measuring extravesicular free [Ca2+] changes in a fluorescence spectrophotometer (JOBIN YVON-SPEX FluoroMax-2). Uptake, initiated by adding a small volume of Mg-ATP (2 mM ATP, 3 mM MgCl2) was completed in <20 min. After cessation of uptake, vesicles retained the accumulated Ca2+ for periods ≥30 min. To assess the effect of intravesicular Ca2+ load on Ca2+ release, vesicles were actively loaded in three different conditions: with no CaCl2 added, with 50 μM CaCl2 or with 100 μM CaCl2. The average free [Ca2+] of uptake solutions with no added CaCl2 was 5 μM, and increased to 9 μM after addition of 1 mg/ml SR vesicles, as determined by measuring the free [Ca2+] of the filtrate as described below, using Calcium Green 5N (Kd = 27.5 μM). To assess the calcium content acquired by SR vesicles after active loading, vesicles were incubated as above, except that solutions contained 45CaCl2 (specific activity 0.5 μCi/μmol). Vesicular calcium was determined by manual filtration through Millipore filters (AA 0.8 μm), as described in detail previously (Donoso et al., 2000). Vesicles (1 mg/ml) incubated in solutions with 50 μM or 100 μM free [Ca2+] displayed significant nonspecific calcium binding, which amounted to 25% of the total amount of calcium actively accumulated. To determine the free [Ca2+] remaining in the uptake solution after cessation of uptake, vesicles (1 mg/ml) were removed from the solution by manual filtration through MFS-25 filters (0.20 μm, Advantec MFS, Pleasanton, CA). The free [Ca2+] of the filtrate was determined fluorometrically with Calcium Green 2 (Kd = 0.44 μM).
Calcium release kinetics
To follow Ca2+ release kinetics, a SX.18MV fluorescence stopped-flow spectrometer from Applied Photophysics Ltd. (Leatherhead, UK) was used. The increase in extravesicular [Ca2+] was determined with different fluorescent Ca2+ indicators. Fluo 3 was used in the pCa range 6.9–6.3, Calcium Green 2 at pCa 6.3–6, and Calcium Green 5N in the pCa range 6–4.8. Whenever possible, two indicators with different Kd values were used for a given pCa value. Dye fluorescent emission was measured through a 515-nm cutoff long-pass filter, using an excitation wavelength of 488 nm. Calcium release was initiated by mixing 10 volumes of releasing solution with 1 volume of a solution containing Ca2+-loaded SR vesicles. Releasing solutions were designed to generate after mixing a constant free [ATP] of 1.2 mM and different free [Ca2+] to cover the pCa range 6.9–4.8. Higher free [Ca2+] were not investigated because the signal generated by the released Ca2+ was obscured when the free [Ca2+] of the releasing solution was ≥15 μM. All releasing solutions contained (in mM): 100 KCl, 20 imidazole-MOPS, pH 7.2, 1 μM fluorescent Ca2+ indicator and variable free concentrations of Ca2+ and Mg2+. The procedures followed to calculate free [Ca2+] and [Mg2+] and to determine the resulting free [Ca2+] of releasing solutions were described in detail previously (Donoso et al., 2000).
Other procedures
[3H]-Ryanodine binding was measured at pCa 5 as described (Bull et al., 1989), except that we used lower ionic strength to compare Ryanodine binding and release kinetics in similar conditions. The composition of the solution was (in mM): 150 KCl, 0.5 AMP-PNP, 20 MOPS-Tris, pH 7.2, and variable concentrations of free [Ca2+] and [Mg2+]. Total binding was measured in the presence of 10 nM [3H]-Ryanodine (New England Nuclear, Boston, MA). Nonspecific binding was measured in the presence of 10 nM [3H]-Ryanodine plus 10 μM Ryanodine. Protein concentrations were determined as described (Hartree, 1972), using commercial bovine serum albumin as standard.
Materials
All reagents used were of analytical grade. Thimerosal, dithiothreitol, ATP, AMP-PNP, Ryanodine, bovine serum albumin, and protease inhibitors (Leupeptin, Pepstatin A, benzamidine, and phenylmethylsulfonyl fluoride) were obtained from Sigma Chemical Company (St. Louis, MO). All fluorescent calcium indicators were from Molecular Probes (Eugene, OR).
RESULTS
Ryanodine binding
The SR vesicles used in this study had a significant density of Ryanodine binding sites, with values ranging ∼4 pmol per mg of protein when measured in 10 μM free [Ca2+]. These values are comparable to the highest values measured in similar ionic conditions in cardiac microsomal SR preparations isolated with equivalent procedures to the one described in this work (Xu et al., 1999; Yano et al., 2000; Doi et al., 2002). [3H]-Ryanodine binding was negligible at free [Ca2+] <0.1 μM and exhibited a bell-shaped response to Ca2+, reaching the highest binding in a wide free [Ca2+] range, from 10 μM to 1 mM (Fig. 1, solid circles). Increasing free [Ca2+] to 10 mM led to 70% inhibition in [3H]-Ryanodine binding. Addition of 0.8 mM free [Mg2+] decreased [3H]-Ryanodine binding at pCa 6 but did not modify binding in the pCa range 5–2 (Fig. 1, open circles).
FIGURE 1.

Calcium dependence of Ryanodine binding in cardiac SR vesicles; specific [3H]-Ryanodine binding was determined by incubating cardiac SR vesicles with 10 nM [3H]-Ryanodine in solutions containing the indicated pCa and the compositions detailed in the text. (Solid circles) control; (open circles) free [Mg2+] = 0.8 mM. Data represent mean ± SE from independent determinations in three different preparations.
Active calcium accumulation and total calcium released
Isolated cardiac SR vesicles (1 mg/ml) incubated in 50 μM CaCl2 accumulated on average 41 ± 2 nmol Ca2+ per mg protein (mean ± SD, n = 5). Addition of 1.5 μM thapsigargin produced complete inhibition of active Ca2+ transport; in contrast, 5 mM sodium azide had no effect. These results indicate that active Ca2+ uptake was carried out by the cardiac SERCA2 pump and not by a mitochondrial contaminant. Vesicles actively loaded in 100 μM CaCl2 increased their calcium load to 80 ± 6 nmol/mg protein (mean ± SD, n = 11). After cessation of calcium uptake with no added CaCl2, or with 50 μM or 100 μM CaCl2, the extravesicular free [Ca2+] was 0.35 ± 0.4 μM (mean ± SE, n = 6). These results indicate that when loaded at 1 mg/ml vesicles accumulated all the free [Ca2+] present in the extravesicular solution. In particular, these results indicate that vesicles loaded with no added CaCl2 (9 μM free [Ca2+]) accumulated ∼7 nmol/mg of protein (considering 2 nmol/mg of nonspecific binding).
After dilution in releasing solutions, vesicles actively loaded in the presence of 50 or 100 μM CaCl2 released, on average, 50% of the total amount of calcium taken up.
Ca2+ release kinetics
All release records followed exponential functions with rate constants k. The release records obtained at pCa 6 or at pCa 5 followed double exponential functions, characterized by a fast rate constant k1 and a slower rate constant k2, which differed in magnitude by at least fivefold (see below). Accordingly, in these conditions we considered the higher k1 values as an indication of the channel response to a sudden [Ca2+] change. In conditions that promoted very low release activity, such as pCa ≥6.5 or pCa <5 or in the presence of high free [Mg2+] (see below), some release records seemingly followed single exponential functions. Results obtained at pCa 6 or at pCa 5 are detailed below.
pCa 6
The time courses of Ca2+ release at pCa 6, collected at time frames of 0.1, 0.5, and 5 s, are shown in Fig. 2. On average (n = 6) the initial fast exponential component, with k1 = 113 ± 30 s−1, was responsible for 50 ± 16% of the total fluorescence change, whereas the second exponential component, which accounted for the remaining 50%, had a rate constant k2 = 10.0 ± 0.3 s−1 (Fig. 2, A and B). In about half the vesicular preparations, the double exponential increase in fluorescence with time observed at pCa 6 was followed by a fluorescence decrease, which was more evident at longer times of data acquisition (Fig. 2 C). These findings suggest that release channels closed after fast activation by 1 μM [Ca2+], allowing the Ca2+ pump to recapture the released Ca2+ back into the SR.
FIGURE 2.
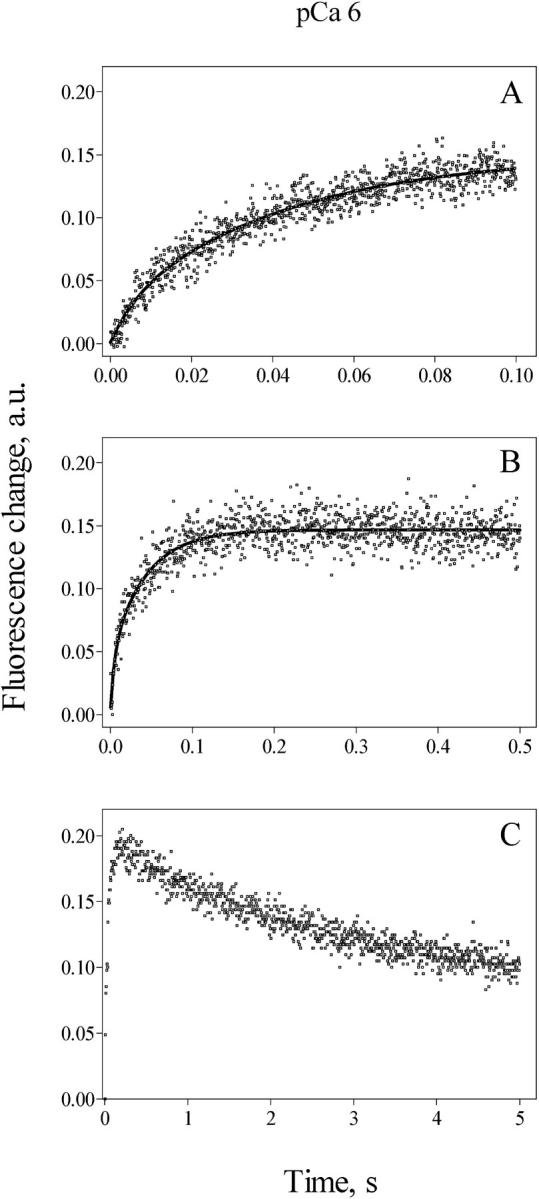
Calcium release kinetics determined at pCa 6. Cardiac SR vesicles (1 mg/ml) actively loaded with calcium were mixed (1:10) in a stopped flow fluorescence spectrometer with a solution designed to obtain after mixing 1.2 mM free [ATP] and pCa 6, as detailed in the text. Calcium release, measured as the change in fluorescence of Calcium Green 5N, followed a double exponential function. Values of k1 = 110 s−1 and k2 = 20 s−1 were obtained in the 0.1 s record illustrated in A. Values of k1 = 110 s−1 and k2 = 22 s−1 were obtained from the 0.5 s record illustrated in B. (C) Calcium release and recapture are shown in a record lasting 5 s. Records represent the average of five to eight determinations.
We investigated next whether partial calcium depletion from the vesicular lumen originated the double exponential kinetic behavior of Ca2+ release. For this purpose, we compared release kinetics at pCa 6 from vesicles actively loaded in solutions with different free [Ca2+]. When incubated in 50 μM, 100 μM, or with no CaCl2 added, vesicles reached significantly different luminal calcium contents, as indicated above. Yet, in all three cases vesicles exhibited double exponential Ca2+ release kinetics (Fig. 3). The values of k1 and k2 were 98 s−1 and 7 s−1, respectively, for vesicles incubated in 100 μM CaCl2 (Fig. 3, upper panel). The values of k1 and k2 were 111 s−1 and 8.3 s−1, respectively, for vesicles incubated in 50 μM CaCl2 (Fig. 3, middle panel). The first release exponential obtained in vesicles loaded either in 50 μM or in 100 μM CaCl2 tended to zero in 40 ms, when the respective luminal calcium contents were 27 nmol/mg or 55 nmol/mg protein (see Discussion). Vesicles incubated with no added CaCl2 presumably attained much lower luminal calcium contents, in the range of only 7 nmol/mg, and had values of k1 and k2 of 62 s−1 and 4 s−1, respectively (Fig. 3, lower panel). Yet in all three cases the first exponential ceased in <60 ms, whereas the second exponential was virtually ended at 500 ms (Fig. 3 B). These results suggest strongly that a decrease in luminal calcium content is not responsible for the double exponential behavior of Ca2+ release.
FIGURE 3.
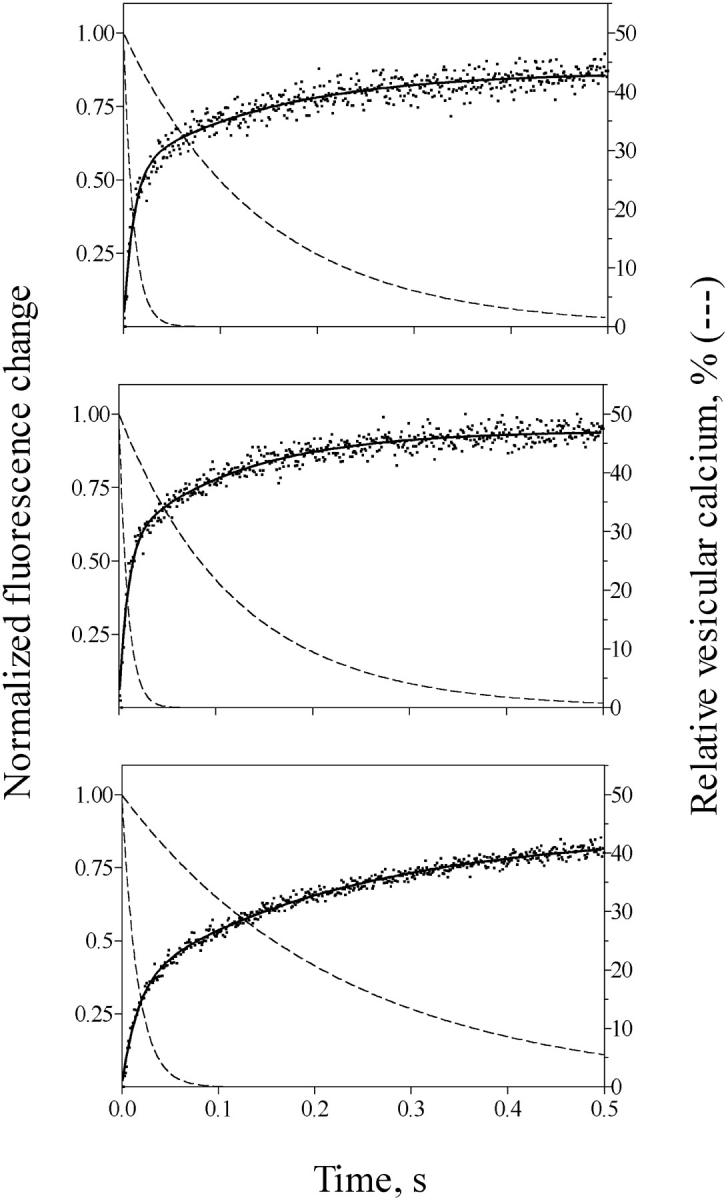
Effect of intravesicular Ca2+ load on Ca2+ release kinetics. Cardiac SR vesicles were actively loaded by incubation in solution with 100 μM CaCl2 (upper panel), with 50 μM CaCl2 (middle panel), or with no added CaCl2 (lower panel). Calcium release was induced at pCa 6 and 1.2 mM free ATP. Release records exhibited double exponential kinetics in all cases, with rate constants k1 = 98 s−1 and k2 = 7 s−1 for vesicles incubated in 100 μM CaCl2. The values were k1 = 111 s−1 and k2 = 8.3 s−1 for Ca2+ release by vesicles incubated in 50 μM CaCl2, and k1 = 62 s−1 and k2 = 4.4 s−1 for vesicles incubated in solution with no added CaCl2. Records represent the average of six to eight determinations. The dashed lines illustrate the corresponding theoretical first and second exponential curves of intravesicular calcium content decay (in %) with time, considering each component contributed 50% to the total.
pCa 5
A double exponential increase in fluorescence with time was also evident at pCa 5. Fig. 4 A shows a 1 s record obtained at pCa 5, with k1 = 12.1 s−1 and k2 = 2.8 s−1. On average (n = 8), the initial fast exponential component had a rate constant k1 = 11 ± 4 s−1 and contributed 43.4 ± 12.3% to the total fluorescence change, whereas the second exponential component had an average rate constant k2 = 1.8 ± 1.3 s−1. In contrast to the behavior observed at pCa 6, the double exponential increase in fluorescence observed at pCa 5.0 was never followed by a fluorescence decrease, even after registering for times as long as 20 s. These results indicate that channels activated at pCa 5 decreased their activity but did not close completely after 20 s, in contrast to the behavior observed at pCa 6. The 20 s Ca2+ release record illustrated in Fig. 4 B, obtained at pCa 5, shows a slower steady increase in fluorescence with a third rate constant of 0.06 s−1. Such low values of rate constants were not taken into account in subsequent experiments.
FIGURE 4.
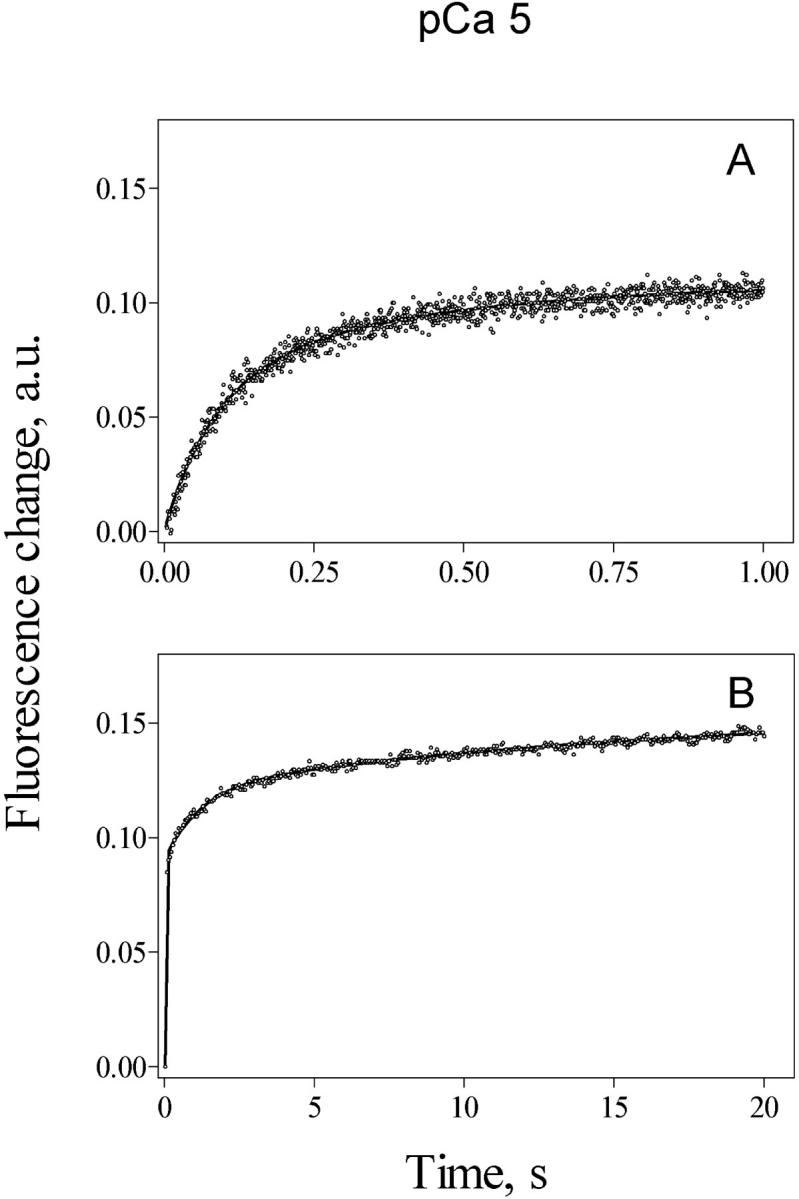
Calcium release kinetics determined at pCa 5. Cardiac SR vesicles (1 mg/ml) actively loaded with calcium were mixed (1:10) in a stopped flow fluorescence spectrometer with a solution designed to obtain, after mixing, 1.2 mM free [ATP] and pCa 5, as stated under Methods. Calcium release, measured as the change in fluorescence of Calcium Green 5N with time, followed a double exponential function with k1 = 12.1 s−1 and k2 = 2.8 s−1 in the 1 s record illustrated in A. The longer fluorescent record illustrated in B exhibited a third rate constant of 0.06 s−1 that contributed in 3.2% to the total fluorescence change illustrated in the record. Records represent the average of five to eight determinations.
Calcium dependence of Ca2+ release
A bell-shaped curve characterized the Ca2+ dependence of the release rate constants (Fig. 5 A). The highest k1 value, 113 ± 30 s−1 (n = 6), was obtained at pCa 6; a marked decrease in release rate constants was observed at lower or higher pCa values. Thus k had an average value of 5.1 s−1 at pCa 6.8 (n = 2), whereas at pCa 4.8, the highest free [Ca2+] that could be reliably studied, k was 15.4 s−1 (n = 2). The bell-shaped curve illustrated in Fig. 5 A was generated by a combination of two independent functions (Marengo et al., 1998), a cooperative Ca2+ activation function with Ka = 0.6 μM and na = 2.8, and a cooperative Ca2+ inactivation function, with Ki = 0.9 μM and ni = 1.6. In experiments where the fluorescence followed a double exponential increase, the rate constant of the slower phase also showed a bell-shaped dependence on extravesicular [Ca2+], reaching its highest values at pCa 6 (data not shown).
FIGURE 5.

Calcium dependence of Ca2+ release rate constants. Cardiac SR vesicles (1 mg/ml) actively loaded with calcium were mixed (1:10) in a stopped flow fluorescence spectrometer with solutions designed to obtain, after mixing, 1.2 mM free [ATP] and the indicated pCa values. Fluo 3 was used for determinations at pCa 6.8 and 6.5, Calcium Green 2 for pCa 6.3 and 6.2, and Calcium Green 5N for pCa ≤6. Release rate constants values were obtained from Ca2+ release records as described in the text as (A) in the presence of nominally free (10 μM) [Mg2+] or (B) in 0.8 mM free [Mg2+]. Data represent Mean ± SE from independent determinations in two to six different preparations. The solid line through the data was obtained with the equation k = kmax/(1+(Ka/[Ca2+])na + ([Ca2+]/Ki)ni). The best fit parameters for the curve shown in A were: kmax = 300 s−1, Ka = 0.6 μM, Ki = 0.9 μM, na = 2.8, ni = 1.6. The best fit parameters for the curve of B were: kmax = 300 s−1, Ka = 1.9 μM, Ki = 0.25 μM, na = 2, ni = 1.
Effects of Mg2+ on Ca2+ release kinetics
Previous reports have shown that RyR2 channels are less sensitive to Mg2+ inhibition than skeletal RyR1 channels. However, these results have been obtained by methods that essentially represent equilibrium or steady-state measurements, and studies on the effects of Mg2+ in the time range of physiological Ca2+ release are lacking.
To investigate the effects of Mg2+ on the calcium dependence of release kinetics, we carried out experiments in the presence of 1.2 mM free [ATP] plus 0.8 mM free [Mg2+] to approach physiological conditions. The results illustrated in Fig. 5 B indicate that 0.8 mM free [Mg2+] had a marked effect on the calcium dependence of the release rate constant. The maximal values of k1 obtained at pCa 6 decreased markedly, from 113 s−1 to 13 s−1, approaching k values at pCa 5. It has been reported that Mg2+ competes effectively with Ca2+ for the high affinity activation sites of both RyR1 and RyR2 channels when measured in steady state conditions (Laver et al., 1997). The present kinetic results support this report because 0.8 mM free [Mg2+] essentially abolished the strong stimulatory effect of 1 μM free [Ca2+] on release kinetics. Addition of Mg2+ also produced a shift to the right in the calcium-dependence curve; Ka increased from 0.6 μM to 1 μM and Ki from 0.9 μM to 2.6 μM. However, the significant dispersion of the experimental points precludes a more detailed analysis of this shift.
To investigate the effects of varying free [Mg2+] on release kinetics, we carried out experiments at pCa 6 or pCa 5 while increasing free [Mg2+] up to 0.8 mM. Further increase in free [Mg2+] was not feasible while maintaining constant free [Ca2+] and free [ATP].
The records illustrated in Fig. 6, A and B show that increasing free [Mg2+] from 10 μM to 300 μM in the presence of 1.2 mM free [ATP] slowed down release kinetics at pCa 6, reducing k1 to 40 s−1, and k2 to 3.7 s−1. The experiment illustrated in Fig. 6 C shows a single exponential record obtained in 0.8 mM [Mg2+], with a rate constant k = 5.3 s−1. Fig. 7 illustrates the decrease in k values caused by increasing free [Mg2+] from 10 μM to 0.8 mM. Half-maximal inhibition of k values was obtained at free [Mg2+] = 63 ± 28 μM, showing that increasing free [Mg2+] strongly inhibited RyR2 channels activated by 1 μM free [Ca2+].
FIGURE 6.
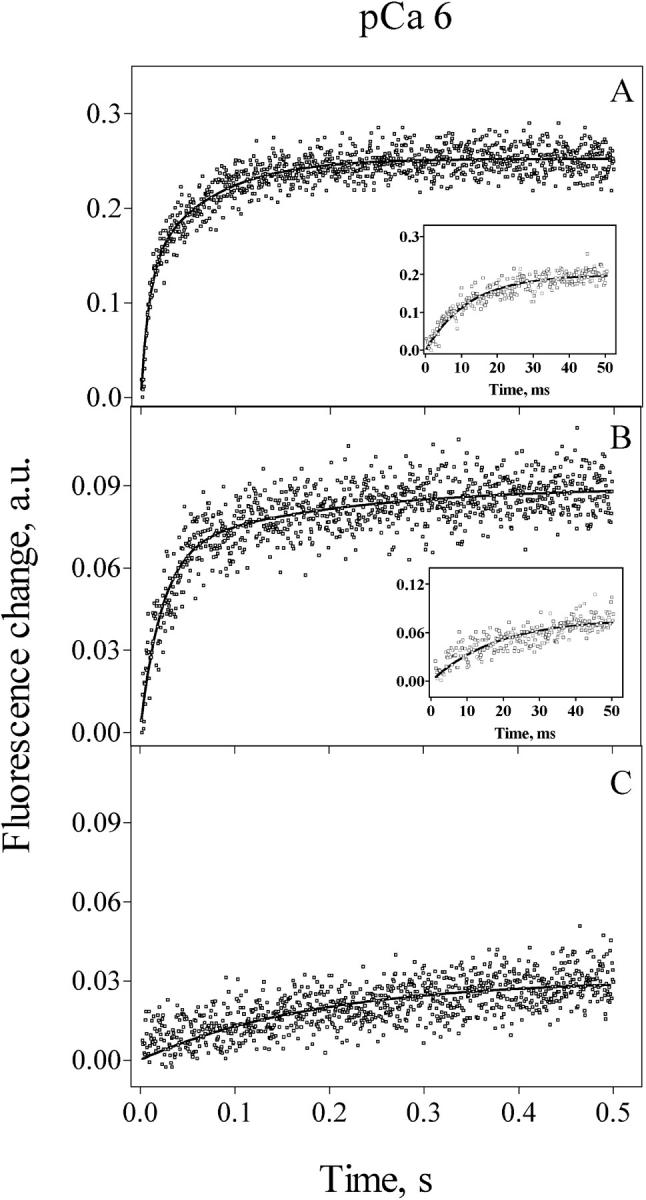
Effect of [Mg2+] on Ca2+ release kinetics. Calcium release was induced by mixing 1 volume of actively loaded cardiac SR vesicles with 10 volumes of releasing solutions designed to obtain, after mixing, 1.2 mM free [ATP] and various [Mg2+] at pCa 6. (A) Calcium release in 13.6 μM [Mg2+] exhibited rate constants of 111 s−1 and 14.5 s−1. The inset shows the initial 50 ms of Ca2+ release. (B) Calcium release in 305 μM [Mg2+] exhibited rate constants of 40 s−1 and 3.7 s−1. The inset shows the initial 50 ms of Ca2+ release. (C) Calcium release in 0.8 mM [Mg2+] followed a single exponential increase in fluorescence with a rate constant of 5.3 s−1. Records represent the average of six to eight determinations.
FIGURE 7.

Mg2+ inhibition of Ca2+ release rate constants at pCa 6 and pCa 5. Rate constants of Ca2+ release of the faster exponential obtained in the presence of different [Mg2+] were calculated from fluorescence records such as the records shown in Fig. 6. Data represent Mean ± SE from independent determinations in three to seven different preparations.
It is interesting to note that, even in 0.3 mM free [Mg2+], very fast Ca2+ release could still be elicited at pCa 6 by addition of 6 mM caffeine. Fig. 8 shows the double exponential release records obtained at pCa 6, 0.3 mM free [Mg2+], with or without 6 mM caffeine. Caffeine addition increased k1 from 30 s−1 to 83 s−1, and k2 from 0.2 s−1 to 4.3 s−1. The k1 value obtained in 0.3 mM free [Mg2+] plus 6 mM caffeine, 83 s−1, is comparable within experimental error to the k1 values of 113 ± 30 s−1 obtained at pCa 6 in the absence of Mg2+ (see Fig. 7). Thus caffeine was able to overcome the strong inhibition exerted by Mg2+ on RyR2 channels activated by 1 μM free [Ca2+].
FIGURE 8.

Effect of Caffeine on Ca2+ release kinetics in the presence of Mg2+. Calcium release was induced by mixing 1 volume of actively loaded cardiac SR vesicles with 10 volumes of a releasing solution designed to obtain, after mixing, 1.2 mM free [ATP] and 305 μM [Mg2+] at pCa 6. Releasing solutions had no caffeine (lower record) or contained 6 mM caffeine (upper record). Release rate constants in the absence of caffeine were k1 = 30 s−1 and k2 = 0.2 s−1. After addition of 6 mM caffeine the values of release rate constants were k1 = 83 s−1 and k2 = 4.3 s−1. Records represent the average of six to eight determinations.
At pCa 5 and in the free [Mg2+] range from 10 μM up to 0.8 mM, all preparations displayed Ca2+ release time courses that followed a double exponential function. In contrast to the results obtained at pCa 6, increasing free [Mg2+] up to 0.8 mM had little effect at pCa 5 on the values of the faster rate constants k1 (Fig. 7) or k2 (data not shown). These results are in agreement with previous reports indicating that at pCa 5 Mg2+ mostly binds to the low affinity inhibitory site of RyR2 channels without interfering with the Ca2+ activation site that at pCa 5 is saturated with Ca2+ (Laver et al., 1997).
Effects of thimerosal on Ca2+ release kinetics
Thimerosal, which presumably affects the redox state of RyR2 channels through reaction with free sulfhydryl residues, significantly enhances Ca2+ activation of single RyR2 channels in the absence of Mg2+ (Marengo et al, 1998) and diminishes the inhibitory effects of Mg2+ on release kinetics in skeletal SR (Donoso et al., 2000). To investigate the effects of thimerosal on RyR2-mediated calcium release, we measured release kinetics at pCa 6 or 5 at different free [Mg2+].
pCa 6
Incubation of cardiac SR vesicles with thimerosal did not modify release kinetics at pCa 6 when measured in free [Mg2+] <100 μM, yielding k values undistinguishable from those obtained in control vesicles (Fig. 9). However, we observed a modest stimulation of release kinetics at higher free [Mg2+], reflected in an increase in k values when compared to controls (Fig. 9). Incubation with thimerosal increased twofold the K0.5 for Mg2+ inhibition of k values, from 63 ± 28 μM to 136 ± 34 μM. These results suggest that thimerosal, presumably through oxidation, diminishes the strong inhibitory effect of Mg2+ on channel activation by 1 μM [Ca2+].
FIGURE 9.

Effect of Mg2+ on Ca2+ release rate constants in oxidized vesicles. Cardiac SR vesicles actively loaded with Ca2+ were incubated for 5 min with 250 μM thimerosal and release was induced at different [Mg2+] and 1.2 mM free [ATP] at pCa 5 and 6. Release rate constant values were obtained from Ca2+ release records as described in the text. Data represent Mean ± SE from independent determinations in three to five different preparations.
pCa 5
On average, vesicles incubated with thimerosal displayed at pCa 5 and in the absence of Mg2+ higher rate constants of Ca2+ release, 34.9 ± 6.3 s−1 (n = 3), than control vesicles. Furthermore, incubation of vesicles with thimerosal produced an increase in the release rate constant k1 over the whole range of Mg2+ concentrations tested, with no signs of inhibition (Fig. 9).
We also found that the effect of thimerosal was reversible provided the time of exposure to thimerosal was limited. Fig. 10 illustrates an experiment where native channels displayed, at pCa 5, a k1 value of 12 s−1. After incubation of vesicles for 5 min with 0.25 mM thimerosal, k1 increased to 29 s−1. A second 5 min incubation with 5 mM dithiothreitol produced a marked decrease in k1, to a value of 7 s−1. These results show that reducing agents not only reversed the activation of release kinetics by thimerosal but also produced a strong inhibition of CICR. Similar inhibition of RyR2 channels by reducing agents has been described in single channel experiments (Marengo et al., 1998).
FIGURE 10.
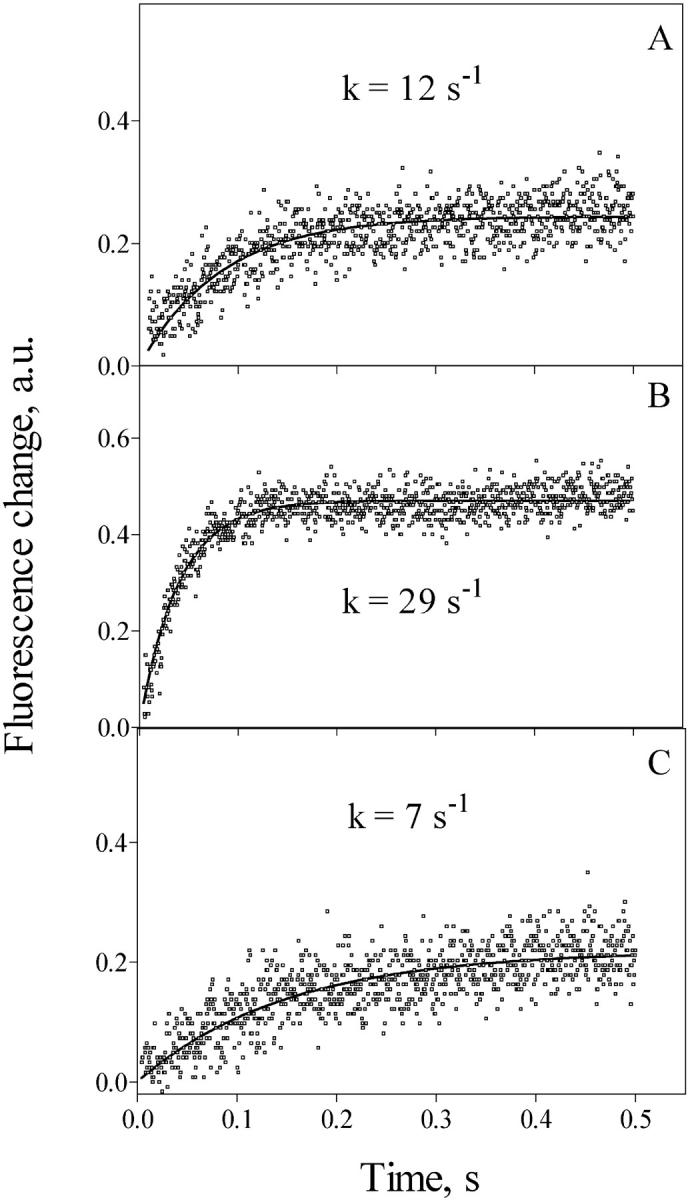
Reversibility of the effects of thimerosal. Calcium release was induced at pCa 5, 1.2 mM free ATP, 0.8 mM free [Mg2+]. (A) Native cardiac SR vesicles. (B) Vesicles incubated with 250 μM thimerosal for 5 min. (C) Vesicles incubated with 250 μM thimerosal for 5 min and then for 5 min with 5 mM dithiothreitol.
DISCUSSION
Ryanodine binding studies and determinations of the activity of single channels incorporated in lipid bilayers have been the methods most commonly used to characterize the Ca2+ dependence of RyR2 channels. These methods, however, are not equivalent to the determination of fast Ca2+ release fluxes in isolated vesicles because they measure the response of the channels in slower time frames. Ryanodine binding measurements are done at equilibrium, and steady-state conditions are used most often to characterize the activity of single channels. However, steady-state measurements may not reveal all the aspects of cellular channel regulation because RyR2 channels function in a dynamic calcium signaling environment where cytoplasmic free [Ca2+] increases and decreases in the ms time scale. Determinations of fast Ca2+ release kinetics provide information on how a channel population responds in the ms temporal scale to changes in the composition of the extravesicular solution. Accordingly, we used this technique to investigate how Ca2+, Mg2+ and channel redox state influence release kinetics from SR vesicles isolated from cardiac muscle. The results obtained suggest that RyR2 channel-mediated release is subject to two different types of regulation: fast regulation of channel activity by Ca2+ and Mg2+, followed by time-dependent regulation, which is also modulated by Ca2+ and Mg2+. We will analyze time-dependent regulation first.
Time-dependent changes in Ca2+ release fluxes
At pCa 6 or 5, all release records followed double exponential functions. In all cases, the first exponential contributed ∼50% to total calcium release. This behavior is not a consequence of inhibition of channel activity by an extravesicular [Ca2+] increase. All releasing solutions contained 1.2 mM free ATP, which, in our experimental conditions, limited the total increase in free [Ca2+] to less than 0.3 μM. (Please note that the final protein concentration in all release experiments was 90 μg per ml. Vesicles actively loaded with 100 μM [Ca2+], the highest concentration used, had released 27 nmol/mg of protein after the completion of the first exponential; see Fig. 11. This release would cause an increase in total extravesicular [Ca2+] of only 2.4 nmol/ml. Inasmuch as all releasing solutions contained 1.2 mM free [ATP], the calculated increase in free [Ca2+] was <0.3 μM.) We can also dismiss the combined presence, in equal proportions, of vesicles containing channels highly activated by Ca2+ with vesicles containing less responsive channels as the source of the double exponential release kinetics. Cardiac SR vesicles contain only RyR2 channels, and it is very unlikely that all preparations contained exactly one-half each channel kind.
FIGURE 11.
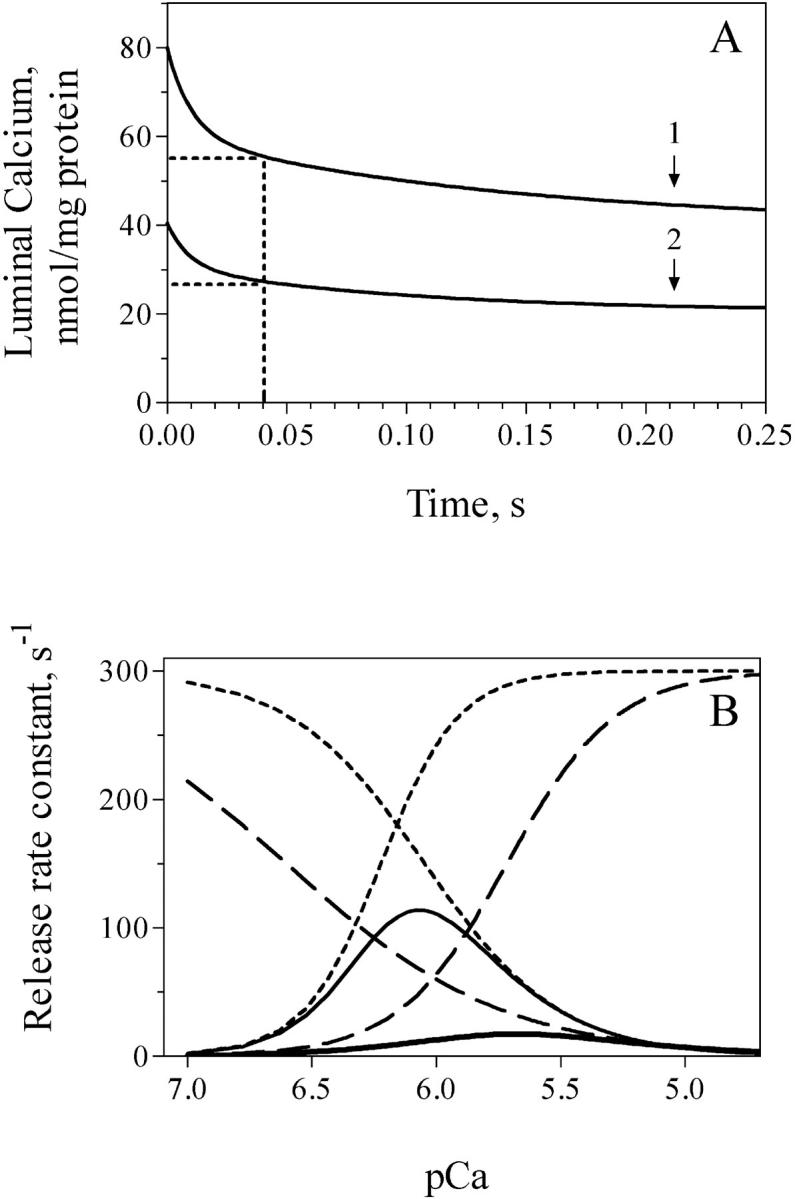
(A) Theoretical curves depicting the variation in luminal calcium content as a function of time. A function representing the sum of two exponential decay functions (N = N1 exp(−k1 t) + N2 exp(−k2 t) + basal) was generated. The following parameters correspond to vesicles actively loaded in 100 μM CaCl2 (curve 1) or in 50 μM CaCl2 (curve 2). (Curve 1) k1 = 98 s−1, k2 = 7 s−1, N1 = N2 = 20 nmol/mg protein, basal = 40 nmol/mg protein. (Curve 2) k1 = 110 s−1, k2 = 10 s−1, N1 = N2 = 10 nmol/mg protein, basal 20.5 nmol/mg protein. (B) Theoretical calcium-dependence curves of calcium release rate constants. The bell-shaped curves (solid lines) correspond to the calcium dependencies of k measured in the absence or in the presence of 0.8 mM free [Mg2+] shown in Fig. 5, A and B. (Dotted lines) Illustrate the individual activation and inactivation functions generated in the absence of [Mg2+]. (Dashed lines) The functions corresponding to 0.8 mM [Mg2+]. For further details, see article text and Fig. 5 legend.
A decrease in luminal (trans) [Ca2+] inhibits calcium release mediated by skeletal RyR1 channels (Donoso et al., 1995) and the activity of single RyR2 channels in bilayers (Sitsapesan and Williams, 1995). Accordingly, to investigate if the double exponential behavior was caused by luminal calcium decrease, we measured Ca2+ release kinetics at pCa 6 in vesicles with different calcium contents. Vesicles loaded with no added CaCl2, which at most attained total luminal calcium contents of 7 nmol/mg of protein, had by 60 ms completed the first exponential, which exhibited a reduced k1 value (two-thirds) when compared to vesicles loaded at higher [CaCl2]. The lower intravesicular calcium content attained by these vesicles could account for this reduction in k1. Vesicles actively loaded in 50 μM or 100 μM [Ca2+], which attained much higher calcium contents, exhibited first exponential components that ceased by 40 ms (Fig. 3). Theoretical simulations give intravesicular calcium contents of 27 nmol/mg or 55 nmol/mg of protein, respectively, at this time (Fig. 11 A). Hence, we can rule out luminal calcium decrease as the cause of the double exponential behavior, because release kinetics changed even in vesicles that contained significant luminal calcium. We can extrapolate this conclusion to experiments done at pCa 5, or at different pCa values and varying free [Mg2+], because in all cases the first exponential ceased when vesicles contained significant luminal calcium contents.
We propose that a time-dependent decrease in RyR2 channel open probability as the most likely explanation for the double exponential kinetic behavior of release fluxes. Single RyR2 channels display spontaneous changes in channel activity, a process known as modal gating. Periods of high open probability (H-mode) alternate with periods of low (L-mode) or no activity (I-mode) (Zahradníková and Zahradník, 1995, 1996; Armisén et al., 1996). Modal gating could account for the phenomenon of adaptation exhibited by RyR2 channels in bilayers in response to a step increase in [Ca2+] generated by flash photolysis of DM-nitrofen (Györke and Fill, 1993; Valdivia et al., 1995). After a fast increase in free [Ca2+], channels would open first in the H-mode and would adopt at later times the L-mode to reach equilibrium between both modes in the steady state (Zahradníková et al., 1999). Our findings are compatible with an adaptation process. Furthermore, channel experiments in bilayers indicate that Mg2+ accelerates channel adaptation after a step increase in [Ca2+] from 0.1 μM to 1 or 10 μM (Valdivia et al., 1995). We found that increasing free [Mg2+] caused a progressive decrease in the rate constant of the first release exponential at pCa 6 but not at pCa 5 (see below). These results suggest that at pCa 6 increasing free [Mg2+] would accelerate the conversion of channels from a high to a lower open probability mode. However, adaptation has been questioned (Sitsapesan et al., 1995; Lamb et al., 2000) and other reports are consistent with inactivation rather than adaptation of Ca2+ release channels. Inactivation of RyR2 channels, which is responsible for termination of Ca2+ release in isolated cardiac myocytes (Sham et al., 1998), would also explain our results. Furthermore, Ca2+ release fluxes, mediated either by skeletal RyR1 channels or by IP3 receptor channels, exhibit double exponential kinetics (Champeil et al., 1989; Mészáros et al., 1998). To explain this behavior, a model based on the tetrameric structure of the channels was proposed whereby channels would open regardless of how many sites of the activating ligand (Ca2+) are occupied, but the tendency to inactivate would be higher at lower occupancy (Mészáros et al., 1998). In principle, this model would explain why at lower activating [Ca2+] (pCa 6), but not at higher [Ca2+] (pCa 5), RyR2 channels closed completely after activation.
Calcium dependence of fast Ca2+ release fluxes
The calcium dependence of the fast release rate constants represents the immediate response of RyR2 channels to a sudden free [Ca2+] increase. We found that in 1.2 mM free [ATP] and in the absence of [Mg2+], the Ca2+ dependence of release rate constants followed a bell-shaped curve, with maximal k values in the order of 110 s−1 at pCa 6. This bell-shaped curve can be generated by a combination of two independent functions, a Ca2+ activation function and a Ca2+ inactivation function (Marengo et al, 1998). A more detailed analysis of these two functions is given below, when discussing the effects of Mg2+ on Ca2+ release fluxes.
Release rate constants increased 20-fold when raising free [Ca2+] from pCa 6.8 to pCa 6, showing that even in 1.2 mM [ATP], RyR2 channels show Ca2+-dependent activation. A similar Ca2+-dependent increase in rate constants in the presence of β,γ-methyleneadenosine-5′-trisphosphate has been reported previously for cardiac SR vesicles (Meissner and Henderson, 1987). These results show conclusively that ATP by itself is not sufficient for maximal activation of RyR2 channels. Increasing free [Ca2+] beyond 1 μM caused a marked decrease in channel activity. The present findings differ from previous reports indicating that, in the absence of Mg2+, RyR2 channels remain maximally activated even at pCa <4 (Laver et al., 1995; Xu et al., 1996; Liu et al., 1998; Marengo et al., 1998; Fruen et al., 2000). A similar discrepancy between results obtained using different methodologies was reported by Chu et al. (1993), who found that Ca2+ release from cardiac vesicles was inhibited at μM free [Ca2+] whereas RyR2 channels in bilayers were not. We interpret these results as an indication that RyR2 channels respond differently when activated by a sudden increase in free [Ca2+] than when exposed to the same [Ca2+] for longer periods, as in Ryanodine binding experiments. It is worth noting in this regard that the rate of calcium release in skinned cardiac Purkinje fibers depends not only on trigger [Ca2+] but also on the time taken to reach trigger [Ca2+] (Fabiato, 1985). A model was proposed whereby RyR2 channels inactivation sites bind Ca2+ with higher affinity but with lower association rates than activating sites (Fabiato, 1985). This model would explain why methods with different time resolution yield different results. After fast calcium activation, channels would initially display high activity, switching to lower activity after calcium binding to inhibitory sites. Only the lower activity mode would remain in steady-state conditions. This model may explain as well the observed time-dependent changes in channel activity discussed above.
A comparison of the calcium-dependence curve of rate constants obtained in cardiac SR vesicles (Fig. 5 A) with the calcium dependence of skeletal SR vesicles (Donoso et al., 2000) shows significant differences. Maximal activation of k was observed at pCa 5 in skeletal SR, whereas cardiac SR displayed maximal activation at pCa 6. Cardiac SR vesicles presented significant reduction in k values at pCa 5, whereas only at pCa 4 skeletal SR vesicles show a significant decrease in k. This comparison suggests that, if the present results reflected the physiological response of RyR2 channels, Ca2+ activation of Ca2+ release in cardiac muscle would be most efficient in 1 μM free [Ca2+]. However, Ca2+ release in vivo occurs in the presence of 0.7–1 mM free [Mg2+]. Accordingly, to have a better approximation to the physiological situation, it was necessary to measure CICR kinetics mediated by RyR2 channels in the presence of physiological free Mg2+ concentrations.
Effects of Mg2+ on Ca2+ release fluxes
Ryanodine-binding experiments and determinations of single channel activity in planar lipid bilayers indicate that cardiac RyR2 channels are less sensitive to inhibition by Mg2+ than their skeletal muscle counterparts (Meissner and Henderson, 1987; Chu et al., 1993; Laver et al., 1997). Presumably, this lower sensitivity to Mg2+ inhibition allows CICR to operate efficiently in the heart but not in mammalian skeletal muscle (Lamb and Laver, 1998, Lamb, 2000). In native vesicles we found that Mg2+ was a very effective inhibitor of Ca2+ release at pCa 6, with K0.5 < 100 μM. The present K0.5 values agree with reported μM Ki values for Mg2+ inhibition of channel open probability at pCa 6 and 5 mM ATP (Kawano, 1998). In contrast, even 0.8 mM free [Mg2+] had no effect at pCa 5. These results agree with other reports showing that at free [Ca2+] >15 μM, free [Mg2+] <1 mM does not inhibit RyR2 channel open probability or [3H]-Ryanodine binding to isolated cardiac SR vesicles (Xu et al., 1996; Laver et al., 1997). Our data support the proposal that Mg2+ and Ca2+ compete for channel activation sites at pCa 6, whereas at pCa 5 Mg2+ does not bind to Ca2+-saturated activation sites (Laver et al., 1997).
Additionally, we found that although the calcium dependence of release rate constants changed significantly by addition of 0.8 mM free [Mg2+] (Fig. 5 B), Ryanodine binding was much less affected (Fig. 1). These results show once again that RyR2 channels respond differently to activation by a sudden increase in free [Ca2+] than when exposed to the same [Ca2+] for a sustained period. As discussed above, the bell-shaped calcium-dependence curve can be generated by a combination of two independent functions, a Ca2+ activation function and a Ca2+ inactivation function (Marengo et al., 1998). We have constructed a model assuming that Mg2+ competes with Ca2+ for the high affinity channel activation sites (Laver et al., 1997) and shifts to the right the activation function. We propose, in addition, that increasing free [Mg2+] progressively enhances channel inactivation by Ca2+ and effectively shifts to the left the inactivation function. Thus, by increasing Ka from 0.6 μM to 1.9 μM and decreasing Ki from 0.9 μM to 0.25 μM this model can generate the calcium-dependence curve obtained experimentally in the presence of 0.8 mM free [Mg2+]. A comparison of the individual activation and inactivation functions and the resulting calcium-dependence curves is illustrated in Fig. 11 B. In the absence of Mg2+, this theoretical model predicts maximal activation of release rate constants at pCa 6, due to the relative predominance of the activation over the inactivation function. Likewise, in the presence of Mg2+ the model predicts the observed decrease in k values at pCa 6, due to the predominance of the inactivation over the activation function. In contrast, regardless of the presence of Mg2+, at pCa 5 both functions would have attained their maximal values, yielding significantly decreased yet similar k values at pCa 5. This model accounts well for the experimentally observed lack of inhibition by Mg2+ at pCa 5. According to this model, caffeine would shift markedly to the left the activation function allowing high release rates even in the presence of 0.3 mM free [Mg2+] (Fig. 8).
It is important to point out that the model described in this work is conceptually different from the model proposed by Fabiato (1985), according to which RyR2 channels inactivation sites bind Ca2+ with higher affinity but with lower association rates than activating sites. In contrast, the model we propose postulates that RyR2 channels have calcium activation and inhibitions sites with equally fast binding kinetics.
Effect of thimerosal on Ca2+ release fluxes
The redox status of the channel protein affects RyR channel activity, as determined with different experimental approaches. Changes in redox state of RyR channels from skeletal or cardiac muscle modifies the open probability of single channels in planar bilayers (Abramson et al., 1995; Favero et al., 1995; Eager et al., 1997; Marengo et al., 1998), as well as Ryanodine binding (Abramson et al., 1995; Favero et al., 1995; Aghdasi et al., 1997; Suko and Hellman, 1998). Changes in redox state also affect Ca2+ release from cardiac SR vesicles (Prabhu and Salama, 1990).
We found that incubation of RyR2 channels with thimerosal—that presumably alkylates free sulfhydryl residues of the RyR2 protein—did not modify release rate constants at pCa 6 in the absence of Mg2+. These results suggest that the sites of the RyR2 channel protein involved in high affinity Ca2+ activation lack thimerosal-responsive sulfhydryl groups. However, thimerosal reduced the inhibitory effect of 0.8 mM free [Mg2+] at pCa 6. Likewise, at pCa 5 thimerosal induced a threefold increase in release rate constants either in the absence or in the presence of up to 0.8 mM free [Mg2+] (Figs. 7 and 9). In this regard, release through RyR2 channels behaves as release through skeletal RyR1 channels, which even in the presence of 1 mM free [Mg2+] is significantly activated by thimerosal at pCa 5 (Donoso et al., 2000). We propose that modification of free sulfhydryl residues present in Ca2+/Mg2+ inhibitory site(s) of the cardiac RyR2 channels would decrease the affinity of the site for both Ca2+ and Mg2+. In terms of our model (Fig. 11 B), we propose that thimerosal reduces the left shift of the inactivation function induced by Mg2+.
Taken together, these results suggest that oxidation of RyR2 channels would produce significant enhancement of CICR in vivo. Redox modulation of RyR2 channels could be a physiologically relevant mechanism in the heart, especially in circumstances where there is an increase in free radical production, as in ischemia/reperfusion situations (Ambrosio and Tritto, 1999; Menshikova and Salama, 2000).
Physiological implications
ATP in mM concentrations is a well-known endogenous activator of RyR channels. Accordingly, in an attempt to approach the conditions present in the cytoplasm of heart cells, all kinetic experiments described in this study were done in the presence of 1.2 mM ATP. In such conditions, Mg2+ was an effective inhibitor of CICR at pCa 6, with K0.5 <100 μM, but not at pCa 5. We explain this preferential inhibition by Mg2+ with a model that proposes fast binding of Ca2+ and Mg2+ to independent activation and inhibition sites of the RyR2 channel protein. In the presence of 0.8 mM [Mg2+] release rate constants at pCa 6 were comparable to those obtained at pCa 5, although in both cases the first exponential component of calcium release ceased when vesicles still contained significant luminal calcium. These results suggest that under physiological conditions a sudden increase in cytoplasmic [Ca2+], from its resting level to 1 μM or 10 μM, would suffice to activate RyR2 channels maximally, although transiently. According to the present results, after activation by a sudden free [Ca+2] increase, RyR2 channels would change with time from a high activity to a low activity mode. We propose that this response is a reflection of an intrinsic time-dependent change in RyR2 channel open probability. Assuming our results from release experiments in vitro reflect the behavior of cardiac Ca2+ release channels in vivo, we propose that a time-dependent decrease in channel activity after activation by Ca2+ might facilitate channel closure during each cardiac contraction/relaxation cycle.
Acknowledgments
The authors thank Dr. Ricardo Bull for his help with curve analysis and Dr. Andrew Quest for his critical reading of the manuscript.
This study was supported by Fondo Nacional de Investigación Científica y Tecnológica grants 8980009 and 1000642. The institutional support to the Centro de Estudios Científicos by a group of Chilean companies (Compañía del Cobre, Dimacofi, Empresas CMPC, MASISA, and Telefónica del Sur) is also acknowledged. The Centro de Estudios Científicos is a Millennium Institute.
References
- Abramson, J. J., A. C. Zable, T. G. Favero, and G. Salama. 1995. Thimerosal interacts with the Ca2+ release channel ryanodine receptor from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 270:29644–29647. [DOI] [PubMed] [Google Scholar]
- Aghdasi, B., M. B. Reid, and S. L. Hamilton. 1997. Nitric oxide protects the skeletal muscle Ca2+ release channel from oxidation induced activation. J. Biol. Chem. 272:25462–25467. [DOI] [PubMed] [Google Scholar]
- Ambrosio, G., and I. Tritto. 1999. Reperfusion injury: experimental evidence and clinical implications. Am. Heart J. 138:S69–S75. [DOI] [PubMed] [Google Scholar]
- Armisén, R., J. Sierralta, P. Vélez, D. Naranjo, and B. A. Suárez-Isla. 1996. Modal gating in neuronal and skeletal muscle ryanodine-sensitive Ca2+ release channels. Am. J. Physiol. 271:C144–C153. [DOI] [PubMed] [Google Scholar]
- Bers, D. M. 2001. Excitation-Contraction Coupling and Cardiac Contractile Force, 2nd Ed. Kluwer Academic Publishers, Dordrecht, Netherlands.
- Bers, D. M. 2002. Cardiac excitation-contraction coupling. Nature. 415:198–205. [DOI] [PubMed] [Google Scholar]
- Beuckelmann, D. J., and W. G. Wier. 1988. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. J. Physiol. 405:233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatter, L. A., J. Hüser, and E. Rios. 1997. Sarcoplasmic reticulum Ca2+ release flux underlying Ca2+ sparks in cardiac muscle. Proc. Natl. Acad. Sci. USA. 94:4176–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull, R., J. J. Marengo, B. A. Suárez-Isla, P. Donoso, J. L. Sutko, and C. Hidalgo. 1989. Activation of calcium channels in sarcoplasmic reticulum from frog muscle by nanomolar concentrations of ryanodine. Biophys. J. 56:749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champeil, P., L. Combettes, B. Berthon, E. Doucet, S. Orlowski, and M. Claret. 1989. Fast kinetics of calcium release induced by myo-inositol trisphosphate in permeabilized rat hepatocytes. J. Biol. Chem. 264:17665–17673. [PubMed] [Google Scholar]
- Chu, A., M. Fill, E. Stefani, and M. L. Entman. 1993. Cytoplasmic Ca2+ does not inhibit the cardiac muscle sarcoplasmic reticulum ryanodine receptor Ca2+ channel, although Ca2+-induced Ca2+ inactivation of Ca2+ release is observed in native vesicles. J. Membr. Biol. 135:49–59. [DOI] [PubMed] [Google Scholar]
- Coronado, R., J. Morrissette, M. Sukhareva, and D. M. Vaughan. 1994. Structure and function of ryanodine receptors. Am. J. Physiol. Cell Physiol. 266:C1485–C1504. [DOI] [PubMed] [Google Scholar]
- Doi, M., M. Yano, S. Kobayashi, M. Kohno, T. Tokuhisa, S. Okuda, M. Suetsugu, Y. Hisamatsu, T. Ohkusa, M. Kohno, and M. Matsuzaki. 2002. Propranolol prevents the development of heart failure by restoring FKBP12.6-mediated stabilization of ryanodine receptor. Circulation. 105:1374–1379. [DOI] [PubMed] [Google Scholar]
- Donoso, P., H. Prieto, and C. Hidalgo. 1995. Luminal calcium regulates calcium release in triads isolated from frog and rabbit skeletal muscle. Biophys. J. 68:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso, P., P. Aracena, and C. Hidalgo. 2000. SH oxidation overrides Mg2+ inhibition of calcium induced calcium release in skeletal muscle triads. Biophys. J. 79:279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, G. G., G. Xinghua, V. K. Khanna, and D. H. MacLennan. 2001. Ryanodine sensitizes the cardiac Ca2+ release channel (ryanodine receptor isoform 2) to Ca2+ activation and dissociates as the channel is closed by Ca2+ depletion. Proc. Natl. Acad. Sci. USA. 98:13625–13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eager, K. R., L. D. Roden, and A. F. Dulhunty. 1997. Actions of sulfhydryl reagents on single ryanodine receptor Ca2+-release channels from sheep myocardium. Am. J. Physiol. Cell Physiol. 272:C1908–C1918. [DOI] [PubMed] [Google Scholar]
- Fabiato, A. 1985. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 85:247–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero, T. G., A. C. Zable, and J. J. Abramson. 1995. Hydrogen peroxide stimulates the Ca release channel from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 270:25557–25563. [DOI] [PubMed] [Google Scholar]
- Fruen, B. R., J. M. Bardy, T. M. Byrem, G. M. Strasburg, and C. F. Louis. 2000. Differential Ca2+ sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am. J. Physiol. Cell Physiol. 279:C724–C733. [DOI] [PubMed] [Google Scholar]
- Györke, S., and M. Fill. 1993. Ryanodine receptor adaptation: control mechanism of Ca2+-induced Ca2+ release in heart. Science. 260:807–809. [DOI] [PubMed] [Google Scholar]
- Hartree, E. F. 1972. Determination of protein: a modification of the Lowry method that gives a linear photometric response. Anal. Biochem. 48:422–427. [DOI] [PubMed] [Google Scholar]
- Ikemoto, N., M. Yano, R. El-Hayek, B. Antoniu, and M. Morii. 1994. Chemical depolarization-induced SR calcium release in triads isolated from rabbit skeletal muscle. Biochemistry. 33:10961–10968. [DOI] [PubMed] [Google Scholar]
- Inui, M., S. Wang, A. Saito, and S. Fleischer. 1988. Characterization of junctional and longitudinal sarcoplasmic reticulum from heart muscle. J. Biol. Chem. 263:10843–10850. [PubMed] [Google Scholar]
- Kawano, S. 1998. Dual mechanisms of Mg2+ block of ryanodine receptors Ca2+ release channels from cardiac sarcoplasmic reticulum. Recept. Channels. 5:405–416. [PubMed] [Google Scholar]
- Lamb, G. D. 2000. Excitation-contraction coupling in skeletal muscle: comparisons with cardiac muscle. Clin. Exp. Pharmacol. Physiol. 27:216–224. [DOI] [PubMed] [Google Scholar]
- Lamb, G. D., and D. R. Laver. 1998. Adaptation, inactivation and inhibition in ryanodine receptors. In The Structure and Function of Ryanodine Receptors. R. Sitsapesan, and A. J. Williams, editors. Imperial College Press, UK. pp. 269–290.
- Lamb, G. D., D. R. Laver, and D. G. Stephenson. 2000. Questions about adaptation in ryanodine receptors. J. Gen. Physiol. 116:883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laver, D. R., L. D. Roden, G. P. Ahern, K. R. Eager, P. R. Junankar, and A. F. Dulhunty. 1995. Cytoplasmic Ca 2+ inhibits the ryanodine receptor from cardiac muscle. J. Membr. Biol. 147:7–22. [DOI] [PubMed] [Google Scholar]
- Laver, D. R., T. M. Baynes, and A. F. Dulhunty. 1997. Magnesium inhibition of ryanodine-receptor calcium channels: evidence for two independent mechanisms. J. Membr. Biol. 156:213–229. [DOI] [PubMed] [Google Scholar]
- Liu, W., D. A. Pasek, and G. Meissner. 1998. Modulation of Ca2+-gated cardiac muscle Ca2+-release channel (ryanodine receptor) by mono- and divalent ions. Am. J. Physiol. 274:C120–C128. [DOI] [PubMed] [Google Scholar]
- Marengo, J. J., C. Hidalgo, and R. Bull. 1998. Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophys. J. 74:1263–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner, G., E. Darling, and J. Eveleth. 1986. Kinetics of rapid Ca2+ release by sarcoplasmic reticulum. Effects of Ca2+, Mg2+, and adenine nucleotides. Biochemistry. 25:236–244. [DOI] [PubMed] [Google Scholar]
- Meissner, G., and J. S. Henderson. 1987. Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. J. Biol. Chem. 262:3065–3073. [PubMed] [Google Scholar]
- Menshikova, E. V., and G. Salama. 2000. Cardiac ischemia oxidizes regulatory thiols on ryanodine receptors: captopril acts as a reducing agent to improve Ca2+ uptake by ischemic sarcoplasmic reticulum. J. Cardiovasc. Pharmacol. 36:656–668. [DOI] [PubMed] [Google Scholar]
- Mészáros, L. G., A. Zahradníková, and P. Volpe. 1998. Kinetic basis of quantal calcium release from intracellular calcium stores. Cell Calcium. 23:43–52. [DOI] [PubMed] [Google Scholar]
- Miller, C. 1984. Ion channels in liposomes. Annu. Rev. Physiol. 46:549–558. [DOI] [PubMed] [Google Scholar]
- Ono, K., M. Yano, T. Ohkusa, M. Kohno, T. Hisaoka, T. Tanigawa, S. Kobayashi, M. Kohno, and M. Matsuzaki. 2000. Altered interaction of FKBP12.6 with ryanodine receptor as a cause of abnormal Ca(2+) release in heart failure. Cardiovasc. Res. 48:323–331.11054478 [Google Scholar]
- Otsu, K., H. F. Willard, V. K. Khanna, F. Zorzato, N. M. Green, and D. H. Maclennan. 1990. Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J. Biol. Chem. 265:13472–13483. [PubMed] [Google Scholar]
- Prabhu, S. D., and G. Salama. 1990. Reactive disulfide compounds induce Ca2+ release from cardiac sarcoplasmic reticulum. Arch. Biochem. Biophys. 282:275–283. [DOI] [PubMed] [Google Scholar]
- Rousseau, E., J. S. Smith, J. S. Henderson, and G. Meissner. 1986. Single channel and 45Ca2+ flux measurements of the cardiac sarcoplasmic reticulum calcium channel. Biophys. J. 50:1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham, J. S. K., L.-S. Song, Y. Chen, L.-H. Deng, M. D. Stern, E. G. Lakatta, and H. Cheng. 1998. Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes. Proc. Natl. Acad. Sci. USA. 95:15096–15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan, R., and A. J. Williams. 1995. The gating of the sheep skeletal sarcoplasmic reticulum Ca2+-release channel is regulated by luminal Ca2+. J. Membr. Biol. 146:133–144. [DOI] [PubMed] [Google Scholar]
- Sitsapesan, R., R. A. P. Montgomery, and A. J. Williams. 1995. New insights into the gating mechanisms of cardiac ryanodine receptors revealed by rapid changes in ligand concentration. Circ. Res. 77:765–772. [DOI] [PubMed] [Google Scholar]
- Suko, J., and G. Hellman. 1998. Modification of sulfhydryls of the skeletal muscle calcium release channel by organic mercurial compounds alters Ca2+ affinity of regulatory Ca2+ sites in single channel recordings and [3H]ryanodine binding. Biochem. Biophys. Acta. 1404:435–450. [DOI] [PubMed] [Google Scholar]
- Valdivia, H. H., J. H. Kaplan, G. C. R. Ellies-Davies, and W. J. Lederer. 1995. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 267:1997–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher, D. R., R. J. Kovacs, H. Schulman, D. C. Cefali, and L. R. Jones. 1991. Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J. Biol. Chem. 266:11144–11152. [PubMed] [Google Scholar]
- Xu, L., G. Mann, and G. Meissner. 1996. Regulation of cardiac Ca2+ release channel (Ryanodine receptor) by Ca2+, H+, Mg2+, and adenine nucleotides under normal and simulated ischemic conditions. Circ. Res. 79:1100–1109. [DOI] [PubMed] [Google Scholar]
- Xu, L., A. Tripathy, D. A. Pasek, and G. Meissner. 1999. Ruthenium Red modifies the cardiac and skeletal muscle Ca2+ release channels (Ryanodine Receptors) by multiple mechanisms. J. Biol. Chem. 274:32680–32691. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, N., K. Igami, and M. Kasai. 1997. Kinetics of depolarization-induced calcium release from skeletal muscle triads in vitro. J. Biochem. (Tokyo). 121:432–439. [DOI] [PubMed] [Google Scholar]
- Yano, M., T. Yamamoto, M. Kohno, T. Hisaoka, K. Ono, T. Tanigawa, T. Ueyama, T. Ohkusa, and M. Matsuzaki. 1998. Polylysine-induced rapid Ca2+ release from cardiac sarcoplasmic reticulum. J. Cardiovasc. Pharmacol. 32:96–100. [DOI] [PubMed] [Google Scholar]
- Yano, M., K. Ono, T. Ohkusa, M. Suetsugu, M. Kohno, T. Hisaoka, S. Kobayashi, Y. Hisamatsu, T. Yamamoto, M. Kohno, N. Noguchi, S. Takasawa, H. Okamoto, and M. Matsuzaki. 2000. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation. 102:2131–2136. [DOI] [PubMed] [Google Scholar]
- Zahradníková, A., and I. Zahradník. 1995. Description of modal gating of the cardiac calcium release channel in planar lipid membranes. Biophys. J. 69:1780–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradníková, A., and I. Zahradník. 1996. A minimal gating model for the cardiac calcium release channel. Biophys. J. 71:2996–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradníková, A., M. Dura, and S. Gyorke. 1999. Modal gating transitions in cardiac ryanodine receptors during increases of Ca2+ concentration produced by photolysis of caged Ca2+. Pflugers Arch. Eur. J. Physiol. 438:283–288. [DOI] [PubMed] [Google Scholar]
- Zucchi, R., and S. Ronca-Testoni. 1997. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol. Rev. 49:1–51. [PubMed] [Google Scholar]