

Abstract
Nystatin is a membrane-active polyene antibiotic that is thought to kill fungal cells by forming ion-permeable channels. In this report we have investigated nystatin interaction with phosphatidylcholine liposomes of different sizes (large and small unilamellar vesicles) by time-resolved fluorescence measurements. Our data show that the fluorescence emission decay kinetics of the antibiotic interacting with gel-phase 1,2-dipalmitoyl-sn-glycero-3-phosphocholine vesicles is controlled by the mean number of membrane-bound antibiotic molecules per liposome, 〈A〉. The transition from a monomeric to an oligomeric state of the antibiotic, which is associated with a sharp increase in nystatin mean fluorescence lifetime from ∼7–10 to 35 ns, begins to occur at a critical concentration of 10 nystatin molecules per lipid vesicle. To gain further information about the transverse location (degree of penetration) of the membrane-bound antibiotic molecules, the spin-labeled fatty acids (5- and 16-doxyl stearic acids) were used in depth-dependent fluorescence quenching experiments. The results obtained show that monomeric nystatin is anchored at the phospholipid/water interface and suggest that nystatin oligomerization is accompanied by its insertion into the membrane. Globally, the experimental data was quantitatively described by a cooperative partition model which assumes that monomeric nystatin molecules partition into the lipid bilayer surface and reversibly assemble into aggregates of 6 ± 2 antibiotic molecules.
INTRODUCTION
Amphotericin B (AmB) and nystatin are two structurally similar polyene macrolide antibiotics characterized by a very low antibacterial activity and a potent broad-spectrum antifungal action (Bolard, 1986). After more than 40 years after its discovery, AmB still is the most common antifungal agent used to treat systemic fungal infections (Hartsel and Bolard, 1996). However, several side effects, especially nephrotoxicity, have restricted its use and promoted the development of various liposomal formulations to minimize these problems (Schaffner, 1984). Nystatin, on the other hand, is only used topically, as it is ineffective orally and severely toxic when administered intravenously (Schaffner, 1984). To be able to develop rationale strategies to circumvent the severe secondary effects caused by these antibiotics, it is essential to obtain a detailed picture of their mode of action at the molecular level.
The most widely accepted model for the mechanism of action of these antibiotic molecules considers that their cellular target is the plasma membrane of the antibiotic-sensitive organisms where they act by forming aqueous pores (Bolard, 1986; Hartsel et al., 1993). Several electrophysiological studies have shown that bilayer-spanning potassium-selective channels are formed upon one-sided addition of nystatin or AmB to different lipid membranes (Akaike and Harata, 1994). These pores have size-discriminating properties, being permeable only to solutes not larger than glucose or sucrose for nystatin and AmB, respectively (Andreoli et al., 1969; Holz and Finkelstein, 1970). The disturbance of the cellular electrochemical gradients caused by the increase of cell permeability to ions and small molecules ultimately leads to cell lysis and death (Bolard, 1986). Despite the general consensus about the antibiotic mode of action, there is still some controversy regarding the molecular architecture of the ion-permeable channels formed by these membrane-active compounds. In particular, the role and even the indispensability of sterols in the antibiotic self-assembly process that occurs within the lipid bilayers are still matters of debate (Hartsel et al., 1993). Some authors explain the sterol requirement frequently found in nystatin and AmB activity measurements by invoking the formation of transmembrane pores constituted by antibiotic-sterol complexes composed of alternated antibiotic and sterol molecules (Andreoli, 1974; de Kruijff and Demel, 1974; van Hoogevest and de Kruijff, 1978). The distinct antibiotic sensitivities presented by cholesterol-containing mammalian cells relative to ergosterol-rich fungal cells are therefore justified by the different affinities of the polyene antibiotics toward the sterol present in the plasma membranes of the antibiotic-sensitive organism (Bolard, 1986). On the other hand, other studies suggested that the key factor controlling the ability of large polyene antibiotic molecules to pack together is the overall membrane organization. In this way, the formation of active antibiotic species may be facilitated by the presence of an ordered state in the lipid bilayer. This can be achieved either by the progressive incorporation of a sterol in the membrane (Marty and Finkelstein, 1975) or by keeping the lipid bilayers in a gel phase (Hsu-Chen and Feingold, 1973).
The presence of a conjugated double-bond system in the chemical structure of the polyene antibiotics has enabled the application of several optical spectroscopic techniques to the study of the molecular structure of the channels formed by these molecules in the lipid bilayers. The main spectroscopies applied so far to AmB (with a heptanes group) were ultraviolet-visible absorption and circular dichroism (CD) (Bolard, 1986; Hartsel et al., 1993). The great potentialities of these techniques derive from their ability to detect the formation of several different antibiotic species in the membranes, which are identified by a characteristic signature spectrum. In addition, it is possible to evaluate the influence that several parameters have on their occurrence, e.g., the membrane-lipid composition (type of sterol and mole fraction) and antibiotic-to-lipid ratios used (Bolard et al., 1980; Vertut-Croquin et al., 1983). However, until recently a quantitative analysis of the data obtained in most of these studies was impaired by the complex equilibria present in the samples that involved antibiotic aggregation in the aqueous solution (Mazerski et al., 1982, 1990), partition into the lipid bilayers (Szponarski et al., 1988) and self-association in the lipid environment (Balakrishnan and Easwaran, 1993). Fujii et al. (1997) were the first to overcome this problem by guaranteeing a complete association of AmB with the liposomes used in all experimental conditions tested. From a combination of functional assays with spectroscopic measurements, these authors were able to show unequivocally that a transition from the ion-impermeable to the ion-permeable state occurred concomitantly with significant spectroscopic changes, indicating that two distinct forms of AmB were present in the membranes, namely a monomeric and an aggregated form (Fujii et al., 1997).
Regarding the application of optical techniques to the study of polyene antibiotics mode of action, much less attention has been paid to fluorescence so far. However, from the anticipated sensitivity of several fluorescence parameters to the properties of the fluorophore microenvironment, this technique is also expected to be able to report changes of antibiotic location/aggregation state in the lipid bilayer that are associated with the formation of active species (Strom et al., 1976; Castanho and Prieto, 1992, Coutinho and Prieto, 1995). In a previous work, we began to exploit the intrinsic fluorescence properties of the tetrane-containing antibiotic nystatin (Coutinho and Prieto, 1995). We carried out a general characterization of the photophysical behavior of nystatin both in homogeneous media and in interaction with phospholipid vesicles and established a comparison with the photophysical properties of the well-studied tetrane fluorescent fatty-acid trans-parinaric acid. Before addressing more complex, and yet more biologically relevant lipid mixtures—namely ergosterol- and cholesterol-containing lipid bilayers—we have decided to extend the previous investigation of nystatin interaction with single component liposomes to studies with small unilamellar vesicle (SUV) and large unilamellar vesicle (LUV) prepared with DPPC and 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC). To evaluate the influence exerted by the lateral packing of phospholipids on nystatin interaction with the lipid vesicles, several experiments were carried out with the lipid vesicles kept in a gel phase. The creation of a lipid-ordered state by adding sterol to a phospholipid at temperatures above the phase transition would be a better in vitro model of a biomembrane in a liquid-ordered phase. However, we envisaged that the use of a gel phase would eventually allow us to distinguish a general influence (the presence of lipid ordered phase-gel phase, in this case) from a possible specific effect (formation of specific antibiotic-cholesterol complexes) on nystatin interaction with the lipid vesicles.
By measuring the fluorescence decay curves of the lipid-bound antibiotic molecules, nystatin was shown to form strongly fluorescent antibiotic aggregates in gel-phase membranes that were dependent upon the antibiotic and lipid concentrations used. These results were ascribed to a self-association process undergone by the monomeric antibiotic molecules within the lipid bilayer. Since it is believed that the assembly of the large polyene antibiotics into conducting pores plays a key role in their biological activity (Bolard, 1986), we decided to further characterize the factors controlling nystatin oligomerization in the lipid vesicles. By carrying out experiments with DPPC liposomes with different outer diameters (SUV and LUV), we were able to show that the emission decay kinetics of the antibiotic was governed by the mean number of membrane-bound nystatin molecules per lipid vesicle and not strictly by its surface density.
MATERIALS AND METHODS
Reagents
Nystatin (pharmaceutical grade) was a gift from Squibb Farmacêutica Portuguesa and was used without further purification. DPPC and POPC were purchased from Avanti Polar Lipids (Alabaster, AL). The spin labels 5- and 16-doxyl-stearic acids (5-DS and 16-DS, respectively) were obtained from Molecular Probes (Eugene, OR). All other chemicals were of analytical or spectroscopic reagent grade.
The antibiotic was stored in the dark at −20°C. A stock solution of nystatin in methanol (spectroscopic grade) (∼1 mM) was also stored at −20°C in the dark before being used. Its concentration was determined by UV spectroscopy using an absorption coefficient of 7.4 × 104 M−1 cm−1 at 304 nm (Coutinho and Prieto, 1995). The stock solutions of n-DS were also prepared in methanol (8 and 6 mM for 5-DS and 16-DS, respectively) and kept at −20°C.
Vesicle preparation
SUV were prepared by ultrasonic irradiation using a Branson 250 sonicator with a standard flat tip as described elsewhere (Coutinho and Prieto, 1995). The standard buffer used was HEPES buffer (20 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, 1 mM EDTA) but some lipid suspensions were also prepared using a Tris buffer (50 mM Tris-HCl (pH 7.4), 10 mM NaCl, 0.2 mM EDTA). By means of the extrusion technique (Mayer et al., 1986) we prepared LUV with a mean diameter of 100 nm. Briefly, the lipid was dissolved in chloroform in a round-bottom flask and the solvent was first dried under a stream of N2 and then further evaporated overnight under an oil pump vacuum. The dried lipid was dispersed to the desired concentration by adding the appropriate volume of the standard buffer and by repeated vortexing at 50°C, well above the gel-to-liquid crystalline phase transition temperature (Tm) of the lipids used, until all lipid was removed from the flask wall. Then a freeze-thaw cycle was repeated eight times. Subsequently, the lipid suspension was extruded four and 10 times through polycarbonate membranes of 400- and 100-nm pore size (Nucleopore, Pleasanton, CA), respectively. The resulting stock solution was stored at 4°C. The final lipid concentration was determined by phosphate analysis (McClare, 1971).
Nystatin partitioning experiments
The partitioning of nystatin into the lipid bilayer was determined through the fluorescence intensity increase of the antibiotic's tetrane group with the lipid concentration measured in 0.5-cm cuvettes at 21 or 45°C. Due to the high susceptibility of nystatin to photochemical degradation, several dilutions of a stock solution of SUV or LUV (∼5 mM) were prepared with buffer. Then, a constant volume from nystatin stock solution was injected to each of these samples. The final volume of methanol in solution never exceeded 1.5% of the total volume of each sample. After an incubation time of 1 h in the dark and at room temperature, the fluorescence signal of each sample was monitored as a function of the lipid concentration used. The contribution of the liposomes alone was subtracted from this intensity.
The partitioning experiments were analyzed according to Eq. 1 (White et al., 1998):
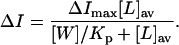 |
(1) |
In this equation, 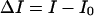 stands for the difference between the steady-state fluorescence intensity of the antibiotic measured in the presence (I) and in the absence of phospholipid vesicles (I0);
stands for the difference between the steady-state fluorescence intensity of the antibiotic measured in the presence (I) and in the absence of phospholipid vesicles (I0); 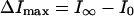 is the maximum value of this difference, since I∞ is the limiting value of I measured upon increasing the lipid concentration, [L], of the solution; Kp is the mole-fraction partition coefficient of the antibiotic between the aqueous and lipid phases and [W] is the molar concentration of water. It was assumed that only 60% and 50% of the overall lipid used was available, [L]av, for the initial partition of the antibiotic in the experiments carried out with SUV and LUV, respectively.
is the maximum value of this difference, since I∞ is the limiting value of I measured upon increasing the lipid concentration, [L], of the solution; Kp is the mole-fraction partition coefficient of the antibiotic between the aqueous and lipid phases and [W] is the molar concentration of water. It was assumed that only 60% and 50% of the overall lipid used was available, [L]av, for the initial partition of the antibiotic in the experiments carried out with SUV and LUV, respectively.
Quenching studies
The experiments were carried out with 3 mM DPPC SUV incubated with either 3.0 or 12.5 μM nystatin for 1 h at room temperature. The antibiotic-labeled lipid suspensions were divided into several samples and variable aliquots from a methanolic stock solution of either spin probe (5- or 16-DS, respectively) were then added to obtain different final quencher concentrations for each sample. After another incubation time of 1 h at room temperature, the steady-state fluorescence intensity of nystatin in each sample was measured at 21°C. The volume of organic solvent did not surpass 3.0% of each sample total volume. On the other hand, the highest mole fraction of spin-labeled fatty acids used never exceeded 4–6% of the phospholipid present in the outer monolayer of the liposomes.
Quenching data were analyzed by the Stern-Volmer plot of I0/I versus [Q]l, where I0 and I stand for the fluorescence intensities in the absence and in the presence of the quencher, respectively, and [Q]l is the quencher (spin label) concentration in the phospholipid phase,
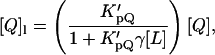 |
(2) |
where 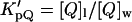 is the Nernst partition coefficient of the quencher between the phospholipid phase and the aqueous phase, [Q] is the bulk molar concentration of the quencher (relative to the total sample volume, Vt (Vt = Vl + Vw)) and Vl and Vw are the volumes of the lipid and aqueous phases, respectively. For 5-DS and 16-DS,
is the Nernst partition coefficient of the quencher between the phospholipid phase and the aqueous phase, [Q] is the bulk molar concentration of the quencher (relative to the total sample volume, Vt (Vt = Vl + Vw)) and Vl and Vw are the volumes of the lipid and aqueous phases, respectively. For 5-DS and 16-DS, 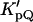 is 12,570 and 3340, respectively, for the lipid vesicles in the gel phase (Wardlaw et al., 1987) and a phospholipid molar volume, γ, of 0.688 dm3 mol−1 was used for DPPC (Marsh, 1990).
is 12,570 and 3340, respectively, for the lipid vesicles in the gel phase (Wardlaw et al., 1987) and a phospholipid molar volume, γ, of 0.688 dm3 mol−1 was used for DPPC (Marsh, 1990).
Absorption and steady-state fluorescence measurements
Absorption spectra were measured at room temperature using a Jasco V-560 spectrophotometer. Fluorescence measurements were made on a Spex F112A Fluorolog photon-counting instrument (Edison, NJ) under the control of a IBM PC equipped with the software DM3000, using a 150-W Xenon light source, as previously described (Coutinho and Prieto, 1995). The spectrofluorometer was equipped with a thermostated cuvette holder (±1°C) and 0.5 cm pathlength quartz cuvettes were used. The conditions used in most measurements were λexc = 315 nm (bandwidth 0.9 nm) and λem = 415 nm (bandwidth 9 nm). Stray light was reduced using an ultraviolet band-pass filter SB-300 (Corion) in the exciting beam and a WG-360 (Corion) cutoff filter in the emission beam. Moreover, background intensities in nystatin-free samples due to the lipid vesicles were subtracted from each recording of fluorescence intensity. All the fluorescence measurements were carried out with a right-angle geometry, and the geometry effect was taken into account when necessary (Coutinho and Prieto, 1995).
Nystatin aggregation in Tris and HEPES buffers and the detection of vesicle aggregation or fusion induced by the antibiotic were studied by measuring the light-scattered intensity by the antibiotic solutions or vesicle suspensions, respectively, at λexc = λem = 450 nm (detected at 90°).
Time-resolved fluorescence measurements
Fluorescence lifetimes were determined by the single-photon timing technique using a nitrogen-filled flashlamp (Edinburgh Instruments, 119F) as described previously (Coutinho and Prieto, 1995). The measurements were done using λexc = 316 nm and λem = 415 nm with bandwidths of 4 and 8 nm, respectively. Fluorescence intensity decay curves were analyzed by nonlinear least-squares regression, fitting to the data a sum of exponentials,
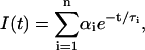 |
(3) |
where αi and τi are the normalized amplitude (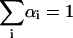 ) and lifetime of the ith decay component, respectively. The reduced chi-squared value (χ2) and weighted residuals with their autocorrelation were used as best fit criteria. The range of chi-squared values obtained was 1.0–1.3.
) and lifetime of the ith decay component, respectively. The reduced chi-squared value (χ2) and weighted residuals with their autocorrelation were used as best fit criteria. The range of chi-squared values obtained was 1.0–1.3.
The intensity-weighted mean fluorescence lifetime, 〈τ〉, is given by Eq. 4:
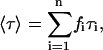 |
(4) |
where fi, the fractional intensity of the ith decay component, is:
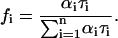 |
(5) |
On the other hand, 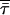 is the amplitude-weighted mean fluorescence lifetime,
is the amplitude-weighted mean fluorescence lifetime,
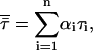 |
(6) |
or the lifetime-weighted quantum yield, since it is directly proportional to the area under the decay curve (Lakowicz, 1999).
Parameters that describe the occupation of lipid vesicles with nystatin
To identify the controlling parameter of nystatin self-association in the lipid vesicles, two different quantities were calculated for each sample studied. The first one was the phospholipid-to-antibiotic surface molar ratio, RS,
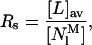 |
(7) |
where 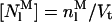 , the concentration of monomeric membrane-bound nystatin, is given by
, the concentration of monomeric membrane-bound nystatin, is given by
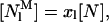 |
(8) |
[N] is the bulk molar concentration of antibiotic and xl is the membrane-bound mole fraction of monomeric antibiotic molecules:
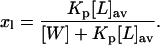 |
(9) |
It should be noted that in these calculations it is implicitly assumed that 1), only monomeric antibiotic molecules partition between the aqueous phase and the external lipid monolayer of the lipid vesicles; and 2), the mole fraction of antibiotic molecules involved in oligomer formation is so small that it can be ignored.
When liposomes with variable diameters are used, it is also useful to compute 〈A〉, the mean number of antibiotic molecules associated with a lipid vesicle. 〈A〉 is readily derived from RS and μ, the average number of lipid molecules per vesicle:
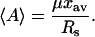 |
(10) |
xav is the fraction of lipid molecules in the outer half of a lipid vesicle. For SUV and LUV we estimated μ = 6 018 and μ = 82 045, respectively (Table 1).
TABLE 1.
Summary of the physical characteristics considered for the lipid vesicles used
| Model system of membranes | Mean diameter (nm) | μ |
|---|---|---|
| SUV | 30 | 6.0 × 103 |
| LUV | 100 | 8.2 × 104 |
The calculation of the average number of lipids in a single liposome, μ, was based upon a 4.5-nm bilayer thickness, an average surface area occupied by each phospholipid molecule of 0.7 nm2 (fluid phase) (Marsh, 1990; Nagle and Wiener, 1988) and assuming an homogeneous population of spherical lipid vesicles. It was also considered that there were no phospholipid packing constraints in the smaller vesicles and that the temperature did not influence the average number of lipids per liposome (Cornell et al., 1981).
Simulations of antibiotic binding to the lipid vesicles
As indicated in the Discussion section, it was assumed that two basic steps were involved in the interaction of nystatin with the lipid vesicles. First, monomeric nystatin associates with the membrane. This first step can be described by a partition equilibrium of nystatin between the aqueous and the membrane phases:
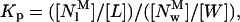 |
(11) |
where Kp is the mole-fraction partition coefficient of the antibiotic, 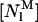 and
and 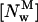 are the bulk molar concentrations of monomeric antibiotic in the lipid (i = l) and aqueous (i = w) phases, respectively, and [L] and [W] are the molar concentrations of lipid and water, respectively (White et al., 1998).
are the bulk molar concentrations of monomeric antibiotic in the lipid (i = l) and aqueous (i = w) phases, respectively, and [L] and [W] are the molar concentrations of lipid and water, respectively (White et al., 1998).
After partitioning into the lipid vesicles, the membrane-bound antibiotic molecules may reversibly assemble into aggregates. The association constant for the conversion of monomers into an aggregated form, a z-mer, is given by
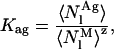 |
(12) |
where 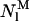 and
and 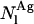 represent the two different kinds of fluorescent antibiotic species that may be present in the liposomes, namely monomeric and oligomeric nystatin. Each aggregate is formed by z antibiotic molecules. The concentration unit chosen to describe this equilibrium was the mean number of membrane bound antibiotic molecules per liposome.
represent the two different kinds of fluorescent antibiotic species that may be present in the liposomes, namely monomeric and oligomeric nystatin. Each aggregate is formed by z antibiotic molecules. The concentration unit chosen to describe this equilibrium was the mean number of membrane bound antibiotic molecules per liposome.
The mass conservation law for the antibiotic is
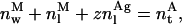 |
(13) |
where 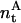 represents the total amount (i.e., number of moles) of antibiotic,
represents the total amount (i.e., number of moles) of antibiotic, 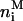 are the amount of monomeric nystatin molecules present in each phase (i = w, aqueous phase; i = l, lipid phase, respectively), and
are the amount of monomeric nystatin molecules present in each phase (i = w, aqueous phase; i = l, lipid phase, respectively), and 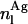 is the amount of aggregates formed by nystatin molecules in the lipid vesicles.
is the amount of aggregates formed by nystatin molecules in the lipid vesicles.
For any lipid or antibiotic concentration used, both the partition (Eq. 11) and nystatin association equilibria (Eq. 12) must be simultaneously obeyed. Combination of Eqs. 11 and 12 with the mass balance equation (Eq. 13) results in:
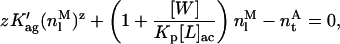 |
(14) |
where
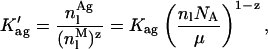 |
(15) |
with NA, [L]av, nl, and μ being the Avogadro's number, the lipid concentration available to the antibiotic binding, the amount of lipid molecules in a sample and the number of lipids forming a single liposome, respectively. Generally, this polynomial of zth-order (Eq. 14) has no analytical resolution and therefore the simulation of nystatin binding to the lipid vesicles had to be performed by an iterative numerical technique. For the initial simulations, the antibiotic was assumed to undergo a cooperative aggregation to an hexamer (z = 6) and the partition coefficient of nystatin was set equal to its experimental value. After assigning a numerical value to Kag, the mass balance equation was solved using the routine Solve for from Quattro Pro Version 5.0 to calculate the mole fractions of each antibiotic population present in the system
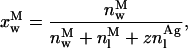 |
(16) |
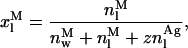 |
(17) |
and
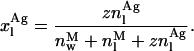 |
(18) |
RESULTS
Nystatin interaction with DPPC and POPC LUV
In a previous study of nystatin interaction with gel phase DPPC SUV anomalous binding curves were obtained for this antibiotic when high antibiotic concentrations (6.5, 7.8, and 13.5 μM) were used in some experiments (Coutinho and Prieto, 1995). It was then shown that the steady-state fluorescence intensity of the antibiotic leveled off toward a plateau, or even decreased sharply upon increasing the lipid concentration, while nystatin overall concentration was kept constant. The deviations presented by these experimental binding curves from the expected hyperbolic behavior (Eq. 1) were then tentatively ascribed to the fusion/aggregation of these lipid vesicles induced by the antibiotic molecules (Coutinho and Prieto, 1995). Therefore, in this work DPPC and POPC LUV were used in nystatin binding studies to avoid these problems. In fact, it is well known that an important characteristic of the model membrane system used is its diameter since the curvature radius of the lipid vesicles modulates its phospholipid packing, thereby conditioning its interfacial properties and stability (Chrzeszczyck et al., 1977; Brouillette et al., 1982). Considering that LUV prepared by the extrusion procedure through filters with pores of 100 nm have an outer diameter that is almost 3–4 times larger than SUV prepared by sonication, we expected these vesicles to be more stable and not so prone to undergo morphological changes.
The relative increase in the steady-state fluorescence intensity of nystatin in the presence of DPPC LUV was used to quantify its phospholipid/water partition coefficient (Fig. 1). It was considered that only the external monolayer of the lipid vesicles was accessible to the antibiotic incorporation because Finkelstein and co-workers (Marty and Finkelstein, 1975; Kleinberg and Finkelstein, 1984) have shown that the pores formed from one-sided and two-sided addition of nystatin to black lipid membranes had different properties. If an equilibrium distribution of antibiotic molecules among the two halves of the lipid bilayer was rapidly reached due to a fast flip-flop rate, the method used in antibiotic delivery should not influence the final results, contrary to what was observed experimentally. From a two-parameter fitting procedure (ΔImax and Kp, Eq. 1), a Kp = (1.1 ± 0.3) × 104 was obtained for nystatin interacting with these lipid vesicles in the gel phase (Table 2). This partition coefficient was independent of the antibiotic concentration used (Fig. 1) at variance with what was previously reported for the experiments carried out with DPPC SUV (Coutinho and Prieto, 1995). In addition, light-scattering measurements provided no evidence for vesicle fusion/aggregation induced by the antibiotic binding (results not shown). Similar partition coefficients for fluid lipid vesicles, either DPPC LUV at 45°C (Kp = (0.9 ± 0.1) × 104) or POPC LUV at 21°C (Kp = (1.4 ± 0.2) × 104, were also determined for nystatin (Table 2).
FIGURE 1.

Determination of the partition coefficient of nystatin from its fluorescence intensity increase upon incorporation into LUV of DPPC at 21°C. The nystatin concentrations used were (○) 3.2 and (•) 7.9 μM. The solid line is the best fit of Eq. 1 to the experimental data with Kp = (1.1 ± 0.3) × 104.
TABLE 2.
Mole-fraction partition coefficients of nystatin, Kp, for different phospholipid vesicles and temperatures
| Model system of membranes | Phospholipid | T (°C) | Buffer | Kp (×104) |
|---|---|---|---|---|
| SUV | DPPC | 21 | Tris | 6.5 ± 0.8* |
| HEPES | 5.8 ± 0.7 | |||
| 45 | Tris | 1.4 ± 0.3 | ||
| HEPES | 1.6 ± 0.3 | |||
| LUV | DPPC | 21 | HEPES | 1.1 ± 0.3 |
| 45 | HEPES | 0.9 ± 0.1 | ||
| POPC | 21 | HEPES | 1.4 ± 0.2 |
Partition coefficients were obtained from steady-state fluorescence intensity data as described in Materials and Methods. It was considered that only 60% and 50% of the total lipid of SUV and LUV could interact with the antibiotic, respectively.
Regarding the photophysical behavior of nystatin, the fluorescence decay curves of the antibiotic obtained in the presence of DPPC and POPC LUV at 21°C were both well described by a sum of three exponentials as judged from the fitting criteria used (χ2 value, random-weighted residuals and autocorrelation plots; see also Table 3). In the presence of gel-phase lipid vesicles, the fluorescence emission decay kinetics of nystatin was dominated by two long-lived decay components ∼14 and 43 ns that, together, accounted for nearly 95% of the antibiotic fluorescence intensity (Table 3). In addition, the fluorescence decay parameters of nystatin were found to be independent of the antibiotic concentration used (Table 3), the mean fluorescence lifetime of the antibiotic (〈τ〉 ≈ 36 ns) being close to the largest values previously measured for nystatin in interaction with gel-phase DPPC SUV (Coutinho and Prieto, 1995). When the lipid vesicles were in a fluid phase (1 mM POPC LUV), the short-lived fluorescent antibiotic species detected (〈τ〉 = 5.9 ns) was also found to be concentration independent (Table 3). In this case, however, a strong correlation between the fitting parameters (pre-exponentials and lifetimes) prevented an accurate description of each individual fluorescence decay curve. Therefore, a global analysis of the experimental data was carried out through linking the lifetimes between all fluorescence intensity decay curves. The global fit was considered satisfactory due to the low individual and global χ2 values obtained as well as by the random distributions of the weighted residuals for each fluorescence decay curve (data not shown), and three lifetime components of 1.2, 5.2, and 9.5 ns were recovered for nystatin (Table 3). The fast decaying fluorescent antibiotic species formed in these conditions had a mean fluorescence lifetime close to the one obtained for nystatin in interaction with DPPC SUV at 45°C (〈τ〉 = 2.8 ns; see Coutinho and Prieto, 1995).
TABLE 3.
Fluorescence intensity decay parameters for nystatin in interaction with 1 mM LUV prepared with different phospholipids (T = 21°C)
| Phospholipid | f1 | τ1 (ns) | f2 | τ2 (ns) | f3 | τ3 (ns) | 〈τ〉 (ns) |
|---|---|---|---|---|---|---|---|
| DPPC* | 0.06 | 1.9 | 0.15 | 14 | 0.79 | 43 | 36 |
| POPC† | 0.20 | 1.2 | 0.45 | 5.2 | 0.35 | 9.5 | 5.9 |
fi and τi are the fractional fluorescence intensity and lifetime, respectively, of each decay component. 〈τ〉 is the intensity-weighted mean fluorescence lifetime of the antibiotic. The liposomes were prepared in HEPES buffer.
The decay components were independent of nystatin concentration between 1.9 and 12.5 μM. f1, ±0.01; τ1, ±0.6; f2, ±0.02; τ2, ±1.0; f3, ±0.03; τ3, ±1.0; 〈τ〉, ±1.0.
The fluorescence intensity decays were independent of nystatin concentration between 3.5 and 12.5 μM. They were globally analyzed by linking the lifetimes of each fluorescence intensity decay (n = 7), a  of 1.1 being obtained. f1, ±0.01; f2, ±0.02; f3, ±0.04; 〈τ〉, ±0.2.
of 1.1 being obtained. f1, ±0.01; f2, ±0.02; f3, ±0.04; 〈τ〉, ±0.2.
Detection of nystatin oligomers in gel-phase lipid vesicles by time-resolved fluorescence measurements
The photophysical properties of nystatin in interaction with gel phase DPPC SUV were previously found to be very interesting because its intensity-weighted mean fluorescence lifetime increased sharply with the antibiotic concentration used (Coutinho and Prieto, 1995). This behavior was interpreted as due to a cooperative oligomerization of the antibiotic in the phospholipid bilayer. If this assumption is correct, then the fluorescence emission decay kinetics of nystatin should be critically dependent upon the surface density of antibiotic molecules adsorbed to the lipid vesicles. To conclusively check this point, some additional time-resolved fluorescence experiments were carried out using different antibiotic and phospholipid concentrations. But first, to avoid a significant aggregation of the antibiotic in the aqueous solution that would unnecessarily complicate the interpretation of the experimental data, we exchanged the buffer system used. Light-scattering measurements of nystatin aqueous solutions prepared with HEPES buffer were found to be essentially independent of the antibiotic concentration used within 0–30 μM in contrast to what was previously found with the Tris buffer (Fig. 2 B). Furthermore, a sharp increase in the fluorescence intensity of the antibiotic above a concentration ∼3 μM was detected only when nystatin aqueous solutions were prepared in Tris buffer (Fig. 2 A). This data indicates that large fluorescent antibiotic aggregates are not formed in HEPES buffer, and therefore this was the buffer system chosen to carry out all the subsequent experiments. The higher ionic strength of the HEPES buffer solution must screen the electrostatic interactions between the charged groups of the antibiotic, thereby preventing an extensive self-association of the antibiotic in aqueous solution, in agreement with what was previously shown for AmB (Mazerski et al., 1990). On the other hand, it was also found that nystatin partition coefficients for DPPC SUV were essentially independent of the buffer used at both temperatures studied (T = 21 and 45°C; see Table 2).
FIGURE 2.

Variation of (A) the steady-state fluorescence intensity, If (λexc = 304 nm; λem = 410 nm) and (B) light scattering intensity, Id (λexc = λem = 450 nm), with nystatin concentration in (•) Tris and (○) HEPES buffers.
The fluorescence decay parameters obtained for nystatin in interaction with different phospholipid concentrations of DPPC SUV are presented in Table 4. The main result of this experiment was that upon increasing the lipid concentration used there was a pronounced shift in the antibiotic critical concentration where nystatin photophysics switched from a fast to a slow decaying fluorescent species (from ∼5 to 12 μM nystatin at 1.1 and 3.5 mM DPPC SUV, respectively; see Table 4). In either case, the alteration in nystatin photophysics was mainly caused by an enhancement in the fractional fluorescence emission intensity of the longest-lived decay component of the antibiotic upon an increase of its bulk molar concentration in solution (Table 4), in conformity with what was previously reported (Coutinho and Prieto, 1995).
TABLE 4.
Fluorescence intensity decay parameters for nystatin in interaction with SUV of DPPC as a function of phospholipid and antibiotic concentration (T = 21°C)
| [DPPC] (mM) | [Nystatin] (μM) | f1 ± 0.07 | τ1 (ns) ± 0.8 | f2 ± 0.05 | τ2 (ns) ± 1.3 | f3 ± 0.05 | τ3 (ns) ± 3 | 〈τ〉 (ns) ± 2 |
|---|---|---|---|---|---|---|---|---|
| 1.1 | 1.9 | 0.20 | 1.4 | 0.66 | 7.0 | 0.14 | 29 | 9.0 |
| 3.8 | 0.24 | 2.7 | 0.54 | 7.9 | 0.22 | 28 | 11 | |
| 4.7 | 0.20 | 2.3 | 0.51 | 8.2 | 0.29 | 33 | 14 | |
| 5.4 | 0.16 | 2.7 | 0.30 | 10 | 0.54 | 38 | 24 | |
| 6.3 | 0.12 | 2.5 | 0.27 | 10 | 0.61 | 38 | 26 | |
| 6.7 | 0.11 | 2.4 | 0.24 | 10 | 0.65 | 38 | 27 | |
| 7.9 | 0.08 | 1.9 | 0.23 | 10 | 0.69 | 39 | 29 | |
| 9.4 | 0.11 | 1.9 | 0.20 | 12 | 0.69 | 40 | 30 | |
| 12.5 | 0.08 | 2.1 | 0.20 | 12 | 0.72 | 40 | 31 | |
| 3.5 | 3.2 | 0.36 | 3.0 | 0.60 | 8.0 | 0.05 | 28 | 7.3 |
| 4.7 | 0.33 | 2.6 | 0.60 | 8.1 | 0.07 | 28 | 7.7 | |
| 6.3 | 0.29 | 2.6 | 0.61 | 7.9 | 0.10 | 27 | 8.3 | |
| 7.9 | 0.24 | 2.7 | 0.56 | 8.2 | 0.19 | 32 | 11 | |
| 9.4 | 0.24 | 2.8 | 0.54 | 8.3 | 0.23 | 33 | 13 | |
| 11.0 | 0.26 | 2.9 | 0.48 | 8.7 | 0.25 | 34 | 13 | |
| 12.5 | 0.09 | 2.9 | 0.29 | 9.9 | 0.62 | 37 | 26 | |
| 14 | 0.08 | 2.4 | 0.22 | 11 | 0.70 | 39 | 30 |
fi and τi are the fractional fluorescence intensity and lifetime, respectively, of each decay component. 〈τ〉 is the intensity-weighted mean fluorescence lifetime of the antibiotic. The liposomes were prepared in HEPES buffer.
To test the former hypothesis, i.e., that the controlling parameter of nystatin assembly in the lipid vesicles was the surface density of membrane-bound antibiotic molecules, it was essential to correct the experimental data for the variable mole fraction of nystatin partitioned into the liposomes (which is dependent upon the phospholipid concentration used; see Eq. 9). Fig. 3 shows that the computation of the phospholipid-to-antibiotic surface molar ratio, RS, according to Eq. 7 allowed for a unified description of the experimental data obtained with DPPC SUV because the appearance of a long-lived fluorescent antibiotic species is now shown to occur within a rather narrow range of nystatin surface densities, 150 < RS < 280. Furthermore, the alterations detected in nystatin fluorescence decay kinetics are also shown to be essentially independent of the method used to prepare the samples, i.e., either keeping the phospholipid concentration constant and varying the antibiotic concentration (Fig. 3 A) or vice versa (Fig. 3 B). Therefore, it seems that the photophysical behavior of nystatin is controlled by the surface concentration of monomeric antibiotic molecules partitioned into the small unilamellar vesicles, and it is not dependent upon the presence of preformed antibiotic aggregates in the aqueous solution.
FIGURE 3.

Relationship between the mean fluorescence lifetime of nystatin, 〈τ〉, and the phospholipid-to-antibiotic surface molar ratio, RS, obtained with SUV of DPPC at 21°C. (A) The phospholipid concentration was kept constant (○) 1.1 mM, (•) 1.6 mM, (□) 2.3 mM, (▵) 2.4 mM, and (▴) 3.5 mM DPPC) while the antibiotic concentration was varied. (B) The nystatin concentration was kept constant (▾) 7.9 μM, (▪) 9.4 μM, and (▿) 11.0 μM antibiotic) while the phospholipid concentration was varied in each experiment. The dashed vertical lines define the range of RS values (150 < RS < 280) correspondent to the transition region between a long and short mean fluorescent lifetimes of nystatin.
To compare the data obtained with gel-phase DPPC SUV and LUV it was further necessary to take into account the different partition coefficients obtained for each model lipid bilayer system used (Table 2). After taking this point into consideration, one would expect that the mean fluorescence lifetime of nystatin as a function of RS should follow the same general trend in both cases if nystatin photophysical behavior was solely determined by the surface density of the adsorbed antibiotic molecules. However, contrary to the expectations, Fig. 4 A shows that it was not enough to keep the same phospholipid-to-antibiotic surface molar ratio in the two membrane systems to get a similar mean fluorescence lifetime for nystatin. In fact, even when very diluted antibiotic surface concentrations were reached with DPPC LUV (RS ≫ 280), there still was no significant decrease in 〈τ〉.
FIGURE 4.
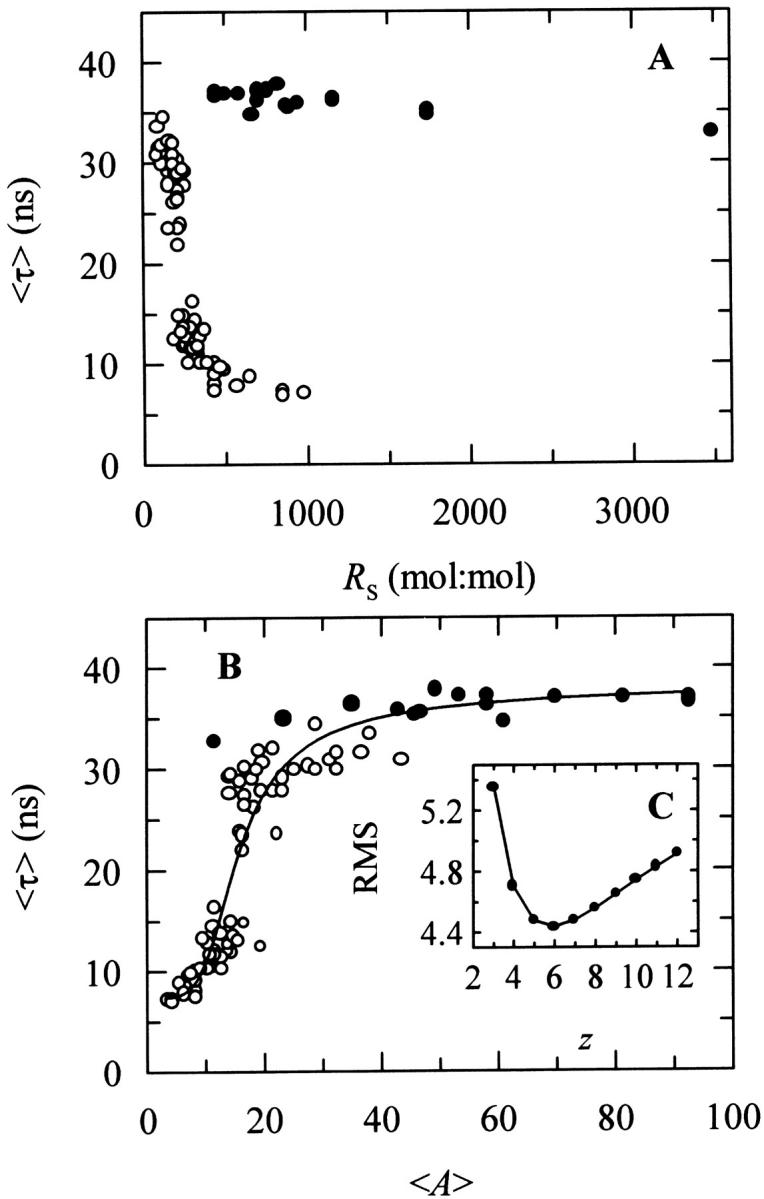
Mean fluorescence lifetime of nystatin, 〈τ〉, at different (A) surface molar ratios of phospholipid-to-antibiotic, RS, and (B) mean number of membrane bound antibiotic molecules per liposome, 〈A〉. The model membrane systems used were DPPC SUV (○) or LUV (•) at 21°C. In some experiments the phospholipid concentration was kept constant and the antibiotic concentration was varied and vice versa (see Fig. 3, legend). The solid line is the best fit of the cooperative partition model of the antibiotic to the experimental data (see the text and Fig. 8, legend, for details). (C, inset) Plot showing the relationship between the lowest value of the root-mean-square deviations (RMSD), and the aggregation number of nystatin, z. K′ag was allowed to vary in each fitting while z was kept constant at different integer numbers (see Table 5).
Why is the photophysical behavior of nystatin distinct when the antibiotic interacts with LUV instead of SUV? A possible explanation is that its fluorescence emission decay kinetics is controlled by the antibiotic-to-liposome ratio instead of its surface concentration. In fact, a SUV with an average outer diameter of 30 nm consists of ∼6000 lipid molecules compared to ∼82,000 for a LUV with an average outer diameter of 100 nm (Table 1). Consequently, although the total surface area of all the SUV in a lipid suspension is only 1.2 times larger than that of LUV at the same lipid concentration, there is ∼14 times more SUV than LUV in solution (note that the outer surface area of a LUV is ∼11 times larger than the one from a SUV). Therefore, and as a test for the former hypothesis, we replotted the data as 〈τ〉 versus the mean number of membrane bound antibiotic molecules per liposome, 〈A〉 (Eq. 10). Fig. 4 B supports the view that indeed the transition from a monomeric to an aggregated state of the antibiotic is essentially controlled by the mean occupation number of a lipid vesicle since the experimental data now describes a curve that is essentially independent of the model membrane system used. However, due to the complexity of the system, it cannot be ruled out that other factors, e.g., the curvature of the lipid vesicles, are also influencing nystatin oligomerization, as it will be discussed below. Fig. 4 B further shows that there is a threshold value for the mean occupation number of the lipid vesicles, 〈A〉* ≈ 10, above which the long-lived fluorescent antibiotic species starts to be formed in the gel-phase lipid vesicles. This data is also a definitive demonstration that the inclusion of sterols in the composition of a lipid vesicle is not an absolute prerequisite for nystatin oligomerization to take place.
Fluorescence quenching studies of nystatin by 5- and 16-DS
To estimate the antibiotic location in the lipid vesicles, a fluorescence quenching study of nystatin by two lipophilic probes, 5- and 16-DS, was carried out. These two spin-labeled fatty acids are known to insert predominantly vertical to the bilayer surface of the lipid vesicles (Ellena et al., 1988). Therefore their doxyl groups are located at different depths in the lipid bilayer (Fig. 5; see also Ellena et al., 1988; Feix et al., 1984), allowing information about the transversal position of the antibiotic fluorophore in the lipid vesicles to be obtained. The fluorescence quenching study of nystatin was carried out with DPPC SUV at 21°C using two different antibiotic concentrations, 3.0 and 12.5 μM. As these antibiotic samples have very different mean fluorescence lifetimes (∼10 and 28 ns, respectively), it was anticipated that they might provide information regarding the influence that the antibiotic aggregation state—monomer or oligomer—had on its transversal position in the lipid bilayer.
FIGURE 5.

Schematic representation of the expected transversal positions for the lipophilic probes 5- and 16-DS in a lipidic membrane. The dashed line represents the boundary between the polar and apolar regions of the membrane. Nystatin is represented perpendicular to the membrane surface just to illustrate that, upon internalization, its fluorophore group should get closer to both spin probes.
The Stern-Volmer plots for the steady-state fluorescence quenching study of nystatin by 5- and 16-DS in 3 mM SUV of DPPC at 21°C are depicted in Fig. 6. The actual quencher concentrations in the phospholipid phase, [Q]l, were computed from Eq. 2 and it was found that ∼94% and 81% of the 5-DS and 16-DS probes, respectively, were partitioned into the lipid vesicles. It should also be noted that with the lipid concentration used, ∼30% of the antibiotic remained in the aqueous phase. However, this antibiotic mole fraction does not influence the interpretation of the quenching experiments inasmuch as nystatin quantum yield in this phase is negligibly small compared to its value in the lipid phase (Coutinho and Prieto, 1995). Thus, its contribution to the total fluorescence intensity of the antibiotic sample, and therefore to the measured fluorescence quenching, is not significant. Fig. 6 A shows that the shallower spin-labeled fatty acid 5-DS was a more effective quencher of 3 μM nystatin than 16-DS, indicating a superficial location for the tetrane fluorophore of the antibiotic. On the other hand, upon increasing the antibiotic concentration to 12.5 μM, both spin-labeled fatty acids became equally effective in quenching nystatin fluorescence (Fig. 6 B). Due to the probable existence of more than one antibiotic population in these samples, the interpretation of this quenching data is not so straightforward as before and it will be discussed in more detail in the Discussion section.
FIGURE 6.

Stern-Volmer plots for the steady-state fluorescence quenching study of nystatin by 5- and 16-DS in 3 mM SUV of DPPC at 21°C. The nystatin concentrations used were (A) 3.0 and (B) 12.5 μM. Liposomes were prepared in Tris buffer. Lines are drawn just to guide the eye.
DISCUSSION
Nystatin interaction with different model lipid bilayer systems
The main objective of this work was to evaluate the influence that both the physical state of one-component phospholipid bilayers and the type of lipid vesicle used had on nystatin binding to the liposomes and its subsequent ability to reversibly assemble into aggregates. In a following study, the effects exerted by sterols—cholesterol and ergosterol—on both these processes will be described.
In the first part of this work, the intrinsic fluorescence emission properties of the polyene antibiotic nystatin were explored to characterize its binding to DPPC and POPC vesicles. Two main differences were found between nystatin interaction with DPPC SUV and LUV in the gel phase: 1), nystatin was only able to induce the fusion/aggregation of the smaller lipid vesicles (Coutinho and Prieto, 1995); and 2), the partition coefficient of this antibiotic was ∼5–7 times larger for SUV than for LUV in the gel phase (Table 2). The most probable explanation for these results lies in the nonequilibrium structure of SUV due to its small curvature radius. In fact, it is well known that the size of a liposome has a lower limit below which the curvature radius of its internal monolayer is so small that packing constraints between the phospholipid headgroups arise, preventing the formation of smaller lipid vesicles and making the outer monolayer of a SUV more expanded than the one obtained with a planar lipid bilayer (Chrzeszczyck et al., 1977; Brouillette et al., 1982). These structural characteristics of the SUV make them thermodynamically unstable (Suurkuusk et al., 1976) and more prone to undergo fusion/aggregation induced by an active molecule (van Dijck et al., 1978). The interfacial packing defects, which are more important in gel phase SUV, also justify the larger partition coefficient found for nystatin toward DPPC SUV compared to DPPC LUV (Table 2) because they are expected to be preferential partitioning sites for the antibiotic. In contrast, when the lipid vesicles were in a fluid phase an antibiotic partition coefficient Kp ≈ (1.3 ± 0.3) × 104 was always obtained (Table 2), which was essentially independent of the type of phospholipid (DPPC or POPC) or liposome (SUV or LUV) employed.
Bolard and co-workers had previously obtained similar results with vacidin A (Bolard et al., 1984), an aromatic polyene antibiotic, and with AmB (Milhaud et al., 1989). These authors found that these polyene antibiotics had a higher affinity for gel phase relative to fluid phase lipid membranes only when the model lipid bilayer system used were SUV. Furthermore, Bolard and Cheron (1982) showed by gel filtration and ultracentrifugation techniques that AmB was only able to induce the fusion or aggregation of small vesicles in the gel phase. When the lipid vesicles were in the liquid-crystalline phase or when larger lipid vesicles were used (although in the gel state), these processes did not occur.
Fluorescence decay kinetics reports on nystatin self-association in gel-phase lipid vesicles
Considering that the main factor governing nystatin photophysics is the aggregation state of the antibiotic in the lipid vesicles (Coutinho and Prieto, 1995), one would expect its mean fluorescence lifetime to be controlled by the surface density of membrane-bound antibiotic molecules in the phospholipid vesicles. Although the first experiments performed to test this hypothesis seemed to confirm this prediction (Fig. 3), they were unable to explain why the mean fluorescence lifetime of the antibiotic interacting with DPPC LUV was essentially independent of the phospholipid-to-antibiotic surface molar ratio used, always presenting a large value characteristic of the long-lived/aggregated antibiotic species, even when very diluted samples were prepared (Fig. 4 A). Further analysis of these results (Fig. 4 B) gave support to a different hypothesis, namely that the formation of highly fluorescent oligomers by nystatin was controlled by the mean occupation number of a lipid vesicle, 〈A〉, and not strictly by its phospholipid-to-antibiotic surface molar ratio. For this to be possible, the rate of aggregate formation must be fast compared to the incubation time (≥1 h) that preceded the time-resolved fluorescence measurement of each sample. Tachyia (1987) deduced a relationship between the mean reaction time, τm, of a pair of randomly distributed molecules in the surface of a sphere (e.g., a micelle or a lipid vesicle) and the sum of the radii of the sphere and the reactant, r, and the sum of the diffusion coefficients of both reactants, D:
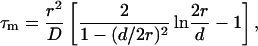 |
(19) |
with d being the sum of the radii of the reactants. If one considers the worst situation of our studies, i.e., a lipid vesicle with a radius of 50 nm (LUV) and D ≈ 10−14 m2 s−1 (lipids in the gel phase; Sackmann, 1983), we get τm ≈ 1 s. Therefore, it is safe to conclude that even if the aggregates formed are larger than a dimer, the incubation time used for each lipid and antibiotic mixture must have been sufficiently long for nystatin oligomerization reaction to attain its final equilibrium value even when the larger lipid vesicles were used.
The time-resolved fluorescence data also show that the minimal number of monomeric-bound antibiotic molecules required to be present in a lipid vesicle for the long-lived fluorescent antibiotic species to be formed is 〈A〉* ≈ 10 (Fig. 4 B). It is important to note, however, that this parameter corresponds to the antibiotic aggregation number in the liposomes only if nystatin self-association in the lipid vesicles is an irreversible process, which is not the case (Coutinho and Prieto, 1995). According to St. Pierre-Chazalet and co-workers (St. Pierre-Chazalet et al., 1988) and Seoane et al. (1997) the superficial area occupied by AmB in an antibiotic monolayer at the air/water interface is 1.8–1.9 nm2/molecule when the molecules are horizontal, i.e., when their polar faces are anchored in the water and their hydrophobic polyene chains are in contact with the air. On the other hand, upon switching to an orientation perpendicular to the interface (vertical form), the AmB molecules occupy ∼0.55 nm2/molecule. Considering that the surface area of a DPPC molecule is 0.5 nm2 (gel phase; see Marsh, 1990), the performance of a simple calculation shows that for this critical occupation number (〈A〉* = 10), less than 1% of the external surface area of a small unilamellar vesicle is occupied by horizontally adsorbed nystatin molecules.
This hypothesis also provides an explanation for the anomalous binding curves obtained for this antibiotic when small unilamellar gel-phase DPPC vesicles with high antibiotic concentrations were used (see Coutinho and Prieto, 1995; see also Results section, this article). In fact, the formation of long-lived and highly fluorescent antibiotic oligomers in the liposomes at low liposome concentrations (〈A〉 ≫ 10) must be the cause for this puzzling behavior. Upon increasing the lipid concentration in each sample, there must have been a rapid exchange of antibiotic molecules between the lipid vesicles causing the dissociation of these aggregates (〈A〉 ≪ 10). Since the monomeric membrane-bound antibiotic molecules have a lower quantum yield than the oligomers, the steady-state fluorescence intensity of the antibiotic samples stabilized, or even decreased, upon increasing the lipid concentration in solution, contrary to the theoretical expectation. These deviations were not noticed with the lowest antibiotic concentration used in the partitioning experiments (4.2 μM nystatin; Coutinho and Prieto, 1995) because in this case the alterations in the fluorescence emission decay kinetics of the antibiotic occurred at a very low phospholipid concentration (≈ 0.5 mM). This interpretation of the experimental data is further supported by other studies that show the polyene antibiotics ability's to undergo a rapid distribution between the aqueous phase and the lipid bilayers, either black lipid membranes (Cass et al., 1970) or liposomes (Witzke and Bittman, 1984). The spectroscopic CD studies carried out by Bolard et al. (1981) have also shown the capacity of AmB to rapidly exchange between DPPC SUV in the gel phase. Vertut-Croquin et al. (1984) even took advantage of this property to change the incorporation procedure of AmB into the lipid vesicles from an injection of an antibiotic stock solution prepared with an organic solvent to the utilization of liposomes preloaded with AmB.
However, two major criticisms may be raised against the hypothesis discussed above. First, a rigorous demonstration of its validity would require testing if a slow-decaying antibiotic species is obtained when nystatin samples are prepared with DPPC LUV containing a mean antibiotic occupation number below the determined critical value, i.e., 〈A〉 <10. Unfortunately, these measurements were extremely difficult to perform with our experimental setup because they would either require the use of a very low antibiotic concentration (implying a large acquisition time for the time-resolved fluorescence measurement) or of a very concentrated lipid suspension (that would originate an important scattering contribution to the experimental data). Therefore, it cannot be ruled out that the curvature radius of the lipid vesicles used may play some role in nystatin oligomerization. In fact, since this parameter determines the lipid packing of the external monolayer of LUV and SUV, it may also influence the penetration of the membrane surface by nystatin molecules and, consequently, their ability to form aggregates.
Secondly, it should be noted that the calculation of the mean occupation number of the liposomes by the antibiotic molecules was carried out using two major approximations: 1), it was admitted that both liposome suspensions, SUV and LUV, consisted of an homogeneous population of lipid vesicles; and 2), nystatin ability to induce the aggregation/fusion of SUV was ignored. Both these factors must be responsible, at least to some extent, for the scattering of 〈τ〉 values displayed in Figs. 3 and 4, particularly the one presented by the samples prepared with the highest concentrations of antibiotic. However, their relative importance on the final result must not have been too severe, otherwise it would not have been possible to normalize all the experimental data by computing 〈A〉 for each sample, as it is shown in Fig. 4 B.
Nystatin location in the lipid vesicles
Depth-dependent fluorescence quenching studies of nystatin with two lipophilic probes, 5- and 16-DS, were carried out using two different antibiotic concentrations, namely 3.0 and 12.5 μM. These samples displayed distinct mean fluorescence lifetimes (∼10 and 28 ns, respectively) and therefore they represented two different experimental situations: in the first case (3.0 μM nystatin; 〈A〉 = 4.1), the bound antibiotic molecules were predominantly monomeric in the lipid bilayer, whereas in the second case (12.5 μM nystatin; 〈A〉 = 17.0) a significant fraction of the bound antibiotic molecules was already engaged in the formation of oligomers with a long-lived fluorescence lifetime. Hence, it was anticipated that these fluorescence quenching studies could eventually provide some information about nystatin assembly being accompanied by an internalization of the antibiotic molecules from the bilayer surface toward its hydrophobic interior.
In the first situation studied (〈A〉 = 4.1), it was concluded that the monomeric antibiotic molecules reside near the phospholipid/water interface since the Stern-Volmer plot showed that the shallower spin-labeled fatty acid 5-DS was a more effective quencher of the tetrane fluorophore than the16-DS probe (Fig. 6 A). This location is compatible with the fast fluorescence decay kinetics presented by this antibiotic species (〈τ〉 ≈ 10 ns) because the surface-adsorbed antibiotic molecules must not experience a very rigid environment upon membrane binding.
Since the same experimental methodology was also used to determine the transverse location of filipin (Castanho and Prieto, 1995), another polyene antibiotic, it is interesting to compare both studies. Contrary to the present data, it was reported that the spin-labeled fatty acid 16-DS was a more efficient quencher of the fluorescence from the filipin pentane group than the 5-DS probe. Therefore, it was concluded that filipin location was in the inner region of the membrane, near the phospholipid bilayer center. The difference between the preferred locations found for these two polyene antibiotics must be related to their distinct chemical structures; whereas nystatin has a mycosamine residue attached to its macrolide ring, the small polyene macrolide antibiotic filipin does not. Hence, this sugar must be essential in anchoring nystatin at the membrane surface due to its polar character.
The analysis of the quenching data obtained in the second situation studied (〈A〉 = 17.0) is necessarily more complex, because two different antibiotic populations—monomers and oligomers—co-exist in equilibrium in the lipid vesicles. In addition to different fluorescence quantum yields, these two antibiotic populations most probably also have distinct transverse locations in the lipid membrane as well as a variable exposure of their tetrane groups to the quenchers used. Ladokhin (1997) and Asuncion-Punzalan et al. (1998) have already addressed some of these problems by considering the effect of two populations of fluorophores at different depths in the lipid bilayer in the analysis of the quenching results. The authors concluded that the information obtained about the center of the transverse distributions of the molecules in the liposomes reflected their average depth in the lipid bilayer weighted by their respective fluorescence quantum yields. Considering that the oligomers formed by nystatin exhibit a lifetime-weighted quantum yield ∼5× higher than the membrane-bound monomeric molecules (see next section), the fluorescence emitted by the 12.5 μM nystatin sample must be dominated to a large extent by the long-lived aggregates formed by nystatin. Therefore, it seems reasonable to assume that the quenching results must reflect predominantly the average location of the tetrane fluorophore in these antibiotic assemblies. Since both spin-labeled fatty acids quenched 12.5 μM nystatin fluorescence nearly to the same extent (Fig. 6 B), it seems that nystatin oligomerization in the liposomes is accompanied by a coordinate translocation of the antibiotic molecules from the bilayer surface toward its center (Fig. 7). On the other hand, it is expected that the self-association undergone by nystatin in the liposomes must have shielded to some extent the antibiotic fluorophores from the spin-labeled fatty acids used. However, this alteration in the fluorophores accessibility to the quenchers does not influence the interpretation of the quenching data because the masking of the quenching efficiencies is expected to be the same for both spin-labeled fatty acids.
FIGURE 7.

Schematic representation of the main stages of nystatin interaction with gel-phase lipid vesicles. (i) After nystatin addition to a liposome suspension, the monomeric antibiotic molecules partition into the membrane-water interface of the lipid vesicles. (ii) When the mean number of antibiotic molecules per liposome reaches a critical value (∼10 for gel-phase lipid vesicles), nystatin starts to self-associate. This process is accompanied by an increase in nystatin mean fluorescence lifetime from 7–10 ns to ∼35 ns. Concurrently, nystatin molecules translocate from the surface toward the interior of the lipid vesicles. The nystatin molecules that form an oligomer must shield their polar groups from contact with the acyl chains of the phospholipids.
Cooperative partition model of nystatin
According to the previous discussion, the following model is proposed for nystatin interaction with gel phase liposomes (Fig. 7):
When nystatin is added to the aqueous solution at a sufficient low concentration, monomeric antibiotic molecules adsorb predominantly at the phospholipid/water interface. This surface location of the antibiotic does not impose severe restrictions to the vibrational movements of the macrolide ring of the antibiotic molecules, and therefore their fluorescence emission decay kinetics is relatively rapid, i.e., the antibiotic presents a short mean fluorescence lifetime (〈τ〉 ≈ 7–10 ns).
Upon increasing the antibiotic concentration added to each sample, the membrane-bound antibiotic molecules start to oligomerize. Since the antibiotic molecules are putatively more rigidified in the antibiotic assemblies formed in the lipid bilayer than in its monomeric form, its nonradiative deactivation pathways decrease and its fluorescence emission decay kinetics becomes slower. Therefore, the mean fluorescence lifetime of nystatin progressively increases from 7–10 ns to ∼35 ns.
The quenching experiments support the hypothesis that nystatin aggregation in the lipid vesicles is accompanied by an internalization of the antibiotic molecules from the lipid bilayer surface toward its center (Fig. 7). This re-orientation of nystatin in the membrane must increase the local perturbation exerted by the antibiotic molecules upon the lipid bilayer structure. When SUV are used, this effect is translated into an increased liposome aggregation/fusion induced by the antibiotic molecules.
The above model raises two important questions, both of which deserve further consideration. The first question is, what is the effect of the interdependence between both equilibria—nystatin partition and oligomerization—on nystatin partitioning into the lipid vesicles? In fact, if both equilibria must be obeyed simultaneously, they may influence each other significantly depending on the experimental conditions employed (i.e., antibiotic and phospholipid concentrations used). The second issue that must be considered is whether this model has the ability to reproduce the general trends of the fluorescence emission decay kinetics of nystatin, even if only in a qualitative way. In particular, it is important to assess if this model can predict the occurrence of a critical value for the mean occupation number of liposomes by nystatin, 〈A〉*, as it was experimentally demonstrated in this study (Fig. 4 B).
The first problem was addressed by simulating the binding of nystatin to gel-phase SUV as described in the Materials and Methods section. Fig. 8 displays the results obtained in one simulation where the lipid concentration was kept constant (1 mM) while the antibiotic concentration was varied between 1 and 14 μM. Kp was set equal to 9.2 × 104, the experimentally determined partition coefficient for 4.2 μM nystatin (gel-phase SUV, HEPES buffer; see Table 1; see also Coutinho and Prieto, 1995). In addition, it was assumed that nystatin reversibly assembled in the lipid bilayer into hexamers (z = 6) with an association constant 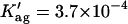 μmol−5 (Kag = 3.7 × 10111; Eq. 15). Fig. 8 A shows that in these conditions there was an increasing formation of oligomers by nystatin above an antibiotic concentration of ∼4 μM. Concomitantly, there was a reduction in the monomeric antibiotic mole fractions present both in the aqueous solution and in the lipid vesicles (Fig. 8 A). The main result from this simulation is therefore the demonstration that nystatin self-association in the lipid bilayer drives a progressive recruitment of monomeric antibiotic molecules from the aqueous phase, generating a cooperative partition of the antibiotic molecules into the lipid vesicles. In fact, if we define an apparent partition coefficient for nystatin,
μmol−5 (Kag = 3.7 × 10111; Eq. 15). Fig. 8 A shows that in these conditions there was an increasing formation of oligomers by nystatin above an antibiotic concentration of ∼4 μM. Concomitantly, there was a reduction in the monomeric antibiotic mole fractions present both in the aqueous solution and in the lipid vesicles (Fig. 8 A). The main result from this simulation is therefore the demonstration that nystatin self-association in the lipid bilayer drives a progressive recruitment of monomeric antibiotic molecules from the aqueous phase, generating a cooperative partition of the antibiotic molecules into the lipid vesicles. In fact, if we define an apparent partition coefficient for nystatin, 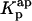 , that takes into account the z-mers in addition to the monomers present in the lipid bilayer:
, that takes into account the z-mers in addition to the monomers present in the lipid bilayer:
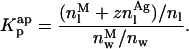 |
(20) |
It can be shown that its value gradually increases with the antibiotic concentration used from the moment that nystatin aggregation in the lipid bilayer starts to become important (Fig. 8, A and B). This effect has already been demonstrated experimentally in a different system by Wimley et al. (1998) when studying the partition of an hydrophobic peptide into LUV.
FIGURE 8.
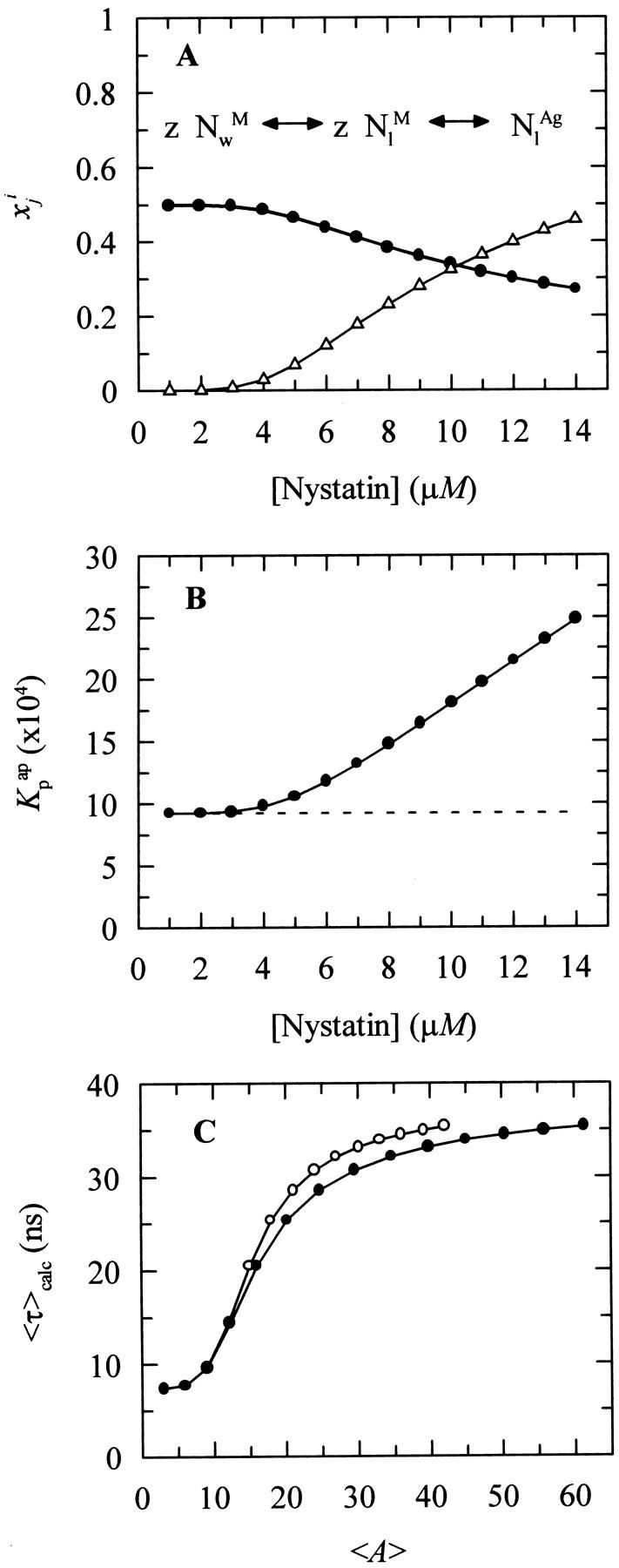
Simulation of the effect of nystatin self-association upon the antibiotic binding to the lipid vesicles and its fluorescence emission decay kinetics. (A) Antibiotic mole fractions as a function of the bulk molar nystatin concentration added to the SUV suspension: (○) 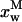 , (•)
, (•) 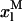 , and (▵)
, and (▵) 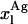 (calculated according to Eqs. 16–18).
(calculated according to Eqs. 16–18). 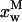 and
and 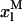 are coincident. (B) Relationship between the partition coefficient of the antibiotic and its total concentration in solution. The dashed line depicts the value used for nystatin partition coefficient in this simulation (
are coincident. (B) Relationship between the partition coefficient of the antibiotic and its total concentration in solution. The dashed line depicts the value used for nystatin partition coefficient in this simulation (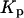 = 9.2 × 104), whereas (•) represent
= 9.2 × 104), whereas (•) represent 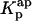 (calculated according to Eq. 20). (C) Mean fluorescence lifetime computed for nystatin, 〈τ〉calc, at different mean number of membrane bound antibiotic molecules per liposome, 〈A〉. This last parameter was calculated according to Eq. 10 using either
(calculated according to Eq. 20). (C) Mean fluorescence lifetime computed for nystatin, 〈τ〉calc, at different mean number of membrane bound antibiotic molecules per liposome, 〈A〉. This last parameter was calculated according to Eq. 10 using either 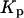 (○) or
(○) or 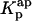 (•) in Eq. 9. The simulation of the cooperative partition model of nystatin was done considering that i), only monomeric antibiotic molecules partition into the liposomes ([L]t = 1 mM; μ = 6,018; T = 21°C); and that ii), nystatin self-associates into hexamers (z = 6) with an association constant Kag = 3.7 × 10111. 〈τ〉calc was computed according to Eq. 27 considering for monomeric nystatin αM1 = 0.63; αM2 = 0.36; αM3 = 0.01; τM1 = 2.6 ns; τM2 = 8.1 ns e τM3 = 28 ns, and for the antibiotic molecules involved in aggregate formation αAg1 = 0.40; αAg2 = 0.10; αAg3 = 0.50; τAg1 = 2.4 ns; τAg2 = 14.0 ns e τAg3 = 42 ns.
(•) in Eq. 9. The simulation of the cooperative partition model of nystatin was done considering that i), only monomeric antibiotic molecules partition into the liposomes ([L]t = 1 mM; μ = 6,018; T = 21°C); and that ii), nystatin self-associates into hexamers (z = 6) with an association constant Kag = 3.7 × 10111. 〈τ〉calc was computed according to Eq. 27 considering for monomeric nystatin αM1 = 0.63; αM2 = 0.36; αM3 = 0.01; τM1 = 2.6 ns; τM2 = 8.1 ns e τM3 = 28 ns, and for the antibiotic molecules involved in aggregate formation αAg1 = 0.40; αAg2 = 0.10; αAg3 = 0.50; τAg1 = 2.4 ns; τAg2 = 14.0 ns e τAg3 = 42 ns.
The calculation of the mean occupation number of a liposome by antibiotic molecules, 〈A〉, is affected by the partition coefficient chosen to compute its value, either the experimental coefficient or the apparent one, Kp or 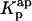 , respectively (Fig. 8 C). As expected, the more extensive the nystatin oligomerization is, the more divergent are the two computed values. For example, in Fig. 8 C the relative deviation between the 〈A〉 values reaches ∼23% and 45% for 10 and14 μM nystatin, respectively. The correct procedure is of course to use
, respectively (Fig. 8 C). As expected, the more extensive the nystatin oligomerization is, the more divergent are the two computed values. For example, in Fig. 8 C the relative deviation between the 〈A〉 values reaches ∼23% and 45% for 10 and14 μM nystatin, respectively. The correct procedure is of course to use 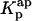 in Eq. 9. However, it should be noted that without having independent knowledge about the aggregation number and the association constant of the antibiotic in the lipid vesicles beforehand, it is neither possible to estimate the mole fraction of antibiotic molecules involved in aggregate formation,
in Eq. 9. However, it should be noted that without having independent knowledge about the aggregation number and the association constant of the antibiotic in the lipid vesicles beforehand, it is neither possible to estimate the mole fraction of antibiotic molecules involved in aggregate formation, 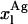 , nor to determine
, nor to determine 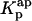 . The use of the partition coefficient, Kp, in every calculation is thus equivalent to admit that nystatin self-association is not significant in any of the experimental conditions used, which may not always be true. Despite this problem, it is important to realize that the experimental determination of 〈A〉* is never affected by the above approximation. In fact, until reaching this critical antibiotic concentration, Kp is essentially equal to
. The use of the partition coefficient, Kp, in every calculation is thus equivalent to admit that nystatin self-association is not significant in any of the experimental conditions used, which may not always be true. Despite this problem, it is important to realize that the experimental determination of 〈A〉* is never affected by the above approximation. In fact, until reaching this critical antibiotic concentration, Kp is essentially equal to 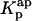 , as shown in Fig. 8 B. In addition, the performance of other simulations (using different total lipid and antibiotic concentrations) also revealed that the use of this approximation in the computation of 〈A〉 did not prevent the description of all the data by a universal curve of 〈τ〉calc versus 〈A〉 (see next section). These simulations showed that the scattering of 〈τ〉calc versus 〈A〉 was similar to the one found experimentally for 〈τ〉 versus 〈A〉.
, as shown in Fig. 8 B. In addition, the performance of other simulations (using different total lipid and antibiotic concentrations) also revealed that the use of this approximation in the computation of 〈A〉 did not prevent the description of all the data by a universal curve of 〈τ〉calc versus 〈A〉 (see next section). These simulations showed that the scattering of 〈τ〉calc versus 〈A〉 was similar to the one found experimentally for 〈τ〉 versus 〈A〉.
The second important question raised by the model presented above is whether it can reproduce or not, even if only in a qualitative fashion, the abrupt increment registered by nystatin mean fluorescence lifetime upon increasing the occupation number of the liposomes by the antibiotic. The testing of this condition requires the assumption of some additional approximations in order for the theoretical calculation of the mean fluorescence lifetime of nystatin to be possible from the mole fractions of antibiotic computed for each sample. In this respect, it was considered that 1), the radiative rate constant and the molar absorption coefficient of nystatin were both independent of its location (either aqueous solution or membrane-bound) and aggregation state (monomer or oligomer); and 2), that the absorbance of nystatin samples was always low. In fact, the tetrane oscillator strength is largely insensitive to the dielectric properties of its environment and this chromophore does not present significant solvatochromatic effects (Coutinho and Prieto, 1995). The decay law of a nystatin sample, Imix(t), should then be equal to:
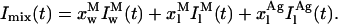 |
(21) |
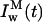 and
and 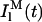 represent the intrinsic fluorescence decay curves of monomeric nystatin in the aqueous phase and membrane-bound, respectively, and
represent the intrinsic fluorescence decay curves of monomeric nystatin in the aqueous phase and membrane-bound, respectively, and 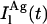 is the fluorescence emission decay kinetics of nystatin included in an oligomer. As previously mentioned, although the phospholipid concentrations used in the experiments carried out with gel-phase SUV (1 < [L]t < 3 mM) implied that there was always a significant fraction of antibiotic molecules in the aqueous solution (0.50 – 0.59 >
is the fluorescence emission decay kinetics of nystatin included in an oligomer. As previously mentioned, although the phospholipid concentrations used in the experiments carried out with gel-phase SUV (1 < [L]t < 3 mM) implied that there was always a significant fraction of antibiotic molecules in the aqueous solution (0.50 – 0.59 > 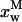 > 0.25 – 0.32), their contribution to the fluorescence intensity decay curve of each sample may be ignored because they have only a very fast decay component (Petersen et al., 1987). In this way, the previous equation reduces to:
> 0.25 – 0.32), their contribution to the fluorescence intensity decay curve of each sample may be ignored because they have only a very fast decay component (Petersen et al., 1987). In this way, the previous equation reduces to:
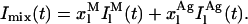 |
(22) |
According to the experimental data, it was admitted that both the membrane-bound nystatin monomers and oligomers had complex fluorescence emission decay curves described by a three-exponential sum:
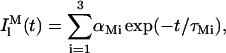 |
(23) |
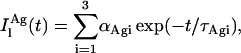 |
(24) |
with
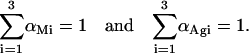 |
(25 and 26) |
In this regard, it should be noted that all the fluorescence decay curves obtained for nystatin were analyzed by empirically fitting a sum of three and not six exponentials to the experimental data. This simplification results from the impossibility to recover too many meaningful parameters from the analysis of a single decay curve.
From the previous Eqs. 4, 6, 23, and 24, the following expression is derived for the theoretical mean fluorescence lifetime presented by a nystatin sample:
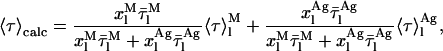 |
(27) |
where 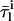 and
and 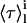 are the lifetime-weighted quantum yield and intensity-weighted mean fluorescence lifetimes, respectively, of monomeric (i = M) and oligomeric (i = Ag) nystatin. This equation shows that 〈τ〉calc is not a simple average of
are the lifetime-weighted quantum yield and intensity-weighted mean fluorescence lifetimes, respectively, of monomeric (i = M) and oligomeric (i = Ag) nystatin. This equation shows that 〈τ〉calc is not a simple average of 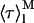 and
and 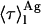 weighted by the antibiotic mole fractions since
weighted by the antibiotic mole fractions since 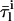 , which is proportional to the species fluorescence quantum yield, also appears in the weighting factors. Therefore, if the fluorescence quantum yield of nystatin aggregates is much larger than the one characteristic of the monomeric antibiotic molecules, the relative contribution of
, which is proportional to the species fluorescence quantum yield, also appears in the weighting factors. Therefore, if the fluorescence quantum yield of nystatin aggregates is much larger than the one characteristic of the monomeric antibiotic molecules, the relative contribution of 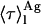 for 〈τ〉calc may be significant even if very few antibiotic molecules are self-assembled in the lipid bilayer, and 〈τ〉calc rapidly reaches its limiting value
for 〈τ〉calc may be significant even if very few antibiotic molecules are self-assembled in the lipid bilayer, and 〈τ〉calc rapidly reaches its limiting value 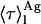 . This general conclusion is exemplified in Fig. 8 C. The amplitudes and decay times chosen for monomeric (αM1 = 0.63; αM2 = 0.36; αM3 = 0.01; τM1 = 2.6 ns; τM2 = 8.1 ns and τM3 = 28 ns;
. This general conclusion is exemplified in Fig. 8 C. The amplitudes and decay times chosen for monomeric (αM1 = 0.63; αM2 = 0.36; αM3 = 0.01; τM1 = 2.6 ns; τM2 = 8.1 ns and τM3 = 28 ns;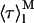 = 7.4 ns) and aggregated nystatin (αAg1 = 0.40; αAg2 = 0.10; αAg3 = 0.50; τAg1 = 2.4 ns; τAg2 = 14.0 ns; and τAg3 = 42 ns;
= 7.4 ns) and aggregated nystatin (αAg1 = 0.40; αAg2 = 0.10; αAg3 = 0.50; τAg1 = 2.4 ns; τAg2 = 14.0 ns; and τAg3 = 42 ns; 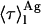 = 38.7 ns) were based upon the limiting values experimentally found for very diluted and concentrated nystatin samples (〈A〉 ≪ 10 and 〈A〉 ≫10, respectively). This choice implied that
= 38.7 ns) were based upon the limiting values experimentally found for very diluted and concentrated nystatin samples (〈A〉 ≪ 10 and 〈A〉 ≫10, respectively). This choice implied that 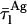 ≈ 5
≈ 5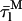 and therefore, in the simulation conditions used in Fig. 8 C, the participation of only 30% of the antibiotic molecules in hexamer formation was sufficient for 〈τ〉calc to reach asymptotically
and therefore, in the simulation conditions used in Fig. 8 C, the participation of only 30% of the antibiotic molecules in hexamer formation was sufficient for 〈τ〉calc to reach asymptotically 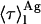 = 38.7 ns.
= 38.7 ns.
Eq. 27 also shows indirectly that for each model membrane system used, the variation of 〈τ〉calc between its limiting values, 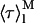 and
and 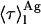 , is essentially conditioned by the choice made for z and Kag, the parameters that describe the antibiotic oligomerization process within the lipid bilayer. As in any other co-operative process, the larger the z value is, the more abrupt the transition of 〈τ〉calc between its limiting values is expected to be. Therefore, and within all the constraints discussed above, the model proposed for nystatin interaction with the lipid vesicles can be fitted to the experimental data of 〈τ〉 versus 〈A〉 through finding a pair of values for z and Kag that simultaneously minimize the root-mean-square (RMS) deviations between the experimental and calculated values for the intensity-weighted mean fluorescence lifetime of nystatin, 〈τ〉exp and 〈τ〉calc, respectively:
, is essentially conditioned by the choice made for z and Kag, the parameters that describe the antibiotic oligomerization process within the lipid bilayer. As in any other co-operative process, the larger the z value is, the more abrupt the transition of 〈τ〉calc between its limiting values is expected to be. Therefore, and within all the constraints discussed above, the model proposed for nystatin interaction with the lipid vesicles can be fitted to the experimental data of 〈τ〉 versus 〈A〉 through finding a pair of values for z and Kag that simultaneously minimize the root-mean-square (RMS) deviations between the experimental and calculated values for the intensity-weighted mean fluorescence lifetime of nystatin, 〈τ〉exp and 〈τ〉calc, respectively:
 |
(28) |
where n is the number of data points used in each fitting procedure. Because of the high correlation between z and Kag, z (integer) was fixed and Kag (or 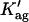 ) was varied in each RMS minimization. Table 5 and Fig. 4 C show that nystatin reversibly assembles into aggregates of 6 ± 2 antibiotic molecules when interacting with gel-phase DPPC SUV.
) was varied in each RMS minimization. Table 5 and Fig. 4 C show that nystatin reversibly assembles into aggregates of 6 ± 2 antibiotic molecules when interacting with gel-phase DPPC SUV.
TABLE 5.
Determination of nystatin aggregation number, z, in gel-phase DPPC vesicles
| z* | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||
| K′ag* (μmol)1−z | 9.6 × 10−3 | 3.0 × 10−3 | 1.0 × 10−3 | 3.7 × 10−4 | 1.4 × 10−4 | 5.3 × 10−5 | 2.0 × 10−5 | 8.0 × 10−6 | 3.1 × 10−6 | 1.2 × 10−6 | |
| RMS | 5.36 | 4.70 | 4.48 | 4.44 | 4.48 | 4.56 | 4.65 | 4.74 | 4.83 | 4.91 | |
The cooperative partition model of nystatin in interaction with the lipid vesicles was fitted to the experimental data of 〈τ〉 versus 〈A〉 (n = 86) according to the procedure described in the text. K′ag was allowed to vary in each minimization of the root-mean-square deviations (RMSD), while z was kept constant at different integer numbers. These results are presented in the inset of Fig. 4.
Kag can be obtained from Eq. 11.
Comparison with the literature
The above model proposed for nystatin interaction with the phospholipid vesicles is also supported by several spectroscopic and calorimetric studies carried out mainly with AmB. From the experimental data obtained using CD and differential scanning calorimetry (DSC), it was proposed that AmB underwent an oligomerization process in the lipid bilayer as well (Bolard et al., 1980; Vertut-Croquin et al., 1983; Balakrishnan and Easwaran, 1993; Fournier et al., 1998). For example, using SUV prepared from egg-yolk lecithin, Vertut-Croquin et al. (1983) found that the CD spectrum of AmB was typical of a monomer adsorbed at the lipid bilayer interface only when very low antibiotic-to-phospholipid ratios, RA:L, were used. Upon increasing RA:L, the CD spectrum of the samples progressively changed showing some new characteristics, which were ascribed to the insertion of the antibiotic molecules in a more rigid environment. Later, using DPPC SUV in the fluid phase, Balakrishnan and Easwaran (1993) showed unequivocally that the changes detected in the CD spectra of AmB upon varying the antibiotic-to-phospholipid molar ratio used were due to an aggregation process of the antibiotic in the lipid bilayer.
On the other hand, Grant et al. (1989) and Hamilton et al. (1991) determined partial phase diagrams for AmB incorporated in liposomes with a variable lipid composition (prepared with saturated or unsaturated phospholipids) using electron spin resonance and DSC, respectively. Regarding nystatin, Milhaud et al. (1997) also obtained a partial phase diagram for this antibiotic incorporated in dilauroylphosphatidylcholine liposomes by carrying out DSC measurements. All the partial phase diagrams obtained, except for the dimyristoylphosphatidylcholine: dimyristoylphosphatidylglycerol (DMPC:DMPG) 7:3 system employed, suggested that within the antibiotic concentration ranges used (0–17 or 33 mol% of antibiotic) there was an immiscibility of components in the solid phase since the solidus line was approximately horizontal. Hence, the antibiotic molecules were not uniformly distributed in the solid matrix of phospholipid; rather they segregated and formed a distinct phase. This segregation may be explained by the distinct structural characteristics of the polyene antibiotics compared to the surrounding phospholipids. On the other hand, the liquidus line showed a positive slope because the temperature corresponding to the ending of the phospholipid phase transition increased with the antibiotic content of the lipid vesicles. This result suggests that antibiotic-enriched domains are formed in the lipid bilayers which have a higher transition temperature compared with the one obtained with pure phospholipids. This effect was confirmed for nystatin and filipin (Bolard and Milhaud, 1996), as well as for AmB (Fournier et al., 1998).
It should be noted that there is no apparent contradiction between these studies and the model developed here for nystatin, although the last one admits that membrane-bound antibiotic molecules self-associate into aggregates with a well-defined stoichiometry. In fact, due to the distinct sensitivity of the techniques employed in each study (electron spin resonance, deuterium nuclear magnetic resonance (2H-NMR), and DSC compared to fluorescence), the phospholipid-to-antibiotic ratios used were very different. Therefore, it is possible that upon increasing the antibiotic mole fraction in the liposomes, there is an increased formation of aggregates that may progressively coalesce, ultimately leading to a phase separation, as it is observed in the DSC studies. The phase separation may be complete when the lipid vesicles are in a gel phase or only partial, when the liposomes are kept in a fluid phase.
Finally, 2H-NMR studies carried out with 30 mol% of AmB (Dufourc et al, 1984) or ∼5 and 33 mol% nystatin (Milhaud et al., 1997) demonstrated the ability of these polyene antibiotics in rigidifying their lipid microenvironment. The first study suggested that AmB was able to induce an all-trans conformation in the acyl chains of the phospholipids. On the other hand, the 2H-NMR experiments carried out with nystatin showed that this antibiotic increased the lipid order only at temperatures immediately above Tm, causing an increase in the phospholipid bilayer width of ∼0.2 nm. In addition, nystatin produced an increased order of all deuterated positions of an acyl chain, with this effect being more pronounced in the center of the lipid bilayer.
CONCLUSIONS
The sensitivity of the fluorescence emission properties of nystatin to the characteristics of its microenvironment was explored here to study its interaction with liposomes of different phospholipid composition, phase, and curvature radius. The main conclusions obtained were:
The larger partition coefficient found for nystatin toward gel-phase DPPC SUV compared to DPPC LUV was ascribed to the presence of a large number of interfacial packing defects in the smaller vesicles, which must facilitate nystatin incorporation. For the liposomes kept in a fluid phase (SUV or LUV prepared with DPPC or POPC), and for gel-phase DPPC LUV, an antibiotic partition coefficient of (1.3 ± 0.3) × 104 was always obtained.
The fluorescence emission decay kinetics of nystatin in DPPC SUV and LUV in the gel phase was found to be controlled by the mean number of membrane bound antibiotic molecules per lipid vesicle, 〈A〉. Above a critical concentration, 〈A〉* ≈ 10, there was a pronounced increase in the antibiotic mean fluorescence lifetime from 7–10 to ∼35 ns. This change in the decay kinetics of nystatin was attributed to interactions between the antibiotic molecules that drove its oligomerization in the lipid vesicles. Since the antibiotic molecules in the aggregates formed experienced a more rigid environment, there was a decrease in its nonradiative rate constant which produced the reported variation in nystatin mean fluorescence lifetime.
The depth-dependent quenching studies carried out with the spin-labeled fatty acids (5- and 16-doxyl stearic acids) showed that monomeric nystatin was anchored at the phospholipid/water interface, and suggested that nystatin assembly into aggregates was followed by their translocation into the membrane.
Altogether, these results could be adequately explained by a mathematical model which assumes that monomeric nystatin molecules partition into the lipid bilayer and reversibly aggregate to form channels consisting of 6 ± 2 antibiotic molecules. Furthermore, our data shows that the utilization of membrane-containing sterols is not an essential condition for nystatin to assemble into aggregates.
The influence that more biologically relevant lipid mixtures, namely ergosterol- and cholesterol-containing lipid bilayers, have on nystatin oligomerization, will be presented in a future work.
Acknowledgments
This study was supported by project POCTI/36389/FCB/2000 (Fundação para a Ciência e Tecnologia, Portugal).
References
- Akaike, N., and N. Harata. 1994. Nystatin perforated patch recording and its applications to analyses of intracellular mechanisms. Jpn. J. Physiol. 44:433–473. [DOI] [PubMed] [Google Scholar]
- Andreoli, T. E. 1974. The structure and function of amphotericin B-cholesterol pores in lipid bilayer membranes. Ann. N.Y. Acad. Sci. 235:448–468. [DOI] [PubMed] [Google Scholar]
- Andreoli, T. E., V. W. Dennis, and A. M. Weigl. 1969. The effect of amphotericin B on the water and nonelectrolyte permeability of thin lipid membranes. J. Gen. Physiol. 53:133–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuncion-Punzalan, E., K. Kachel, and E. London. 1998. Groups with polar characteristics can locate at both shallow and deep locations in membranes: the behavior of dansyl related probes. Biochemistry. 37:4603–4611. [DOI] [PubMed] [Google Scholar]
- Balakrishnan, A. R., and K. R. K. Easwaran. 1993. Lipid-amphotericin B complex structure in solution: a possible first step in the aggregation process in cell membranes. Biochemistry. 32:4139–4144. [DOI] [PubMed] [Google Scholar]
- Bolard, J. 1986. How do the polyene macrolide antibiotics affect the cellular membrane properties? Biochim. Biophys. Acta. 864:257–304. [DOI] [PubMed] [Google Scholar]
- Bolard, J., A. Vertut-Croquin, B. Cybulska, and C. M. Gary-Bobo. 1981. Transfer of the polyene antibiotic amphotericin B between single-walled vesicles of dipalmitoylphosphatidylcholine and egg-yolk phosphatidylcholine. Biochim. Biophys. Acta. 647:241–248. [Google Scholar]
- Bolard, J., and J. Milhaud. 1996. Interaction of the anti-Candida amphotericin B (and other polyene antibiotics) with lipids. In Lipids of Pathogenic Fungi. R. Prasad and M. A. Ghannoum, editors. CRC Press, Boca Raton. pp. 253–274.
- Bolard, J., and M. Cheron. 1982. Association of the polyene antibiotic amphotericin B with phospholipid vesicles: perturbation by temperature changes. Can. J. Biochem. 60:782–789. [DOI] [PubMed] [Google Scholar]
- Bolard, J., M. Cheron, and J. Mazerski. 1984. Effect of surface curvature on the interaction of single lamellar phospholipid vesicles with aromatic and nonaromatic heptaene antibiotics (vacidin A and amphotericin B). Biochem. Pharmacol. 33:3675–3680. [DOI] [PubMed] [Google Scholar]
- Bolard, J., M. Seigneuret, and G. Boudet. 1980. Interaction between phospholipid bilayer membranes and the polyene antibiotic amphotericin B: lipid state and cholesterol content dependence. Biochim. Biophys. Acta. 599:280–293. [DOI] [PubMed] [Google Scholar]
- Brouillette, C. G., J. P. Segrest, T. C. Ng, and J. L. Jones. 1982. Minimal size of phosphatidylcholine vesicles: effects of radius of curvature on head group packing and conformation. Biochemistry. 21:4569–4575. [DOI] [PubMed] [Google Scholar]
- Cass, A., A. Finkelstein, and V. Krespi. 1970. The ion permeability induced in thin lipid membranes by the polyene antibiotics nystatin and amphotericin B. J. Gen. Physiol. 56:100–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanho, M. A., and M. J. Prieto. 1992. Fluorescence study of the macrolide pentaene antibiotic filipin in aqueous solution and in a model system of membranes. Eur. J. Biochem. 207:125–134. [DOI] [PubMed] [Google Scholar]
- Castanho, M., and M. Prieto. 1995. Filipin fluorescence quenching by spin-labeled probes: studies in aqueous solution and in a membrane model system. Biophys. J. 69:155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzeszczyck, A., A. Wishnia, and C. S. Springer, Jr. 1977. The intrinsic structural asymmetry of highly curved phospholipid bilayer membranes. Biochim. Biophys. Acta. 470:161–169. [DOI] [PubMed] [Google Scholar]
- Cornell, B. A., G. C. Fletcher, J. Middlehurst, and F. Separovic. 1981. Temperature dependence of the size of phospholipid vesicles. Biochim. Biophys. Acta. 642:375–380. [DOI] [PubMed] [Google Scholar]
- Coutinho, A., and M. Prieto. 1995. Self-association of the polyene antibiotic nystatin in dipalmitoylphosphatidylcholine vesicles: a time-resolved fluorescence study. Biophys. J. 69:2541–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kruijff, B., and R. A. Demel. 1974. Polyene antibiotic-sterol interactions in membranes of Acholeplasma laidlawii cells and lecithin liposomes. III. Molecular structure of the polyene antibiotic-cholesterol complexes. Biochim. Biophys. Acta. 339:57–70. [DOI] [PubMed] [Google Scholar]
- Dufourc, E. J., I. C. Smith, and H. C. Jarrell. 1984. Interaction of amphotericin B with membrane lipids as viewed by 2H-NMR. Biochim. Biophys. Acta. 778:435–442. [DOI] [PubMed] [Google Scholar]
- Ellena, J. F., S. J. Archer, R. N. Dominey, B. D. Hill, and D. S. Cafiso. 1988. Localizing the nitroxide group of fatty acid and voltage-sensitive spin-labels in phospholipid bilayers. Biochim. Biophys. Acta. 940:63–70. [DOI] [PubMed] [Google Scholar]
- Feix, J. B., C. A. Popp, S. D. Venkataramu, A. H. Beth, J. H. Park, and J. S. Hyde. 1984. An electron-electron double-resonance study of interactions between [14N]- and [15N]stearic acid spin-label pairs: lateral diffusion and vertical fluctuations in dimyristoylphosphatidylcholine. Biochemistry. 23:2293–2299. [DOI] [PubMed] [Google Scholar]
- Fournier, I., J. Barwicz, and P. Tancrède. 1998. The structuring effects of amphotericin B on pure and ergosterol- or cholesterol-containing dipalmitoylphosphatidylcholine bilayers: a differential scanning calorimetry study. Biochim. Biophys. Acta. 1373:76–86. [DOI] [PubMed] [Google Scholar]
- Fujii, G., J.-E. Chang, T. Coley, and B. Steere. 1997. The formation of amphotericin B ion channels in lipid bilayers. Biochemistry. 36:4959–4968. [DOI] [PubMed] [Google Scholar]
- Grant, C. W. M., K. S. Hamilton, K. D. Hamilton, and K. R. Barber. 1989. Physical biochemistry of a liposomal amphotericin B mixture used for patient treatment. Biochim. Biophys. Acta. 984:11–20. [DOI] [PubMed] [Google Scholar]
- Hamilton, K. S., K. R. Barber, J. H. Davis, K. Neil, and C. W. Grant. 1991. Phase behaviour of amphotericin B multilamellar vesicles. Biochim. Biophys. Acta. 1062:220–226. [DOI] [PubMed] [Google Scholar]
- Hartsel, S. C., C. Hatch, and W. Ayenew. 1993. How does amphotericin B work? Studies on model membrane systems. J. Liposome Res. 3:377–408. [Google Scholar]
- Hartsel, S., and J. Bolard. 1996. Amphotericin B: new life for an old drug. Trends Pharmacol. Sci. 17:445–449. [DOI] [PubMed] [Google Scholar]
- Holz, R., and A. Finkelstein. 1970. The water and nonelectrolyte permeability induced in thin lipid membranes by the polyene antibiotics nystatin and amphotericin B. J. Gen. Physiol. 56:125–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Chen, C. C., and D. S. Feingold. 1973. Polyene antibiotic action on lecithin liposomes: effect of cholesterol and fatty acyl chains. Biochem. Biophys. Res. Commun. 51:972–978. [DOI] [PubMed] [Google Scholar]
- Kleinberg, M. E., and A. Finkelstein. 1984. Single-length and double-length channels formed by nystatin in lipid bilayer membranes. J. Membr. Biol. 80:257–269. [DOI] [PubMed] [Google Scholar]
- Ladokhin, A. S. 1997. Distribution analysis of depth-dependent fluorescence quenching in membranes: a practical guide. Methods Enzymol. 278:462–473. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Plenum Press, New York.
- Marsh, D. 1990. Handbook of Lipid Bilayers. CRC Press, Boca Raton.
- Marty, A., and A. Finkelstein. 1975. Pores formed in lipid bilayer membranes by nystatin. Differences in its one-sided and two-sided action. J. Gen. Physiol. 65:515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer, L. D., M. J. Hope, and P. R. Cullis. 1986. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta. 858:161–168. [DOI] [PubMed] [Google Scholar]
- Mazerski, J., J. Bolard, and E. Borowski. 1982. Self-association of some polyene macrolide antibiotics in aqueous media. Biochim. Biophys. Acta. 179:11–17. [Google Scholar]
- Mazerski, J., J. Grzybowska, and E. Borowski. 1990. Influence of net charge on the aggregation and solubility behaviour of amphotericin B and its derivatives in aqueous media. Eur. Biophys. J. 18:159–164. [DOI] [PubMed] [Google Scholar]
- McClare, C. W. 1971. An accurate and convenient organic phosphorous assay. Anal. Biochem. 39:527–530. [DOI] [PubMed] [Google Scholar]
- Milhaud, J., J. Berrehar, J. M. Lancelin, B. Michels, G. Raffard, and E. J. Dufourc. 1997. Association of polyene antibiotics with sterol-free lipid membranes. II. Hydrophobic binding of nystatin to dilauroylphosphatidylcholine bilayers. Biochim. Biophys. Acta. 1326:54–66. [DOI] [PubMed] [Google Scholar]
- Milhaud, J., M.-A. Hartmann, and J. Bolard. 1989. Interaction of the polyene antibiotic amphotericin B with model membranes: differences between small and large unilamellar vesicles. Biochimie. 71:49–56. [DOI] [PubMed] [Google Scholar]
- Nagle, J. F., and M. C. Wiener. 1988. Structure of fully hydrated bilayer dispersions. Biochim. Biophys. Acta. 942:1–10. [DOI] [PubMed] [Google Scholar]
- Petersen, N. O., R. Gratton, and E. M. Pisters. 1987. Fluorescence properties of polyene antibiotics in phospholipid bilayer membranes. Can. J. Chem. 65:238–244. [Google Scholar]
- Sackmann, E. 1983. Physical foundations of the molecular organization and dynamics of membranes. In Biophysics. W. Hoppe, W. Lohmann, H. Marke, and H. Ziegler, editors. Springer-Verlag, Berlin. pp. 425–457.
- St. Pierre-Chazalet, M., C. Thomas, M. Dupeyrat, and C. M. Gary-Bobo. 1988. Amphotericin B-sterol complex formation and competition with egg phosphatidylcholine: a monolayer study. Biochim. Biophys. Acta. 944:477–486. [DOI] [PubMed] [Google Scholar]
- Schaffner, C. P. 1984. Polyene macrolides in clinical practice: pharmacology and adverse and other effects. In Macrolide Antibiotics. Chemistry, Biology, and Practice. S. Ómura, editor. Academic Press, New York. pp. 457–507.
- Seoane, J. R., N. Vila Romeu, J.J. Miñones, O. Conde, P. Dynarowicz, and M. Casas. 1997. The behavior of amphotericin B monolayers at the air/water interface. Prog. Coll. Polym. Sci. 105:173–179. [Google Scholar]
- Strom, R., W. E. Blumberg, R. E. Dale, and C. Crifo. 1976. The interaction of the polyene antibiotic lucensomycin with cholesterol in erythrocyte membranes and in model systems. III. Characterization of spectral parameters. Biophys. J. 16:1297–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suurkuusk, J., B. R. Lentz, Y. Barenholz, R. L. Biltonen, and T. E. Thompson. 1976. A calorimetric and fluorescent probe study of the gel-liquid crystalline phase transition in small, single-lamellar dipalmitoylphosphatidylcholine vesicles. Biochemistry. 15:1393–1401. [DOI] [PubMed] [Google Scholar]
- Szponarski, W., J. Wietzerbin, E. Borowski, and C. M. Gary-Bobo. 1988. Interaction of 14C-labelled amphotericin B derivatives with human erythrocytes: relationship between binding and induced K+ leak. Biochim. Biophys. Acta. 938:97–106. [DOI] [PubMed] [Google Scholar]
- Tachyia, M. 1987. Stochastic and diffusion models of reactions in micelles and vesicles. In Kinetics of Nonhomogeneous Processes. G. R. Freeman, editor. Wiley, New York. pp. 575–650.
- van Dijck, P. W. M., B. de Kruijff, P. A. M. M. Aarts, A. J. Verkleij, and J. de Gier. 1978. Phase transitions in phospholipid model membranes of different curvature. Biochim. Biophys. Acta. 506:183–191. [DOI] [PubMed] [Google Scholar]
- van Hoogevest, P., and B. de Kruijff. 1978. Effect of amphotericin B on cholesterol-containing liposomes of egg phosphatidylcholine and didocoseneoylphosphatidylcholine. A refinement of the model for the formation of pores by amphotericin B in membranes. Biochim. Biophys. Acta. 511:397–407. [DOI] [PubMed] [Google Scholar]
- Vertut-Croquin, A., J. Bolard, and C. M. Gary-Bobo. 1984. Enhancement of amphotericin B selectivity by antibiotic incorporation into gel state vesicles. A circular dichroism and permeability study. Biochem. Biophys. Res. Commun. 125:360–366. [DOI] [PubMed] [Google Scholar]
- Vertut-Croquin, A., J. Bolard, M. Chabbert, and C. Gary-Bobo. 1983. Differences in the interaction of the polyene antibiotic amphotericin B with cholesterol- or ergosterol-containing phospholipid vesicles. A circular dichroism and permeability study. Biochemistry. 22:2939–2944. [DOI] [PubMed] [Google Scholar]
- Wardlaw, J. R., W. H. Sawyer, and K. P. Ghiggino. 1987. Vertical fluctuations of phospholipid acyl chains in bilayers. FEBS Lett. 223:20–24. [DOI] [PubMed] [Google Scholar]
- White, S. H., W. C. Wimley, A. S. Ladokhin, and K. Hristova. 1998. Protein folding in membranes: determining energetics of peptide-bilayer interactions. Methods Enzymol. 295:62–87. [DOI] [PubMed] [Google Scholar]
- Wimley, W. C., K. Hristova, A. S. Ladokhin, L. Silvestro, P. H. Axelsen, and S. H. White. 1998. Folding of β-sheet membrane proteins: a hydrophobic hexapeptide model. J. Mol. Biol. 277:1091–1110. [DOI] [PubMed] [Google Scholar]
- Witzke, N. M., and R. Bittman. 1984. Dissociation kinetics and equilibrium binding properties of polyene antibiotic complexes with phosphatidylcholine/sterol vesicles. Biochemistry. 23:1668–1674. [DOI] [PubMed] [Google Scholar]