

Abstract
Sequence-function analysis of K+-selective channels was carried out in the context of the 3.2 Å crystal structure of a K+ channel (KcsA) from Streptomyces lividans (Doyle et al., 1998). The first step was the construction of an alignment of a comprehensive set of K+-selective channel sequences forming the putative permeation path. This pathway consists of two transmembrane segments plus an extracellular linker. Included in the alignment are channels from the eight major classes of K+-selective channels from a wide variety of species, displaying varied rectification, gating, and activation properties. Segments of the alignment were assigned to structural motifs based on the KcsA structure. The alignment's accuracy was verified by two observations on these motifs: 1), the most variability is shown in the turret region, which functionally is strongly implicated in susceptibility to toxin binding; and 2), the selectivity filter and pore helix are the most highly conserved regions. This alignment combined with the KcsA structure was used to assess whether clusters of contiguous residues linked by hydrophobic or electrostatic interactions in KcsA are conserved in the K+-selective channel family. Analysis of sequence conservation patterns in the alignment suggests that a cluster of conserved residues is critical for determining the degree of K+ selectivity. The alignment also supports the near-universality of the “glycine hinge” mechanism at the center of the inner helix for opening K channels. This mechanism has been suggested by the recent crystallization of a K channel in the open state. Further, the alignment reveals a second highly conserved glycine near the extracellular end of the inner helix, which may be important in minimizing deformation of the extracellular vestibule as the channel opens. These and other sequence-function relationships found in this analysis suggest that much of the permeation path architecture in KcsA is present in most K+-selective channels. Because of this finding, the alignment provides a robust starting point for homology modeling of the permeation paths of other K+-selective channel classes and elucidation of sequence-function relationships therein. To assay these applications, a homology model of the Shaker A channel permeation path was constructed using the alignment and KcsA as the template, and its structure evaluated in light of established structural criteria.
INTRODUCTION
Potassium channels are integral membrane proteins that exist in the plasma membrane of the majority of cells in all life forms. They stabilize and determine the membrane resting potential, control the shape and timeframe of action potentials in excitable cells, and play a role in osmoregulation, neurotransmitter release, secretion, and enzyme activity (Hille, 1992). In response to an environmental stimulus such as change in membrane potential or pH, or changes in concentration of Ca2+, cyclic nucleotides, or ATP, they open, allowing K+ ions to traverse the membrane down their electrochemical potential gradient. A wide variety of channels selective for K+ have been discovered and classified according to functional characteristics. These include: delayed rectifiers and transiently activating A channels (DRK/A), small and large conductance calcium-sensitive channels (SK and BK), inward rectifiers (Kir), K+-selective hyperpolarization-activated channels gated by cyclic nucleotides (HYP), plant inward rectifiers (AKT), ether-a-go-go and related types (EAG), and two-domain subunit channels (designated Two-Pore). In addition, potassium channels have been cloned from several prokaryotes. The enormous variety of potassium channels is manifest in the fact that the genome of Caenorhabditis elegans apparently codes for at least 80 potassium channels (Bargmann, 1998). Analysis of cloned sequences of voltage-gated and cyclic nucleotide-binding channels suggests that ancient duplications gave rise to a superfamily of three groups, consisting of Na+- and Ca2+-selective channels, cyclic nucleotide-gated channels, and K+-selective channels (Jan and Jan, 1997; Strong et al., 1993). Subsequent gene duplications and divergence are responsible for K+-selective channels' wide variation in physiological function.
Although they perform a wide variety of physiological functions, K-selective channels are similar to each other in overall structure and function. The vast majority of K+-selective channels contain four identical subunits that form a central pore that serves as the ion permeation pathway. Each subunit contains two, six, or ten transmembrane (TM) segments, depending on channel class. Six is the most common number. Although they exhibit single channel conductances over a range from 4 to 300 pS (Grissmer et al., 1994; Lancaster et al., 1991), they all have similar permeation characteristics (Hille, 1992). Selectivity for K+ over Na+ and Li+ is universal in this class. Many members of this class are almost perfectly selective for potassium over lithium and sodium, but K+/Na+ selectivities as low as 3:1 have been measured in a few cases (Schrempf et al., 1995; Gauss et al., 1998; Ludwig et al., 1998). Weak selectivity is characteristic of a well-defined class of channels that we have called HYP, and have also been termed Ih or pacemaker channels (Kaupp and Seifert, 2001). K+-selective channels are similar with respect to the part of their sequence that has been found to confer the permeation and selectivity properties unique to them. An extracellular segment between the 5th and 6th TM segments of the transiently activating Shaker A channel has been deduced from numerous experiments to form the outer part of the permeation path (Yool and Schwartz, 1991; Hartmann et al., 1991; Kavanaugh et al., 1991; Yellen et al., 1991; Heginbotham et al., 1994; Guy and Durell, 1995; Gross and Mackinnon, 1996). This segment, termed the P region (P for pore), forms a tight loop that extends partly back into the membrane, toward the center of the tetramer. It resides between the 5th and 6th TM segments of 6- and 10-TM subunits and between the sole two in Kir and the 2-TM prokaryotic channels. Within each loop is the sequence GYG, sometimes GFG, found to be the K+-selectivity signature sequence (Heginbotham et al., 1994); the four G[YF]G's come together toward the channel axis to form the selectivity filter (MacKinnon, 1995).
A potassium channel from Streptomyces lividans (KcsA; pdb 1bl8) was crystallized and its structure resolved to 3.2 Å by Doyle et al. (1998). It is composed of a tetramer of 2-TM subunits, joined by a linker segment of 36 residues, containing the P region (Fig. 1). The structure provides insights into the mechanisms of selectivity, extracellular peptide toxin binding, and rapid K+ permeation through its long pore (Doyle et al., 1998; MacKinnon et al., 1998). The structure shows four well-delineated motifs in the linker region: a 10 residue random coil (termed the turret) on the C-terminal side of the prepore TM helix, a 13 amino acid α-helix extending inward from the extracellular pore periphery, the selectivity filter loop end, and an extended segment connecting the selectivity filter and the postpore TM helix. The pore α-helix and selectivity filter comprise the P region.
FIGURE 1.

A cartoon schematic of the KcsA channel from Streptomyces lividans highlighting its structural motifs. (a) Extracellular view of all four subunits, displaying the permeation path in the center. (b) Side view of two of the four subunits comprising the channel, with the extracellular side on top. The permeation path runs vertically through the center. The motifs shown are the prepore (M1) TM segment (blue), turret region (red), pore α-helix (magenta), selectivity filter containing the signature G[YF]G sequence (green), an extended segment (yellow), and the postpore (M2) TM segment (cyan).
Because of the high level of similarity in global topology, function, and the universal selectivity filter signature sequence among K+-selective channels, it is reasonable to postulate that the 6-TM subunit K+ channels (DRK/A, HYP, EAG, SK, and AKT), the 10-TM BK channels, and the 2-TM Kir's form permeation pathways with sequence and structural motifs similar to that of KcsA's. To provide a framework upon which to investigate this postulate, we have generated a comprehensive alignment of the permeation pathway from K+-selective channels. In conjunction with this, we determined sets (clusters) of residues in hydrogen bond or hydrophobic contact in the KcsA structure. Using the alignment and the identity of these clusters of residues, we investigated whether their homologous clusters in other K+-selective channels had similar electrostatic and/or size properties. Using sequence-structure correlations as leitmotifs for the channel, we identified key residues that are responsible for pore selectivity and ion permeation. Further to validate our results, a homology model of the permeation pathway of the Shaker A-type channel was constructed using the alignment and the KcsA structure as the template, and the structure validated against an array of experimental results.
METHODS
Alignment construction
The alignment was constructed in a two-stage manner: 1), For each K channel class, the two TM segments on either side of the extracellular linker were located using a sequence profiling method; and 2), using the location of these segments as constraints, the linker regions were aligned using the CLUSTAL algorithm, first within the class, then between the classes. The first stage was implemented by Perl programs that constructed the profiles and used them to locate the TM segments. The second stage was implemented using the NCSA Biology Workbench (Subramaniam, 1998), which integrates sequence databases and analysis tools into a web-based user interface.
Selection of isoforms
Selection of K+ channels to include in the alignment was done by a BLAST 2.0 (Altschul et al., 1997; BLOSUM 62 matrix, probe sequence filtered, gap open penalty = 11, gap extension = 1; these values used unless stated otherwise)) of the National Center for Biotechnology Information nonredundant database using a representative sequence from each of the eight K+-selective classes (Table 1). Complete channel sequences were chosen based on comprehensiveness of annotation in the database and degree of published experimental investigation. The set of channels from each class was chosen to include all known subclasses in as small a set as possible. For example, in the DRK/A class, members of the four major subclasses of Drosophila channels and their homologs (Shaker, Shab, Shaw, and Shal) were chosen, along with their mammalian counterparts (Kv1–Kv4) and additional mammalian radiations. A total of 155 sequences were chosen for alignment: 64 DRK/A, 4 SK, 8 BK, 45 Kir, 7 AKT, 8 HYP, 13 EAG, and 6 Two-Pore (3 sets of 2 subunits).
TABLE 1.
The eight major K+-selective channel classes used in the study
| Class name | Mnemonic(s) | Typical range of conductance (pS) | Physiological function | No. of TM segments/subunit |
|---|---|---|---|---|
| Delayed rectifer/ transiently activating | DRK/A; Shaker, Shab, Shal, Shaw, Kv, KCNA | 4–27 | Shorten action potential shortening; act as stabilizers in signaling | 6 |
| Small conductance Ca+ sensitive | SK | 4–80 | Create slow afterhyperpolarizations regulated by cytoplasmic Ca2+ | 6 |
| Large conductance Ca+ sensitive | BK; slo | 200–300 | Regulate secretion in endocrine and exocrine glands; Na+/K+ coupling in kidney tubules; regulated by cytoplasmic Ca2+ | 10 |
| Inward rectifier | Kir; IRK, GIRK, KATP, ROMK | 20–80 | Maintain resting potential; regulated by hormones, neurotransmitters, ATP, G-proteins | 2 |
| Plant inward rectifiers | AKT; KAT, KST | 6–28 | Uptake of K+ from soil solution; mediation of K+ flux in stomatal opening; regulated by cGMP | 6 |
| Hyperpolarization- activated cyclic nucleotide-sensitive | HYP; KCNG, SPIHCH, HAC | <10 | K+-selective, but permeable to Na+, and K+; regulation of flagellar beating in sperm, pacemaking; activated by hyperpolarization, regulated by cyclic nucleotides | 6 |
| Ether-a-go-go | EAG; erg, herg | 10–25 | Mediation of the expression of voltage- and Ca2+-activated K currents | 6 |
| Two domain subunit | Two-Pore; TWIK, TOK, TRAAK, TASK | 20–80 | Setting of the background membrane K+ conductance in many cell types | 4 |
In bold is the mnemonic used to refer to the class as a whole.
Transmembrane segment localization
A sequence specific profiling method adapted to TM sequence localization is employed in this work. There are two parts to this method: 1), a sequence profile is constructed from an aligned set of putative TM sequences in an integral transmembrane protein family; and 2), this profile is employed as a probe on a more distantly related protein sequence to locate its TM segments, using a scoring formula. The particular type of sequence profile used in this work consists of all the observed residues at each location in the TM alignment. The result is similar to a PROSITE (www.genebio.com/prosite.html) query string; each position in the alignment has a list of residues associated with it. In the application cited above (Ramarathnam and Subramaniam, 2000), the method utilized putative aligned TM's from opsins to generate profiles that could be used to probe other membrane proteins for their TM segments A particularly interesting feature of the S6/TM2 inner helix is the completely conserved glycine exactly in the middle; i.e., in position 13 of 25. This is the glycine that is implicated in the glycine hinge mechanism of K channel opening, which has been postulated based on the crystal structure of the open MthK channel (Jiang et al., 2002a,b).
Construction of TM profiles for K+-selective channels
In the present application, profiles were constructed from the putative S5 and S6 TM segments of members of the DRK/A class of K channels and used to locate the homologous TM segments bracketing the P region for the other seven K+-selective channel classes. DRK/A was chosen because members of this class of channels are most similar to KcsA and because the Shaker/Kv1 subclass produces an ungapped alignment with KcsA along its permeation path. This permits unambiguous assignment of the DRK/A S5 (prepore) and S6 (postpore) segments, based on alignments with KcsA's TM segments M1 and M2. This method assumes that the profile derived from the DRK/A's can be extended to K channels in general. The patterning of the results, to be described below, bear out the validity of the assumption. In particular, we will see that for inward rectifier channels, which have been seen to have distinct patterns of conservation in the M1 and M2 region relative to a variety of other K channels including the voltage-gated channels (Minor et al., 1999), our assignment for the transmembrane region agrees reasonably closely with an assignment based on analyses of mutations and sequence minimization experiments (Minor et al., 1999). Since the inward rectifier channels are distinctive in this regard, it seems likely that the fits for other classes of potassium channels will be equally good, or better.
To initiate the collection of DRK/A sequences, five sequences were chosen from the four major mammalian subclasses of DRK/A (Rattus norvegicus Kv1.1 from Kv1 (CIK1_RAT; GenPept 116421), R. norvegicus Kv1.4 from Kv1 (RNRCK4; GenPept 116431), Homo sapiens Kv2.1 from Kv2 (HUMKV2CH-1; GenPept 1168948), R. norvegicus Kv3.1 from Kv3 (RATKV4-1; GenPept 205107), and Mus musculus Kv4.1 from Kv4 (MUSMSHAL; GenPept 199813)). These five sequences were aligned with the entire KcsA sequence (GenPept 2127577) using CLUSTALW version 1.26b (Higgins et al., 1992; BLOSUM matrix series, gap open = 10, gap extension = 0.1). The resulting alignment yielded ungapped segments aligned with KcsA's prepore and postpore TM segments. These segments were taken to be the estimated extents of S5 and S6 in the DRK/A sequences. Each of the five mammalian probe sequences was then BLASTed against the NCBI nonredundant database, and the top 25 hits from each BLAST were retained. Redundant sequences (those that were returned from more than one BLAST) were eliminated from all but one set, so that there were no sequences common to more than one set. The resultant five sets of sequences, plus the set of five mammalian probe sequences along with KcsA, plus a set of Drosophila equivalents of the four mammalian subclasses (Shaker, GenPept 85110; Shab, GenPept 158459; Shaw, GenPept 158461; and Shal, GenPept 158457 were aligned separately by CLUSTALW. Each alignment yielded ungapped S5 and S6 TM segments in all sequences. The putative TM segment endpoints were determined in each of the seven alignments. All sequences from the seven sets of alignments consisting of the putative TM segments (25 residues each) plus 10 extra residues on each side were merged using CLUSTALW in profile alignment mode one at a time, starting with the mammalian probe alignment. The resultant combined alignment, consisting of 94 sequences, was used to construct the profiles, by making a list of the amino acids occurring at each position in these TM alignments corresponding to the M1 and M2 segments in KcsA. This process of profile construction is shown in Fig. 2 a], using a representative set of DRK's for illustration purposes. Table 2 displays both the prepore (DRK S5/KcsA M1) and postpore (DRK S6/KcsA M2) TM profiles.
FIGURE 2.

Process of TM localization in K+ channels. (a) In this illustrative example, an alignment of 13 DRK/A prepore (S5) TM sequences is used to generate a profile, which is used to localize the prepore TM segment in a rat SK channel. In actuality, 94 DRK/A sequences were used to generate K+ channel TM profiles. The amino acid residue list at each profile position is shown below the alignment. (b) The profile is shown aligned at a particular point in the SK sequence. A binary score is generated for each position in the profile: a 1 is scored at profile position i if the aligned SK residue is a member of the profile's residue set at that position. The set of binary scores is summed and divided by profile length to yield the profile score for this given alignment. (c) The sequence is shown advanced one residue, with the scoring process repeated. The segment yielding the highest score is taken to be the candidate TM segment. This technique is used to localize the pre- and postpore TM's of each studied isoform of the eight K+ channel classes (see Table 1). Table 2 presents the full profiles generated by this method.
TABLE 2.
Profiles of the two transmembrane segments in the permeation path of DRK/A channels
| Transmembrane position | DRK/A prepore (S5) profile | DRK/A postpore (S6) profile |
|---|---|---|
| 1 | {A,F,M,Q,T,W,Y} | {A,F,G,I,L,M,P,Q,S,T,V,W} |
| 2 | {E,H,K,N,R,S} | {G,S} |
| 3 | {A,D,E,Q} | {K,M,Q,R} |
| 4 | {A,F,L,V} | {F,I,L,S,V} |
| 5 | {G,I,L,M,Q,T} | {F,I,N,T,V} |
| 6 | {A,F,L,M} | {A,G} |
| 7 | {A,I,L,M} | {A,F,G,L,S,T,V} |
| 8 | {I,L,M,T,V} | {A,I,L,M,S,T,V} |
| 9 | {I,F,L,M,V} | {A,C,S,V} |
| 10 | {F,L,M,S,V,Y} | {A,C,F,I,M,S,V} |
| 11 | {L,Y} | {I,L,M,V} |
| 12 | {A,C,F,I,L,S,T,V} | {A,C,M,S} |
| 13 | {I,L,M,N,T,V} | {G} |
| 14 | {A,F,G,V} | {I,V} |
| 15 | {I,L,M,V} | {I,L,T} |
| 16 | {A,F,I,L,M,S,V} | {A,L,S,T,V} |
| 17 | {A,F,I,L,M,V} | {F,I,L,M,V} |
| 18 | {F,G} | {A,G,V} |
| 19 | {A,S} | {F,L,M} |
| 20 | {A,P,S,T,V,Y} | {P,V} |
| 21 | {A,C,I,L,M,V} | {I,T,V} |
| 22 | {A,E,G,I,L,M,V} | {A,H,P,T} |
| 23 | {F,V,Y} | {A,I,L,P,S,V} |
| 24 | {F,L,S,T,V,Y} | {I,L} |
| 25 | {A,I,L,M,V} | {A,F,I,L,N,Q,V} |
The KcsA transmembrane segments are included in this profile. Each profile is 25 residues long, based on analysis of the TM helices in the KcsA structure.
Location of TM segments using the constructed profiles
The potassium channel isoforms chosen to be in the alignment were segregated into the eight classes as defined in Table 1, and each class was aligned using CLUSTALW. A set of two or three representative sequences from each class was chosen as those in which to locate TM segments. To locate the prepore TM segment (denoted alternatively as M1 or S5, depending on channel class) in each such sequence, a subsequence of 100 residues ending with the G[YF]G signature sequence was extracted from the sequence. This subsequence was searched for the most likely TM segment by aligning the prepore TM profile at the beginning of the subsequence, scoring the alignment with a binary score, moving the profile one residue downstream, scoring again, and continuing in this fashion for the 100 residue length of the subsequence (see Fig. 2, b and c) for an illustration of binary scoring. (The key point is that each of the 25 positions in the profile is scored with either a 0 or a 1: a 0 if the residue at that point is not one of the residues in the profile at that position, a 1 if the residue at that position is represented at the profile at that position.) The extent of the prepore TM segment was identified as the segment that when aligned with the profile's extent produces the highest score. In the case where there were two or more placements yielding the highest score, each placement was rescored using a scoring of 0 or 1/ni (instead of 0 or 1), where ni is the number of different amino acids in position i in the profile, and the highest scoring placement was used. This procedure gives a higher weight to matching positions that are more highly conserved. The same procedure was used to locate the postpore TM segment (denoted M2 or S6, depending on class), using an extracted subsequence starting at G[YF]G and continuing downstream for 100 residues.
Linker alignment
Alignment of the segment between the pre- and postpore TM segments, denoted the linker, was done using CLUSTALW in a two-step procedure, first using sequence and then profile alignment mode. Linker sequences were first extracted from all candidate sequences, and arranged in order of length. Each group, defined by its linker length, was aligned in sequence alignment mode. The group with the shortest linker (the Shaker/Kv1 subclass of DRK/A's) was aligned with the group with the next shortest linker (Shal/Kv4 subclass of DRK/A) using profile alignment mode. Successive groups were merged in this manner, in increasing order of linker length.
Shaker homology model construction
A homology model of the permeation path of Shaker (GenPept 85110) was built using the KcsA structure (PDB Accession 1bl8; side chains for R27, I60, E64, E71, and R117 added and optimized using CHARMm 23.2, Brooks et al., 1983) as the template. The alignment of Shaker and KcsA over its permeation path (residues 23–119) produced no gaps, making it an ideal candidate for homology modeling. The model building was performed in three steps: 1), construction of an unoptimized Shaker model containing the stereochemical and geometric properties of KcsA; 2), optimization of the resulting model; and 3), symmetrization of the four subunits in the model.
In the first step, MODELLER version 4 (Sali and Blundell, 1993) was used, employing KcsA as the template and the permeation pathway of Shaker as the target. Ten Shaker variant models were generated. From these models, the one with the smallest RMS difference with KcsA in the tyrosine of the signature GYG sequence (KcsA Y78) was chosen. Polar hydrogens were added to this model using CHARMm. The three K+ ions and the single water molecule in the KcsA pdb structure were also added.
In the second step, the resulting model was minimized in CHARMm using backbone harmonic constraints weighted proportionally to the inverse of the B-factors from the KcsA crystallographic analysis (constants = 500/B kcal mole−1 Å−2), omega dihedral constraints (constant = 100) with reference positions set to the existing dihedral values, and the crystallographic H2O and K+ ions held fixed. (The exception to this constraint is that any omega dihedral whose value is less than 169° will have its reference value set to 169°; and any omega dihedral whose value exceeds 191° will have a reference value of 191°. A deviation of 11° from 180° is two standard deviations from the mean omega dihedral found in a large set of protein structures.) A distance-dependent dielectric was employed. Two thousand steps of steepest descent minimization was done, followed by conjugate gradient until 0.05 kcal/mole convergence occurred.
In the third step, the subunit with the smallest RMSD between its Y78 and that of the equivalent KcsA subunit was used as the one to use for all four subunits to symmetrize the model. This subunit was superposed onto the main chain positions of the other three Shaker subunits, and the resulting model is again minimized in CHARMm using fixed constraints on the H2O and K+ and harmonic constraints with a constant of 100 kcal mole−1 Å−2 on the backbone. Five hundred steepest descent steps were done. The resulting structure is the Shaker A permeation path model.
RESULTS AND DISCUSSION
Evaluation of alignment and topology
Placement and composition of transmembrane segments
Spot checking of transmembrane extent using Kyte-Doolittle hydropathy plots and annotations of transmembrane location in SWISSPROT showed agreement in transmembrane location with that found by the TM localization profile method within a few amino acids. Within each class, the transmembrane locations found by the profiling algorithm were aligned. Transmembrane locations for the remaining candidate sequences were taken from these alignments. For the 156 sequences in the alignment, an analysis was done of aromatic and charged residue distribution within and around the putative transmembrane segments, as shown in Fig. 3, a and b. It is seen that charged residues plus tryptophans and tyrosines tend to be situated near the ends of the transmembrane segments, corresponding to the membrane region where the phospholipid head groups and the water interpenetrate each other. These tendencies are common to all membrane proteins (Reithmeier, 1995), and the details of the interactions leading to this tendency are being studied in model peptide systems (de Planque et al., 1999). Fig. 3 c shows the distribution of glycine and alanine residues in the S6/M2 inner helix. The figure shows the universality of the glycine hinge mechanism suggested by Jiang et al. (2002a,b) for K channel opening. A majority of the sequences have a glycine in the middle of the inner helix and the others have a glycine within a few residues of the middle. We also note a frequent occurrence of alanine at position 18, which has been pointed out to be structurally significant (Jiang et al., 2002b). In the MthK open channel structure, the residue at position 18 projects its side chain into the channel lumen at the narrowest point. The small side chain of the alanine maximizes the open cross section of the channel.
FIGURE 3.


Relative frequency of aromatic and charged amino acid occurrence along the (a) prepore and (b) postpore segments of the 156 sequences in the alignment, plus seven residues on each side of them. Proportions of W, Y, or H (dark gray), D or E (medium gray), and R or K (light gray) at each position were computed for the eight channel classes, and then averaged, to prevent bias due to overrepresentation of any single class. Phenylalanine was not included in the aromatic tabulation because it often occurs in the middle of transmembrane helices, and thus is not thought to be a particular marker for the end region of such helices. Most charged and non-Phe aromatic residues appear at the edges of and beyond the TM segments derived in this analysis, with the notable exception of a conserved W in Kir's near the middle of the prepore TM segment. (c) shows the distribution of glycine and alanine in the S6/M2 inner (postpore) helix, revealing the widespread conservation of glycine and alanine at positions 13 and 18 respectively, and a highly conserved glycine at position 2.
It should be noted that all of our alignments were done before we became aware of the MthK channel structure, and we did no adjustments of the alignments afterwards. Therefore the alignments anticipated the results of Jiang et al. (2002b) in identifying the conserved glycine in the center of the inner helix. Another feature of Fig. 3 c is a glycine at position 2 in the inner helix, almost as strongly conserved as the glycine at position 13. The flexibility provided by a glycine at position 2 might have the role of reducing stress on the P region when the inner helix is pulled open to gate the channel, preserving the structural integrity of the extracellular vestibule and the selectivity filter.
Features of the alignment
Fig. 4 shows an alignment of 116 of the entire 156 channel sequences chosen for alignment. The 40 most redundant sequences have been removed. Three general observations can be made on the alignment as a whole (see Fig. 4). First, linker lengths are similar within each class of K+-selective channels, but vary significantly across the classes. Second, the prepore TM segment, linker, and postpore TM segments show strong conservation within each class of channels. Arguably this high conservation within each class is tied to its specific functional properties. Third, all gaps that emerged as the alignment was built by increasing linker length reside in two KcsA-defined structural regions: the turret and the post-G[YF]G extended region (Fig. 4). The selectivity filter and the pore α-helix regions were naturally aligned with no gaps in this process. This suggests that the pore helix/selectivity filter structural motif may be conserved throughout the family of K+-selective channels. The specific patterns of gaps in the turret region have little significance, since the turret region is so variable in composition as well as length between classes. For example, our gap pattern in comparing the turret regions of KscA and Kir is different from that shown in Lu et al. (2001), but the turret sequences in KcsA and Kir are so different from each other that the comparison is of little value. On the other hand, our alignments for the pore helix and selectivity filter agree.
FIGURE 4.
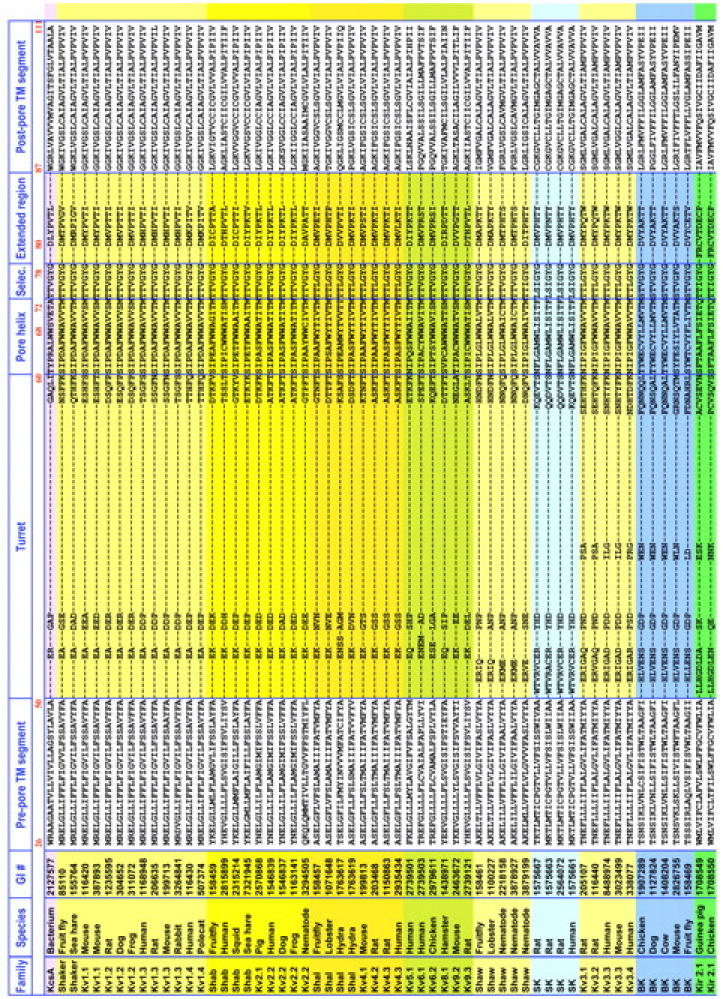
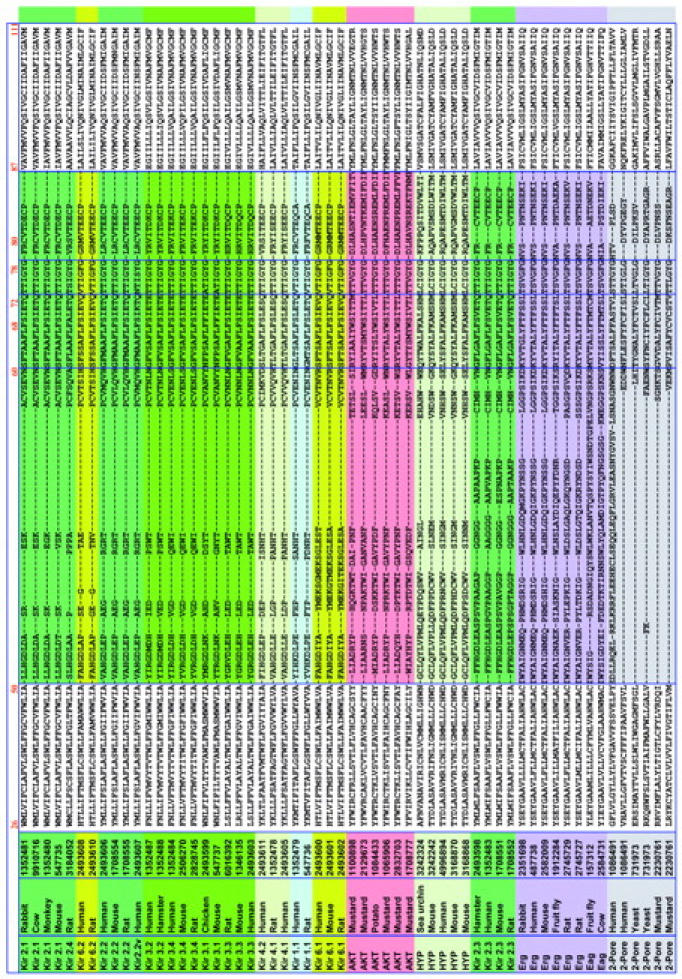

Alignment of the permeation paths of a comprehensive set of channels from the eight major K+-selective channel classes. Displayed are the 116 unique sequences collapsed from the 156 sequences chosen for the study. From top to bottom are delayed rectifier and transiently activating channels (DRK/A; yellow), Ca2+-activated small conductance channels (SK; light blue), Ca2+-activated large conductance channels (BK; medium blue), inward rectifiers (Kir; green), plant-specific inward rectifiers (AKT; red), hyperpolarization-activated cyclic nucleotide-gated channels (HYP; teal), and two-subunit domain channels (Two-Pore; gray). Subclasses are highlighted in varying shades of the classes' primary color. Residue numbers are those of KcsA at the beginning of each structural motif. Pore α and selectivity filter sequences displayed no gaps through the entire alignment. Although turret and extended region alignments were aligned using CLUSTALW (see Methods), since the lengths are highly variable, no structural homology at a specific locus in these regions should be inferred from the alignment.
A particularly intensive analysis of the M1 and M2 regions, combined with substantial mutation data, has been carried out for inward rectifier channels by Minor et al. (1999). For Kir 2.1, they assign 22 residues to each TM. We assign 25 residues to each TM in each sequence, after the KcsA crystal structure. For M1, their 1–22 corresponds to our 5–26. For M2, their 1–22 corresponds to our 2–23. In comparing the specific KcsA-Kir 2.1 alignment presented by Minor et al. with our alignment in Fig. 4, we find that their inward rectifier M1 is shifted two residues to the left relative to ours, and that their inward rectifier M2 is shifted three residues to the right compared to ours. We note that our analysis does not utilize the type of class-specific data that they have generated for the inward rectifier, nor do we consider the issue of helix-helix interactions.
It is of particular interest to consider the M2/S6 inner helix alignment in Fig. 4 in the light of the glycine hinge that is postulated to be the mechanism for K channel opening based on the crystal structure of the open MthK channel (Jiang et al., 2002 a,b). For 91 out of the 116 sequences in the Fig. 4, there is a glycine in the middle (position 13th out of 25) of the putative inner helix segment. For another 21 out of the 116 sequences, there is a glycine near the middle, in positions ranging from 9 to 15. In only four sequences is the putative glycine hinge missing. They are the three Kir 4.x sequences and the second pore sequence in the mustard TWIK sequence. The first pore sequence from the mustard TWIK contains the glycine.
The second distinctive inner helix residue that Jiang et al. (2002a,b) postulated as significant for the open pore structure is an alanine five residues in the C-terminal direction from the glycine hinge. They found that in the MthK open channel structure, that alanine was at the narrowest part of the intracellular end of the channel, and postulated that it was selected for the small size of the side chain, leaving the intracellular vestibule minimally occluded for ion entry. In the alignments of Fig. 4, we find an alanine five residues from the hinge glycine in 88 of the 112 sequences that have the glycine. The alanine is missing in all four examples of Kir 2.2 (serine instead of alanine), all four examples of Kir 2.3 (serine instead of alanine), all five plant AKT's (leucine instead of alanine), and TWIK's (various residues). There are also a few other isolated cases where the alanine is missing, such as in KcsA (where it is replaced by a glycine), one (out of five) of the Ih channels, where it is replaced by an asparagines, and in mouse Kv9.2, where it is replaced by a valine. It should be noted that some of the inward rectifiers (for example all of the Kir channels 2.1, 3.1, 3.2, 3.3, and 3.4) do have both the glycine hinge and the following alanine.
Based on the cross section of sequences analyzed in this study, which were chosen to be as representative as possible of all the types of K channels, it appears likely that the glycine hinge mechanism is almost universal for opening K channels, and that the strategy of following with an alanine to maximize access is general, albeit not as universal as the glycine hinge.
The second position where a glycine is almost completely conserved is at position 2 in the inner helix. The subfamilies where this residue is consistently different from glycine include the Kir 1.1, 2.x, 4.x, and 6.x, where the 2 residue is alanine, the Ih and the Erg channels, where the 2 residue is a serine, and the TWIK's, where the 2 residue is variable.
A possible role for the glycine at position 2 would be to provide additional flexibility for the inner helix to change its orientation during gating without transmitting conformational stresses to the P region, and thereby ensuring that the vestibule and selectivity filter conformations remain relatively rigid and unchanged during channel opening. Alanine, the second most common residue at position 2, has the second smallest side chain after glycine, and could display a similar effect. Consistent with this hypothesis, we note that for Erg channels, which have a serine at this position, there is evidence that structure of the extracellular vestibule changes when the channel opens (Pardo-Lopez et al., 2002), whereas for Shaker channels that have a glycine in this position the vestibule shape appears to remain unchanged when the channel opens (Terlau et al., 1999). Liu and Siegelbaum (2000) provided evidence for gating-induced change of extracellular vestibule conformation in a bovine rod CNG channel, which is closely related to Ih but has a different selectivity filter. Alignment of that sequence with the Ih's in our alignment of Fig. 4 revealed that where the Ih's had a serine in the 2 position in the inner helix, the bovine rod CNG channel had an aspartic acid. Like the Ih's, it did not have either a glycine or an alanine in the few positions nearest the extracellular end of the inner helix.
We note also that the glycine in position 2 in the KcsA structure inner helix is a point of helix-helix contact, in particular with the alanine at position 22 in the outer helix. It is reasonable to postulate that the nature of the helix-helix contact at this point could be significant for modulating the gating mechanism as the channel opens. It can be seen from the profile for the outer helix in Table 2 that the composition of position 22 in the outer (prepore) helix is extremely permissive; i.e., many different residues are found in that position in the sequence alignment. An inspection of the full alignment confirms that, as even other residues are seen in that position than those in the profile, which is derived from a representative sampling of the various subfamilies of K channels. There are some patterns among the subfamilies; for example Shaker channels generally have a valine at that position. It is possible that the variable composition of residues at this position in the outer helix is correlated with variations in gating properties among different K channels; this will be a subject of future investigation.
Identification of selectivity motifs from the comprehensive alignment
Identification of spatial clusters significant for function
Structural analysis of the KcsA structure in the α-helix/selectivity region has revealed two residue clusters, of three residues each, with Y78 (the Y in GYG) as a primary element in both.
One cluster is characterized by hydrophobic interactions. In KcsA, the Y78 side chain points away from the pore, allowing its backbone carbonyl oxygen to protrude into it. Two hydrophobic interactions of the Y78 phenyl group stabilize this twisting of the backbone: with V76 in the selectivity filter (entire sidechain) and with Y82 (Cβ only). Both interactions are between neighboring subunits. The position homologous to V76 contains only V, I, or L (with the single exception of a T at this position in AKT's), lending support that this hydrophobic interaction is important. The position homologous to Y82 is not conserved. In the KcSA structure, the β-carbon from Y82 fits into the aromatic ring in Y78. This interaction can be satisfied by any residue with a β-carbon, which interacts with the ring of Y78, or the F that is in the homologous position in some K+ channels.
A second cluster is defined by hydrogen bonding patterns in KcsA, and is illustrated in Fig. 5. Two hydrogen bonds are formed between Y78 and residues of the pore α-helix: between Y78 Oη and W68 Nɛ1's hydrogen, and between Y78 Hη and T72 Oγ. Again, both interactions are between neighboring subunits. The first was mentioned as a stabilizing interaction in Doyle et al. (1998), and considered as a building component of the cuff-like sheet forming the filter. In channels with a Y at the position homologous to Y78, there is a residue whose side chain can serve as a hydrogen bond donor or acceptor in at least one of the two positions corresponding to 68 and 72. In all cases except that of the weakly selective HYP channels (see discussion below), there is invariably an S or T at 72 if there is a Y at 78, suggesting that a hydrogen bond formed between the Oγ of 72 and Hη of Y78 is more crucial for selectivity than that formed by an Nɛ1 of W68 and Oη of Y78.
FIGURE 5.

Hydrogen bonding pattern of Y78 and its neighbors from the KcsA structure, and the associated residue clusters from a representative sample of the K+-selective channels in the comprehensive alignment. In KcsA, there are no other H, O, or N within 4.25 Å of Oη in Y78 except for the Nɛ1 of W68 and Oγ of T72. Y78 presumably needs hydrogen bond donors and acceptors to stabilize the buried oxygen of its phenyl group of Y78. In all isoforms with Y at the homologous position, there is either a T or S at position 72 or a W at position 68, supplying the hydrogen bond partners. In isoforms with an F at this position, W68 is replaced by F. Assuming that the structural motif for these isoforms is similar to that of KcsA, this substitution implies that the stabilizing interaction in these cases is hydrophobic, rather than via hydrogen bonding.
In contrast, in channels with an F78 at this position, there tend to be hydrophobic residues at both of these positions (refer to table in Fig. 5), suggesting that the stabilizing interaction in this case is hydrophobic rather than hydrogen bonding. The single exception is in EAG channels, where position 72 is a serine and 78 is a phenylalanine. However, position 68 is also an F in these channels, and in general, with an F at 78, there are never polar residues at both 68 and 72. The presence of these residue clusters in KcsA and the sequence correlation suggests that the clusters are preserved in a large subset of K+-selective channels. Further evidence comes from an independent calculation that is more purely sequence based. We made a profile for selectivity and pore-helix region of a set of 94 potassium channels from the classes DRK/A, EAG, BK, SK, and AKT that are known to be very highly selective and ran that profile across the sequences of fifteen hyperpolarization-activated GYG channels (HYP/Ih) that are only weakly selective for potassium over sodium (Gauss et al., 1998; Ludwig et al., 1998; Kaupp and Seifert, 2001). The logic of this calculation is that each position in the weakly selective channel sequence that does not fit the profile is a candidate for a residue critically involved in establishing a very high selectivity for potassium. From this analysis it emerged that both positions 68 and 72 are candidate residues for being critical for high selectivity, since those analogous positions are K and H respectively in all 15 of the weakly selective channels, residues that are never seen in those positions in the highly selective channels. It should be noted that the 15 weakly selective channels span an enormous range of organisms, including human, rat, rabbit, mouse, sea urchin, silkmoth, and fruitfly. This range of organisms spans the protostome-deuterostome divide that occurred very early in animal evolution, during the Cambrian explosion over 500 million years ago (Erwin et al., 1997). This finding supports and complements the significance of the structural cluster analysis illustrated in Fig. 5. It suggests that a very strong selective pressure for weakly selective potassium channels that have a reversal potential a few millivolts depolarized from the resting potential, combined with a very specific sequence of residues could lead to this property.
Other sequence-function correlations
Table 3 shows that the selectivity filter region is highly conserved. Positions 75 and 76, the two directly before G[YF]G, only have three and four residues in their profile, respectively. Position 76 is in each of the two residue clusters discussed above. Position 75 is either a serine or threonine in all K+-selective isoforms except the HYP/Ih class. In the KcsA structure, the T75 side chain points into the interior cavity of the channel pore, just below the narrow part of the filter. It presumably is a binding site for monovalent cations. There appears to be a slight tendency for residues toward the amino end of the pore helix to be less conserved than those closer to the selectivity filter (Table 3). The residues toward the amino end are further from the pore axis (see Fig. 1) and primarily interact with prepore TM residues, which are not conserved highly. The positions 67 and 68 near the C-terminal end of the helix, which interact with Y78 in KcsA, show the strongest conservation.
TABLE 3.
Profile of the 13 residue pore α-helix and 6 residue selectivity filter, based on the 116 nonredundant sequences used in the alignment
| Motif | KcsA Position | Profile | MthK Sequence |
|---|---|---|---|
| Pore α-helix | 61 | {S,G,N,T,K,R,Q,L;v,d,m} | S |
| 62 | {I,F,Y,L,V;m} | W | |
| 63 | {P,V,T,S,L,W,I,M;f,g} | T | |
| 64 | {A,S,D,T,I,G,E,Y,L,F;c,n,w,r,q} | V | |
| 65 | {A,S,G,C;t} | S | |
| 66 | {F,L,V,M,W,I,Y} | L | |
| 67 | {W,L,Y,F} | Y | |
| 68 | {F,W,L,Y,K} | W | |
| 69 | {A,S,T,L,C,I,F;v} | T | |
| 70 | {I,V,M,T,F,L,S;a,g} | F | |
| 71 | {V,E,I,S,T;c,a,k,l} | V | |
| 72 | {T,S,H,V;c} | T | |
| 73 | {M,Q,L,E,F;a,t} | I* | |
| Selectivity filter | 74 | {T,L,V,S,A;n} | A |
| 75 | {T,S,C} | T | |
| 76 | {V,I,L,T} | V | |
| 77 | {G} | G | |
| 78 | {Y,F;l} | Y | |
| 79 | {G} | G |
Amino acids at each position of the profile are ordered by frequency of occurrence; a lowercase letter indicates that the residue occurred in exactly one or two sequences only. The right-hand column shows, for comparison, the MthK sequence that was not included in the alignment but for which the high-resolution open structure was determined. The format coding for the MthK sequence is as follows: Italics signifies a location where the MthK sequence has the single most common residue in the alignment at that position (8 out of 19 positions); normal typeface signifies a location where more than two sequences out of the full alignment have the same sequence as MthK (8 out of 19 positions); Boldface signifies a position where the location where none of the sequences in the full alignment have the same residue as the MthK sequence (3 out of 19 positions); and an asterisk added to a boldface position signifies that, although the MthK is different from all the native aligned sequences in that position, the residue in question was shown by Heginbotham et al. (1994) to be consistent with maintenance of K selectivity (one of the three boldface positions).
It is of interest to discuss experimentally induced mutations in the K channel signature sequence in the context of the profile in Table 3 and the structure and alignment presented in Fig. 5. Heginbotham et al. (1994) did an extensive series of such mutations from positions 72 through 78 (our numbering scheme as in this paper) in Shaker channels. Some particular results and relationship to our analysis are described below. Heginbotham et al. found that mutation T72S retains potassium selectivity. Based on Fig. 5 we would expect that, since serine and threonine both have identically positioned hydroxyl groups for the interaction with Y78, and indeed in Table 3 we see that S is one of the residues permissible in position 72. We also find that V is one of the permissible residues in 72, which initially appears to contradict Heginbotham et al. until we note from Fig. 5 that V at 72 appears always in conjunction with F replacing Y at 78. (We have verified this for all the 156 sequences in the alignment.) Our inference is that in these cases it is likely that a hydrophobic interaction between V72 and F78 replaces the polar interaction between T72 and Y78 in KcsA and Shaker.
Heginbotham et al. found that position 73 was quite permissive, and that is suggested also by Table 3. At position 74 there is one inconsistency between our Table 3 and the Heginbotham et al. results; our Table 3 suggests that N is allowed at this position, but T74N mutant failed to be expressed in their experiments. Inspection of our alignment of 156 sequences reveals N74 in just one sequence out of the 156, one of the inward rectifiers. Because this occurrence is so singular, we are not considering its implications in detail. It is conceivable that there is a sequencing error that is causing this anomaly.
Our results for positions 75–78 are completely consistent with Heginbotham et al.; i.e., none of the substitutions that destroy expression or selectivity in their experiments appear in our Table 3 at those positions.
The fourth column in Table 3 shows, for comparison, the sequence for the MthK channel for which the structure was recently determined (Jiang et al., 2002a). This sequence was not included in our alignment and analysis, so it is of interest to see how it compares to those that were, especially in the most highly conserved region of the channel sequence. We see in Table 3 that in 8 of the 19 positions (61, 71, 72, and 75 through 79, coded by italics) MthK has the most typical residue at that position in the entire alignment. In another 8 positions (63, 65 through 70, and 74, coded by normal face) MthK has one of the typical residues but not the most typical residue. In one position (73, coded by bold face plus asterisk), MthK has a an isoleucine residue that does not appear in any of the native states in the alignment but was shown by Heginbotham et al. (1994, Fig. 5) to be a substitution that could be made in Shaker that would still preserve potassium selectivity. In two other positions (62 and 64, coded by boldface), MthK has a residue that is not in any of the native channels. Position 64 is very permissive; the 116 native channels in the alignment show 15 different amino acids in that location, so our judgment is that if MthK introduces a 16th possibility, the significance is just confirmation that almost anything is permissible in that position. At position 62, the native channels typically have either aliphatic (V, I, L) or aromatic (F, Y) residues at that position. In the context, the W62 in MthK seems like a reasonably conservative substitution although it was not represented in the original alignment. In looking at the overall similarity of MthK to the other sequences, it is remarkable how well its sequence in the pore helix/selectivity filter region conforms to the conservation patterns inferred from the alignment in this paper and from prior mutation experiments by Heginbotham et al. (1994).
Among the K+-selective channel linkers, the highest degree of variation in length appears in the turret region (10 residues in DRK/A to 46 in EAG). Variability in the turret is likely a primary factor in differential toxin sensitivity, based on a large number of electrophysiological experiments. A three-point mutation study of this segment in the KcsA channel has verified that it plays a role in toxin sensitivity (MacKinnon et al., 1998). Turret length is much less variable within each K+ channel class than across classes, implying that it may contribute to specific function within class. However, significant turret length variations occur in two large classes of K+-selective channels, the DRK/A's and Kir's. The four major DRK/A subclasses show a length variation between 10 and 19 residues in the turret. Each subclass has distinctly different properties (Chandy and Gutman, 1995). Kir's have even larger turret variation than the DRK/A's.
In KcsA M2 forms the cytoplasmic end of the pore. Residues I100, F103, G104, T107, A111, and V115 are exposed to the pore, forming a hydrophobic interior where presumably cations and water can pass quickly without forming hydrogen bonds, allowing higher permeation (Doyle et al., 1998). The homologous residues in the set of K+-selective classes are primarily hydrophobic; notably, position 107 is hydrophobic in all classes except EAG and in KcsA, and 103 is hydrophobic in all classes except SK, Kir, and AKT, where it is polar or acidic. The other residues are mostly hydrophobic. These patterns of conservation lend support to the supposition that the intracellular end of the channel lumen is strongly nonpolar throughout the K channels, as it is in KcsA.
Evaluation of Shaker model
Two lines of evidence made the construction of a Shaker permeation model straightforward and suggest its validity. First, in alignments by various workers (MacKinnon et al., 1998; others), it was found that no gapping occurred along the entire length of the permeation path when KcsA and Shaker A was aligned. The present alignment verifies this condition (Fig. 4, top two lines). Secondly, experiments by Gross and MacKinnon (1996) made before the determination of the KcsA structure indicated that the N-terminal part of the pore loop in the Shaker channel is an α-helix, and the C-terminal an extended region. The permeation path model has these motifs.
The model (including all four monomers assembled into the tetrameric channel) was evaluated using atom pairwise distance probability density functions (PDF's; Rojnuckarin and Subramaniam, 1999) and the full range of side chain and backbone robustness criteria from PROCHECK 3.5 (Laskowski et al., 1993). Although both PDF's and Procheck criteria are derived from soluble proteins, they appear to be essentially equally applicable to membrane proteins as exemplified for example by using these criteria to score the refined KcsA channel itself, and seeing that the scores are quite good.
The PDF for each atom pair type (e.g., Leu CB-Ile CA) is constructed from all such distances in a set of 461 disparately chosen x-ray protein structures. Because the PDF's are atomically detailed, they include detailed information on side chain interactions, including implicit information on side chain as well as backbone conformations. For each atom pair in the subject protein, a p-value is computed from its PDF. All atom pair p-values in the subject protein are combined into a composite PDF score. Residue PDF scores are computed from a subset of those pairs belonging singly or jointly to a particular residue. In addition, particular atom pair distances with a PDF score below a specified value are listed as improbable distances. The set of improbable distances fell well within the expected number of a protein structure of similar size and resolution in the Protein Data Bank. PROCHECK analysis showed that the steric health of the structure within normal limits. In ongoing and future work we will do detailed electrostatic analysis and multiscale simulation of current flow through Shaker and other K channel structures homology-modeled by the same process (Mashl et al., 2001).
The presence of a PVP segment in the Shaker S6 has led some researchers (Durell et al., 1998) to postulate the existence of a kink at this position in the helix, ∼3.5 turns from the S6 C-terminal end. The KcsA crystal structure shows no kink at this position. MODELLER, using this structure as the template, did not produce a kink at that location in the Shaker model. Evaluation of the Φ/Ψ angles of the two prolines shows that they are −39°/−60° and −36°/−44°, respectively, which are in an admissible region of observed proline residues, according to PROCHECK 3.5 analysis. Whether or not there is actually a kink in the Shaker S6 at the proline site, it does not seem essential for voltage sensitivity, since a KcsA permeation pathway-Shaker voltage sensor chimeric construct gates in a similar voltage-sensitive fashion to a native Shaker channel. (Lu et al., 2001).
CONCLUSIONS
The availability of a three-dimensional model structure for a K+ channel and sequences of hundreds of voltage-gated K+-selective channel proteins enables a comprehensive sequence-structure-function alignment that will provide valuable information for mechanistic and functional studies of K+ channels. The comprehensive alignment was constructed to allow examination of sequence patterns within K+-selective channel classes and correlate them with the residue clusters in KcsA, establishing structural conservation in the areas responsible for selectivity and permeation. The diverse families of K+-selective channels show significant sequence similarity in the pore region, yet maintain their distinctive class structure through select sequence conservation. This conservation lends itself to sequence profiling that in turn allows classification and alignment of novel potassium channel sequences. Further, a robust and accurate alignment is the first step toward building a three-dimensional structural homology model. There is a vast amount of structure-function data on potassium channel mutants. A structural model can thus be validated against these data and can provide further hypothesis for experimental testing through mutagenesis. Most importantly, a comprehensive alignment provides a wealth of information on ion selectivity and nature of the ion pore in terms of its function.
One significant insight to come from this initial combined sequence-structure analysis is a testable hypothesis for the basis of weak versus strong selectivity in the potassium channel homologs. There are many specific differences in sequence between the weakly selective potassium channel homologs (HYP/Ih) and the strongly selective potassium channels, so that sequence analysis alone does not provide strong evidence for the basis of the selectivity difference. The structural analysis, showing that the positions corresponding to KcsA 68 and 72 are likely to stabilize the selectivity filter by strongly interacting with the aromatic residue in the G(Y,F)G triad, directs attention to those residues for comparison. Focusing on those, we find without exception that members of the weakly selective family have Y in the central position of the triad and K and H respectively at the 68 and 72 positions. The strongly selective channels never have either K at 68 or H at 72. We hypothesize that this combination of residues is the basis for the particular selectivity properties of this class of channels. It would be of interest for experimentalists to mutate residues in the 68 and 72 positions specifically to observe the effect on the degree of K/Na selectivity.
We note that this is in a context of a particular physiological role for the weakly selective channels. In addition to being weakly selective, they are activated by hyperpolarization and the gating is modulated by cyclic nucleotides. This array of properties provides the basis for enabling cyclic nucleotides to modulate rhythmic behavior of cells. The usefulness of this capability is attested to by the extraordinary evolutionary persistence of this class of channels (Kaupp and Seifert, 2001).
The comprehensive alignment confirms the generality of the glycine hinge mechanism for opening K channels as postulated by Jiang et al. (2002a,b) and their related hypothesis that there is usually an alanine at the inner helix position marking the narrowest point of the intracellular region of the pore. The alignment also shows a second highly conserved glycine near the extracellular end of the inner helix, which could have the effect of providing flexibility for isolating the extracellular vestibule structure from stresses caused by the channel opening. In support of this hypothesis, we note that for channels lacking the glycine near the extracellular end of the inner helix, there is evidence that the extracellular vestibule changes conformation when the channel opens (Liu and Siegelbaum, 2000; Pardo-Lopez et al., 2002). On the other hand for Shaker channels, which have the glycine near the extracellular end, the extracellular vestibule does not seem to change shape when the channel opens (Terlau et al., 1999).
Acknowledgments
We acknowledge funding from National Science Foundation grants NSF DBI-0003231, NSF MCB-9873384, and NSF MCB-9873199 and computational resources at the National Center for Supercomputing Applications and the San Diego Supercomputer Center.
Robin T. Shealy's present address is Dept. of Crop Sciences, University of Illinois at Urbana-Champaign, Urbana, IL 61801.
References
- Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann, C. I. 1998. Neurobiology of the Caenorhabditis elegans genome. Science. 282:2028–2033. [DOI] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Chandy, K. G., and G. A. Gutman. 1995. Voltage-gated K+ channels. In Ligand- and Voltage-Gated Ion Channels. R. A. North, editor. CRC Press, Boca Raton, FL. 1–71.
- de Planque, M. R. R., J. A. W. Kruijtzer, R. M. J. Liskamp, D. Marsh, D. V. Greathouse, R. E. Koeppe II, B. de Kruijff, and J. A. Killian. 1999. Different membrane anchoring positions of tryptophan and lysine in synthetic transmembrane alpha-helical peptides. J. Biol. Chem. 274:20839–20846. [DOI] [PubMed] [Google Scholar]
- Doyle, D., J. C. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Durell, S. R., Y. Hao, and H. R. Guy. 1998. Structural models of the transmembrane region of voltage-gated and other K+ channels in open, closed, and inactivated conformations. J. Struct. Biol. 121:263–284. [DOI] [PubMed] [Google Scholar]
- Erwin, D., J. Valentine, and D. Jablonski. 1997. The origin of animal body plans. Am. Sci. 85:126–137. [Google Scholar]
- Gauss, R., R. Seifert, and U. B. Kaupp. 1998. Molecular identification of a hyperpolarization-activated channel in sea urchin sperm. Nature. 393:583–587. [DOI] [PubMed] [Google Scholar]
- Grissmer, S., A. Nguyen, A. Jayashree, D. Hanson, R. Mather, G. Gutman, M. Karmilowicz, D. Auperin, and K. G. Chandy. 1994. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 45:1227–1234. [PubMed] [Google Scholar]
- Gross, A., and R. MacKinnon. 1996. Agitoxin footprinting the Shaker potassium channel pore. Neuron. 16:399–406. [DOI] [PubMed] [Google Scholar]
- Guy, H. R., and S. R. Durell. 1995. In Ion Channels and Genetic Diseases. D. C. Dawson, editor. Rockefeller University Press, New York. 1–16.
- Hartmann, H. A., G. E. Kirsch, J. A. Drewe, M. Taglialatela, R. H. Joho, and A. M. Brown. 1991. Exchange of conduction pathways between two related K+ channels. Science. 251:942–944. [DOI] [PubMed] [Google Scholar]
- Heginbotham, L., Z. Lu, T. Abramson, and R. MacKinnon. 1994. Mutations in the K+ channel signature sequence. Biophys. J. 66:1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins, D. G., A. J. Bleasby, and R. Fuchs. 1992. CLUSTAL V: improved software for multiple sequence alignment. Comput. Appl. Biosci. 8:189–191. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1992. Ionic Channels of Excitable Membranes, 2nd ed. Sinauer and Associates, Sunderland, MA.
- Jan, L. Y., and Y. N. Jan. 1997. Cloned potassium channels from eukaryotes and prokaryotes. Annu. Rev. Neurosci. 20:91–123. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002a. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B. Chait, and R. MacKinnon. 2002b. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- Kaupp, U. B., and R. Seifert. 2001. Molecular diversity of pacemaker ion channels. Annu. Rev. Physiol. 63:235–257. [DOI] [PubMed] [Google Scholar]
- Kavanaugh, M. P., M. D. Varnum, P. B. Osborne, M. J. Christie, A. E. Busch, J. P. Adelman, and R. A. North. 1991. Interaction between tetraethylammonium and amino acid residues in the pore of cloned voltage-dependent potassium channels. J. Biol. Chem. 266:7583–7587. [PubMed] [Google Scholar]
- Lancaster, B., R. A. Nicoll, and D. J. Perkel. 1991. Calcium activates two types of potassium channels in rat hippocampal neurons in culture. J. Neurosci. 11:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291. [Google Scholar]
- Liu, J., and S. A. Siegelbaum. 2000. Change of pore helix conformational state upon opening of cyclic nucleotide-gated channels. Neuron. 28:899–909. [DOI] [PubMed] [Google Scholar]
- Lu, Z., A. M. Klem, and Y. Ramu. 2001. Ion conduction pore is conserved among potassium channels. Nature. 413:809–813. [DOI] [PubMed] [Google Scholar]
- Ludwig, A., X. Zong, M. Jeglitsch, F. Hofmann, and M. Biel. 1998. A family of hyperpolarization-activated mammalian cation channels. Nature. 393:587–591. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R. 1995. Pore loops: an emerging theme in ion channel structure. Neuron. 14:889–892. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., S. L. Cohen, A. Kuo, A. Lee, and B. T. Chait. 1998. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 280:106–108. [DOI] [PubMed] [Google Scholar]
- Mashl, R. J., Y. Tang, J. Schnitzer, and E. Jakobsson. 2001. Hierarchical approach to predicting permeation in ion channels. Biophys. J. 81:2473–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor, D. L., S. J. Masseling, Y. N. Jan, and L. Y. Jan. 1999. Transmembrane structure of an inwardly rectifying potassium channel. Cell. 96:879–891. [DOI] [PubMed] [Google Scholar]
- Pardo-Lopez, L., M. Zhang, J. Liu, M. Jiang, L. D. Possani, and G. N. Tseng. 2002. Mapping the binding site of a human ether-a-go-go-related gene-specific peptide toxin (ErgTx) to the channel's outer vestibule. J. Biol. Chem. 19:16403–16411. [DOI] [PubMed] [Google Scholar]
- Ramarathnam, R., and S. Subramaniam. 2000. A novel microarray strategy for detecting genes and pathways in microbes with unsequenced genomes. Microb. Comp. Genomics. 5:153–161. [DOI] [PubMed] [Google Scholar]
- Reithmeier, R. A. 1995. Characterization and modeling of membrane proteins using sequence analysis. Curr. Opin. Struct. Biol. 5:491–500. [DOI] [PubMed] [Google Scholar]
- Rojnuckarin, A., and S. Subramaniam. 1999. Knowledge-based interaction potentials for proteins. Proteins. 36:54–67. [DOI] [PubMed] [Google Scholar]
- Sali, A., and T. Blundell. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815. [DOI] [PubMed] [Google Scholar]
- Schrempf, H., O. Schmidt, R. Kummerlen, S. Hinnah, D. Muller, M. Betzler, T. Steinkamp, and R. Wagner. 1995. A prokaryotic potassium ion channel with two predicted transmembrane segments from Streptomyces lividans. EMBO J. 14:5170–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong, M., K. G. Chandy, and G. A. Gutman. 1993. Molecular evolution of voltage-sensitive ion channel genes: on the origins of electrical excitability. Mol. Biol. Evol. 10:221–242. [DOI] [PubMed] [Google Scholar]
- Subramaniam, S. 1998. The biology workbench—a seamless database and analysis environment for the biologist. Proteins. 32:1–2. [PubMed] [Google Scholar]
- Terlau, H., H. Boccaccio, B. M. Olivera, and F. Conti. 1999. The block of Shaker K+ channels by kappa-conotoxin PVIIA is state dependent. J. Gen. Physiol. 114:125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen, G., M. Jurman, T. Abramson, and R. MacKinnon. 1991. Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+ channel. Science. 251:939–941. [DOI] [PubMed] [Google Scholar]
- Yool, A. J., and T. L. Schwartz. 1991. Alteration of ionic selectivity of a K+ channel by mutation of the H5 region. Nature. 349:700–704. [DOI] [PubMed] [Google Scholar]