

Abstract
The energy transfer processes between carotenoids and Chls have been studied by femtosecond transient absorption in the CP29-WT complex, which contains only two carotenoids per polypeptide located in the L1 and L2 sites, and in the CP29-E166V mutant in which only the L1 site is occupied. The comparison of these two samples allowed us to discriminate between the energy transfer pathways from the two carotenoid binding sites and thus to obtain detailed information on the Chl organization in CP29 and to assign the acceptor chlorophylls. For both samples, the main transfer occurs from the S2 state of the carotenoid. In the case of the L1 site the energy acceptor is the Chl a 680 nm (A2), whereas the Chl a 675 nm (A4–A5) and the Chl b 652 nm (B6) are the acceptors from the xanthophyll in the L2 site. These transfers occur with lifetimes of 80–130 fs. Two additional transfers are observed with 700-fs and 8- to 20-ps lifetimes. Both these transfers originate from the carotenoid S1 states. The faster lifetime is due to energy transfer from a vibrationally unrelaxed S1 state, whereas the 8- to 20-ps component is due to a transfer from the S1,0 state of violaxanthin and/or neoxanthin located in site L2. A comparison between the carotenoid to Chl energy transfer pathways in CP29 and LHCII is presented and differences in the structural organization in the two complexes are discussed.
INTRODUCTION
In photosynthetic organisms, the light energy is absorbed by the antenna complexes and transferred to the reaction center where charge separation takes place. The supramolecular antenna system of Photosystem II from higher plants is composed of at least four different complexes all belonging to the Lhc antenna family (Jansson, 1999): LHCII, the major light-harvesting complex present in trimeric form in the thylakoid membrane, and three minor complexes named CP29, CP26, and CP24. These three complexes, present as monomers in the supercomplex, are located between the reaction center and the peripherally located major LHCII complex (Bassi and Dainese, 1992; Boekema et al., 1998, 1999; Harrer et al., 1998; Hankamer et al., 1997). The minor complexes appear to be the sites of different photoprotective mechanisms, which allow regulating the flow of energy to the reaction center depending on light and other conditions (Bassi and Caffarri, 2000). All these complexes bind noncovalently two species of chromophores: Chls and carotenoids. The pigment composition of the higher plant photosynthetic apparatus is very well conserved in different organisms: chloroplast-encoded photosynthetic reaction center complexes bind β-carotene and Chl a, whereas nuclear-encoded, light-harvesting proteins bind Chl a, Chl b, and three xanthophylls: lutein, violaxanthin, and neoxanthin. Besides these xanthophylls, which are always present in higher plant antennae, excess illumination leads to the appearance of two additional xanthophylls, i.e., antheraxanthin and zeaxanthin, deriving from violaxanthin by de-epoxidation of either one or both rings (Demmig et al., 1987).
The partial resolution of the structure of the LHCII complex has revealed the presence of two carotenoid binding sites, named L1 and L2, located in the center of the structure forming a hydrophobic core (Kühlbrandt et al., 1994). Biochemical analysis showed that two additional xanthophyll binding sites, i.e., N1 and V1, which are, respectively, resistant and labile to the extraction with detergent and low pH (Ruban et al., 1999; Caffarri et al., 2001), are present in the LHCII complex as well. The N1 site is located near the C-helix (Croce et al., 1999a), whereas the positioning of the V1 site is still unknown. All the other Lhcb proteins bind only two carotenoid molecules. In addition, each member of the family has a unique carotenoid composition. Thus neoxanthin, which is bound only in the N1 site in LHCII, occupies the L2 site in both CP29 and CP26, and is not present in CP24 and in the antenna complexes of Photosystem I (LHCI) (Bassi et al., 1993; Schmid et al., 1997). The amount of violaxanthin varies strongly in the different complexes. Whereas in the case of CP29 and CP26 this carotenoid is located in the L2 site, in LHCII it is positioned in the labile V1 site (Caffarri et al., 2001). The L1 site seems the most conserved one and it exhibits a high affinity for lutein in all the complexes (Bassi et al., 1999; Croce et al., 1999b). Carotenoids play many roles in higher plants: 1), structure stabilization; 2), light harvesting; 3), photoprotection via Chl triplet and singlet oxygen scavenging; and 4), nonphotochemical quenching NPQ via the xanthophyll cycle.
Due to the difference in carotenoid composition between the Lhc proteins, it is worthwhile to ask the question if the different carotenoids do play different roles in these complexes on the basis of their physicochemical properties and their localization. Such an analysis was made possible using recombinant Lhc proteins refolded in vitro with purified pigments (Plumley and Schmidt, 1987; Paulsen, 1997). This procedure, which yields antenna complexes indistinguishable from the native protein when the total mixture of the thylakoids pigments is added to an apoprotein carrying the wt sequence (Giuffra et al., 1996), allowed us to modify either the primary structure of the protein and/or the pigment composition. The approach was successfully used for the study of Chl and carotenoid organization in both CP29 and LHCII (Bassi et al., 1999; Remelli et al., 1999; Yang et al., 1999; Rogl and Kühlbrandt, 1999; Hobe et al., 2000; Formaggio et al., 2001), and more recently has been used for a detailed study of the carotenoid-to-Chl energy transfer in LHCII (Croce et al., 2001). In that case, two complexes with different carotenoid compositions were used to discriminate between the two central sites, L1 and L2, and the external one, i.e., N1. The data indicate for LHCII an ultrafast transfer (<100 fs) from carotenoids to both Chl a and Chl b. A similar study by Gradinaru et al. (2000) performed on native LHCII and CP29 concluded that only the neoxanthin located in the N1 site of LHCII should be able to transfer energy to Chl b whereas the xanthophylls located in the L1 and L2 sites were proposed to transfer energy only to Chl a in both complexes. This conclusion is at variance with the results of our own studies, which indicate energy transfer from all three carotenoids to Chl b in LHCII, albeit with different efficiencies (Croce et al., 2001; Connelly et al., 1997). There thus still exists a pronounced disagreement about the ability of the carotenoids in the central sites to transfer energy to Chl b, an issue that is important for the determination of the Chl organization in the antenna system of higher plants in view of the lack of structural data to distinguish between Chl a and Chl b. The problem is thus a central one for the understanding of the structure/function relationships in these complexes.
The subject of this work is to extend our studies on carotenoid-to-Chl energy transfer to the CP29 complex. We expect in particular to gain further insight into the function of the Chls and their localization relative to the carotenoids, thus complementing our recent study of the Chl-Chl energy transfer in this complex (Croce et al., 2003). Among all the antenna complexes of higher plants, CP29 has the lowest pigment content, i.e., eight Chls and two carotenoid molecules located in the L1 and L2 sites. A mutant which contains only one carotenoid, located in the L1 site, is available for CP29 as well, which allows for critical tests of our assignments of individual energy transfer steps.
The sequence comparison between LHCII and CP29 shows a high homology and the mutation analysis performed on both CP29 and LHCII confirmed that all the Chl binding sites present in CP29 are Chl binding sites also in LHCII (Pichersky and Green, 1990; Bassi et al., 1999; Remelli et al., 1999). CP29 can thus be used as a simplified model to study the energy transfer process in the Lhc complexes. Previous studies of steady-state and time-resolved energy transfer in CP29 have been published by various groups (Gradinaru et al., 1998; Pieper et al., 2000; Cinque et al., 2000; Iseri et al., 2000; Voigt et al., 2002). From the qualitative point of view, CP29 binds the same xanthophyll molecules as are present in LHCII, but in different ratios and in different sites. The comparison between the results obtained for LHCII and CP29 will thus provide valuable information about the role played in the energy transfer process by identical xanthophylls located in different sites and may help to understand the possible reasons for the different carotenoid composition throughout the Lhc family.
MATERIAL AND METHODS
Sample preparation and analysis
The constructs overexpressing maize Lhcb4 and the E166V mutant were obtained as described earlier (Giuffra et al., 1996; Bassi et al., 1999) except for a sequence coding for a His6 tail inserted at the 3′ end before the stop codon.
Reconstitution of complexes was performed as already reported (Giuffra et al., 1996) using a Chl a/b ratio of 4.0 in the pigment mixture. Purification of the reconstituted complex was performed by affinity chromatography on an Ni column followed by ultracentrifugation on a glycerol gradient (15–40% glycerol, 0.06% β-dodecylmaltoside (DM), 10 mM Hepes pH 7.6, 12 h at 55.000 rpm in a SW60 rotor; Beckman).
The pigment composition was determined by HPLC analysis (Gilmore and Yamamoto, 1991) and analysis of the absorption spectrum of the acetone extracts was performed with the spectra of the individual pigments (Croce et al., 2002).
Steady-state spectroscopy
Absorption spectra were recorded at RT using a SLM-Aminco DW-2000 spectrophotometer. Corrected fluorescence excitation and emission spectra were obtained by using a Jasco FP-777 spectrofluorimeter, with a Chl concentration of 0.01 μg/ml. All samples were in 10 mM Hepes, pH 7.6, 0.03% DM, and 20% glycerol.
Femtosecond transient absorption measurements
Femtosecond transient absorption measurements were performed as previously reported and were analyzed as lifetime density maps (Croce et al., 2001). The excitation pulses had a spectral FWHM of ∼4 nm and a pulse width of ∼70 fs. The intensity used was in the range of ∼3×1013 to 1×1014 photons/(cm2 pulse) depending upon the excitation and detection conditions. All measurements were performed at RT in a 1-mm cuvette that was moved in two different directions simultaneously to yield a circular rotation, to avoid overexciting a particular region of the sample. The absorption and fluorescence of the samples before and after the measurements was essentially the same within the error limits.
Data analysis
Lifetime density maps have been described in detail in a previous article (Croce et al., 2001). Such lifetime density plots present the amplitudes of the lifetime components in a quasicontinuous lifetime range. White-yellow spots represent positive amplitudes and reflect either decay of an absorption or rise of a bleaching. Blue to black spots represent negative amplitudes and reflect either decay of the bleaching or rise of absorption.
To obtain information equivalent to a decay-associated difference spectrum (DADS) for a particular lifetime (Holzwarth, 1996) from the lifetime density maps, we integrated the signals in the lifetime density maps, which have a quasicontinuous lifetime scale, over defined time intervals as indicated in each case. Lifetime distribution plots at particular detection wavelengths were calculated as cuts through the lifetime density map surface at the indicated wavelengths.
The descriptions and analysis of the absorption spectra in terms of individual pigment absorption forms was performed as previously described (Croce et al., 2000).
RESULTS
The energy transfer process from carotenoids to chlorophylls has been studied in CP29 using two reconstituted complexes. The CP29-WT complex used binds 5.6 Chls a, 2.4 Chls b, and two carotenoid molecules per polypeptide. The stoichiometry was 0.87 ± 0.05 lutein, 0.64 ± 0.06 neoxanthin, and 0.48 ± 0.04 violaxanthin. The carotenoids are located in the L1 and L2 sites in the center of the structure, with L1 accommodating lutein and L2 either violaxanthin or neoxanthin (Bassi et al., 1999). The nonstoichiometric amount of xanthophylls clearly indicates heterogeneity in the population. The second complex used in this study was the CP29-E166V mutant, in which the glutamic acid which coordinates the Chl in site B6 is substituted by a valin, an amino acid which is not able to coordinate the central Mg of the Chl. This mutant complex contains only six Chl molecules (4.7 Chls a and 1.3 Chls b) and only one carotenoid per polypeptide with a stoichiometry of 0.66 ± 0.05 molecules of lutein, 0.33 ± 0.05 violaxanthin, and 0.04 ± 0.03 neoxanthin. It has been proposed that the carotenoids in this mutant are located exclusively in the L1 site, whereas the L2 site is supposed to be empty (Bassi et al., 1999).
The absorption spectra of the two samples in the 350- to 520-nm absorption range are shown in Fig. 1. The absorption in the 480- to 500-nm region is lower in the mutant than in the WT complex, indicating the lower carotenoid content of this sample.
FIGURE 1.

Room temperature absorption spectra of CP29-WT (solid line) and CP29-E166V mutant (dotted line). The spectra are normalized to the maximum.
Pump-probe experiments with femtosecond resolution were performed on both samples with excitations in the 450- to 500-nm absorption band. Two excitation wavelengths (490 nm and 500 nm) were used for the WT and three for the mutant (475 nm, 490 nm, and 500 nm) to excite as selectively as possible the various carotenoids. However, overlap of the spectra in all regions prevents exclusive excitation of specific carotenoids. The detailed transient absorption kinetics has been measured over two wavelength intervals, 450–570 nm (carotenoid S2 absorption region) and 610–730 nm (Qy Chl absorption region), in all cases.
Kinetics of the CP29-E166V mutant
In Fig. 2 the lifetime density maps of the CP29-E166V mutant excited at 500 nm are shown for the 480- to 570-nm (left) and the 610- to 730-nm (right) detection ranges. In the red region (Chl Qy), three different bleaching rise components for Chl a are observed with lifetimes of 90–130 fs, 500–600 fs, and 15–16 ps. The fastest component probably comprises at least two different components, as can be deduced from the large width of the lifetime range. The decay of the overall Chl-a absorption bleaching is in the ns time range. In the Chl-b Qy region no bleaching rise signal, which would have positive amplitude, is observed. Rather, the 600- to 660-nm and the above 700-nm regions are dominated by the rise of excited-state absorption (ESA), which shows a rise time of 80–130 fs and a decay time of ∼350–400 fs. In the blue region (480–570 nm) the S2 bleaching decay and the stimulated emission decay of the carotenoids (both weak signals) and the rise in the Sn-S1 ESA of the carotenoid (strong signal) are detected with a 100- to 130-fs lifetime component in the 500- to 570-nm range. The latter makes the most dominant contribution to the signal in the ultrafast time range ∼100 fs. A lower amplitude decay with 450- to 800-fs lifetime is present in the 560- to 570-nm region. Furthermore, the main decay of the Sn-S1 carotenoid ESA, reflecting the S1-decay, is present as a relatively broad lifetime distribution band in the 510- to 570-nm range with lifetimes ranging from 8–20 ps. The distribution of the ESA decay is much wider than expected for such a well-characterized strong lifetime component, which hints to the presence of at least two contributions. To better present the corresponding spectra, DADS were calculated by integration over narrow lifetime ranges from the lifetime density maps as described in the Methods section. They are shown in Fig. 3. For the Qy range, the spectra of components centered on lifetimes of 100 fs, 600 fs, and 16 ps are shown in Fig. 3, A–C. The absorption maxima for these three components are 679, 682, and 682 nm, respectively, and the corresponding amplitudes are 84%, 8.5%, and 7% of the total Chl a bleaching. The spectrum in the 100-fs range shows negative amplitude attributed to rise of ESA over almost the whole detection range except ∼680 nm. The whole spectrum can be understood as a pronounced Chl a Qy bleaching rise at 680 nm, sitting on a very broad and structureless background, reflecting a rise in ESA. This spectrally broad ESA rise in excited-state absorption is neither from its spectrum nor its large intensity (relative to the Qy bleaching rise) characteristic for Chl. It must thus be assigned to its largest part to carotenoid ESA. In the blue region, the spectral broad ∼130 fs rise in ESA (negative component) extending from ∼520–570 nm corresponds to the subsequent decay of this ESA in the time range above 8 ps. This broad ESA decay can be decomposed into two components, which show that the DADS of the shorter lifetime range are quite different from that of the longer lifetime component. The faster component (center lifetime ∼11 ps) has a maximum at 552 nm whereas the slower one (center at ∼16 ps) has a maximum at 530 nm. The two spectra are shown in Fig. 3 D. Essentially identical results are obtained for 490-nm excitation (data not shown), suggesting that the excitation probabilities at the two wavelengths should be very similar. Similar results are also obtained for 475-nm excitation, where essentially one additional component in the ps time range from the Chl b to Chl a energy transfer is present. In Table 1 a summary of the observed lifetimes and wavelength ranges of the signals is presented.
FIGURE 2.

Lifetime density maps for CP29-E166V for the 500-nm excitation in two detection ranges: 480–570 nm (left) and 610–730 nm (right). Note: The red background denotes the zero level. Positive amplitudes are shown in yellow/white color, whereas negative amplitudes are shown in blue/black color. It is important to note that in transient absorption spectra a positive amplitude in a DAS can mean either a decay of excited-state absorption or a rise in a bleaching signal. Likewise, a negative amplitude can either be a decay of the bleaching or the rise of an excited-state absorption signal. Which possibility applies must be decided upon analysis of the lifetime density map and the whole kinetics. The lifetime scale shown is a logarithmic scale. It is important to note that these color maps represent a qualitative or at best semiquantitative picture of the kinetics in a very condensed form. The density of the color has been chosen proportional to the amplitude, but due to printer and reproduction quality limitations, one should be careful in the quantitative interpretation of the amplitudes based on these maps. Any quantitative information should only be deduced from the actual numerical amplitude distribution function underlying this surface.
FIGURE 3.

Analysis of the lifetimes density maps for the CP29-E166V mutant excited at 500 nm. DADS calculated by integrating the map (Fig. 2, right) over different time intervals: (A) 50–170 fs; (B) 500 fs–2 ps; and (C) 11–23 ps. (D) DADS of the 11-ps (open circle) and 16-ps (circle) components obtained from the map in Fig. 2 A. Please note that the plots have different scales.
TABLE 1.
Summary of observed lifetimes, wavelength ranges of observation and assignment of components for CP29-E166V complex upon excitation at 490 and 500 nm
| Lifetimes | Carotenoids (L1: 66% Lutein, 33% Violaxanthin) | Chl b | Chl a | ||
|---|---|---|---|---|---|
| 490–555 nm | 555–570 nm | 610–660 and >700 nm | 640–655 nm | 670–680 nm | |
| 90–130 fs | Bleaching decay and SE decay and ESA rise S2,v→S2,0 + S1,v | ESA rise S2,0, S1,v | (No activity) | Bleaching rise EET from S2 | |
| 350–800 fs | ESA decay S2,0, S1,v | Bleaching rise EET from S1,v | |||
| 800 fs–1.6 ps >530 nm | ESA rise S1,0 | ||||
| 8–20 ps | Bleaching and ESA decay (distributed, compare with 11 and 16 ps) | ||||
| 11 ps (max. 555 nm) | S1 ESA decay (lutein) | ||||
| 15–16 ps (max. 530 nm) | S1 ESA decay (viola and neo) | Bleaching rise EET from S1 | |||
| >3 ns | Bleaching decay | ||||
Kinetics of CP29-WT
In Fig. 4 the lifetime density maps for the 480–570 nm and the Chl Qy detection regions of CP29-WT excited at 490 nm (top) and 500 nm (bottom) are shown. The overall kinetic features observed for CP29-WT are quite similar to those observed for the E166V mutant, but for the WT the picture is somewhat more complex due to the presence of several Chl b to Chl a energy transfer components which are not observed in the mutant. For 490-nm excitation (Fig. 4, top) an energy transfer component from Chl b-640 nm to a Chl a is observed with a 600-fs lifetime. The transfer from Chl b-652 occurs in ∼1.2–1.3 ps, whereas an additional transfer step from Chl b-640 is observed with 4-ps lifetime and with 6.5 ps the equilibration between Chl a-670 nm and Chl a-682 nm is detected. For 500-nm excitation (Fig. 4, bottom) the picture is still similar, although the transfer with 600-fs lifetime from Chl b-640 is not present, whereas a transfer from Chl b-640 with a 4-ps lifetime is still present, but with substantially reduced amplitude. In the short time range at both excitation wavelengths a rise in the Chl a Qy region is observed in the 80- to 250-fs range, comprising at least two components with the faster one related to a rise at 675 nm (centered ∼80 fs) and the slower one at 680 nm (centered ∼150 fs lifetime). These two components are better resolved in the 490-nm excitation experiment, but they are clearly also present in the 500-nm excitation. A slow rise of Chl a bleaching is present with a 12- to 15-ps lifetime (Fig. 4, bottom). For 490-nm excitation this component is also present but it is not completely separated from the 6.3-ps Chl-a equilibration. Its presence can, however, be deduced also from the fact that the amplitude of the rise in the 680-nm Chl a is higher than what would be expected for a simple equilibration with the 670-nm Chl a.
FIGURE 4.

Lifetime density maps for CP29 WT for the two different excitation wavelengths of 490 nm (top) and 500 nm (bottom).
Due to the limited color resolution of the lifetime density maps some of the components discussed above are better seen in the DADS representation (which is derived directly from the information in the density maps). To this end, we again calculated DADS from the corresponding lifetime density maps by integration. The resulting spectra for 490-nm excitation are shown in Fig. 5. For 490-nm excitation, the total bleaching decay in Chl b calculated from the decay components centered at 600 fs, 1.2 ps, and 4 ps (Fig. 5, C–E), is ∼28% of the total bleaching in Chl a. For the calculations we assumed a ratio of extinction coefficients for Chl b to Chl a of 0.7 to 1. Out of this Chl b to Chl a decay, ∼10% derives from Chl b-640 nm (6% with the 600-fs lifetime and 4% with the 4-ps lifetime), whereas the other 18% derives from Chl b-652 nm. The total Chl b contribution of Chl b to Chl a transfer drops to 12% for 500-nm excitation, and almost all the energy in Chl b is now in the 652-nm form.
FIGURE 5.

DADS obtained from the lifetime density maps of CP29-WT. A–E for 490-nm excitation in the 610- to 730-nm detection range (see Fig. 4 B) and F in the 400- to 570-nm range. The integration ranges were (A) 50–100 fs; (B) 100–500 fs; (C) 500 fs–1 ps; (D) 1–2.5 ps; (E) 2.4–15 ps; and (F) 8–20 ps. Please note that the plots have different scales.
The short time range is dominated by the rise in ESA. At least two components can be detected in the 610- to 630-nm range and above 700 nm where an ESA rise is observed with lifetimes in the 60- to 80-fs range. An additional smaller ESA rise with a 100- to 150-fs lifetime is observed in the 640- to 660-nm and 710- to 730-nm regions for 500-nm excitation (Fig. 4, bottom) and in the 710- to 730-nm region for the 490-nm excitation (Fig. 4, top). Note that the region between 630 and 660 nm is quite different in the two experiments. For the 500-nm excitation a strong negative amplitude is detected at these wavelengths, whereas in the case of 490-nm excitation only a much smaller negative amplitude is observed at ∼640 nm. The very fast (∼80 fs) rise in ESA is reminiscent of what we observed for isolated lutein in solvents (Müller et al., unpublished results). This component for lutein alone is attributed to S2→S1 internal conversion giving rise to a strong Sn–S1,v ESA. The characteristic spectral features of this component in the protein complex are, in fact, very similar to those for the isolated lutein, whereas the lifetimes are somewhat shorter in the protein complex, indicating some lifetime shortening due to energy transfer.
In the blue region, the decay of S2 SE (around the excitation wavelength) from the carotenoids is present together with the rise of the Sn-S1 ESA with lifetimes in the 80- to 130-fs range for both excitation wavelengths. A pronounced decay of this S1 ESA is observed at 560–570 nm with a 500- to 800-fs lifetime. This component is clearly due to the decay of the vibrationally hot S1 state. The main decay of the Sn-S1 ESA is associated, however, with the wide (in terms of lifetime distribution) band at 520–560 nm with 8- to 20-ps lifetime for both excitations. The ESA decay spectrum in this lifetime range for 490-nm excitation is shown in Fig. 5 F. A summary of the lifetimes and the observed wavelength ranges is given in Table 2.
TABLE 2.
Summary of observed lifetimes, wavelength ranges of observation and assignment of components for CP29-WT complex upon excitation at 490 and 500 nm
| Lifetimes | Carotenoids (L1: 87% Lutein; L2: 64% Neoxanthin, 48% Violaxanthin) | Chl b | Chl a | ||||
|---|---|---|---|---|---|---|---|
| 490–555 nm | 555–570 nm | 610–630 and >700 nm | 640 nm | 652 nm | 670–675 nm | 682 nm | |
| 80 fs (60–100 fs) | Bleaching decay and SE decay and ESA rise S2,v→S2,0 | ESA rise S2,0 | Bleaching rise compensated by Car ESA rise | Bleaching rise EET from S2 | |||
| 150 fs (110–200 fs) | Bleaching decay and SE decay and ESA rise S2,0→S1,v | Car ESA rise for 500 nm ex (630–660 nm) | Bleaching rise EET from S2,0 (lute) | ||||
| 500 fs (300–900 fs) | ESA rise S1,0 | ESA decay S1,v | Bleaching rise EET from S1,v | ||||
| 600 fs (Ex: 490 nm only) | Bleaching decay (small direct ex.) | ||||||
| 1.2 ps | Bleaching decay | Bleaching rise EET from Chl b-652 | |||||
| 4 ps (Ex: 490 nm only) | Bleaching decay (small direct ex.) | Bleaching rise | |||||
| 6.5 ps | Bleaching decay | Bleaching rise EET from Chl a-670 | |||||
| 8–20 ps (max. 530 nm) | S1 ESA decay (viola and neo) | Part of bleaching rise EET from S1 | |||||
| > 3 ns | Bleaching decay | ||||||
Chl b to Chl a transfer
In the CP29-WT complex the kinetics of the transfer from carotenoids is mixed up with the Chl b to Chl a energy transfer processes which occur on a similar time scale. To interpret correctly the data for the excitation in the blue spectral region, it is important to have reliable information about the kinetics of the Chl b to Chl a transfer. The CP29-WT complex was thus excited in the Qy absorption region of the Chls b and spectra were collected in the 610- to 730-nm range. With respect to Chl b to Chl a transfer, two major ultrafast transfer components from Chl b 652 nm were observed with 150-fs and 1.2-ps lifetimes, in addition to energy transfer steps between the other Chls; in particular, the equilibration between the long-wave Chls and 670-nm Chl with lifetimes of ∼1 ps and 6–7 ps. We do not present these data here, since a detailed analysis is given in a separate article (Croce et al., 2003). We thus refer to that work in the following for the interpretation of the carotenoid to Chl b transfer steps.
Analysis of absorption spectra and estimation of transfer efficiencies
To properly interpret the kinetic data, it is important to know the percentages of direct excitation of the different pigments at the excitation wavelengths used in the experiments. The 400- to 520-nm region of the absorption spectra of the antenna complexes is characterized by the overlap of the absorption of several pigments, i.e., Chl a, Chl b, and xanthophylls. Whereas the region below 435 nm is dominated by the Chl a Bx and By absorption bands, the region between 450 and 500 nm, which is relevant for this work, shows a high degree of overlap between the absorption of the three different xanthophylls and the Chls b. A method to decompose the Soret absorption in terms of the absorption of the isolated pigments has been recently published (Croce et al., 2000). That article discussed, in detail, the spectra of the individual pigments in the protein environment and their extinction coefficients. The fitting of the CP29-WT absorption and excitation spectra is presented in Fig. 6. To obtain the fit of the excitation spectra (Fig. 6, bottom), the position of the maxima of the pigments obtained by the analysis of the absorption spectrum (Fig. 6, top) were fixed and only the amplitudes were used as free parameters to determine the overall energy transfer efficiencies. The comparison of the areas of the individual pigments in the absorption and excitation spectra gives the transfer efficiencies for each pigment. The results for CP29-WT indicate an overall carotenoid to Chl energy transfer of 70%, where neoxanthin and violaxanthin transfer with 60–65% efficiency and lutein with more than 75%. The error is at least ∼±5%, assuming for Chls a 100% transfer efficiency.
FIGURE 6.
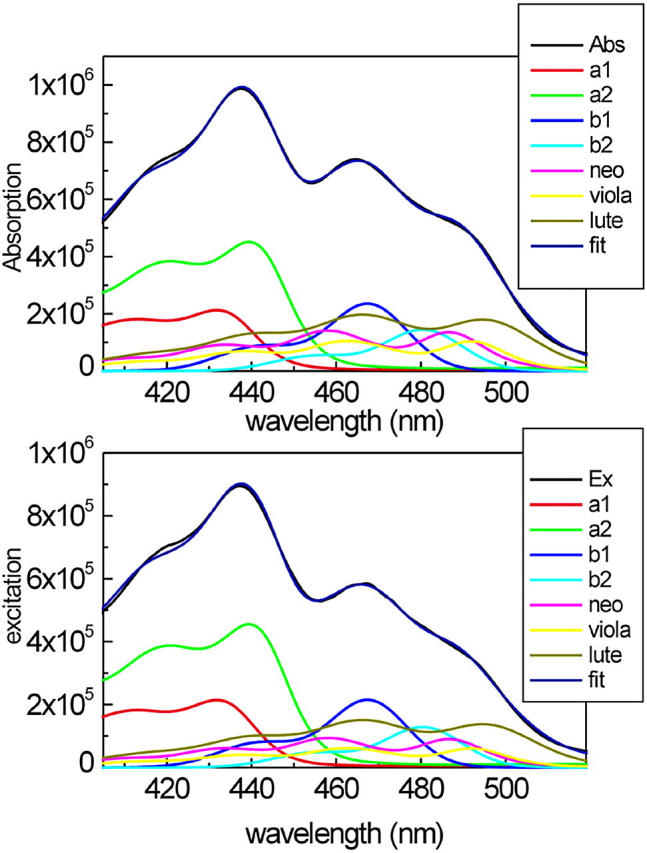
Decomposition of the 400- to 520-nm region of the absorption spectrum (top panel) and the fluorescence excitation spectrum (bottom panel) of CP29-WT in terms of the absorption of single pigments. The absorption and the excitation spectra of both complexes (black) are compared with the reconstituted spectra obtained as a sum of the bands of the single pigments (blue). The spectra of the individual pigments are also shown.
Similar analyses have been performed for the E166V mutant (see Fig. 9 B), showing that in this case the overall transfer efficiency from car to Chl is ∼58%, whereas the Chl b to Chl a transfer was only 70%. These apparently low transfer efficiencies found for the mutant are biased to some extent to the low side due to the fact that this complex with only one carotenoid and lower total Chl content is less stable than the WT (Bassi et al., 1999). As a consequence, some free or not properly bound Chl b is present, which is quite typical for this kind of mutant. However, even a small amount of free Chl b influences the results strongly. The numbers for the transfer efficiencies obtained are thus to be considered as absolute lower limits—in particular, for the Chl b to Chl a transfer.
To determine the distribution of the excitation energy produced by the excitation pulses, the spectral contribution of each pigment was convoluted with the pump profile of the pulses (assuming a FWHM of 4 nm). The results are summarized in Table 3. For CP29-WT excitation at 490 nm allows to excite more neoxanthin, but 21% of the excitation energy still directly reaches Chl b. At 500 nm, ∼87% of the excitation is in the xanthophylls, especially lutein. The excitations at 500 nm and 490 nm for the mutant show 90% of the excitation directly in the carotenoids and almost zero Chl b direct excitation, whereas for the 475-nm excitation more than 45% of the energy is directly absorbed by Chl b. The comparison between these measurements will thus give us the possibility to discriminate between the effects of Chl-b direct excitation into the S3 state and car to Chl transfer, in particular to Chl b.
TABLE 3.
Percentage of the direct excitation of the pigments in the pump-probe experiments on the basis of the Soret absorption spectra description of the samples calculated using Gaussian shape for the pump spectrum
| CP29-WT
|
CP29-E166V
|
||||
|---|---|---|---|---|---|
| 490 nm | 500 nm | 475 nm | 490 nm | 500 nm | |
| Chl a | 2.8% | 4.8% | 4.1% | 5.2% | 8.3% |
| Chl b-467 | 3.4% | 0.8% | 46.6% | 3.3% | 0.6% |
| Chl b-480 | 17.7% | 7.2% | |||
| Neoxanthin | 24.3% | 16.3% | – | – | – |
| Violaxanthin | 18.9% | 20.8% | 15.2% | 33.0% | 31.4% |
| Lutein | 33% | 50.1% | 34.1% | 58.4% | 59.7% |
DISCUSSION
Decomposition of the absorption spectra
Reliable knowledge about the direct excitation of the individual pigments is of utmost importance for the proper interpretation of the time-resolved data. Due to the very fast energy transfer from the carotenoid S2 state to Chls (Connelly et al., 1997; Peterman et al., 1997; Gradinaru et al., 2000; Croce et al., 2001) and the presence of ESA over almost the entire spectral detection region, it is hard to discriminate between direct excitation, especially in the Chl b region, and energy transfer from the time-resolved data alone. We thus have to rely, to a certain extent, on the decomposition of the absorption spectrum as presented above. In the Appendix, the accuracy and implications of this absorption decomposition are discussed in more detail.
Lifetime data for CP29-E166V
The CP29-E166V mutant contains only one carotenoid molecule per polypeptide, located in the L1 site. It is thus perfectly suited to selectively study the energy transfer from the carotenoid in the L1 site to the Chls. We excited the sample at three different wavelengths to discriminate between the effects of the direct Chl b excitation and the carotenoid excitation and subsequent energy transfer. Although there are many common features for all three excitations, the 475-nm excitation clearly shows some additional components associated with Chl b to Chl a transfers, which were not present in the 490- and 500-nm excitations, thus confirming independently that no significant amount of Chl b direct excitation is present at these longer wavelengths (see also Appendix).
The main pathway for the carotenoid L1 to Chl energy transfer starts from the S2 state of the carotenoid as donor and leads to Chl a 680 nm as acceptor. This transfer occurs with a lifetime ∼110 fs and it accounts for more than 80% of all the energy transferred from carotenoid to Chls. With the same lifetime the rise in the Sn-S1 transition is observed in the 500- to 570-nm range, indicating that 110 fs is the lifetime of the S2 state of xanthophyll in this mutant. A 80- to 110-fs rise time in the 600- to 660-nm region and above 700 nm is also observed. This rise is spectrally close to what we observed for lutein in solution and it can be explained as a rise of the Sn–S1,v ESA, due to S2 to S1 internal conversion. This feature was previously attributed to a rise in Chl ESA alone (Croce et al., 2001), but the measurements of lutein in solvents now clearly suggests that the main part of this signal is due to carotenoids. There must be some Chl contribution, although it is much smaller than the xanthophyll signal. This ESA then decays in the 400- to 800-fs lifetime range, as the positive features in the lifetime density map at 560–570 nm and 600–640 nm suggest. A rise in the Chl a bleaching is observed with the same lifetime, indicating energy transfer from higher S1,v carotenoid vibrational states to Chl. This transfer seems to be highly efficient, as indicated by the absence of a new rise in Sn–S1,0 absorption with a similar lifetime.
In the long time range the decay of Sn-S1ESA for carotenoid is present with 11- and 16-ps lifetimes. Two xanthophyll species (lutein and violaxanthin) are present in the sample. The S1 lifetime for lutein in solution has been found to be 14.6 ps (Frank et al., 1997), whereas, for the violaxanthin, a value of 23 ps has been suggested (Frank et al., 2000). In the discussion of these data we have to take in to account that two effects can act in shortening the S1 lifetimes of carotenoids: 1), energy transfer from this state; and 2), an environmental effect. We tentatively attribute the 11-ps S1 decay to lutein, whereas the 16-ps component is proposed to be associated with the S1 decay of violaxanthin. This is supported by the fact that the ESA shapes for the 11-ps and 16-ps lifetimes (compare with Fig. 3 D) show a high similarity with the ESA spectra of these two carotenoids (Frank et al., 1997; Polivka et al., 2002). Considering that in the Chl a absorption region, a pronounced rise of the bleaching at 680 nm is observed with a lifetime of 16 ps, and that this rise cannot be associated to Chl a equilibration because it is not present when the Chls a are directly excited in the Qy region at 670 and 680 nm (data not shown), we attribute this feature to the energy transfer from the violaxanthin S1 state to Chl a.
Normalizing the various transfer components to the total bleaching in Chl a at long times, 84% of the energy is transferred with the 110-fs lifetime from S2, 8.5% in 700 fs from S1,v, and 7.5% in 16 ps from the S1 state of violaxanthin. To use these values in a quantitative way, we have to normalize them, taking into account that not all the energy absorbed in the carotenoids reaches Chl a, but that the overall transfer efficiency in this mutant is ∼60%. This makes the transfer efficiency for the 110-fs component ∼50%, whereas for the 700-fs and 16-ps components the values are respectively 5% and 4.5%. At time zero, 30% of the absorbed energy is in the S2 state of violaxanthin. This indicates that the absolute transfer efficiency from the S1 state of violaxanthin in this mutant is 15% (4.5%/30%). Assuming for violaxanthin the same S1 lifetime found as in solution (23 ps), a decrease of the S1 lifetime from 23 to 16 ps indicates that a maximum of 30% of the energy which reaches S1 is transferred to Chl a, whereas the rest decays nonradiatively to S0. Taking into account that part of the decrease of the violaxanthin S1 lifetimes can be due to an environmental effect, we can conclude that the maximum energy transfer efficiency from the violaxanthin S2 state could be 50%, whereas at least 35% of the energy absorbed by this xanthophyll decays nonradiatively to S0. Concerning the transfer with 700-fs lifetime, we tentatively attribute it to lutein on the basis that the same relaxation and spectral feature, albeit with a lifetime of ∼900 fs, is observed for lutein in solution (Müller et al., unpublished results). At time zero, 60% of the energy absorbed by carotenoids is in the lutein. This indicates that the transfer contribution with 700-fs lifetime is minor, i.e., less than 10% of the energy transferred to Chl a through the hot carotenoid S1 state. The lifetime of the relaxed S1 state of lutein in this complex is ∼11 ps, which is shorter than the 14.6 ps found in solution. The lifetime density map does not show any significant rise in the Chl bleaching with this lifetime, indicating that probably the shortening of the lutein S1 lifetime is not associated primarily with an energy transfer process, but possibly to a large part with a conformational change of this xanthophyll in the complex with respect to solution which induces a larger radiationless rate.
Which are the Chl acceptors?
The data for the mutant show that the main acceptor of energy from the carotenoid in the L1 site is a Chl a, peaking at 680 nm, and that there is no substantial Car to Chl b energy transfer occurring from this xanthophyll. This is not unexpected, since the carotenoid in the L1 site is in close contact with the Chls in sites A1, A2, and A3. Mutational analysis performed on CP29 indicated that sites A1 and A2 bind Chl a and the mixed site A3 binds Chl a with 70% probability and Chl b with only 30% probability (Bassi et al., 1999). This means that a small amount of the energy, if any, is expected to be transferred to Chl b from the L1 site. Moreover, when a Chl a molecule is accommodated in the A3 site, its absorption is tuned to 668 nm (Bassi et al., 1999). Again, it was not possible to observe any rise of the bleaching at this wavelength upon carotenoid excitation, and no transfer from Chl A3 to red Chl a was detected in the measurements. These observations indicate that, at least in the E166V mutant, the Chl in site A3 is not acting as an efficient energy acceptor from the L1 site, independently of the site occupancy by Chl a or Chl b. Rather, the transfer from carotenoid in this mutant is directed mostly to a red Chl a absorbing at 680 nm. According to the mutation analysis, it is now possible to suggest that the Chl a in site A2 is the energy acceptor. The Chl a in site A1 may also be involved in the transfer (Gradinaru et al., 2000) but, as it was recently calculated (Cinque et al., 2000), the energy equilibration between Chls in sites A1 and A2 occurs in ∼70 fs, i.e., faster than the car to Chl a transfer. Thus, even if there is transfer to Chl a in site A1, we will not be able to resolve it due to the fast equilibration between the Chls a in sites A1 and A2.
Kinetics of CP29-WT
The measurements of the WT show a more complex picture than the E166V mutant due to the simultaneous occupancy of two carotenoid binding sites and additional Chl b to Chl a energy transfer processes to be taken into account. The information obtained from the mutant, which allowed studying the transfer from the L1 site exclusively without disturbance by direct Chl b excitation, is thus of utmost importance for the interpretation of the WT results. We do not repeat here the results of the direct Chl excitation data which are presented elsewhere in detail (Croce et al., 2003) but simply refer to these data and interpretations in the following course of the discussion.
Carotenoid to Chl a energy transfer
Two fast transfer components from carotenoids to Chl-a forms from the S2 state are observed with lifetimes of ∼80–90 fs and 110–130 fs. Two different accepting Chls correspond to these lifetimes with bleaching rise observed at 675 and 682 nm, respectively. They are particularly well resolved for 490-nm excitation (Fig. 4 B) but are also seen for 500-nm excitation. Taking into account the analysis of the mutant in which only the carotenoid in site L1 is present and in which the accepting Chl has its maximum at 680 nm, we attribute the transfer to the Chl a-675-nm form to the Car in the L2 site. The difference in the rise of Chl a in the WT compared to the mutant can be better observed in Fig. 7, where the spectra in the 50- to 100-fs lifetime range of the two complexes are compared, showing that the rise at 676 nm is not present in the mutant whereas it dominates the kinetics in the WT. The attribution of the transfer from L2 xanthophyll to Chl a 675 nm is also supported by the measurements of the WT after excitation at 490 nm and 500 nm: more transfer to 680 nm is observed when the sample is excited at 500 nm—considering that at 500 nm the ratio between the excitation in the lutein (in the L1 site) and in the couple neoxanthin/violaxanthin (in the L2 site) is 1.35, whereas it is only 0.77 at 490 nm. This is also in agreement with the mutation analysis performed on CP29 where the absorption at 675–676 nm was found to be associated with the Chls in sites A4 and A5, which are indeed located near the carotenoid in site L2 (Bassi et al., 1999).
FIGURE 7.

Comparison between the DADS of the mutant (squares) and the WT (circles) in the 610- to 730-nm detection range. The integration was in the range 50–100 fs and the spectra are normalized to the maximum.
Carotenoid to Chl b energy transfer
The question of whether carotenoids in CP29-WT can transfer energy to Chl b is more complicated to answer, due to the presence of several processes which occur simultaneously in the Chl b Qy absorption range in the 100-fs time scale. These are: 1), rise of carotenoid ESA in the 610- to 660-nm range (negative feature, broad spectrum); 2), fast energy transfer process between Chl b-652 nm and Chl a with a 100- to 200-fs lifetime (negative feature at 652 nm; Croce et al., 2003), and, finally, 3), a possible energy transfer from Car in the L2 site to Chl b (positive signal).
All these signals will mix up in the same time-wavelength region. However, comparing the different excitation wavelengths and the spectral shapes of the DADS it is possible to answer this question. For the 490-nm excitation, a rise of the Car ESA is observed up to 630 nm; at ∼640 nm a negative feature is still present, whereas the spectrum is almost zero at 652 nm (Fig. 5 A). This suggests that a rise of bleaching (positive amplitude feature) in the 652-nm range must be present, which is compensating the spectrally very broad carotenoid ESA signal (negative feature, see Fig. 2 B) and also the decay of the 652-nm bleaching (also negative feature) due to transfer to Chl a. The latter component has to be present because we are directly exciting Chl b to a significant extent and all the other Chl b to Chl a transfer components in the longer time range are present as expected. Part of this decay is actually visible with a 150- to 200-fs lifetime. Looking at spectrum B in Fig. 5, it is possible to observe that a “negative” feature in the 640- to 652-nm range is superimposed on the positive signal of the underlying carotenoid ESA, which is present as a broad and unstructured band over all the detection range. Moreover, the total energy present in the Chl b forms after 490-nm excitation is 28% of the total Chl-a bleaching, when we only take into account the decay of the bleaching in Chl b in the 600-fs to 4-ps lifetime range (this estimation even ignores that only 70–80% of the energy absorbed by the system is effectively transferred to Chl a and contributes to the total Chl a bleaching). Thus this contribution accounts already for more than the direct Chl b excitation at this wavelength. Considering that, as an effect of the direct excitation, some part of the energy has also to be in the Chl b-652-nm form transferring its energy to Chl a with a 100- to 200-fs lifetime (Croce et al., 2003), we would need ∼50–60% of direct excitation in Chl b to explain the decay in the Chl b bleaching. Such a high percentage of Chl b excitation can be clearly ruled out. Furthermore, if one were willing to accept such a high percentage, we could then not explain why we do not see the expected strong negative feature at ∼652 nm due to the Chl b bleaching decay (Fig. 4, top). The only other reasonable explanation that resolves this discrepancy is thus to assume a rise in Chl b-652-nm bleaching due to the transfer from carotenoid (positive amplitude feature), which would occur in the same lifetime range as the rise of the broad carotenoid S1 ESA signal (the rise time in both signals must be the same as the decay time of the carotenoid S2 population). This Chl b bleaching rise would approximately compensate the rise of carotenoid ESA and can well explain the absence of the otherwise expected strong negative signal ∼652 nm. The fact that the decay lifetime of Chl b bleaching due to transfer to Chl a is only slightly longer than the carotenoid transfer time also acts to keep the positive amplitude signal small. It is nevertheless large enough to compensate the large negative signal, which means that there must exist a very pronounced energy transfer to Chl b from carotenoids.
For 500-nm excitation, the situation is somewhat different. Most of the excitation is in the carotenoid bands, and only a small part in Chl b. This produces an even more intense carotenoid ESA in the 610- to 660-nm range. The direct Chl b excitation is still present, albeit less intense, but it is now all concentrated in the 652-nm Chl b. This is supported by several pieces of evidence. If our assignment of the absorption couples 466/640 480/652 for the two Chl-b absorption forms is correct (see Appendix), this means that at 500 nm we are not exciting the 640-nm Chl b and we expect not to see any transfer components from this Chl b, unless it receives energy from carotenoids, which can be essentially excluded on the basis of the mutant data. This is in agreement with the data (Fig. 4) where, for the 500-nm excitation, no transfer components from Chl b-640 nm are observed with 600-fs lifetime, which represents the major component in the transfer pathway from Chl b-640 nm to Chl a. In the density map the rise in the carotenoid ESA with ∼100 fs lifetime is observed, albeit with two different lifetimes (Fig. 4, bottom left): a faster one up to 630 nm centered ∼80 fs and a seemingly slower one between 640 and 660 nm, centered ∼130 fs. The maximum in the lifetime distribution in the second component is most easily explained due to the presence of a rise in Chl b bleaching (positive amplitude) with ∼80 fs that is compensated by the overlapping contribution with carotenoid ESA (in particular lutein) and ∼150 fs lifetime of the decay of Chl b. However, due to the partial compensation of amplitudes of components with slightly different lifetimes, the peak of the lifetime distribution is shifted to longer lifetimes. The total bleaching in Chl b for 500-nm excitation reaches ∼12% of the total Chl a bleaching, as calculated from integration of the areas in the respective DADS (not shown), when we take into account the decay of the Chl b bleaching in the 1- to 4-ps lifetime range. The direct excitation in Chl b has been calculated to be 8%. The data on Chl b to Chl a transfer show two kinetic components for the Chl b-652-nm form, one ∼1.2 ps and a faster one at 150 fs (Croce et al., 2003). It is thus clear that the fast transfer should be also present in our measurements and that we can expect about equal direct excitation of these two Chl b forms (4% each). Comparing this with the experimental data implies, however, that the total amount of energy in Chl b with a 1.2-ps decay alone is already three times higher than what is expected due to direct excitation. We thus conclude that ∼10% of the total energy absorbed by the carotenoids is transferred to the Chl b with a 1.2-ps lifetime. Considering that the L1 site does not transfer to Chls b, ∼20% of the energy absorbed by the carotenoid in the L2 site is transferred to this Chl b. We also arrive at similar results in analyzing the 490-nm excitation, where the contribution associated with the “1.2 ps-Chl b-652 nm” is ∼18%. This cannot be the result of direct excitation only, since, otherwise, we would expect to have the same amount of energy directly in the 150-fs Chl b-652-nm form in addition to the energy which is transferred by carotenoids, which would have to be high to compensate not only the Chl b decay but also the rise of the carotenoid ESA. In this case, we would, in fact, end up with almost all the energy arriving in Chl b, which is impossible, considering the results obtained in the mutant which indicates that for the L1 site the acceptors are only Chl a molecules.
From the mutation analysis two Chl b forms have been found to have their Qy band ∼652 nm, namely the Chls in the mixed sites B5 and B6, when these sites are occupied by Chl b. Recently, on the basis of the mutation analysis (Bassi et al., 1999) and the determination of the dipole moment orientation for the Chls in CP29 (Simonetto et al., 1999), the expected energy transfer rates for the different Chls were calculated (Cinque et al., 2000). The transfer time from a Chl b in site B5 was estimated to be ∼170 fs, whereas for a Chl b in the B6 site, this transfer was expected with an ∼1.4 ps lifetime. These calculations actually match the experimental energy transfer times associated with the two 652-nm Chl b forms very well (Croce et al., 2003). We can thus propose that the major Chl b acceptor from the L2 carotenoid is the Chl b in site B6.
Site B6 is located near the L2 site, which accommodates either violaxanthin or neoxanthin. If the donor for the Chl b 652 nm was the lutein we would expect to see much more transfer to this Chl b in the case of 500-nm excitation as compared to 490-nm excitation, in agreement with more direct excitation in lutein. This would have produced a more pronounced rise component in the Chl b bleaching ∼650 nm, which is not the case. Moreover, the transfer efficiency to this Chl b is almost the same for the two excitation wavelengths, in agreement with the similar percentage of direct excitation in violaxanthin plus neoxanthin at these wavelengths. Thus, both the biochemical and the spectroscopic data clearly suggest that the donor for the 1.2-ps lifetime Chl b-652-nm form in site B6 are the carotenoids located in site L2, i.e., violaxanthin and neoxanthin.
Does the Chl b-640 nm accept energy from a carotenoid? The excitation at 500 nm shows that no transfer from the 640-nm Chl b is observed in 600 fs. This means that the 600-fs decaying “Chl b-640 nm” is not accepting energy from the carotenoid. A transfer with a 4-ps lifetime from a Chl b-640 nm is still observed in the 500-nm excitation experiment. The amplitude associated with this Chl b is quite low, however, accounting for only ∼2% of the total energy. We can thus essentially exclude a carotenoid transfer to Chl b 640 nm from the L1 site in the WT, in agreement with the mutant data. This is also in agreement with the data of Gradinaru et al. (2000). The 640-nm absorption in CP29 has been attributed to Chl b accommodated in the mixed sites A3 and B3 (Bassi et al., 1999). As already mentioned, the Chl a in site A3 (located nearby carotenoid L1) absorbs at 668 nm. Again, as discussed for the mutant, no rise at 668 nm is observed with a lifetime of carotenoid transfer. This suggests also that in CP29-WT, the xanthophyll in site L1 does not transfer energy to Chl in site A3.
Transfer from the S1 state of carotenoids
In the CP29-WT the Sn-S1 ESA rise in the 500–570 nm region is observed within the lifetime range of 80–130 fs, reflecting the Car S2 state decay by internal conversion. An absorption decay is also observed in the 555- to 570-nm region, with a 500- to 600-fs lifetime, and this is most likely associated with the decay in the Qy range which shows the same lifetime (Fig. 4). With the same lifetime, a rise component in the ESA ∼530–540 nm is observed (Fig. 4, top). We interpret these signals as due to ESA from a S1 vibrationally nonrelaxed state, as discussed already for the mutant. We observed similar processes also for lutein in organic solvents (Müller et al., unpublished results). In the Sn-S1 carotenoid ESA region we observe the main decay of the carotenoid ESA from the vibrationally relaxed S1 state with a broad lifetime distribution ranging from 8–20 ps for both excitations. The maximum of the DADS associated with these decays is at 535 nm. This spectrum is more a violaxanthin-like, than a lutein-like, spectrum (compare to Polivka et al., 2002), suggesting that the majority of the S1 formation is associated with violaxanthin and neoxanthin. Comparing the 490- with the 500-nm excitation, it is possible to see that the ratio between the amplitudes at 535 and 550 to 555 nm is changed and it is higher in the case of 490-nm excitation, where the excitation ratio between lutein and violaxanthin + neoxanthin is 0.77, while it is smaller for the 500-nm excitation where this ratio is 1.35. This is in agreement with the shape of the ESA decay, showing a more blue peak (535 nm) associated with neoxanthin and violaxanthin and a more red-shifted peak associated with lutein (555 nm). The S1 lifetimes of violaxanthin and neoxanthin in solution are 23 ps and 35 ps, respectively (Frank et al., 1997, 2000). These lifetimes are both longer than observed in our experiments, suggesting that the shortening of the lifetimes is due to the presence of a competing energy transfer process from the respective S1 states. In fact, in the Chl a absorption region, a rise in the bleaching is observed with a similar lifetime, especially for 500-nm excitation, where the low amount of the 6 ps Chl a equilibration allowed to resolve better the two components. This longer-lived component (12–15 ps) accounts for ∼10% of the total bleaching in Chl a due to car to Chl transfer at both excitation wavelengths. At the two excitation wavelengths the sum of the excitations in neoxanthin and violaxanthin is quite similar, whereas for lutein the ratio between the 490- and 500-nm excitation is 1:1.5. If lutein was involved in this slow transfer from S1 we would expect a higher amplitude associated with the bleaching of Chl a at this lifetime in the case of 500-nm excitation, which is not the case (Fig. 4). We thus conclude that the transfer from the carotenoid S1 state with 12- to 15-ps lifetime originates from violaxanthin and/or neoxanthin located in site L2. At both excitation wavelengths the total percentage of energy directly absorbed by these two xanthophylls is ∼40%. This allows us to estimate that the total transfer efficiency from the S1 state (as a percentage of the total transfer to Chl) is ∼20–30% for these two carotenoids. Lutein, in contrast, seems to transfer mostly from the S2 state. For 500-nm excitation, more energy is associated with the 110-fs rise of Chl a bleaching than for 490-nm excitation, supporting this hypothesis. A transfer with a 700-fs lifetime to Chl a is also present in the case of the WT, accounting for another 10% of the total carotenoid to Chl transfer energy. In agreement with the discussion above, we attribute this tentatively to the transfer from vibrationally unrelaxed S1,v states of lutein, as in the case of the mutant.
The summary of this discussion of energy transfer steps in CP29 is given in schematic presentation in Fig. 8. Note that in this article we do not assign nor discuss the exact energy levels of the acceptor Chls. We simply observe the energy rise in the Qy Chl bands. The fact that, in Fig. 8 the energy acceptors are the Qy levels, thus has to be considered a simplification.
FIGURE 8.

Qualitative kinetic scheme for energy transfer steps from carotenoids to chlorophylls in the CP29-WT complex.
Comparison of CP29 versus LHCII
We found here that lutein is very ineffective in transferring energy from the S1 state, whereas this transfer pathway is used more efficiently by both neoxanthin and violaxanthin. In the case of LHCII two lifetimes were associated with the decay of the carotenoid S1 state, i.e., ∼15 ps and 3 ps (Croce et al., 2001). In the reconstituted complexes only lutein was present, but quite similar results were also obtained in the case of the WT complex, where one molecule of neoxanthin was present in addition. In both cases, the 15-ps component was associated with the lutein in the L1 site, whereas the 3-ps component was attributed to the decay of the lutein in the L2 site. The results obtained with CP29, where lutein is located in the site L1 only (while in LHCII it is located in both the L1 and L2 sites), seem to indicate that the ability of lutein to transfer energy to Chls from the S1 state is more connected to the interaction with the environments (i.e., the specific spectral characteristics of the Chl acceptors and perhaps also the protein environment) rather than with the physicochemical characteristics of the carotenoid itself. The violaxanthin present in the mutant seems to behave in a different manner, but this can be an effect of a slightly different folding assumed by this complex.
The presented data on CP29 indicate that there is no energy transfer from the carotenoid in the L1 site to the Chl in the A3 site, in contrast to LHCII in which this transfer has been detected (Croce et al., 2001). On the other hand, in CP29 an energy transfer from the L2 xanthophyll to the B6 Chl was observed, whereas in LHCII the B6 site is the energy acceptor primarily from the neoxanthin in N1. This suggests a slightly different structural organization between the two complexes, with the A3 site located possibly more distant from the L1 site or differently oriented, and B6 located closer to L2 in CP29 as compared to LHCII—as suggested also from the mutation analysis, where the mutation at the ligand of Chl B6 (E166V) excludes the L2 binding in CP29 whereas it does not in LHCII.
CONCLUSIONS
In the present article, we present a study of car to Chl energy transfer in the minor antenna complex CP29 and in a mutant of this protein (E166V), which contains the carotenoid only in the site L1. The data clearly indicate that the L1 xanthophyll transfers directly to a Chl a pool absorbing at 680 nm (Chl A2). The main transfer (84% of the total transfer) occurs in 100 fs and it originates from the carotenoid S2 level. Transfer from a high vibronic S1 state in 700 fs and from an S1 relaxed state in 16 ps are also observed, but they account for only 8.5 and 7.5% of the total transfer. The former S1 transfer is attributed to lutein, whereas the latter originates for violaxanthin, which in this mutant is present in the L1 site, somehow different from the WT.
In the WT complex the L1 site is occupied mainly by lutein, whereas the L2 site accommodates both violaxanthin and neoxanthin. The data suggest that the kinetics for lutein in the L1 site is identical to what is observed in the mutant. The L2 site transfers energy to a Chl a pool absorbing ∼675 nm (Chls A4 and A5) and to a Chl b 652 nm (Chl B6). Whereas for the carotenoids in the L2 site most of the energy is transferred from the S2 Car state, a clear transfer component also originates from the S1 state. The data presented here from the femtosecond measurements are in full agreement with the Chl organization obtained by mutation analysis on CP29 (Bassi et al., 1999).
In addition, in this article we present the analysis of the blue region of the absorption spectra of the antenna systems, demonstrating that the use of the spectra of individual pigments in protein environment allowed a correct evaluation of the direct excitation in each chromophore. It is also clearly demonstrated that, in the case of CP29-WT, two Chl-b Soret bands are present with absorption maxima at 467 nm and 480 nm.
Acknowledgments
R.C. was supported by a TMR Marie Curie Fellowship Grant ERBFMBICT983216 from the European Union. We also thank the Deutsche Forschungsgemeinschaft (SFB 189, Heinrich-Heine-Universität Düsseldorf and Max-Planck-Institut für Strahlenchemie) for financial support.
APPENDIX
Whereas some significant differences in the signal-to-noise ratios and the time resolution exist between different studies, most of the discrepancies in the interpretations of carotenoid to Chl energy transfer studies on LHCII and CP29 antennae originates from the largely different values assumed for the direct excitation of Chl b (compare to Croce et al., 2001, and Gradinaru et al., 2000, and the data presented in this article). It is thus important to have a reliable method to describe the absorption spectrum of the complexes in the 400- to 520-nm region and then to calculate the value of the direct excitation. In the following, we analyze in detail the 400- to 520-nm region of CP29 and we check the validity of our description measuring the direct excitation in the CP29-E166V mutant.
Recently we studied the spectral characteristics of the various pigments in a protein environment, i.e., the spectral shapes and the extinction coefficients, which on the basis of these studies we consider to be more or less conserved across the Lhc family (Croce et al., 2000). However, the E0 energy levels of the pigments, which are another important parameter in the fits, can change in different proteins. Whereas the heterogeneity in the Qy absorption band of the complexes due to the presence of Chls in different protein environments is well accepted, and this region is always described in terms of multiple Chl a and Chl b bands, this is not the case for the Soret region—where only one Chl a and one Chl b form are usually used in the description. This is quite surprising and would require a coincidental agreement of the Soret bands of all the Chls a on the one hand and all the Chls b on the other hand. Since the Qy absorptions of Chls react quite sensitively to changes in the environment, such as charges, hydrogen bonds, polarity, etc., it would be highly surprising if the Soret transitions would not. Thus, from a general spectroscopic point of view, the total overlap of the Soret bands for each Chl species appears very unlikely.
If only one Chl b form is used in the CP29 Soret description, it has to be positioned ∼476 nm with the amplitude corresponding to two Chls b for any reasonable fit. In that case it would, however, not be possible to explain the spectrum of CP29 at low temperatures, where a valley is present at 478 nm, for which the second derivative analysis does not show any contribution ∼476 nm, as should be the case if most of the absorption in the 450–500 nm range was associated with only one spectral Chl b form (Pascal et al., 1999). Two Chl b spectral forms are thus needed to describe the spectrum. They have to be located at 467 and 480–482 nm. This suggestion comes from different pieces of evidences: 1), CP29 complex reconstituted with a lower Chl a/b ratio shows an enhancement of the peak at 466–467 nm (Giuffra et al., 1996). 2), The difference spectra in the Soret region between CP29-WT and mutant A3 and B3, which lose the Chl b- 638/640 nm form, show a major peak in the Chl b region ∼466–467 nm. 3), The CP29-E166V mutant analyzed here, which has a reduced content in both Chl b and carotenoids, shows the contribution of the remaining Chl b at 466–467 nm. These results thus provide enough evidence for assigning Chl b to the 466- to 467-nm band. However, this form cannot account for both Chls b present. Assuming that the second peak of the carotenoids absorbs at similar wavelengths (∼455–465 nm), the sum of these two effects would produce a much more intense absorption in this wavelength range than in the 480- to 500-nm region. This is not the case, neither in the RT nor in the low temperature spectra (Pascal et al., 1999). Consequently a second Chl-b form is needed to describe the spectrum of CP29. The analysis of the B6 and B5 mutants, which lack the 650/652 nm Chl-b form, shows in the WT minus mutant difference spectra a main peak ∼484 nm. Unfortunately these mutants are also affected in the carotenoid content and thus this maximum cannot be directly attributed to the second Chl b form. However, the stringency and internal consistency of the fits of the absorption of the two complexes strongly suggest that the second Chl b form has its maximum ∼480–482 nm. On the basis of the results on the CP29 mutants we relate the Chl b absorption at 638/640 nm with the 466- to 467-nm band (see above) and the Chl b absorption at 650/652 nm with the 480- to 482-nm band. Only this assignment leads to an internally consistent picture, taking into account all available data.
The second point worth discussing here concerns the shape of the Chl b absorption band in the Soret description. From the analysis of the LHCII and CP29 mutant we suggested that the Chl b spectrum in protein environment is narrower than the spectrum in acetone solvent (Croce et al., 2000). We investigated this, particularly in using the CP29-E166V mutant, which, due to the low number of pigments with respect to the WT, makes the Soret description simpler. Moreover, for a CP29-E166V mutant lacking Chl b altogether, the Soret absorption spectrum has been successfully described previously (Croce et al., 2000), thus demonstrating that the carotenoid spectral forms used are appropriate. To demonstrate that a narrower Chl-b spectrum than ordinarily used is required, we performed the analysis of the CP29-E166V mutant absorption with an identical procedure, but using, in one case, the spectral shape of Chl b in solvent (A), and in the other case, the spectral shape of Chl b in protein environment (B) (Croce et al., 2000). The results of these two fits are presented in Fig. 9. In the case of procedure A, we notice that it is not possible to reproduce the stoichiometry between the pigments with a free fit. The best fit, which still does not allow describing well the 450- to 475-nm region, gives a violaxanthin/lutein ratio of 13, whereas it is clearly 0.5 from pigment analysis. In particular, the correct ratio between violaxanthin and lutein cannot be obtained in this fit independently of the positioning of the bands of the two carotenoids. If we use the stoichiometric data as constraint in fitting case A, the Chl b spectral shape in solvent is much too wide to reproduce well the peak at 464 nm. In case B the fitting was free-running, and it reproduces well (violaxanthin/lutein 0.45) the stoichiometric ratio between the pigments without using any constraints.
FIGURE 9.

Decomposition of the 400- to 520-nm region of the absorption spectrum of the CP29-E166V mutant in terms of the absorption of single pigments. (A) Using, for Chl b, the Chl-b spectral shape in solution, and (B) using the narrower spectral shape for Chl b in protein. (Insets) Errors for the two fittings are shown.
Starting from the two contrasting descriptions, we calculated the direct excitations in Chl b at the three wavelengths used in our experiments. In the case of the fitting A, the values of direct excitation in Chl b at 475, 490, and 500 nm would be 54.3%, 18.2%, and 9.7%, respectively, whereas in the case of the fitting B, they are 46.6%, 3.3%, and 0.6%. The validity of our approach can be directly tested from the time-resolved data. The kinetic traces for the excitation of the CP29-E166V mutant at 490, 500, and 475 nm with detection at 650 nm are shown in Fig. 10. They show that, for both longer wavelength excitations, no direct bleaching in Chl b is produced, whereas a strong bleaching is present when the sample is excited at 475 nm, in agreement with the prediction by the fitting B.
FIGURE 10.

Experimental transient absorption kinetics for 650-nm detection wavelength for the CP29-E166V mutant. (Solid line) Excitation at 490 nm; (dashed line), excitation at 475 nm; and (dotted line), excitation at 500 nm.
In summary, we take these data as a clear indication that the Chl b Soret absorption spectral shape we are using in the fitting is appropriate and leads not only to internal consistency of the absorption analysis but yielding also the proper direct Chl b excitation probabilities that yield an agreement with the kinetic data. In contrast, the use of the Chl b spectral shape in solution would lead to a severe overestimation of the Chl b absorption at long wavelengths in the Soret region of the antenna complexes. In that case, the quite-high Chl b excitation probability predicted for the 490-nm excitation, and also, to some extent, for the 500-nm excitation from this decomposition, can in no way be reconciled with the time-resolved data (Fig. 10).
Roberta Croce's present address is Istituto di Biofisica, CNR sezione di Milano, Via Celoria 26, 20133 Milano, Italy.
Abbreviations used: Car, carotenoid; Chl, chlorophyll; DADS, decay-associated difference spectra; DM, dodecylmaltoside; ESA, excited state absorption; EET, excitation energy transfer; SE, stimulated emission; RT, room temperature.
References
- Bassi, R., and S. Caffarri. 2000. Lhc proteins and the regulation of photosynthetic light harvesting function by xanthophylls. Photosynth. Res. 64:243–256. [DOI] [PubMed] [Google Scholar]
- Bassi, R., R. Croce, D. Cugini, and D. Sandona. 1999. Mutational analysis of a higher plant antenna protein provides identification of chromophores bound into multiple sites. Proc. Natl. Acad. Sci. USA. 96:10056–10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassi, R., and P. Dainese. 1992. A supramolecular light-harvesting complex from chloroplast photosystem-II membranes. Eur. J. Biochem. 204:317–326. [DOI] [PubMed] [Google Scholar]
- Bassi, R., B. Pineau, P. Dainese, and J. Marquardt. 1993. Carotenoid-binding proteins of photosystem-II. Eur. J. Biochem. 212:297–303. [DOI] [PubMed] [Google Scholar]
- Boekema, E. J., H. van Roon, F. Calkoen, R. Bassi, and J. P. Dekker. 1999. Multiple types of association of photosystem II and its light-harvesting antenna in partially solubilized photosystem II membranes. Biochemistry. 38:2233–2239. [DOI] [PubMed] [Google Scholar]
- Boekema, E. J., H. van Roon, and J. P. Dekker. 1998. Specific association of photosystem II and light-harvesting complex II in partially solubilized photosystem II membranes. FEBS Lett. 424:95–99. [DOI] [PubMed] [Google Scholar]
- Caffarri, S., R. Croce, J. Breton, and R. Bassi. 2001. The major antenna complex of photosystem II has a xanthophyll binding site not involved in light harvesting. J. Biol. Chem. 276:35924–35933. [DOI] [PubMed] [Google Scholar]
- Cinque, G., R. Croce, A. R. Holzwarth, and R. Bassi. 2000. Energy transfer among CP29 chlorophylls: calculated Förster rates and experimental transient absorption at room temperature. Biophys. J. 79:1706–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly, J. P., M. G. Müller, R. Bassi, R. Croce, and A. R. Holzwarth. 1997. Femtosecond transient absorption study of carotenoid to chlorophyll energy transfer in the light harvesting complex II of photosystem II. Biochemistry. 36:281–287. [DOI] [PubMed] [Google Scholar]
- Croce, R., G. Canino, F. Ros, and R. Bassi. 2002. Chromophores Organization in the Higher Plant Photosystem II Antenna Protein CP26. Biochemistry. 41:7334–7343. [DOI] [PubMed] [Google Scholar]
- Croce, R., G. Cinque, A. R. Holzwarth, and R. Bassi. 2000. The soret absorption properties of carotenoids and chlorophylls in antenna complexes of higher plants. Photosynth. Res. 64:221–231. [DOI] [PubMed] [Google Scholar]
- Croce, R., M. G. Müller, R. Bassi, and A. R. Holzwarth. 2001. Carotenoid-to-chlorophyll energy transfer in recombinant major light-harvesting complex (LHCII) of higher plants. I. Femtosecond transient absorption measurements. Biophys. J. 80:901–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce, R., M. G. Müller, R. Bassi, and A. R. Holzwarth. 2003. Chlorophyll b to Chlorophyll a energy transfer kinetics in the CP29 antenna complex. A comparative femtosecond absorption study between native and reconstituted proteins. Biophys. J. 84:2508–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce, R., R. Remelli, C. Varotto, J. Breton, and R. Bassi. 1999a. The neoxanthin binding site of the major light harvesting complex (LHC II) from higher plants. FEBS Lett. 456:1–6. [DOI] [PubMed] [Google Scholar]
- Croce, R., S. Weiss, and R. Bassi. 1999b. Carotenoid-binding sites of the major light-harvesting complex II of higher plants. J. Biol. Chem. 274:29613–29623. [DOI] [PubMed] [Google Scholar]
- Demmig, B., K. Winter, A. Krüger, and F.-C. Czygan. 1987. Photoinhibition and zeaxanthin formation in intact leaves. A possible role of the xanthophyll cycle in the dissipation of excess light energy. Plant Physiol. 84:218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formaggio, E., G. Cinque, and R. Bassi. 2001. Functional architecture of the major light-harvesting complex from higher plants. J. Mol. Biol. 314:1157–1165. [DOI] [PubMed] [Google Scholar]
- Frank, H. A., J. A. Bautista, J. Josue, Z. Pendon, R. G. Hiller, F. P. Sharples, D. Gosztola, and M. R. Wasielewski. 2000. Effect of the solvent environment on the spectroscopic properties and dynamics of the lowest excited states of carotenoids. J. Phys. Chem. B. 104:4569–4577. [Google Scholar]
- Frank, H. A., V. Chynwat, R. Z. B. Desamero, R. Farhoosh, J. Erickson, and J. Bautista. 1997. On the photophysics and photochemical properties of carotenoids and their role as light-harvesting pigments in photosynthesis. Pure Appl. Chem. 69:2117–2124. [Google Scholar]
- Gilmore, A. M., and H. Y. Yamamoto. 1991. Zeaxanthin formation and energy-dependent fluorescence quenching in pea chloroplasts under artificially mediated linear and cyclic electron transport. Plant Physiol. 96:635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffra, E., D. Cugini, R. Croce, and R. Bassi. 1996. Reconstitution and pigment-binding properties of recombinant CP29. Eur. J. Biochem. 238:112–120. [DOI] [PubMed] [Google Scholar]
- Gradinaru, C. C., A. A. Pascal, F. van Mourik, B. Robert, P. Horton, R. van Grondelle, and H. Van Amerongen. 1998. Ultrafast evolution of the excited states in the chlorophyll a/b complex CP29 from green plants studied by energy-selective pump-probe spectroscopy. Biochemistry. 37:1143–1149. [DOI] [PubMed] [Google Scholar]
- Gradinaru, C. C., I. H. M. van Stokkum, A. A. Pascal, R. van Grondelle, and H. Van Amerongen. 2000. Identifying the pathways of energy transfer between carotenoids and chlorophylls in LHCII and CP29. A multicolor, femtosecond pump-probe study. J. Phys. Chem. B. 104:9330–9342. [Google Scholar]
- Hankamer, B., J. Barber, and E. J. Boekema. 1997. Structure and membrane organization of photosystem II in green plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 48:641–671. [DOI] [PubMed] [Google Scholar]
- Harrer, R., R. Bassi, M. G. Testi, and C. Schäfer. 1998. Nearest-neighbor analysis of a photosystem II complex from Marchantia polymorpha L. (liverwort), which contains reaction center and antenna proteins. Eur. J. Biochem. 255:196–205. [DOI] [PubMed] [Google Scholar]
- Hobe, S., H. Niemeier, A. Bender, and H. Paulsen. 2000. Carotenoid binding sites in LHCIIb—relative affinities towards major xanthophylls of higher plants. Eur. J. Biochem. 267:616–624. [DOI] [PubMed] [Google Scholar]
- Holzwarth, A. R. 1996. Data analysis of time-resolved measurements. In Biophysical Techniques in Photosynthesis. Advances in Photosynthesis Research. J. Amesz, and A. J. Hoff, editors. Kluwer Academic Publishers, Dordrecht. 75–92.
- Iseri, E., D. Albayrak, and D. Gülen. 2000. Electronic excited states of the CP29 antenna complex of green plants: a model based on exciton calculations. J. Biol. Phys. 26:321–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson, S. 1999. A guide to the Lhc genes and their relatives in Arabidopsis. Trends Plant Sci. 4:236–240. [DOI] [PubMed] [Google Scholar]
- Kühlbrandt, W., D. N. Wang, and Y. Fujiyoshi. 1994. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 367:614–621. [DOI] [PubMed] [Google Scholar]
- Pascal, A., C. Gradinaru, U. Wacker, E. Peterman, F. Calkoen, K.-D. Irrgang, P. Horton, G. Renger, R. van Grondelle, B. Robert, and H. Van Amerongen. 1999. Spectroscopic characterization of the spinach Lhcb4 protein (CP29), a minor light-harvesting complex of photosystem II. Eur. J. Biochem. 262:817–823. [DOI] [PubMed] [Google Scholar]
- Paulsen, H. 1997. Pigment ligation to proteins of the photosynthetic apparatus in higher plants. Physiol. Plant. 100:760–768. [Google Scholar]
- Peterman, E. J. G., R. Monshouwer, I. H. M. van Stokkum, R. van Grondelle, and H. Van Amerongen. 1997. Ultrafast singlet excitation transfer from carotenoids to chlorophylls via different pathways in light-harvesting complex II of higher plants. Chem. Phys. Lett. 264:279–284. [Google Scholar]
- Pichersky, E., and B. R. Green. 1990. The extended family of chlorophyll a/b-binding proteins of PS I and PS II. In Current Research in Photosynthesis, III. M. Baltscheffsky, editor. Kluwer Publishers, Dordrecht. pp. 553–556.
- Pieper, J., K. D. Irrgang, M. Ratsep, J. Voigt, G. Renger, and G. J. Small. 2000. Assignment of the lowest Q(Y)-state and spectral dynamics of the CP29 chlorophyll a/b antenna complex of green plants: a hole- burning study. Photochem. Photobiol. 71:574–581. [DOI] [PubMed] [Google Scholar]
- Plumley, F. G., and G. W. Schmidt. 1987. Reconstitution of chloroform a/b light-harvesting complexes: xanthophyll-dependent assembly and energy transfer. Proc. Natl. Acad. Sci. USA. 84:146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polivka, T., D. Zigmantas, V. Sundstrom, E. Formaggio, G. Cinque, and R. Bassi. 2002. Carotenoid S(1) state in a recombinant light-harvesting complex of Photosystem II. Biochemistry. 41:439–450. [DOI] [PubMed] [Google Scholar]
- Remelli, R., C. Varotto, D. Sandona, R. Croce, and R. Bassi. 1999. Chlorophyll binding to monomeric light-harvesting complex. A mutation analysis of chromophore-binding residues. J. Biol. Chem. 274:33510–33521. [DOI] [PubMed] [Google Scholar]
- Rogl, H., and W. Kühlbrandt. 1999. Mutant trimers of light-harvesting complex II exhibit altered pigment content and spectroscopic features. Biochemistry. 38:16214–16222. [DOI] [PubMed] [Google Scholar]
- Ruban, A. V., P. J. Lee, M. Wentworth, A. J. Young, and P. Horton. 1999. Determination of the stoichiometry and strength of binding of xanthophylls to the photosystem II light harvesting complexes. J. Biol. Chem. 274:10458–10465. [DOI] [PubMed] [Google Scholar]
- Schmid, V. H. R., K. V. Cammarata, B. U. Bruns, and G. W. Schmidt. 1997. In vitro reconstitution of the photosystem I light-harvesting complex LHCI-730: heterodimerization is required for antenna pigment organization. Proc. Natl. Acad. Sci. USA. 94:7667–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetto, R., M. Crimi, D. Sandona, R. Croce, G. Cinque, J. Breton, and R. Bassi. 1999. Orientation of chlorophyll transition moments in the higher-plant light-harvesting complex CP29. Biochemistry. 38:12974–12983. [DOI] [PubMed] [Google Scholar]
- Voigt, B., K. D. Irrgang, J. Ehlert, W. Beenken, G. Renger, D. Leupold, and H. Lokstein. 2002. Spectral substructure and excitonic interactions in the minor photosystem II antenna complex CP29 as revealed by nonlinear polarization spectroscopy in the frequency domain. Biochemistry. 41:3049–3056. [DOI] [PubMed] [Google Scholar]
- Yang, C. H., K. Kosemund, C. Cornet, and H. Paulsen. 1999. Exchange of pigment-binding amino acids in light-harvesting chlorophyll a/b protein. Biochemistry. 38:16205–16213. [DOI] [PubMed] [Google Scholar]