

Abstract
In this paper we address the question of how a protein environment can modulate the absorption spectrum of a chromophore during a molecular dynamics simulation. The effect of the protein is modeled as an external field acting on the unperturbed eigenstates of the chromophore. Using a first-principles method recently developed in our group, we calculated the perturbed electronic energies for each frame and the corresponding wavelength absorption during the simulation. We apply this method to a nanosencond timescale molecular dynamics simulation of the light-harvesting peridinin-chlorophyll-protein complex from Amphidinium carterae, where chlorophyll was selected among the chromophores of the complex for the calculation. The combination of this quantum-classical calculation with the analysis of the large amplitude motions of the protein makes it possible to point out the relationship between the conformational flexibility of the environment and the excitation wavelength of the chromophore. Results support the idea of the existence of a correlation between protein conformational flexibility and chlorophyll electronic transitions induced by light.
INTRODUCTION
Light-harvesting (LH) complexes, which play the important role of enhancing the absorption cross section of the photosynthetic apparatus and of transferring the captured energy efficiently to the reaction center, have been in the last years at the center of a very active interest. The crystallization of several of these antenna complexes and the determination of their structures at high resolution using x-ray or electron diffraction techniques (Hofmann et al., 1996; Matthews et al., 1979; Kuhlbrandt et al., 1994; McDermott et al., 1995; Koepke et al., 1996) have been stimulating for the research in this field. The most recent studies, essentially based on highly sophisticated methodologies (Croce et al., 1999; Jankowiak et al., 2000; Pascal et al., 1999; Salverda et al., 2000; Zucchelli et al., 2002) and high level of electronic structure-based calculations (Ghosh, 1997; Hasegawa et al., 1998; Linnanto and Korppi-Tommola, 2000; Nonomura et al., 1997; Sundholm, 1999), provided important advances in terms of spectroscopical characterization of the LH complexes, theoretical characterization of the pigment electronic structures, and the implication of the different pigments in the energy transfer mechanism (Alden et al., 1997; Hu et al., 1997; Sundström et al., 1999). Understanding of the function of the light-harvesting proteins requires also a description of how the protein structure tunes the electronic properties of the pigments, such as the excited-state energy. In most of the models used so far to treat electronic properties in biosystems, the presence of the protein environment was taken into account just as a static (Sakuma et al., 1997) or averaged perturbation (Siegbahn and Blomberg, 2000) whereas the effects of protein conformational flexibility were not investigated. Only recently the effect of protein dynamics on the chromophore electronic state has been explicitly included in the calculations (Damjanovic et al., 2002). In this paper the authors perform short classical molecular dynamics (MD) simulations on a LH-II complex to obtain a set of the chromophores configurations to be used in the quantum chemical calculations. In this way the excitation energy was calculated in time, reproducing with a good agreement the experimental data. However, these calculations were mainly focused on the local excitation dynamics of the chromophore rather than on the coupling between electronic properties and protein conformational fluctuations, occurring on longer timescale and involving long-range interactions. The relationship between conformational fluctuations and biological activity in proteins, excluding photochemical processes, has been extensively investigated in the last years, and it was shown that large concerted backbone motions can be involved in the activity of the enzymes (van Aalten et al., 1995a,b; Chillemi et al., 1997; de Groot et al., 1996). In this paper we address specifically the question of how the protein conformational flexibility affects the electronic properties of a selected chromophore. The occurrence of such a coupling would confirm the relevance of the large amplitude backbone motions in modulating the biological activity of proteins, and more specifically in the system studied, it could suggest possible regulation mechanism for photosynthetic processes. The system we have chosen for this investigation is the water-soluble external antenna complex of the photosynthetic apparatus of dinoflagellate Amphidinium carterae: the peridinin-chlorophyll-protein complex (A-PCP). This complex is a good model system because its crystal structure was determined by x-ray at 2.0 Å resolution (Hofmann et al., 1996) and a significant body of spectroscopic work has been reported on the PCP complex of A. carterae and related species (Koka and Song, 1997; Bautista et al., 1999; Kleima et al., 2000a,b; Carbonera et al., 1999a,b; Akimoto et al., 1996). This antenna is made of a 32 kD polypeptide that encloses two pigment clusters, related to each other by a twofold pseudosymmetry axis, comprising two chlorophyll a (Chl a) and eight peridinins. The pseudosymmetry axis was recently used in a theoretical investigation of the excitation transfer in the crystal structure of the A-PCP (Damjanovic et al., 2000). Here the pathways of the excitation transfer process between the peridinins and the chlorophyll was clearly described using only the pigment in the NH2-terminal half of the crystal structure and then extending the conclusions to the COOH-terminal half where the same pigment cluster is present. The A-PCP water solubility, its small dimension, and the possibility of reducing further the system dimension, using the twofold pseudosymmetry axis, are important properties that make this protein suitable for studying the coupling between conformational dynamics and electronic properties. Using a first-principles method, perturbed matrix method (PMM) (Aschi et al., 2001), that we recently developed, it is possible to obtain the electronic and hence spectroscopic properties at each configuration generated by a MD computer simulation. Chl a has been selected as the active chromophore because its semirigid structure simplifies the application of the PMM method. Furthermore, whereas in most of the peripheral light-harvesting complexes, for which the crystallographic structure is known, strong interactions are present among the bacteriochlorophylls (BChls), a single Chl a molecule is found in each A-PCP cluster and the electronic coupling information is not needed. The aim of this paper is not to reproduce exactly the absorption spectrum of the chromophore in the protein, which can be affected by the various approximations used in the calculations, but rather to show a correlation, if it exists, between the large concerted (essential) protein internal motions and the electronic properties of an internal chromophore.
METHODS
System definition
A-PCP consists of a trimer of monomers each containing a 312-residue peptide with two molecules of digalactosyldiacylglycerol (DGDG) being an integral part of the structure together with a number of resident water molecules. The NH2- and COOH-terminal halves of the monomer polypeptide form almost identical domains related by a twofold pseudosymmetry axis, each enclosing a pigment cluster. The NH2- and COOH-terminal pigment clusters are related by the same local twofold symmetry axis at the apoprotein. Each cluster contains one Chl a and four peridinins (Per1, Per2, Per3, and Per4) surrounding the chlorophyll (see Fig. 1). The closest distances between pigments belonging to different clusters are greater than those between pigments within a single cluster. Intracluster edge-to-edge distances between peridinins are in the range 4–11 Å, whereas the distance between the Chl a of the two clusters is 17.4 Å. The interchromophoric distances between units of the trimer are all much larger. The surface of the chlorophyll is covered by the peridinins for one half, by the protein for one third, and the remaining by the fatty acid of the lipids. The closest protein-Chl a contacts occur through a van der Waals interaction of the imidazole rings of two histidine residues, His66 and His229, with the NH2- and COOH-terminal chlorophyll rings, respectively (the distances between the Nɛ2 of both histidines and the magnesium atom of the Chl a is 4 Å). A water molecule, which is on one side hydrogen-bonded to these residues, provides the fifth coordination site.
FIGURE 1.

Active center graphic representation: chlorophyll (in light gray) and peridinins (labeled Per1, Per2, Per3, and Per4).
Classical molecular dynamics
The starting configuration of the A-PCP was obtained from the Protein Data Bank, entry code 1PPR (Hofmann et al., 1996). The twofold pseudosymmetry axis present in the system allows consideration of only one half of the system in the MD simulation (see Fig. 2). Hence the whole simulated subsystem consists of 156 residues, an active center (shown in Fig. 1), made of one water-coordinated Chl a and four peridinins, labeled Per1, Per2, Per3, and Per4, and a DGDG molecule. The histidine residues in the vicinity of the chlorophyll, i.e., His66 and His67, were simulated according to the GROMACS optimized force field, as neutral with proton in Nδ1 and Nɛ2, respectively. All the crystallographic water molecules close to the subsystem considered were included. The simulation was performed in the NVT ensemble, using the GROMACS simulation software packages (van der Spoel et al., 1995), implemented in a parallel architecture. The canonical ensemble was chosen because of the simplicity and stability of the algorithms, which provide a rigorous statistical mechanical behavior. A modification of the GROMOS87 force field was used with additional terms for aromatic hydrogens (van Gunsteren et al., 1996) and improved carbon-oxygen interaction parameters (van Buuren et al., 1993). For the cofactors (Chl a, Pers, and DGDG), the force field parameters were estimated in the following way: reference bond distances and angles were taken from the crystal structure, and the force constants were taken in the GROMACS library, whereas the atomic charges were calculated ab initio, i.e., with Hartree-Fock self-consistent field (HF/SCF) calculations using a “split valence” 6-31G basis set (Ditchfield et al., 1971). Based on ab initio calculations (see details in the next subsection), we considered a water molecule to be bound to the magnesium atom of the chlorophyll ring in the classical MD simulation. The SHAKE algorithm (Ryckaert et al., 1977) and the roto-translational constraint algorithm (Amadei et al., 2000) were used. The starting structure was immersed in a rectangular box of SPC water molecules (Berendsen et al., 1981). The simulation was performed with periodic boundary conditions at a temperature of 300 K, which was kept constant with the iso-Gaussian method (Brown and Clarke, 1986). Nonbonded cutoffs of 1.0 nm and 1.2 nm for Lennard-Jones and Coulomb potentials, respectively, were used. The pair lists were updated every 10 steps. A time step of 1 fs was used to integrate numerically the classical equations of motion. The solvent was relaxed by energy minimization, followed by 15 ps of MD at 300 K while restraining protein atomic positions with a harmonic potential. The system was then minimized without restraints and its temperature brought to 300 K in a stepwise way: 15-ps long MD runs were carried out at 50, 100, 200, and 250 K before the run was started at 300 K, generating a 4.0-ns long trajectory. The structures of the trajectory were collected every 0.1 ps. The classical trajectory obtained was analyzed using the standard analysis such as root mean-square deviation (RMSD) and radius of gyration (RGyr). Moreover, the characterization of the protein internal motion was performed by studying the essential dynamics of the system (Amadei et al., 1993). The covariance matrix was built from the equilibrated portion of the trajectory, and its diagonalization provided the principal direction of the large-amplitude concerted motions, i.e., the eigenvectors with largest eigenvalues, that characterize the essential subspace of the internal dynamics of a protein.
FIGURE 2.

Graphic representation of the simulated system obtained from the x-ray structure of PCP from Amphidinium carterae (Hofmann et al., 1996). Picture generated using MolScript (Kraulis, 1991) and Raster3D (Merritt and Bacon, 1997) software.
Quantum chemical calculations
Electronic ground-state calculations of the various pigments in the protein were performed to evaluate the atomic charges to be used in the Coulombic nonbonded interactions of the classical MD force field, via an HF/6-31G Mulliken population analysis (Hehre et al., 1972). To have a rigid molecular system, for a more straightforward application of PMM, a structurally reduced Chl a (SRChl a) was selected for determining unperturbed electronic eigenstates. More precisely, all the flexible substituents of the ring have been removed and substituted with H atoms. For evaluating the quality of the above calculations, the SRChl a vertical ionization potential (IP) was calculated at an HF/SCF level of theory using two methods: the SCF energy difference between the neutral and the ionized molecule in their electronic ground state (ΔSCF), and the method based on the Koopmans' theorem, i.e., the energy of the highest energy electron in the molecular orbitals. To estimate the binding energy, Deq, of the water molecule to the magnesium atom of the chlorophyll ring, HF/SCF calculations with a double ζ-basis set including first polarization functions (3-21G*) (Pietro et al., 1982; Gordon et al., 1982) were carried out for water-coordinated-SRChl a, SRChl a, and water. The estimated binding energy of ∼132 kJ/mol allowed us to consider, in the 300 K MD simulation, the water molecule as covalently bound to the Chl a. All these ground-state calculations were done using GAMESS US package (Schmidt et al., 1993). To evaluate the unperturbed excited states of the SRChl a needed for PMM procedure, we carried out ab initio calculations on the SRChl a ring including the coordinated water. The above calculations of the SRChl a were carried out using the configuration interaction method with only the single excitations (CIS) with a 3-21G basis set (Pietro et al., 1982) for the Mg atom and 4-31G basis set (Gordon, 1980) for the other atoms, the active space consisting of the 10 highest occupied and the 10 lowest unoccupied molecular orbitals. The choice of the basis set was essentially driven by its ability in reproducing the ground to first excited-state energy gap for this system. From these calculations we obtained the unperturbed energies of ground and first 10 excited states. Moreover, at the same level of theory, all the unperturbed electric dipoles of the eigenstates and the transition electric dipoles between them were considered. All the CIS calculations were performed using Gaussian98 package (Frisch et al., 1998). Note that the limitations of the CIS calculations are basically due to the dimension of the SRChl a atomic basis set used; in any case, given the dimension of the SRChl a system, the use of a significantly larger atomic basis set is computationally not feasible at the present state of the computer power. Moreover, recent computational studies (Linnanto and Korppi-Tommola, 2000) have pointed out the inadequacy of CIS calculations in quantitatively reproducing the entire spectrum of a chlorophyll. However, in our case, the use of the ZINDO approach, shown by the Linnanto study to better behave in reproducing electronic excited states energies, would be inappropriate for the application of PMM (Aschi et al., 2001) which explicitly needs the calculation of transition dipole moments that, in this case, revealed to be severely inaccurate when calculated with semiempirical methods.
Mixed calculations
To obtain the electronic properties of the Chl a into the protein environment during the simulation, we modeled the protein perturbation as an electric field, changing according to the protein's motions and acting on the Chl a system. The electric potential felt by the Chl a molecule can be expanded around the chromophore geometrical center. If a first order expansion of the electric potential is used, for an uncharged chromophore, the protein's perturbation reduces simply to the effect of a homogeneous electric field interacting with the dipole operator (we disregard the magnetic interactions). Such a perturbing electric field was assumed to be independent of the chromophore; hence any polarization of the atomic electronic density due to the chromophore interaction and excitation was neglected. It would be possible, in principle, to include these higher order effects, but in this paper where we deal with a large and complex system we disregard them, i.e., we assume that at least the chromophore excitation energy is basically independent of these effects. The electric field acting on the chromophore was approximated using the GROMOS87 atomic charges. Note that the interaction between the Chl a and bulk SPC water molecules was not included in the calculations, as their effect on the electric field acting on the chromophore, calculated as large as 1–10% of the protein electric field, was negligible during the simulation. By the use of the PMM method, given the unperturbed energies, dipoles, and transition dipoles, it is possible to calculate the electronic properties of the perturbed system during the simulation, i.e., for each stored configuration of the MD simulation. The method is based on the matrix expression of the time-independent Schroedinger's equation, that is for a perturbed system:
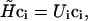 |
(1) |
where 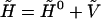 , ci is the ith eigenvector of the perturbed Hamiltonian matrix
, ci is the ith eigenvector of the perturbed Hamiltonian matrix 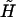 ,
, 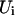 is the corresponding Hamiltonian eigenvalue,
is the corresponding Hamiltonian eigenvalue, 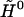 is the unperturbed Hamiltonian matrix, and
is the unperturbed Hamiltonian matrix, and 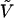 is the perturbation energy matrix. By expressing the Hamiltonian matrix and its eigenvectors in the basis set defined by the unperturbed Hamiltonian matrix eigenvectors, the generic element l,l′ of the Hamiltonian matrix is:
is the perturbation energy matrix. By expressing the Hamiltonian matrix and its eigenvectors in the basis set defined by the unperturbed Hamiltonian matrix eigenvectors, the generic element l,l′ of the Hamiltonian matrix is:
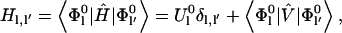 |
(2) |
where Φl0 is the lth eigenfunction of the unperturbed Hamiltonian operator, 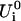 the corresponding energy eigenvalue, δl,l′ the Kroenecker's delta, and
the corresponding energy eigenvalue, δl,l′ the Kroenecker's delta, and 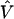 the perturbation energy operator. To obtain the eigenvectors and eigenvalues, and hence any needed property, of the perturbed Hamiltonian matrix, we have only to diagonalize the matrix
the perturbation energy operator. To obtain the eigenvectors and eigenvalues, and hence any needed property, of the perturbed Hamiltonian matrix, we have only to diagonalize the matrix 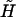 . For a system interacting with an external electric field, we can express in general the perturbation operator in Eq.2 in terms of the electric potential V (Spezia et al., 2002) as
. For a system interacting with an external electric field, we can express in general the perturbation operator in Eq.2 in terms of the electric potential V (Spezia et al., 2002) as
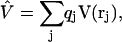 |
(3) |
with rj the coordinates of the jth charged particle and qj the corresponding charge. Expanding at the second order V around a given position r0 we have
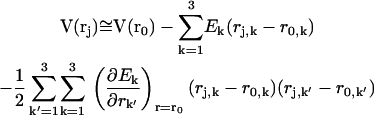 |
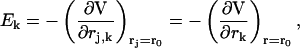 |
where k and k′ define the three components of a vector in space and r is the generic position vector. From these equations, defining with qT the total charge, we readily obtain
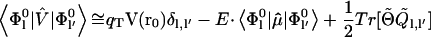 |
(4) |
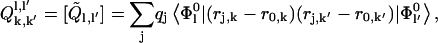 |
(5) |
where
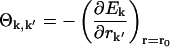 |
(6) |
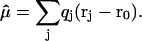 |
(7) |
Hence the complete perturbed Hamiltonian matrix is
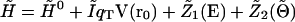 |
(8) |
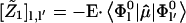 |
(9) |
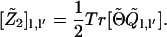 |
(10) |
From the last equations, it is evident that a second order expansion of the electric potential, able to describe electric fields up to linear behavior over the molecular size, requires the knowledge of the total charge and the unperturbed dipoles and quadrupoles. Higher order expansions can be in principle worked out in the same way but would require information on higher order multipoles, which are typically very difficult to obtain. Moreover, it is rather unusual that an applied electric field, at least evaluated from the Coulombic part of a usual molecular force field, is beyond the linear approximation over a molecular size. This was actually verified in our simulation, where the electric field, due to the Coulombic interactions, was for most of the configurations obtained by the MD simulation virtually constant over the chlorophyll. However, it is worth noting that such approximation does not hold in general for short-range atomic interactions, typically described in MD force field by the Lennard-Jones potential, which may modify significantly the electric field over a single atom. However, these interactions can be assumed, as far as the valence excited states are considered, to be basically independent of the Hamiltonian eigenstates, providing only an additive constant for the diagonal elements of the perturbation matrix. Hence the excitation energy may be well evaluated using only the Coulombic interactions. In the present paper, we used the Born-Oppenheimer approximation to describe the chromophore, i.e., its unperturbed eigenfunctions are purely electronic, and we disregard any perturbation effect beyond the dipolar term in Eq. 8. The previous equations could be in principle exact and general only if infinite dimensional matrices and vectors are involved. However, if we focus our attention on the ground and first excited states, the use of a finite dimensional matrix can provide an accurate approximation (Aschi et al., 2001). To build up the matrix 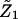 defined by Eq. 9, we need the chromophore unperturbed eigenstates, i.e., the unperturbed energies of a finite set of eigenstates, and the corresponding electric dipoles and the transition electric dipoles. Provided that the chromophore remains basically rigid during the simulation, it is possible to use the unperturbed states calculated at CIS level of theory at a given reference configuration, and build for each stored configuration of the trajectory a perturbed matrix to be diagonalized. Note that the use of the PMM method is possible also in the presence of flexible molecules, although in this case the unperturbed eigenstates should be calculated over a set of different configurations. From each diagonalization of the perturbed matrix, constructed on the unperturbed ground and first 10 excited states, i.e., an 11 × 11 matrix, we obtained the perturbed eigenvalues and eigenvectors. The difference between the ground and the first excited eigenvalues gives us the energy of the first (vertical) electronic transition and therefore the corresponding wavelength.
defined by Eq. 9, we need the chromophore unperturbed eigenstates, i.e., the unperturbed energies of a finite set of eigenstates, and the corresponding electric dipoles and the transition electric dipoles. Provided that the chromophore remains basically rigid during the simulation, it is possible to use the unperturbed states calculated at CIS level of theory at a given reference configuration, and build for each stored configuration of the trajectory a perturbed matrix to be diagonalized. Note that the use of the PMM method is possible also in the presence of flexible molecules, although in this case the unperturbed eigenstates should be calculated over a set of different configurations. From each diagonalization of the perturbed matrix, constructed on the unperturbed ground and first 10 excited states, i.e., an 11 × 11 matrix, we obtained the perturbed eigenvalues and eigenvectors. The difference between the ground and the first excited eigenvalues gives us the energy of the first (vertical) electronic transition and therefore the corresponding wavelength.
RESULTS AND DISCUSSION
Mechanics and dynamics
Stability and equilibration of the simulated system are shown through the behavior of the RMSD with respect to the crystal structure and of the RGyr in function of time (Fig. 3). The secondary structure was preserved during the simulation (data not shown) as well as the overall folding of the subsystem. The trajectory was considered fully equilibrated after 700 ps and hence the last 3.3 ns were used for equilibrium analysis. The main contribution to the motion of the protein is localized in all the loops connecting the α-helices, as usual in protein dynamics (Ceruso et al., 1999). The dynamics of the chromophores was also analyzed: in Table 1 the average RMSD of Chl a and Pers with respect to their own crystal structure and the standard deviation of RMSD are shown. It is evident from the very small RMSD and its standard deviation that Chl a is basically a rigid body during the simulation, i.e., the internal motions are very small. The four Pers molecules show two different behaviors: three are very flexible, whereas the fourth is more rigid, as shown in the same table. The most rigid Per is the Per4, i.e., the Per packed between the Chl a and the protein structure. In Fig. 4, the eigenvalues spectrum and the cumulative fluctuation (inset) of the principal motions of the protein Cα atoms are shown. As expected, the first eigenvalues provide a large part of the overall fluctuations. The motion was projected on the first and second eigenvectors separately as well as on the plane of the first two eigenvectors (Fig. 5). The atomic motions associated with the first two eigenvectors, described in Fig. 6, correspond to a motion of the loop connecting the two helices N4 (from residue 56 to 70) and N5 (from residue 82 to 97) and a tilting of the helices N2 (from residue 22 to 34), N3 (from residue 37 to 53), and N7 (from residue 119 to 125). The loop motion is the largest in both eigenvectors. In the first eigenvector, the helix N2 has a lager amplitude motion than the helix N7 and the helix N3, whereas in the second eigenvector, the helix N3 has a larger displacement than the helix N2 and the helix N7 has only a small displacement. Note that such large amplitude motions are not probably associated to any unfolding process or instability of the monomer used, as already shown by the stability of the secondary structures and radius of gyration. Panels A and B of the Fig. 5 show the typical large amplitude and slow motion of essential coordinates (Amadei et al., 1993) characterized by a slow diffusive kinetics (Amadei et al., 1999b). Such a slow diffusion provides probably only a partial sampling of the subspace defined by the first two eigenvectors within the simulation time length (panel C), as the relaxation and convergence for the essential coordinates are typically beyond the nanosecond timescale. However, the convergence of the essential subspace (usually the first 10-20 eigenvectors) is reasonably achieved within a few nanoseconds (Amadei et al., 1999a). This means that although the single essential eigenvectors are likely to be not the equilibrium ones, i.e., the eigenvectors obtained by a completely converged statistics, the corresponding atomic collective motions are probably significant as they belong to a good approximation of the equilibrium essential subspace.
FIGURE 3.

(A) Trajectory of the Cα root mean-square deviation (RMSD) with respect to the crystal structure. (B) Trajectory of the Cα radius of gyration (RGyr).
TABLE 1.
Average RMSD of Chl a and Pers with respect to their own crystal structure as obtained from the equilibrated MD trajectory
| Molecule | 〈RMSD〉 (nm) | σ (nm) |
|---|---|---|
| Chl a | 0.086 | 0.021 |
| Per1 | 0.350 | 0.095 |
| Per2 | 0.413 | 0.147 |
| Per3 | 0.433 | 0.170 |
| Per4 | 0.251 | 0.028 |
FIGURE 4.

Eigenvalue spectrum of the first 20 eigenvectors and its cumulative fluctuations (in the inset where the units are the same).
FIGURE 5.

Projection of Cα atom trajectory on the first eigenvector (A), on the second eigenvector (B), and on both the first two eigenvectors (C).
FIGURE 6.
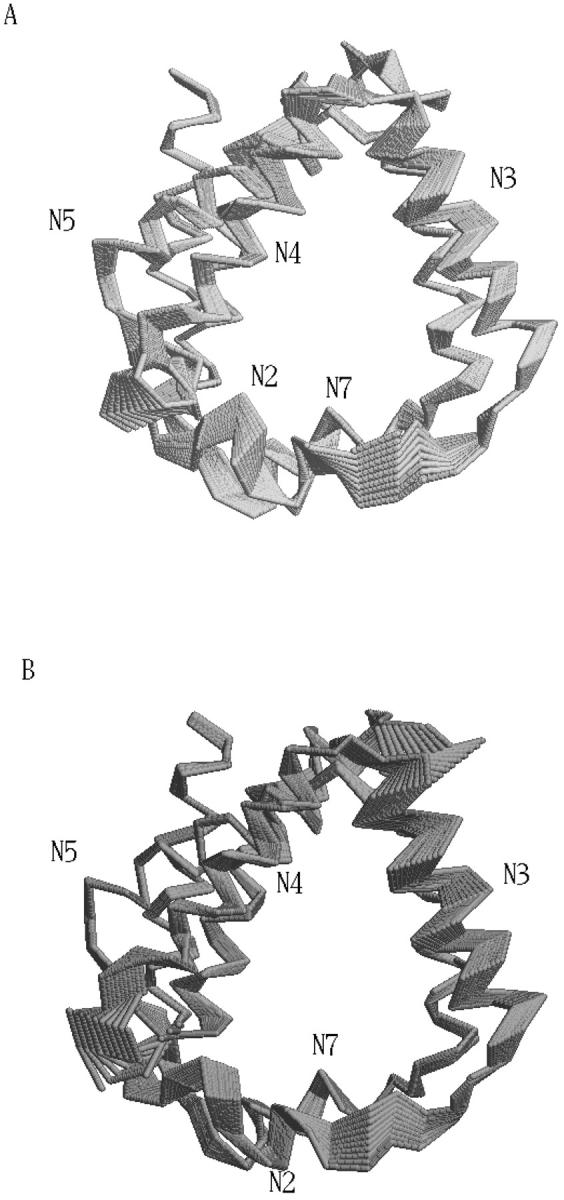
Schematic view of tridimensional backbone motion due to the first (A) and second (B) eigenvectors.
Chlorophyll electronic properties and conformational fluctuations
CIS calculations were carried out on the minimized crystallographic structure of the SRChl a. By comparing our ab initio results with previous findings, we can observe a rather good agreement. The unperturbed transition molecular electric dipole between ground and first excited state of 1.92 a.u. falls in the range of 1.62–2.09 a.u. reported in Kleima et al. (2000b). At the same time, the calculated ΔSCF IP and the Koopmans' theorem IP (5.47 eV and 6.15 eV, respectively) are close to the values reported for Bchl a by Sakuma et al. (1997), i.e., 5.00 eV and 5.76 eV, respectively. This comparison, although performed between Chl and BChl molecules, ensures a good qualitative description of the electronic wave function studied. Finally, despite the use of a simplified molecular structure, the calculated energy difference between the first excited singlet state and the ground state (617 nm) is comparable to the experimental value of 672 for the Qy transition of Chl a in PCP (Koka and Song, 1997). To evaluate the effect of the water molecule bound to SRChl a, we first optimized the geometry of the system without water molecule and then we carried out the CIS calculations. We actually found an increase of the ground to the first excited state excitation energy as large as 121 nm probably due to the geometry change induced by the absence of the water molecule as recently remarked in the spectra of retinal proteins (Ren et al., 2001; Hayashi et al., 2001). Such unperturbed transition dipoles and energies of the ground and first 10 electronic excited states were used to calculate the perturbed energies using the PMM method. The absorption wavelength (λ), calculated from the energy difference between the first excited and ground perturbed eigenvalues, was obtained for each configuration of the trajectory. In Fig. 7 A, the transition wavelengths calculated in the simulation (solid line) and the corresponding unperturbed one (dashed line) are reported. The effect of the environment is a blue shift with an average absorption wavelength of 604 nm with a standard deviation of 8 nm. In Fig. 7 B, we report the unnormalized distribution of the perturbed λ calculated in the equilibrated trajectory. Note that the distribution is not symmetric and hence the maximum, located between 610 and 611 nm, does not coincide with the average λ. Moreover, this dispersion of the λ-values is due only to the flexibility of the surrounding protein, as the inhomogeneous and vibronic contributions to the spectral broadening are not taken into account in this kind of calculations. To evaluate the role played respectively by the protein and Per chromophores in the perturbation and transition energy fluctuation (water molecules are too far and DGDG is a basically apolar molecule), we calculated the effect of the field produced only by the Pers. The average absorption wavelength, and its standard deviation and error, were calculated as a function of the position along the first eigenvector. In Fig. 8 (panel A), we show the absorption (average) λ due to the overall electric field (solid line) and due to the Per electric field (dashed line) as a function of the position along the first eigenvector. In panel B, we also show the corresponding λ standard deviation, provided by the other coordinates fluctuations. It is evident that only in a well-defined range of the protein first eigenvector coordinate, the Per chromophores have a relevant influence on the absorption spectrum. Consistently, the standard deviation of the wavelength due to the Per electric field shows the same behavior. From a more detailed analysis of the MD simulation, we noted that the above behavior corresponds to a translational motion of the three Pers (Per1, Per2, Per3). The absorption wavelength due to the overall electric field is clearly dependent on the first eigenvector coordinate whereas its standard deviation seems to be rather constant. This shows that the essential motions in the protein are important in modulating the photochemical activity of a chromophore. It is worth noting that the average absorption wavelengths evaluated at each position along the first eigenvector are likely to be not fully converged as the motion of the other essential coordinates is characterized by a slow diffusive kinetics (Amadei et al., 1999a,b). To reduce the possible effect of the incomplete convergence on the average λ, we also evaluated the average λ fixing the first two eigenvectors positions. This is likely to provide better converged λ-values as they are averaged on faster relaxing coordinates. The results are reported in Fig. 9, which shows again a clear dependence of the average λ on the first two eigenvectors coordinates.
FIGURE 7.

(A) Trajectory of the transition wavelength (solid line) compared with the unperturbed transition wavelength calculated (dashed line). (B) Spectral distribution of the perturbed λ calculated on the equilibrated trajectory.
FIGURE 8.

Excitation wavelength (λ) with the error expectation value (panel A) and the standard deviation of the excitation wavelength (σ(λ)) (panel B) along the first eigenvector of the protein, taking into account the whole system effect (solid line) and only the Per effect (dashed line).
FIGURE 9.

Excitation wavelength (λ) along the first two eigenvectors of the protein.
CONCLUSIONS
In this paper, we have investigated the influence of the protein conformational flexibility on the electronic properties of the Chl a chromophore in the light-harvesting complex A-PCP. This antenna complex was never simulated in the nanosecond time range by using MD methodologies. The PMM method was applied to a rigidlike chromophore interacting with a stable classical environment (i.e., we neglected any atomic polarization of the protein), and for each configuration, provided by a classical MD simulation, we obtained the corresponding vertical excitation energy due to the “dynamical” perturbation effect of the protein. The combined use of the essential dynamics analysis and PMM clearly showed a coupling between the large concerted motions and the excitation energy, suggesting a possible conformational regulation of the photochemical activity. Although further investigations are needed, e.g., on linear and circular dichroism spectra of the chromophores, we expect this approach to be of great interest in the study of complex biomolecular systems where a “quantum center” interacts with a classicallike environment, as in enzymes and proteins with photochemical activity, or redox biological systems. Moreover, the inclusion of higher order effects neglected in this paper, i.e., the environment polarizability and chromophore flexibility, can be in principle achieved using more sophisticated classical force fields and PMM calculations. The effect of the environment, not only on the absorption wavelength but also on the corresponding transition moments, could be important in enhancing the efficiency of the Förster energy transfer process among the chlorophyll chromophores belonging to different clusters in the natural occurring system. It is worth to note that the approach used in this paper can be extended to treat excitonic systems that are often of great interest in biophysical-biochemical systems. Further calculations on this aspect are among the perspectives of the present work.
Acknowledgments
We acknowledge Prof. G. Giacometti for stimulating discussions and CASPUR (Rome) for computational support and access to the Gaussian98 package.
We acknowledge the Italian Ministry for University and Scientific and Technological Research (PRIN “Structure and Dynamics of Redox Proteins” 2001) and CNR Agenzia2000 for financial support.
References
- Akimoto, S., S. Takaichi, T. Ogata, Y. Nishimura, I. Yamazaki, and M. Mimuro. 1996. Excitation energy transfer in carotenoid-chlorophyll protein complexes probed by femtosecond fluorescence decays. Chem. Phys. Lett. 260:147–152. [Google Scholar]
- Alden, R. G., E. Johnson, V. Nagarajan, W. W. Parson, C. J. Law, and R. G. Cogdell. 1997. Calculations of spectroscopic properties of the LH2 bacteriochlorophyll-brotein antenna complex from Rhodopseudomonas acidophila. J. Phys. Chem. B. 101:4667–4680. [Google Scholar]
- Amadei, A., M.-A. Ceruso, and A. Di Nola. 1999a. On the convergence of the conformational coordinates basis set obtained by the essential dynamics of proteins' molecular dynamics simulations. Proteins. 36:419–424. [PubMed] [Google Scholar]
- Amadei, A., G. Chillemi, M.-A. Ceruso, A. Grottesi, and A. Di Nola. 2000. Molecular dynamics simulations with constrained roto-translational motions: theoretical basis and statistical mechanical consistency. J. Chem. Phys. 112:9–23. [Google Scholar]
- Amadei, A., B. L. de Groot, M.-A. Ceruso, M. Paci, A. Di Nola, and H. J. C. Berendsen. 1999b. A kinetic model for the internal motions of proteins: diffusion between multiple harmonic wells. Proteins. 35:283–292. [PubMed] [Google Scholar]
- Amadei, A., A. B. M. Linssen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Proteins. 17:412–425. [DOI] [PubMed] [Google Scholar]
- Aschi, M., R. Spezia, A. Di Nola, and A. Amadei. 2001. A first-principles method to model perturbed electronic wavefunctions: the effect of an external homogeneous electric field. Chem. Phys. Lett. 344:374–380. [Google Scholar]
- Bautista, J. A., R. G. Hiller, F. P. Sharples, D. Gosztola, M. Wasielewski, and H. A. Frank. 1999. Singlet and triplet energy transfer in the peridinin-chlorophyll a protein from Amphidinium carterae. J. Phys. Chem. A. 103:2267–2273. [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. 1981. Interaction Models for Water in Relation to Protein Hydration. Reidel, Dordrecht, The Netherlands. 331–342.
- Brown, D., and J. Clarke. 1986. Molecular dynamics simulations of polymer fiber microstructure. J. Chem. Phys. 84:2858–2865. [Google Scholar]
- Carbonera, D., G. Giacometti, U. Segre, A. Angerhofer, and U. Gross. 1999a. Model for triplet-triplet energy transfer in natural clusters of peridinin molecules contained in dinoflagellate's outer antenna proteins. J. Phys. Chem. B. 103:6357–6362. [Google Scholar]
- Carbonera, D., G. Giacometti, U. Segre, E. Hofmann, and R. G. Hiller. 1999b. Structure-based calculations of the optical spectra of the Light-harvesting peridinin-chlorophyll-protein complexes from Amphidinium carterae and Heterocapsa pygmaea. J. Phys. Chem. B. 103:6349–6356. [Google Scholar]
- Ceruso, M.-A., A. Amadei, and A. Di Nola. 1999. Mechanics and dynamics of B1 domain of protein G: role of packing and surface hydrophobic residues. Protein Sci. 8:147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chillemi, G., M. Falconi, A. Amadei, G. Zimatore, A. Desideri, and A. Di Nola. 1997. The essential dynamics of Cu, Zn superoxide dismutase: suggestion of intersubunit communication. Biophys. J. 73:1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce, R., R. Remelli, C. Varotto, J. Breton, and R. Bassi. 1999. The neoxanthin binding site of the major light harvesting complex (LHCII) from higher plants. FEBS Lett. 456:1–6. [DOI] [PubMed] [Google Scholar]
- Damjanovic, A., I. Kosztin, U. Kleinekathöfer, and K. Schulten. 2002. Excitons in a photosynthetic light-harvesting system: a combined molecular dynamics, quantum chemistry, and polaron model study. Phys. Rev. E. 65:031919–031963. [DOI] [PubMed] [Google Scholar]
- Damjanovic, A., T. Ritz, and K. Schulten. 2000. Excitation transfer in the peridinin-chlorophyll-protein of Amphidinium carterae. Biophys. J. 79:1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot, B. L., D. M. F. van Aalten, A. Amadei, and H. J. C. Berendsen. 1996. The consistency of large concerted motions in proteins in molecular dynamics simulations. Biophys. J. 71:1707–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditchfield, R., W. J. Hehre, and J. A. Pople. 1971. Self-consistent molecular-orbital method. IX. An extended Gaussian-type basis for molecular orbital studies of organic molecules. J. Chem. Phys. 54:724–728. [Google Scholar]
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle, and J. A. Pople. 1998. Gaussian 98 (Revision A.7). Gaussian, Pittsburgh, PA.
- Ghosh, A. 1997. Theoretical comparative study of free-based porphyrin, chlorin, bacteriochlorin, and isobacteriochlorin: evaluation of the potential roles of ab initio Hartree-Fock and density functional theories in hydrophorphyrin chemistry. J. Phys. Chem. B. 101:3290–3297. [Google Scholar]
- Gordon, M. S. 1980. The isomers of silacyclopropane. Chem. Phys. Lett. 76:163–168. [Google Scholar]
- Gordon, M. S., J. S. Binkley, J. A. Pople, W. J. Pietro, and W. J. Hehre. 1982. Self-consistent molecular orbital methods 22: small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 104:2797–2803. [Google Scholar]
- Hayashi, S., E. Tajkhorshid, E. Pebay-Peyroula, A. Royant, E. M. Landau, J. Navarro, and K. Schulten. 2001. Structural determinants of spectral tuning in retinal proteins-bacteriorhodopsin vs. sensory rhodopsin II. J. Phys. Chem. B. 105:10124–10131. [Google Scholar]
- Hasegawa, J., Y. Ozeki, K. Ohkawa, M. Hada, and H. Nakatsuji. 1998. Theoretical study of the excited states of chlorin, bacteriochlorin, pheophytin a and chlorophyll a by the SAC/SAC-CI method. J. Phys. Chem. B. 102:1320–1326. [Google Scholar]
- Hehre, W. J., R. Ditchfield, and J. A. Pople. 1972. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbitals studies of organic molecules. J. Chem. Phys. 56:2257–2261. [Google Scholar]
- Hofmann, E., P. M. Wrench, F. P. Sharples, R. G. Hiller, W. Welte, and K. Diederichs. 1996. Structural basis of light harvesting carotenoids: peridinin-chlorophyll-protein from Amphidinium carterae. Science. 272:1788–1791. [DOI] [PubMed] [Google Scholar]
- Hu, X., T. Ritz, A. Damjanovic, and K. Schulten. 1997. Pigment organization and transfer of electronic excitation in the photosynthetic unit of purple bacteria. J. Phys. Chem. B. 101:3854–3871. [Google Scholar]
- Jankowiak, R., V. Zazubovich, M. Rätsep, S. Matsuzaki, M. Alfonso, R. Picorel, M. Seibert, and G. J. Small. 2000. The CP43 core antenna complex of photosystem II possesses two quasi-degenerate and weakly coupled Q y-trap states. J. Phys. Chem. B. 104:11805–11815. [Google Scholar]
- Kleima, F. J., E. Hofmann, B. Gobets, I. H. M. van Stokkum, R. van Grondelle, K. Diederichs, and H. van Amerongen. 2000a. Förster excitation energy transfer in peridinin-cholorophyll-a-protein. Biophys. J. 78:344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleima, F. J., M. Wendling, E. Hofmann, E. J. G. Peterman, R. van Grondelle, and H. van Amerongen. 2000b. Peridinin chlorophyll a protein: relating structure and steady-state spectroscopy. Biochemistry. 39:5184–5195. [DOI] [PubMed] [Google Scholar]
- Koepke, J., X. C. Hu, C. Muenke, K. Schulten, and H. Michel. 1996. The crystal structure of the light-harvesting complex II (B800–850) from Rhodospirillum molischianum. Structure. 4:581–597. [DOI] [PubMed] [Google Scholar]
- Koka, P., and P.-S. Song. 1997. The chromophore topography and binding environment of peridinin chlorophyll a protein complexes from marine dinoflagellate algae. Biochim. Biophys. Acta. 495:220–231. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. J. 1991. MolScript: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24:946–950. [Google Scholar]
- Kuhlbrandt, W., D. N. Wang, and Y. Fujiyoshi. 1994. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 367:614–621. [DOI] [PubMed] [Google Scholar]
- Linnanto, L., and J. Korppi-Tommola. 2000. Spectroscopic properties of Mg-chlorin, Mg-porphin and chlorophyll a, b, c1, c2, c3 and d studied by semi-empirical and ab initio MO/CI methods. Phys. Chem. Chem. Phys. 2:4962–4970. [Google Scholar]
- Matthews, B. W., R. E. Fenna, M. C. Bolognesi, M. F. Schmid, and J. M. Olsom. 1979. Structure of a bacteriochlorophyll a-protein from the green photosynthetic bacterium Prosthecochloris aestuarii. J. Mol. Biol. 131:259–285. [DOI] [PubMed] [Google Scholar]
- McDermott, G., S. M. Prince, A. A. Freer, A. M. Hawthornthwaite-Lawless, M. Z. Papiz, R. J. Cogdell, and N. W. Isaacs. 1995. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature. 374:517–521. [Google Scholar]
- Merritt, E. A., and D. J. Bacon. 1997. Raster3D: photorealistic molecular graphics. Methods Enzymol. 277:505–524. [DOI] [PubMed] [Google Scholar]
- Nonomura, Y., S. Igarashi, N. Yoshioka, and H. Inoue. 1997. Spectroscopic properties of chlorophylls and their derivatives. Influence of molecular structure on the electronic state. Chem. Phys. 220:155–166. [Google Scholar]
- Pascal, A., C. Gradinaru, U. Wacker, E. Peterman, F. Calkoen, K.-D. Irrgang, P. Horton, G. Renger, R. van Grondelle, B. Robert, and H. van Amerongen. 1999. Spectroscopic characterization of the spinach Lhcb4 protein (CP29), a minor light-harvesting complex of photosystem II. Eur. J. Biochem. 262:817–823. [DOI] [PubMed] [Google Scholar]
- Pietro, W. J., M. M. Franci, W. J. Hehre, D. J. De Frees, J. A. Pople, and J. S. Binkley. 1982. Self-consistent molecular orbital methods. 21. Supplemented small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 104:5039–5048. [Google Scholar]
- Ren, L., C. H. Martin, K. J. Wise, N. B. Gillespie, H. Luecke, J. K. Lanyi, J. L. Spudich, and R. R. Birge. 2001. Molecular mechanism of spectral tuning in sensory rhodopsin II. Biochemistry. 40:13906–13914. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Phys. 23:327–341. [Google Scholar]
- Sakuma, T., H. Kashiwagi, T. Takada, and H. Nakamura. 1997. Ab initio MO study of the chlorophyll dimer in the photosynthetic reaction center. I. A theoretical treatment of the electrostatic field created by the surrounding proteins. Int. J. Quant. Chem. 61:137–151. [Google Scholar]
- Salverda, J. M., F. van Mourik, G. van der Zwan, and R. van Grondelle. 2000. Energy transfer in the B800 rings of the peripheral bacterial light-harvesting complexes of Rhodopseudomonas acidophila and Rhodospirillum molischianum studied by photon echo techniques. J. Phys. Chem. B. 104:11395–11408. [Google Scholar]
- Schmidt, M. W., K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. J. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis, and J. A. Montgomery. 1993. General atomic and molecular electronic structure system. J. Comput. Chem. 14:1347–1363. [Google Scholar]
- Siegbahn, P. E. M., and M. R. A. Blomberg. 2000. Transition-metal systems in biochemistry studied by high accuracy quantum chemical methods. Chem. Rev. 100:421–438. [DOI] [PubMed] [Google Scholar]
- Spezia, R., M. Aschi, A. Di Nola, and A. Amadei. 2002. Extension of the perturbed matrix method: application to a water molecule. Chem. Phys. Lett. 365:450–456. [Google Scholar]
- Sundholm, D. 1999. Density functional theory calculations of the visible spectrum of chlorophyll a. Chem. Phys. Lett. 302:480–484. [Google Scholar]
- Sundström, V., T. Pullerits, and R. van Grondelle. 1999. Photosynthetic light-harvesting: reconciling dynamics and structure of purple bacterial LH2 reveals function of photosynthetic unit. J. Phys. Chem. B. 103:2327–2346. [Google Scholar]
- van Aalten, M. F., A. Amadei, A. B. M. Linssen, V. G. H. Eijsink, G. Vriend, and H. J. C. Berendsen. 1995a. The essential dynamics of thermolysin: confirmation of the hinge-bending motion and comparison of simulations in vacuum and water. Proteins. 22:45–54. [DOI] [PubMed] [Google Scholar]
- van Aalten, M. F., J. B. C. Findlay, A. Amadei, and H. J. C. Berendsen. 1995b. Essential dynamics of the cellular retinol-binding protein—evidence for ligand-induced conformational changes. Protein Eng. 8:1129–1136. [DOI] [PubMed] [Google Scholar]
- van Buuren, A. R., S. J. Marrink, and H. J. C. Berendsen. 1993. A molecular dynamics study of the decane/water interface. J. Phys. Chem. 97:9206–9212. [Google Scholar]
- van der Spoel, D., H. J. C. Berendsen, A. R. van Buuren, M. E. F. Apol, P. J. Meulenhoff, and A. L. T. M. Sijbers. 1995. GROMACS User Manual. University of Groningen, Groningen, The Netherlands.
- van Gunsteren, W. F., S. R. Billeter, A. A. Eising, P. H. Hunenberger, P. Kuger, A. E. Mark, W. R. P. Scott, and I. G. Tironi. 1996. The GROMOS96 Manual and User Guide. Biomos, Zurich, Switzerland.
- Zucchelli, G., R. C. Jennings, F. M. Garlaschi, G. Cinque, R. Bassi, and O. Cremonesi. 2002. The calculated in vitro and in vivo chlorophyll a absorption bandshape. Biophys. J. 82:378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]