

Abstract
Familial hypertrophic cardiomyopathy is a disease caused by single mutations in several sarcomeric proteins, including the human myosin ventricular regulatory light chain (vRLC). The effects of four of these mutations (A13T, F18L, E22K, and P95A) in vRLC on force generation were determined as a function of Ca2+ concentration. The endogenous RLC was removed from skinned rabbit psoas muscle fibers, and replaced with either rat wildtype vRLC or recombinant rat vRLC (G13T, F18L, E22K, and P95A). Compared to fibers with wildtype rat vRLC, the E22K mutant increased Ca sensitivity of force generation, whereas the G13T and F18L mutants decreased the Ca sensitivity, and the P95A mutant had no significant effect. None of the RLC mutants affected the maximal tension (observed at saturating Ca2+ concentrations), except for F18L, which decreased the maximal tension to 69 ± 10% of the wildtype value. Of the mutant RLCs, only F18L decreased the cooperativity of activation of force generation. These results suggest that the primary cause of familial hypertrophic cardiomyopathy, in some cases, is perturbation in the Ca sensitivity of force generation, in which Ca-sensitizing or Ca-desensitizing effects can lead to similar disease phenotypes.
INTRODUCTION
Familial hypertrophic cardiomyopathy (FHC) is a genetic human heart disease that is phenotypically variable, with features ranging from severe (sudden death) to intermediate to benign (near normal life span). The disease is classically characterized by hypertrophy of the left ventricle and intraventricular septum, myocardial cellular disarray, myofilament disarray, and interstitial myocardial fibrosis. It is typically caused by mutations in cardiac sarcomeric proteins: β-myosin heavy chain, α-tropomyosin, myosin binding protein-C, ventricular essential light chain (vELC), ventricular regulatory light chain (vRLC), troponin I, actin, titin, and troponin T (Roopnarine, 2002; Seidman and Seidman, 2001; Rayment et al., 1995; Bonne et al., 1998; Hernandez et al., 2001; Michele and Metzger, 2000). Most of the mutations are located either in or near functional regions of the mutant proteins (Rayment et al., 1995), suggesting that the molecular mechanism for the FHC disease is related to the dysfunction (either enhanced or diminished function) of the mutant sarcomeric protein. The focus of this paper is to determine the Ca sensitivity of force generation in skeletal muscle fibers containing the myosin RLC mutations that cause FHC. The results in this paper provide evidence that links some of the FHC-RLC mutations to mechanical dysfunction of the muscle fiber, due to impaired Ca sensitivity of force generation.
Myosin is a dimer that is folded symmetrically into two heads and a tail, each monomer containing a myosin heavy chain, an essential light chain, and a regulatory light chain. The myosin head contains the catalytic domain (ATPase site and actin binding sites) and the LC domain (ELC and RLC). Several FHC mutations have been found in vRLC: A13T, E22K, P95A (Poetter et al., 1996), F18L, R58Q (Flavigny et al., 1998), and N47K (Andersen et al., 2001) (Fig. 1). In addition to having the classical FHC disease phenotype, A13T and E22K result in a rare myopathy in the heart (midventricular chamber thickening), and E22K patients exhibit red ragged fiber histology in the slow skeletal muscle (Poetter et al., 1996). These FHC mutations are near the functional regions of the RLC, and the native amino acids are highly conserved across species and isoforms (Poetter et al., 1996; Flavigny et al., 1998). A13T, F18L, and E22K are near the phosphorylation site in vRLC, Ser 15. P95A is in the linker region between the N-terminal and C-terminal domains of RLC. The C-terminal and linker region of RLC interact with the myosin heavy chain (Rayment et al., 1993). N47K and R58Q are near the Ca binding site of RLC.
FIGURE 1.

Crystal structure of chicken skeletal myosin S1 (blue) showing the location of the FHC mutations in RLC (red): E22K, N47K, R58Q, and P95A (yellow). A13T and F18L are not shown because residues 1–19 are missing from the crystal structure (Rayment et al., 1993).
In most current models of muscle contraction, the LC domain of myosin plays a key role in contraction, acting as a lever arm that tilts as myosin changes its actin-bound configuration from weak to strong (Thomas et al., 2002). Electron paramagnetic resonance studies have shown that the myosin LC domain undergoes a dynamic disorder-to-order transition during muscle contraction (Roopnarine et al., 1998), during which the LC domain rotates through a large angle (Baker et al., 1998). In striated muscle contraction, RLC modulates crossbridge contractility by altering myosin's interactions with actin. Phosphorylation of RLC increases the submaximal isometric tension, induces a leftward shift in the tension-pCa curve, but does not change the maximal isometric tension (Persechini et al., 1985), and increases the rate of force development (Metzger et al., 1989). Replacement of the phosphorylatable serines (Ser 14, 15, and 19) with alanines in mouse vRLC induces a variety of negative cardiovascular changes in transgenic mice (Sanbe et al., 1999). Furthermore, the importance of vRLC for normal mammalian cardiac function is demonstrated by the correlations with human diseases. vRLC phosphorylation is decreased during heart failure (Morano, 1992), and in diabetic cardiomyopathic rats (30–40% decrease; see Liu et al., 1997). In contrast, RLC phosphorylation is elevated in rats during increased beat frequency or left ventricular pressure due to exercise or inotropic agents (Fitzsimons et al., 1990).
The molecular mechanisms by which FHC mutations cause cardiac hypertrophy and myocyte disarray are not clearly understood. Previous studies of several FHC-mutant myosins showed decreased myosin ATPase activity (Roopnarine and Leinwand, 1998), actin motility, and force, supporting a hypothesis that a hypocontractile state induces hypertrophy (Bonne et al., 1998). In contrast, other functional studies on FHC mutations show increased Ca sensitivity of force, supporting a hypothesis that a hypercontractile state induces hypertrophy (Bonne et al., 1998). This was observed for the R403Q myosin FHC mutation (Seidman and Seidman, 2001) and several of the troponin T, TnI (Hernandez et al., 2001), and Tm FHC mutations (Michele and Metzger, 2000). In addition, some FHC mutations show insignificant functional defects in vitro, but still have the phenotype of the FHC disease (Bonne et al., 1998), thus complicating the elucidation of molecular mechanisms for the disease.
Functional studies have been done on only one of the FHC-RLC mutations (E22K). Myosin isolated from cardiac biopsies (from a patient with E22K) induced normal actin filament motility (Poetter et al., 1996), but slow skeletal fibers from an E22K-diseased patient increased Ca sensitivity of force (Levine et al., 1998). Transgenic mice expressing E22K in the heart did not show any features of the FHC disease despite almost total replacement of the endogenous wildtype vRLC with mutant vRLC (Sanbe et al., 2000). The Ca-binding affinities of several of the FHC-RLC mutations are diminished in RLC isolated in solution, but are unknown when RLC is bound to myosin in solution or in muscle fibers (Szczesna et al., 2001). In addition, the effect of these mutations on the Ca sensitivity of force within muscle fibers was not previously studied.
In the present report, the Ca sensitivity of force due to the FHC-RLC mutations (A13T, F18L, E22K, and P95A) was studied directly in rabbit psoas skeletal muscle fibers. Rat cardiac vRLC was used as the background for studying the effect of the FHC-RLC mutations, in part because transgenic rodent models for myosin-FHC mutations were being developed in other laboratories (Sanbe et al., 2000) for pathophysiological assessment of the disease. The amino acid similarity between human vRLC and rat vRLC is 95.2%, with 100% similarity in the amino acids flanking the FHC mutation (except for A13T, as explained in Material and Methods). Therefore, the rat vRLC is a good candidate for studying the FHC mutations. Rabbit psoas fibers were used instead of human cardiac muscle fibers because sufficient quantity of healthy human cardiac tissue was not readily available for this study. More importantly, the present study used 5, 5′-dithiobis (2-nitrobenzoic acid) (DTNB) to extract RLC. This procedure works in fast skeletal fibers (Szczesna et al., 1996), but not in soleus or cardiac fibers (Wagner, 1982), probably because fast skeletal RLC, but not vRLC, contains cysteines that can react with DTNB (Huber et al., 1989). The RLC-binding region of myosin heavy chain (808–842) contains only one nonconservative amino acid difference between the rabbit fast skeletal and cardiac β-myosin isoforms, therefore, the binding of vRLC to rabbit fast myosin is unlikely to be impaired. Therefore, it is likely that the functional effects of mutations observed in the present study will provide useful insight into physiological effects of the FHC mutations.
Therefore, in the present study, RLC was extracted from skinned muscle fiber bundles, and then reconstituted with either wildtype rat vRLC or with rat vRLC containing an FHC mutation. The isometric force of single muscle fibers containing wildtype rabbit RLC, vRLC, or mutant vRLC was determined as a function of Ca2+ concentration. The maximal isometric tension and cooperativity were not significantly affected for any mutation, except for F18L. All of the mutations changed the Ca sensitivity of force generation in fibers, except for P95A. These results suggest that some FHC-RLC mutations affect cardiac function by altering the Ca sensitivity of force generation.
MATERIALS AND METHODS
Expression and purification of RLC
Wildtype rat vRLC cDNA was subcloned into the pET-3d expression vector (Novagen, Madison, WI), and the FHC mutations, G13T, F18L, E22K, and P95A (Fig. 1) were introduced using the “QuikChange” mutagenesis kit (Stratagene, La Jolla, CA). In rat vRLC, Ala13 is replaced by Gly, so a G13T mutation was created in rat vRLC. The amino acid sequence identity of rat vRLC (accession #x07314) compared to human vRLC (accession #x66141) is 95.2% in 166 amino acid overlap (96% similarity), with four conserved replacements and three amino acid differences (the human vRLC Ala10 is replaced by Leu, Gly11 is replaced by Glu, Asn14 is replaced by Ser). The mutant RLC genes were sequenced (Microchemical Facility, University of Minnesota) to verify the presence of the engineered mutations, and then transformed into an expression Escherichia coli cell line, JM109DE3, for expression of RLC in luria broth media. RLC expression was induced by addition of 0.4 mM IPTG to cells growing in log phase (OD600 = 0.6–0.7) at 37°C. The cells were then grown for an additional 3 h, and collected by centrifugation at 6000 rpm for 5 min. The RLC was expressed in inclusion bodies, which were released from the cells using a lysis buffer (25 mM Tris, pH 8.0, 5 mM EDTA, 50 mM glucose, 1 mM PMSF, 1 mM DTT) containing 0.15 mg/ml lysozyme, followed by a freeze/thaw cycle, and incubation with 10 mM MgCl2 and 25 μg/ml DNase I for 1 h on ice. The inclusion bodies were isolated by centrifugation at 11,500 rpm in a SS34 rotor in a Sorvall centrifuge for 25 min at 4°C, washed in the lysis buffer +0.1% triton ×100 twice, then in final triton-free lysis buffer. The RLC was released from the inclusion bodies with 7 M urea in column buffer (50 mM MOPS, pH 6.5, 10 mM NaCl, 1 mM DTT), and purified on a DE52 anion exchange column with a linear gradient of 0.01–0.5 M NaCl in 4 M urea in column buffer. After purification, the gradient fractions were analyzed by 15% SDS PAGE for purified RLC, which was then dialyzed six times against 4 liters of 25 mM ammonium bicarbonate and 0.1 mM DTT. The RLC was purified to near homogeneity, then lyophilized, and stored at −20°C until needed. The lyophilized RLC was solubilized and dialyzed before exchanging into muscle fibers as described previously (Roopnarine et al., 1998). Rabbit skeletal troponin C (TnC) was prepared from rabbit muscle as described previously (Potter, 1982).
Preparation of muscle fibers
Skinned rabbit psoas muscle was chosen as the background for studying the effects of the FHC mutations, because this is a well characterized and reproducible preparation in which RLC extraction and reconstitution has been extensively studied. The extraction of vRLC from cardiac fibers has not been reported, and the lack of cysteines in vRLC probably makes DTNB ineffective in enhancing RLC extraction from muscle fiber bundles. Rabbit psoas fibers were prepared as described previously (Roopnarine and Thomas, 1995) with the following modifications: fiber bundles (∼0.4 cm in diameter) were incubated in fiber storage solution (FSB = 50% v/v Rigor solution (RS)/glycerol solution (RS: 130 mM potassium propionate, 2 mM MgCl2, 1 mM EGTA, and 20 mM MOPS, pH 7.0)) for 1 h at 4°C. The solution was then changed three times after 1 h incubations, and fibers stored at −20°C for ∼2 weeks before use.
Extraction of RLC and reconstitution with wildtype RLC or recombinant mutant vRLCs
The untreated fibers (fibers from storage buffer) were dissected into smaller bundles (∼0.2–0.4 mm in diameter), the ends tied with silk thread, placed inside glass capillaries, and then connected in series to a peristaltic pump for extraction of RLC, which consisted of the following five steps. a), The fiber bundles were washed with pre-extraction solution (PES), 20 mM Imidazole (pH 7.0), 20 mM KCl, 10 mM EDTA, 10 mM CDTA, 2 mM EGTA, for 10 min at 4°C; b), then the fiber bundles were washed with extraction solution (ES-PES containing 10 mM DTNB) for 10 min at 25°C; then washed with RS, pH 7; then RS, pH 8, in which MOPS was replaced by EPPS, at 4°C. c), The fiber bundles were incubated in RS (pH 8) with 30 mM DTT for 1.5 h at 4°C to remove 5-thio-2-nitrobenzoic acid (TNB)-modified cysteines, and washed in RS (pH 7) for 10 min. d), TnC (3–5 mg/ml) was re-added to the fiber bundle in RS (pH 7) plus 5 mM MgATP, for 1 h on ice. e), The TnC solution was removed, the fibers washed with RS (pH 7), then reconstituted with either the wildtype or mutant RLCs (3–5 mg/ml) in RS (pH 7) for 3 h on ice. Fibers treated with DTT and TnC were control fibers for experiments with wildtype rabbit RLC (rRLC) or vRLC, whereas the control fibers for comparison of the mutant vRLC contained rat vRLC. The unbound RLC was removed, the fibers were washed with RS (pH 7), washed in FSB solution, and then stored on ice. The complete reversal of TNB-modifed cysteines was verified by measuring the myosin myofibrillar K+- and Ca2+-ATPase activities as described previously (Roopnarine and Thomas, 1994). The mechanical effects of the chemical treatment during each step (a–c) on untreated fibers were determined by measuring the Ca sensitivity of force. The extent of RLC extraction and reconstitution was determined by densitometric analysis of 15% polyacrylamide urea (Tris-borate) gels (Biorad, Richmond, CA). The extent of RLC and TnC extraction and reconstitution were determined from the ratio of RLC/(LC1 + LC3) and TnC/(LC1 + LC3).
Single muscle fiber mechanics
Single fibers were dissected from the fiber bundles containing recombinant RLC for mechanical measurements. The force of a skinned muscle fiber (∼2–3 mm in length) was measured at 25°C using a Muscle Research System (Scientific Instruments, Heidelberg, Germany), varying the pCa (−log[Ca2+]) from 9.0 to 4.5, using Ca-EGTA buffering as described previously (Kerrick et al., 1991). The single fiber segment was mounted between two microtweezers, one of which was attached to a force transducer (Scientific Instruments, model KG3, range of 0–10 mN), which was connected to a bridge amplifier (Scientific Instruments, model BAM4C) with a full scale of 10 V. A stereomicroscope (Nikon) and video camera (Panasonic, model BL200) was mounted above the transducer to aid in mounting of the fiber and in fiber diameter determination. The force of the single fiber was adjusted to zero in rigor (which did not not change during relaxation). The sarcomere length of the single fiber was set to 2.5 μm, and the fiber diameter was measured at four places along the length of the fiber using a calibrated video monitor system, in order to calculate the cross-sectional area. The mounted fiber was then placed in a quartz cuvette (1 cm long; Scientific Instruments, model CU1A) filled with relaxing solution, low Ca solution (pCa 9.0), 7 mM EGTA, 3.025 mM MgO, 85 mM KOH, 2 mM Na2ATP, 163.9 mM proprionic acid (proprionate is the major anion), and 13.2 mM imidazole (pH 7.0). Solution was flowed into the cuvette with a continous flow peristaltic pump, which was connected to a gradient maker, and controlled by a pump unit (Scientific Instruments, model CPER1). The flowthrough solution from the cuvette was immediately removed by gentle suction using a vacuum apparatus. In this paper, Ca always refers to the free calcium ion concentration, [Ca2+]. The activating solution, a high Ca solution (pCa 4.5), consisted of relaxing solution + 6.86 mM CaO. The fiber was pretreated with relaxing solution (pCa 9.0), followed by activating solution (pCa 4.5) for 5–8 seconds, then in relaxing solution until the maximal force was reduced to the orginal relaxed levels. Isometric force was acquired as a function of [Ca], by exposing the fiber segment to a gradient of [Ca] from 10−9 to 10−4.5 using a programmable gradient maker (using the routine AT from the program MUAT, Scientific Instruments) and peristaltic pump. The gradient maker consisted of a flowthrough solution chamber (which is continously stirred), in which fixed volumes of solutions are simultaneously pumped in and out by a computer-controlled perstaltic pump. The activating solution was placed in a separate chamber and was pumped into the chamber containing the relaxing solution. The computer program controlled the peristaltic pump to make a fixed volume solution change (step size of 5) at fixed intervals, with a cuvette incubation time of 5 s to create a linear gradient of [Ca] in the cuvette containing the single fiber. Each pump step corresponded to a specific [Ca], and recorded force measurement. To ensure that the volume of the solution mixed remained the same with each step of the pump for different experiments, the volume of buffer was precisely and periodically measured using different steps.
Data analysis
The resting force (pCa 9–8) was subtracted from the single fiber force pCa curves, and then normalized to the cross-sectional area of the fiber. The program ORIGIN (Microcal Software, ver. 6.0) was used to determine best fits of the tension-pCa curves by means of the Levenberg-Marquardt nonlinear least-squares algorithm. The best fit was determined from the lowest χ2 between the experimental data and fit. The tension P was the force normalized to cross-sectional area of the fiber, and reported as kN/m2. Individual curves of P versus pCa were fitted to the Hill equation in the form P = Po/[1 + 10−n(pCa50−pCa)], Eq. 1, where Po is the maximum tension, n is the Hill coefficient, and pCa50 is the pCa when P = 0.5 Po. The Hill coefficient is a measure of the cooperativity of the activation process and determines the steepness of the tension pCa curve. The tension was normalized to the value of Po obtained from fits to the Hill equation. There was variability in the tension pCa curves amongst different rabbit fiber preparations as previously observed (Hofmann et al., 1990), but the relative changes in pCa50 and n between the different samples remained consistent with different rabbit fiber preparations. As previously observed (Hofmann et al., 1990), the addition of TnC or wildtype rabbit RLC to treated control fibers only restored the mechanical properties of the fiber to that of the untreated fibers within a rabbit fiber preparation. This is illustrated in Figs. 3–5, which contain data sets from different rabbit fiber preparations. The Ca sensitivity (pCa50) and cooperativity (n) is represented as the difference between the FHC-RLC fiber and wildtype vRLC fiber to illustrate the different sensitivities of the mutant RLCs for each fiber preparation.
FIGURE 3.

Effect of TnC reconstitution on the Ca dependence of normalized single fiber tension. Untreated fibers (▪) compared to fibers treated to different experimental steps in the extraction protocol: step c, DTT (○); steps c and d, DTT + TnC (•); steps a and c, pre-extraction solution + DTT (▴); pre-extracted solution + DTT + TnC (▵). Curves show best fits to Eq. 1. The data and curves are averages of four fiber samples from two different fiber preparations.
FIGURE 5.

Ca-dependence of tension of single muscle fibers reconstituted with either wildtype or FHC-vRLC mutants. Wildtype (•) vRLC; compared to FHC-mutant RLC: G13T (⋄); F18L (▵); E22K (□); and P95A (○). (A) Averaged tension (P) versus pCa 9–4.5; (B) mean maximal tension mean ± SE from fits to Eq 1; (C) averaged normalized tension (P/P0) versus pCa; (D) mean tension mean ± SE at pCa 6.0. Curves in A and C are best fits to Eq 1. * = P < 0.05; n.s. = not significantly different from wildtype. The data and curves are averages of the single fiber tension pCa curves from four fibers from two preparations.
Statistical analysis
The parameter values (Po, pCa50, n) for each curve were determined by fitting individual data sets, then the mean ± SE (shown as error bars) were calculated. Each tension pCa curve in Figs. 3–5 is the average obtained from at least two different fiber extractions (at least four single fibers from each preparation were analyzed). The averaged tension pCa curves (in Figs. 3–5) were also fitted to Eq. 1, to verify the validity of the averaging. Therefore, all the experimental data is reported as mean ± SE. The significance was determined by the unpaired Student's t-test using the program ORIGIN. The confidence level P < 0.05 was used for comparison of FHC-RLC fiber to wildtype vRLC fiber.
RESULTS
It was first necessary to optimize the procedure for RLC extraction and reconstitution in rabbit psoas fibers, and to demonstrate that the introduction of cardiac vRLC restores the physiological properties of the muscle fibers. Previously, RLC was extracted from skeletal muscle fibers either at higher temperatures so that reconstitution with rRLC did not fully restore normal maximal tension (Diffee et al., 1995), or by using a DTNB extraction method that did not restore normal Ca sensitivity of the muscle fibers (Szczesna et al., 1996). The protocol developed in this paper for RLC extraction and reconstitution with rRLC results in restoration of maximal tension, Ca sensitivity, and cooperativity of fibers. Reconstitution of skeletal psoas fibers with rat vRLC also restored maximal tension, but conferred Ca sensitivity and cooperativity characteristic of cardiac ventricular muscle fibers. Cardiac muscle fibers were not used for this study because the extraction and exchange of RLC into cardiac muscle fibers have not been reported before, probably because the lack of cysteines in vRLC makes DTNB ineffective in extraction.
Protein composition of extracted and reconstituted fiber bundles
The endogenous rabbit RLC (rRLC) was extracted (50 ± 8%) from fiber bundles, and then replaced with rRLC, wildtype vRLC, or mutant vRLC. TnC was also removed (80 ± 5%) during the extraction procedure (Fig. 2, left, lane 4), so it was subsequently replaced by the addition of purified rabbit skeletal TnC to the fiber bundles. vRLC and mutant vRLCs (except for E22K, which migrated higher than rRLC) migrates faster than the endogenous rRLC on 15% polyacrylamide-urea gels (Fig. 2, left), allowing precise quantitation of the protein bands by densitometric analysis (Fig. 2, right). The extent of RLC and TnC extraction and reconstitution was determined from the ratio of RLC/(LC1 + LC3) and TnC/(LC1 + LC3), which are 0.782 ± 0.05 and 0.317 ± 0.03 for untreated and control fibers (with rRLC), respectively. Reconstitution with purified RLC (rRLC, or vRLC, or mutant vRLC) and TnC restored the RLC and TnC to normal protein levels (Fig. 2), suggesting that the vRLC and mutant vRLC binding to skeletal myosin were comparable to skeletal RLC. The urea gels also confirm that none of the RLCs (either rRLC or vRLC) was phosphorylated in solution or on the fiber (phosphorylated RLC migrate further down the urea gel than the unphosphorylated RLC). Analysis of the reconstituted fiber bundle on 7–16% gradient SDS PAGE showed that the sarcomeric protein composition was similar to the untreated fiber bundle (data not shown). Thus the procedure used in the present study results in a similar extent of protein extraction and reconstitution as obtained with previous procedures (Diffee et al., 1995; Szczesna et al., 1996) but the present procedure results in restoration of physiological performance of the fibers, as described below.
FIGURE 2.

Protein composition of extracted and reconstituted fiber bundles. (Left) 15% polyacrylamide urea gel of muscle fiber proteins; (lane 1) rabbit skeletal myosin; (lane 2) purified rabbit TnC; (lane 3) control muscle fibers; (lane 4) RLC-extracted muscle fibers; and (lane 5) fibers reconstituted with vRLC and TnC. (Right) Densitometric analysis showing the rRLC, vRLC, and TnC content.
Functional reconstitution of fibers
Because the depletion of TnC from muscle fibers decreases the Ca sensitivity (Moss et al., 1985), and DTNB can perturb Ca sensitivity of fibers (Szczesna et al., 1996), the effects of the extraction protocol on the mechanical properties of the fibers were determined at different experimental steps (see Materials and Methods) that had the potential to modify the fiber integrity. Incubation of fibers in step c alone (DTT treatment) decreased Ca sensitivity slightly (Fig. 3, open circles; Table 1, untreated + DTT), compared to untreated fibers (Fig. 3, squares), but addition of TnC (step d) restored the Ca sensitivity (Fig. 3, closed circles; Table 1, untreated + DTT + TnC). Prior to RLC extraction, the fibers were incubated in a pre-extraction solution (PES) in step (a) to enhance removal of divalent cations to weaken the interaction between the RLC and myosin heavy chain to facilitate RLC extraction. Treatment of fibers with both steps a and c decreased Ca sensitivity (Fig. 3, closed triangles; Table 1, untreated + PES + DTT) to a greater extent than with step c alone, but addition of TnC (step d) restored the Ca sensitivity (Fig. 3, open triangles; Table 1, untreated + PES + DTT + TnC). Urea gel analysis of these fiber samples confirmed the partial extraction and complete restoration of TnC (data not shown). Therefore, all fiber bundles were incubated with TnC after the DTT treatment of the fibers to restore normal function.
TABLE 1.
Mechanical parameters for single muscle fibers for Fig. 3
| Fiber sample | Maximal tension | pCa50 | n (Hill coefficient) |
|---|---|---|---|
| Untreated | 255.68 ± 9.02 | 5.83 ± 0.077 | 3.71 ± 0.082 |
| Untreated + DTT | 252.446 ± 5.08 | 5.81 ± 0.009 | 4.02 ± 0.333 |
| Untreated + DTT + TnC (control) | 271.28 ± 15.0 | 5.84 ± 0.029 | 3.00 ± 0.159 |
| Untreated + PES + DTT | 253.848 ± 19.88 | 5.66 ± 0.051 | 2.80 ± 0.378 |
| Untreated + PES + DTT + TnC | 284.69 ± 20.50 | 5.89 ± 0.03 | 4.28 ± 0.12 |
The tension is reported as kN/m2. The values are the mean ± SE for four single fibers from two preparations. Untreated = fibers from storage buffer. The experimental conditions for fibers treated with DTT, TnC, and PES (pre-extraction solution) are given in Materials and Methods. Untreated fibers incubated with DTT and TnC were defined as control fibers for the experiments described below.
Inclusion of 10 mM DTNB in the extraction solution (ES, step b) additionally removed RLC from the fibers (Fig. 4, triangles). Removal of both RLC and TnC from the muscle fiber resulted in ∼50% decrease of maximal isometric tension and severe loss of Ca sensitivity (data not shown). Reconstitution of the RLC/TnC-depleted fibers with purified TnC restored tension to 80 ± 5%, consistent with previous studies (Moss et al., 1985), but not normal Ca sensitivity (Fig. 4, triangles; Table 2, ES + DTT + TnC). Subsequent reconstitution of these fibers with rabbit RLC restored the Ca-dependent force (Fig. 4, squares; Table 2, rRLC) to normal levels of control fibers (fibers treated with DTT and TnC, Fig. 4, circles). This study shows that the procedure used for RLC extraction and reconstitution results in the exchange of ∼50% of the endogenous RLC, and completely restores normal physiological properties, as measured by Ca sensitivity and cooperativity (n), as well as maximal tension. Reconstitution of the RLC fibers with vRLC (Fig. 4, diamonds; Table 2, vRLC) restores maximal tension and increased Ca sensitivity to a level that is even higher than in the original psoas fibers. In fact, the level of Ca sensitivity is similar to that observed for cardiac ventricular fibers (Gordon et al., 2001; Martyn and Gordon 2001), suggesting that Ca sensitivity depends, in part, on the source of RLC. This leftward shift in the Ca sensitivity is not due to phosphorylation of RLC (either the remaining endogenous rabbit RLC or vRLC) since the lower migrating RLC species (typical of phosphorylated RLC) were not observed on urea gels. The amino acid sequence identity of rat vRLC and rabbit skeletal RLC is only 73.2% (87% similarity), so the difference probably contributes to, in part, the less steep tension pCa curve characteristic of cardiac fibers (Gordon et al., 2001).
FIGURE 4.

Effect of RLC extraction and reconstitution on the Ca-dependence of fiber tension. Control (•), RLC-depleted (▵), reconstituted with rRLC (□), and reconstituted with vRLC (⋄). Curves show best fits to Eq. 1. The data and curves are averages of four single fibers from two preparations.
TABLE 2.
Mechanical parameters for single muscle fibers for Fig. 4
| Fiber sample | Maximal tension | pCa50 | n (Hill coefficient) |
|---|---|---|---|
| Untreated + DTT + TnC | 279.67 ± 13.63 | 6.289 ± 0.071 | 4.670 ± 0.47 |
| ES + DTT + TnC | 224.30 ± 26.25 | 6.183 ± 0.004 | 1.601 ± 0.045 |
| rRLC + DTT + TnC | 270.24 ± 15.23 | 6.310 ± 0.048 | 3.154 ± 0.021 |
| vRLC + DTT + TnC | 275.31 ± 18.51 | 6.515 ± 0.015 | 2.244 ± 0.144 |
The tension is reported as kN/m2. The values are the mean ± SE for four single fibers from two preparations. The difference pCa50 for the untreated + DTT + TnC fibers in Table 2 compared with Table 1 is explained in Materials and Methods. ES + DTT + TnC fibers are rRLC-depleted fibers; rRLC + DTT + TnC are fibers reconstituted with rabbit RLC; and vRLC + DTT + TnC are fibers reconstituted with rat vRLC.
Mechanical function of FHC-RLC mutants
The mean maximal tension (determined from fits to Hill equation) was not significantly affected for fibers with G13T, E22K, or P95A, whereas fibers with F18L have substantially decreased tension (69 ± 10%) compared to fibers with wildtype vRLC (Po = 229.42 ± 16.24; see Fig. 5, A and B). The tension at pCa 6.0 (Fig. 5 A, vertical dotted line and Fig. 5 D) was not significantly affected for the fibers with P95A, but was significantly increased for fibers with E22K, and decreased for G13T and F18L (Fig. 5 D).
The pCa50 value, which represents the fiber sensitivity to Ca2+ at half-maximal activation (Fig. 5 C, horizontal dotted line), was affected for all the FHC-RLC mutants except P95A, which was similar to the fibers with wildtype vRLC (pCa50 = 5.96 ± 0.008; see Figs. 5 C and 6, top panel). The pCa50 for E22K mutant increased, suggesting increased Ca sensitivity for this mutation. The G13T mutant induced a decrease in Ca sensitivity (pCa50 is decreased), whereas F18L induced an even further decrease in Ca sensitivity. The Hill coefficient (cooperativity) for fibers with G13T, E22K, and P95A was not significantly different from fibers with wildtype RLC (n = 1.91 ± 0.057; see Fig. 6, lower panel). However, the Hill coefficient for F18L was most severely decreased compared to the other mutations, characteristic of RLC-extracted fibers despite complete restoration of the RLC content of the F18L in the fibers.
FIGURE 6.
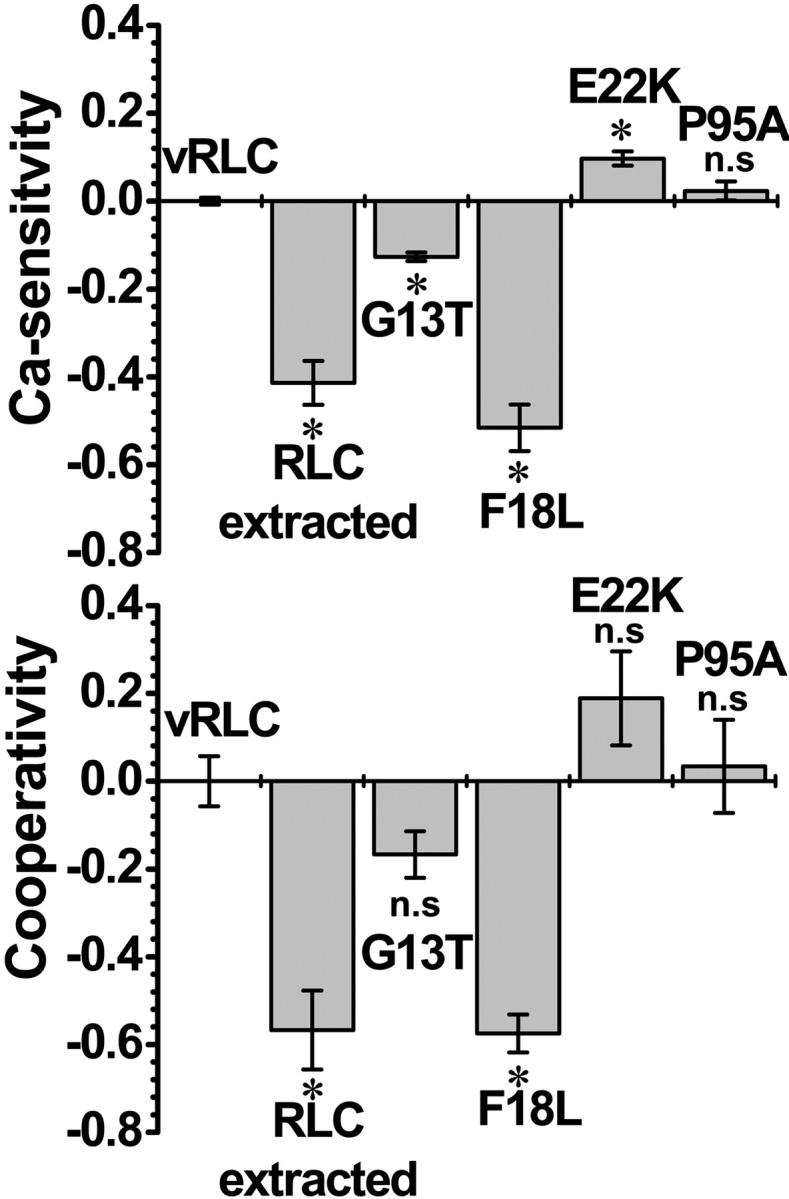
Ca sensitivity (pCa50 = pCa at half-maximal tension) and cooperativity (n) of single fibers containing wildtype vRLC, FHC-vRLC mutants, or 50% rRLC. The Ca sensitivity and cooperativity are plotted as the difference between the FHC-vRLC mutants and wildtype determined from the data in Fig. 5. The data are presented as the mean ± SE. *, P < 0.05; n.s., not significantly different from wildtype vRLC.
DISCUSSION
This is the first study to show that the replacement of rabbit RLC with rat vRLC in rabbit psoas fibers was successful in that the maximal tension of single muscle fibers was restored. This study also shows that myosin FHC-RLC mutants impair the mechanical function of muscle fibers. The RLC mutants display a range of effects in which the Ca sensitivity of contracting fibers were increased (E22K), decreased (G13T and F18L), or not significantly affected (P95A). The maximal tension and cooperativity were not significantly affected for the RLC mutants, except for F18L, which was significantly lowered.
A unique feature of this study is that mutant RLC was introduced into normal muscle fibers, so the effects observed are directly related to the function of myosin, and are not complicated by slowly developing secondary effects (e.g., cellular disarray, fibrosis, necrosis, hypertrophy) that are often associated with cardiac tissue from diseased patients or transgenic animals. Another advantage of this in vitro study is that the amount of mutant RLC in the muscle fiber could be quantitated by gel electrophoresis, in contrast to studies that use diseased human tissue that most likely contains a mixture of mutant and normal vRLC. These results demonstrate that the mutant proteins induce functional defects that are immediate; i.e., that secondary morphological changes do not have to occur before the fiber's functional integrity is impaired by the mutation. A potential limitation of the present study is that a direct correlation of the functional defects of the FHC-RLC mutations in skeletal muscle to that in cardiac muscle must be made cautiously, because although the basic mechanisms for contraction in skeletal and cardiac muscle are similar, there are key differences in the roles of some thick and thin filament proteins.
The amino acid sequence identity of rat vRLC compared to human vRLC is 95.2%, so it is highly probable that the human FHC-RLC mutations in the rat cardiac RLC will faithfully report functional consequences of the FHC-RLC mutations on muscle contraction. Furthermore, transgenic mice with myosin FHC mutations have proven to be useful models of the FHC disease (Seidman and Seidman, 2001; Sanbe et al., 2000). Skeletal muscle was chosen as the background for studying the effects of FHC mutations, because the methodologies for extraction and exchange of mutant RLC, and for mechanical analysis of skinned muscle fibers, have been much more successfully developed in skeletal than in cardiac muscle. This study required a robust preparation of skinned fibers that could stand up to extensive biochemical manipulation and give reproducible mechanical results; rabbit psoas muscle meets these requirements.
Although there are amino acid differences between the skeletal and cardiac RLC isoforms (73% identity), the cardiac vRLC is able to restore Ca-sensitive isometric tension in skeletal muscle fibers (Fig. 4). Indeed, the observed Ca sensitivity of tension appears to be influenced by the RLC source—whereas normal function is restored by addition of rabbit skeletal RLC (Fig. 4), the addition of cardiac vRLC confers increased Ca sensitivity with a lower Hill coefficient (Fig. 4), reminiscent of cardiac muscle (Martyn and Gordon, 2001). Ideally, it would be more appropriate to study the effect of the FHC-RLC mutations in the context of human cardiac muscle, but the availability of sufficient healthy tissue for the functional studies is limited. Although there are some differences in the mechanisms for contraction between cardiac and skeletal muscle, the key elements remain similar. In fact, since vRLC confers physiological properties of cardiac muscle in rabbit psoas fibers, it is likely that the functional perturbations caused by vRLC mutations in the present study reflect their effect in cardiac muscle.
Functional reconstitution of muscle fibers
Removal of 50% RLC from skeletal muscle fibers decreases the maximal tension and Ca sensitivity of force, but reconstitution with skeletal RLC completely restores normal function (Fig. 4), to an extent that has not been previously reported (Diffee et al., 1995; Szczesna et al., 1996). Replacement of skeletal RLC with ventricular RLC also restores maximal tension to normal levels, and induces Ca sensitivity (increased) and cooperativity (decreased) (Fig. 4) similar to that of native cardiac fibers (Gordon et al., 2001; Martyn and Gordon, 2001). This ability of RLC to confer tissue-specific Ca sensitivity in fibers was observed previously for smooth muscle RLC, which increased Ca sensitivity of skeletal muscle fibers (Levine et al., 1998). Similarly, partial replacement of vRLC by skeletal rRLC in the hearts of transgenic mice reduced left ventricular contractility and the rate of relaxation of perfused hearts (Gulick et al., 1997).
G13T decreases Ca sensitivity
The G13T mutation does not significantly affect the maximal isometric tension (at saturating Ca) or the cooperativity, but it reduces force at subsaturating Ca, decreasing the Ca sensitivity (Figs. 5 and 6). This is a physiologically significant defect, since the contracting heart spends most of its time at subsaturating Ca concentrations, where force is submaximal. As mentioned in Materials and Methods, the N-terminus of rat vRLC contains four different amino acids compared to human vRLC: Gly13Ala, Leu10Ala, Glu11Ala, and Ser14Asn (the rat's amino acid precedes the human's). Therefore, it is difficult to make a direct correlation between the mechanical defect in muscle fibers caused by rat G13T-vRLC and the phenotype of the human FHC disease. Nevertheless, it is quite interesting that this mutation (threonine), at the precise location (residue 13) where a mutation causes human disease, causes a significant physiological perturbation.
F18L decreases tension, Ca sensitivity, and cooperativity
The F18L mutation induced a more severe dysfunction, decreasing maximal tension as well as Ca sensitivity and cooperativity (Figs. 5 and 6). Indeed, fibers reconstituted with F18L-RLC have functional properties similar to those of RLC-extracted fibers, indicating that F18L-RLC is the only mutant in this study that fails to fully restore any function. In good correlation with our mechanical measurements, F18L induces the most severe disease phenotype—the percentage of healthy carriers with this mutation is only 20%, substantially lower than for the other mutations studied (Flavigny et al., 1998).
E22K increases Ca sensitivity
E22K increases the Ca sensitivity, without affecting maximal tension and cooperativity of contracting muscle fibers (Figs. 5 and 6). This increased Ca sensitivity is similar to results of a previous study with deltoid muscle fibers from a diseased patient with the E22K mutation (Levine et al., 1998). The latter study shows an increase in Ca sensitivity over the whole range of activation levels, whereas in the present study, the Ca sensitivity increases above pCa10 (10% of maximal tension; see Fig. 5, A and C; Fig. 6, top panel). Similarly, the cooperativity of force activation was not significantly changed (Fig. 6, lower panel) as in the previous study (Levine et al., 1998). These results suggest that the FHC mutation, E22K, in rat vRLC in rabbit psoas muscle shows similar physiological effects as the FHC mutation in human vRLC in slow skeletal muscle. In the present study, 50% E22K-vRLC is exchanged in the muscle fibers, whereas in the previous study with deltoid muscle from a diseased patient, the amount or identity of the RLC mutant in the muscle fiber was not determined. Electron microscopy of myosin filaments extracted from the affected deltoid fibers showed increased filament disorder compared to normal thick filaments, suggesting that E22K promotes head movement away from the thick filament, which might explain the increased activation at low Ca (Levine et al., 1998). Despite these effects of E22K on fiber mechanics and human disease, myosin extracted from diseased deltoid muscle fibers from a patient with the E22K mutation produced normal in vitro actin filament motility (Poetter et al., 1996). In addition, transgenic mice with the E22K-RLC mutation did not display any of the typical phenotypic features of the human disease, despite total replacement of the endogenous vRLC with mutant vRLC (Sanbe et al., 2000). Thus, single-fiber mechanics is more sensitive than some other assays in detecting the functional defect caused by this mutation.
P95A has no significant effect on tension, Ca sensitivity, or cooperativity
P95A does not induce significant changes in the maximal tension, Ca sensitivity, or cooperativity of contracting fibers (Figs. 5 and 6). This mutation in the linker region between the N- and C-terminal domains of RLC is expected to increase flexibility of that region (Rayment et al., 1993). Indeed, it has been shown that deletion of the equivalent proline residue in smooth myosin RLC activates myosin ATPase activity (Ikebe et al., 1998). However, the present study shows clearly that the P95A mutation, which probably increases flexibility of the linker region, is not sufficient to affect the Ca-dependence of isometric tension. Thus, the mechanism whereby this mutation causes FHC must involve some function other than isometric force generation, such as the rate of contraction.
Mechanistic hypotheses
In most current models of muscle contraction, the LC domain acts as a rigid lever arm that undergoes a large rotation during the transition of weakly- to strongly-bound myosin heads on the actin filament. The potential fragility of this lever arm is suggested by the crystal structure of the myosin head, which shows that the LC domain is based on a single long, slightly curved α-helix that extends from the catalytic domain and binds the ELC and RLC (Rayment et al., 1993). It is likely that the mechanical stability of the LC domain depends on the structural integrity of the light chains, so it is plausible that structural perturbations in the RLC can alter isometric tension by changing the number of strong binding heads during contraction. An increase in the number of strong binding heads is itself capable of increasing thin filament activation that will increase both Ca sensitivity and cooperativity (Gordon et al., 2001). The reduction in Ca sensitivity of the G13T and F18L mutations suggests that the number of strong binding crossbridges was reduced during contraction, whereas the increase in Ca sensitivity by E22K increases this fraction.
It was previously shown that a point mutation (D47A) in the Ca2+/Mg2+ binding site of RLC reduced Ca binding, Ca sensitivity, and cooperativity of contracting fibers (Diffee et al., 1995). Furthermore, D47A decreased fiber stiffness, but increased the rate of force redevelopment and rate of relaxation of activated fibers after chelating Ca2+, suggesting that D47A decreases the proportion of cycling crossbridges in the force-generating state by decreasing the rate of formation of force-generating bridges and increasing the rate of detachment (Diffee et al., 1995). Thus the previously observed effects of RLC on the Ca dependence of force (Levine et al., 1998; Diffee et al., 1995; Gulick et al., 1997) are probably due to changes induced by RLC on in the number of strong binding heads.
Several FHC-vRLC mutations used in the present study were examined previously in a study of Ca binding to isolated light chains in solution (Szczesna et al., 2001). Mutations A13T, F18L, and P95A decreased the Ca binding affinity by ∼3-fold, whereas E22K decreased it by 17-fold. These results do not correlate qualitatively with the functional effects on fibers observed in the present study, suggesting that changes in Ca affinity do not provide a simple explanation of our observations. For example, a mutation might affect force by affecting the structural integrity of the LC domain, modifying its effectiveness as a lever arm. However, it is important to note that the conformational order and Ca-binding affinity of isolated RLCs in solution is probably much less than when bound to myosin in fibers, so a simple correlation with fiber function is not really expected.
Mechanisms of FHC disease
The present results show that ∼50% mutant RLC is sufficient to perturb the tension at subsaturating Ca concentration (Fig. 5 D), Ca sensitivity, and cooperativity of force generation of fibers (Fig. 6), supporting the hypothesis that the mechanical dysfunction of the fibers may be directly linked to the mechanism for FHC. The enhanced (E22K) and diminished (G13T and F18L) function of these FHC-RLC mutations fibers suggest that the FHC disease may be caused by fibers that are in either a hypercontractile (E22K) or hypocontractile (G13T and F18L) state. Previous reports also suggest that different FHC mutations can induce either a gain or loss of function (Roopnarine, 2002; Bonne et al., 1998; Seidman and Seidman 2001; Hernandez et al., 2001; Michele and Metzger, 2000). The dysfunction caused by the FHC-RLC mutants suggest that the mutant protein will alter the force generated in myocytes and lead to mechanical stress within the heart, which is likely to cause myocyte disarray and remodeling of the heart via hypertrophy.
This study shows that wildtype ventricular RLC confers Ca-sensitive force in skeletal muscle similar to that of cardiac muscle. Of the four FHC-RLC mutants tested, three caused immediate and substantial changes in the Ca sensitivity of force production, suggesting that these mechanical abnormalities initiate the FHC disease process.
Acknowledgments
The author thanks Dr. Leslie A. Leinwand for the wildtype rat vRLC cDNA; Dr. LaDora Thompson for use of Muscle Research System for the single fiber mechanics; Dr. Dawn Lowe for technical instruction on using the Muscle Research System; and Dr. David D. Thomas for research facilities and scientific advice. The author also thanks Bilal Anwer (University of Minnesota); Richard Timm, Laura Gingras, and Melanie Thomas (Breck High School, Minnesota), for assistance in protein purification and muscle fiber preparation.
This work was supported by grants from the American Heart Association Northland Affiliate, the Muscular Dystrophy Association, the Minnesota Medical Foundation, and the National Institutes of Health (AR32961).
References
- Andersen, P. S., O. Havndrup, H. Bundgaard, J. C. Moolman-Smook, L. A. Larsen, J. Mogensen, P. A. Brink, A. D. Borglum, V. A. Corfield, K. Kjeldsen, J. Vuust, and M. Christiansen. 2001. Myosin light chain mutations in familial hypertrophic cardiomyopathy: phenotypic presentation and frequency in Danish and South African populations. J. Med. Genet. 38:E43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, J. E., I. Brust-Mascher, S. Ramachandran, L. E. LaConte, and D. D. Thomas. 1998. A large and distinct rotation of the myosin light chain domain occurs upon muscle contraction. Proc. Natl. Acad. Sci. USA. 95:2944–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonne, G., L. Carrier, P. Richard, B. Hainque, and K. Schwartz. 1998. Familial hypertrophic cardiomyopathy: from mutations to functional defects. Circ. Res. 83:580–593. [DOI] [PubMed] [Google Scholar]
- Diffee, G. M., M. L. Greaser, F. C. Reinach, and R. L. Moss. 1995. Effects of a non-divalent cation binding mutant of myosin regulatory light chain on tension generation in skinned skeletal muscle fibers. Biophys. J. 68:1443–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons, D. P., P. W. Bodell, and K. M. Baldwin. 1990. Myocardial functional correlates of cardiac myosin light chain 2 phosphorylation. J. Appl. Physiol. 68:2426–2433. [DOI] [PubMed] [Google Scholar]
- Flavigny, J., P. Richard, R. Isnard, L. Carrier, P. Charron, G. Bonne, J. F. Forissier, M. Desnos, O. Dubourg, M. Komajda, K. Schwartz, and B. Hainque. 1998. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J. Mol. Med. 76:208–214. [DOI] [PubMed] [Google Scholar]
- Gordon, A. M., M. Regnier, and E. Homsher. 2001. Skeletal and cardiac muscle contractile activation: tropomyosin “rocks and rolls.” News Physiol. Sci. 16:49–55. [PubMed] [Google Scholar]
- Gulick, J., T. E. Hewett, R. Klevitsky, S. H. Buck, R. L. Moss, and J. Robbins. 1997. Transgenic remodeling of the regulatory myosin light chains in the mammalian heart. Circ. Res. 80:655–664. [DOI] [PubMed] [Google Scholar]
- Hernandez, O. M., P. R. Housmans, and J. D. Potter. 2001. Invited Review: pathophysiology of cardiac muscle contraction and relaxation as a result of alterations in thin filament regulation. J. Appl. Physiol. 90:1125–1136. [DOI] [PubMed] [Google Scholar]
- Hofmann, P. A., J. M. Metzger, M. L. Greaser, and R. L. Moss. 1990. Effects of partial extraction of light chain 2 on the Ca2+ sensitivities of isometric tension, stiffness, and velocity of shortening in skinned skeletal muscle fibers. J. Gen. Physiol. 95:477–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber, P. J., Brunner, U. T., and M. C. Schaub. 1989. Disulfide formation within the regulatory light chain of skeletal muscle myosin. Biochemistry. 28:9116–9123. [DOI] [PubMed] [Google Scholar]
- Ikebe, M., T. Kambara, W. F. Stafford, M. Sata, E. Katayama, and R. Ikebe. 1998. A hinge at the central helix of the regulatory light chain of myosin is critical for phosphorylation-dependent regulation of smooth muscle myosin motor activity. J. Biol. Chem. 273:17702–17707. [DOI] [PubMed] [Google Scholar]
- Kerrick, W. G., J. D. Potter, and P. E. Hoar. 1991. The apparent rate constant for the dissociation of force generating myosin crossbridges from actin decreases during Ca2+ activation of skinned muscle fibres. J. Muscle Res. Cell Motil. 12:53–60. [DOI] [PubMed] [Google Scholar]
- Levine, R. J., Z. Yang, N. D. Epstein, L. Fananapazir, J. T. Stull, and H. L. Sweeney. 1998. Structural and functional responses of mammalian thick filaments to alterations in myosin regulatory light chains. J. Struct. Biol. 122:149–161. [DOI] [PubMed] [Google Scholar]
- Liu, X., N. Takeda, and N. S. Dhalla. 1997. Myosin light-chain phosphorylation in diabetic cardiomyopathy in rats. Metabolism. 46:71–75. [DOI] [PubMed] [Google Scholar]
- Martyn, D. A., and A. M. Gordon. 2001. Influence of length on force and activation-dependent changes in troponin-c structure in skinned cardiac and fast skeletal muscle. Biophys. J. 80:2798–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger, J. M., M. L. Greaser, and R. L. Moss. 1989. Variations in cross-bridge attachment rate and tension with phosphorylation of myosin in mammalian skinned skeletal muscle fibers. Implications for twitch potentiation in intact muscle. J. Gen. Physiol. 93:855–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michele, D. E., and J. M. Metzger. 2000. Physiological consequences of tropomyosin mutations associated with cardiac and skeletal myopathies. J. Mol. Med. 78:543–553. [DOI] [PubMed] [Google Scholar]
- Morano, I. 1992. Effects of different expression and posttranslational modifications of myosin light chains on contractility of skinned human cardiac fibers. Basic Res. Cardiol. 87:129–141. [DOI] [PubMed] [Google Scholar]
- Moss, R. L., G. G. Giulian, and M. L. Greaser. 1985. The effects of partial extraction of TnC upon the tension-pCa relationship in rabbit skinned skeletal muscle fibers. J. Gen. Physiol. 86:585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persechini, A., J. T. Stull, and R. Cooke. 1985. The effect of myosin phosphorylation on the contractile properties of skinned rabbit skeletal muscle fibers. J. Biol. Chem. 260:7951–7954. [PubMed] [Google Scholar]
- Poetter, K., H. Jiang, S. Hassanzadeh, S. R. Master, A. Chang, M. C. Dalakas, I. Rayment, J. R. Sellers, L. Fananapazir, and N. D. Epstein. 1996. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat. Genet. 13:63–69. [DOI] [PubMed] [Google Scholar]
- Potter, J. D. 1982. Preparation of troponin and its subunits. Methods Enzymol. 85:241–263. [DOI] [PubMed] [Google Scholar]
- Rayment, I., H. M. Holden, J. R. Sellers, L. Fananapazir, and N. D. Epstein. 1995. Structural interpretation of the mutations in the beta-cardiac myosin that have been implicated in familial hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA. 92:3864–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment, I., W. R. Rypniewski, K. Schmidt-Base, R. Smith, D. R. Tomchick, M. M. Benning, D. A. Winkelmann, G. Wesenberg, and H. M. Holden. 1993. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 261:50–58. [DOI] [PubMed] [Google Scholar]
- Roopnarine, O. 2002. Familial hypertrophic cardiomyopathic myosin mutations that affect the actin-myosin interaction. Results Probl. Cell Differ. 36:75–86. [DOI] [PubMed] [Google Scholar]
- Roopnarine, O., and L. A. Leinwand. 1998. Functional analysis of myosin mutations that cause familial hypertrophic cardiomyopathy. Biophys. J. 75:3023–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopnarine, O., A. G. Szent-Gyorgyi, and D. D. Thomas. 1998. Microsecond rotational dynamics of spin-labeled myosin regulatory light chain induced by relaxation and contraction of scallop muscle. Biochemistry. 37:14428–14436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopnarine, O., and D. D. Thomas. 1994. A spin label that binds to myosin heads in muscle fibers with its principal axis parallel to the fiber axis. Biophys. J. 67:1634–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopnarine, O., and D. D. Thomas. 1995. Orientational dynamics of indane dione spin-labeled myosin heads in relaxed and contracting skeletal muscle fibers. Biophys. J. 68:1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanbe, A., J. G. Fewell, J. Gulick, H. Osinska, J. Lorenz, D. G. Hall, L. A. Murray, T. R. Kimball, S. A. Witt, and J. Robbins. 1999. Abnormal cardiac structure and function in mice expressing nonphosphorylatable cardiac regulatory myosin light chain 2. J. Biol. Chem. 274:21085–21094. [DOI] [PubMed] [Google Scholar]
- Sanbe, A., D. Nelson, J. Gulick, E. Setser, H. Osinska, X. Wang, T. E. Hewett, R. Klevitsky, E. Hayes, D. M. Warshaw, and J. Robbins. 2000. In vivo analysis of an essential myosin light chain mutation linked to familial hypertrophic cardiomyopathy. Circ. Res. 87:296–302. [DOI] [PubMed] [Google Scholar]
- Seidman, J. G., and C. Seidman. 2001. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 104:557–567. [DOI] [PubMed] [Google Scholar]
- Szczesna, D., D. Ghosh, Q. Li, A. V. Gomes, G. Guzman, C. Arana, G. Zhi, J. T. Stull, and J. D. Potter. 2001. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J. Biol. Chem. 276:7086–7092. [DOI] [PubMed] [Google Scholar]
- Szczesna, D., J. Zhao, and J. D. Potter. 1996. The regulatory light chains of myosin modulate cross-bridge cycling in skeletal muscle. J. Biol. Chem. 271:5246–5250. [DOI] [PubMed] [Google Scholar]
- Thomas, D. D., E. Prochniewicz, and O. Roopnarine. 2002. Changes in actin and myosin structural dynamics due to their weak and strong interactions. Results Probl. Cell Differ. 36:7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, P. D. 1982. Preparation and fractionation of myosin light chains and exchange of the essential light chains. Methods Enzymol. 85:72–81. [DOI] [PubMed] [Google Scholar]