

Abstract
The change in the electrostatic properties on excitation of the cofactor of wild-type photoactive yellow protein (WT-PYP) have been directly determined using Stark-effect spectroscopy. We find that, instantaneously on photon absorption, there is a large change in the permanent dipole moment, 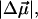 (26 Debye) and in the polarizability,
(26 Debye) and in the polarizability, 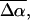 (1000 Å3). We expect such a large degree of charge motion to have a significant impact on the photocycle that is associated with the important blue-light negative phototactic response of Halorhodospira halophila. Furthermore, changing E46 to Q in WT-PYP does not significantly alter its electrostatic properties, whereas, altering the chromophore to prevent it from undergoing trans-cis isomerization results in a significant diminution of
(1000 Å3). We expect such a large degree of charge motion to have a significant impact on the photocycle that is associated with the important blue-light negative phototactic response of Halorhodospira halophila. Furthermore, changing E46 to Q in WT-PYP does not significantly alter its electrostatic properties, whereas, altering the chromophore to prevent it from undergoing trans-cis isomerization results in a significant diminution of 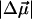 and
and 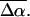 We propose that the enormous charge motion that occurs on excitation of 4-hydroxycinnamyl thioester, the chromophore in WT-PYP, plays a crucial role in initiating the photocycle by translocation of the negative charge, localized on the phenolate oxygen in the ground state, across the chromophore. We hypothesize that this charge motion would consequently increase the flexibility of the thioester tail thereby decreasing the activation barrier for the rotation of this moiety in the excited state.
We propose that the enormous charge motion that occurs on excitation of 4-hydroxycinnamyl thioester, the chromophore in WT-PYP, plays a crucial role in initiating the photocycle by translocation of the negative charge, localized on the phenolate oxygen in the ground state, across the chromophore. We hypothesize that this charge motion would consequently increase the flexibility of the thioester tail thereby decreasing the activation barrier for the rotation of this moiety in the excited state.
INTRODUCTION
Photoactive yellow protein (PYP), a small (14 kDa) water-soluble protein, isolated from Halorhodospira halophila, is implicated in the blue-light negative-phototactic response of the bacterium (Meyer et al., 1987), thereby protecting it from harmful exposure to blue light (Sprenger et al., 1993). This functionality of PYP is intimately linked with a photocycle that it undergoes. Therefore it is essential to thoroughly characterize the spectroscopic properties of PYP to better understand this process. The photocycle shown in Scheme 1 (≈0.1 s to 1 s duration) is characterized by a series of large conformational changes of the protein which are allegedly linked to the physiologically important signal-transduction process (Sprenger et al., 1993). At the heart of this process is the active cofactor, identified as a 4-hydroxycinnamyl cysteine thioester (Scheme 2 a), a p-coumaric acid (Hoff et al., 1994), which triggers these changes in the protein. Photon absorption by the negatively-charged, deprotonated chromophore (Kim et al., 1995) of the ground-state species, pG, initiates the photocycle in PYP (see Scheme 1). Following excitation (λmax = 446 nm), it exhibits a multiexponential fluorescence decay with relaxation to either a product state or to the ground state on timescales of hundreds of femtoseconds to tens of picoseconds (Changenet et al., 1998; Gensch et al., 2002; Meyer et al., 1987). An important event in this photocycle is the trans-cis isomerization around the C C double bond of the chromophore (C7C8 bond in Scheme 2 a) which occurs within 3 ns of excitation of pG (Ujj et al., 1998). Before the formation of the isomerized species, the formation of two other intermediates, I0 and I0#, have also been detected (Ujj et al., 1998). I0, or pR510, is formed in less then 3 ps while the intermediate I0#, formed in less than a nanosecond, was detected before the formation of the cis-isomer, I1. This intermediate, I1 or pR465 (λmax = 465 nm), then undergoes a one-proton uptake (Hendriks et al., 2002), leading to formation of a blue-shifted intermediate, I2 or pB355 (λmax = 355 nm), triggering the partial unfolding of the protein itself. This is considered to be the signaling state (Hendriks et al., 2002; Kort et al., 1996). Deprotonation and reisomerization on a subsecond timescale leads thereafter to reformation of the ground state, pG (Meyer et al., 1987).
C double bond of the chromophore (C7C8 bond in Scheme 2 a) which occurs within 3 ns of excitation of pG (Ujj et al., 1998). Before the formation of the isomerized species, the formation of two other intermediates, I0 and I0#, have also been detected (Ujj et al., 1998). I0, or pR510, is formed in less then 3 ps while the intermediate I0#, formed in less than a nanosecond, was detected before the formation of the cis-isomer, I1. This intermediate, I1 or pR465 (λmax = 465 nm), then undergoes a one-proton uptake (Hendriks et al., 2002), leading to formation of a blue-shifted intermediate, I2 or pB355 (λmax = 355 nm), triggering the partial unfolding of the protein itself. This is considered to be the signaling state (Hendriks et al., 2002; Kort et al., 1996). Deprotonation and reisomerization on a subsecond timescale leads thereafter to reformation of the ground state, pG (Meyer et al., 1987).
SCHEME 1.

Photocycle of WT-PYP.
SCHEME 2.

Molecular structures of the cofactors of (a) WT-PYP, (b) E46Q, and (c) L-PYP.
Because these conformational changes are strongly dependent on small- and large-scale charge motion in the chromophore-protein coupled system, Stark spectroscopy, in which the field-induced change in the absorption (electroabsorption) or emission (electroluminescence) spectrum is measured (Liptay, 1974), can be used to obtain information about the change in the electrostatic properties upon excitation or relaxation, respectively. Indeed, biological pigment-protein complexes and their chromophores in particular have been quite amenable to Stark experiments where, just as in PYP, light-induced charge motion is an integral part of the reaction pathway (Beekman et al., 1997; Boxer et al., 1989; Gottfried et al., 1991). In this investigation, in which we measure the Stark absorption spectra (electroabsorption) of PYP, we focus on the change in electrostatic properties associated with photon absorption by the dark-state species. The study, which is confined to the properties of the initially excited species, provides us with a measure of the extent of charge separation that occurs immediately on excitation of pG. The measured electrostatic properties can then be directly mapped onto the molecular framework of the chromophore thereby allowing us to verify the feasibility of a particular mechanism as a consequence of charge motion. Indeed, light-induced charge motion can play an important role in the photocycle of PYP just as in bacteriorhodopsin (Birge and Hubbard, 1980; Zadok et al., 2002), the protein to which PYP is often compared. Additionally, these measurements are very important because they can be used to verify the robustness and accuracy of electronic-structure calculations (Groenhof et al., 2002; Mollina and Merchán, 2001; Sergi et al., 2001) which, in turn, may then be exploited to obtain information about the electronic properties of the system that are not easily accessible experimentally.
Here, besides wild-type PYP (WT-PYP), we present Stark spectra of two additional derivatives. The first is mutant E46Q (Scheme 2 b), in which the glutamic acid residue of apo-PYP at position 46, identified as the proton donor for the chromophore in the photocycle of WT-PYP (Xie at al., 1996), has been modified to a glutamine (Genick et al., 1997). It should be noted, however, that in other studies it has been concluded that the solvent itself can also be the proton donor, not only in E46Q, but in WT-PYP as well (Genick et al., 1997; Borucki et al., 2002; Devanathan et al., 2000). In the second derivative of WT-PYP, the chromophore is modified to a 7-hydroxy-coumarin3-carboxylic acid chromophore (Scheme 2 c) to prevent the vinyl bond in the native chromophore of WT-PYP (compare to Scheme 2 a) from undergoing the important trans-cis isomerization step (Cordfunke et al., 1998). We shall hereafter refer to this system as locked-PYP (L-PYP). By comparing the change in the electrostatic properties that occurs on excitation of these three different systems, it is possible to discern the importance of the role of charge separation in initiating the photocycle of PYP, and, whether this function is affected by a modification in the chromophore (L-PYP), and/or a modification in the interaction with the protein environment (E46Q).
The changes in electrostatic properties, determined from the Stark experiments, are the magnitude of the change in the permanent dipole moment, 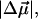 and the (average) change in polarizability,
and the (average) change in polarizability, 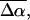 which provide us with a direct measure of the degree of charge motion induced by photon absorption (Liptay, 1974). Therefore, one obtains information about the electronic charge distribution of the excited-state species in the Franck-Condon region of the potential energy surface (PES) on initiating the photocycle. In bacteriorhodopsin, which has a similar photocycle (Mathies et al., 1991), a difference dipole moment on the order of 10 Debye has been measured in the isolated retinal chromophore (Locknar and Peteanu, 1998). Similarly in PYP, the process that initiates the photocycle would be expected to involve a large change in dipole moment (charge separation) on excitation, because the major event in the PYP photocycle is a configurational change of the chromophore, i.e., isomerization. Here, in this direct measurement of the degree of charge separation in PYP, we have found that the Stark signal is dominated by a contribution due to a large change in dipole moment, which is somewhat surprising in such a small chromophore. In L-PYP, containing the locked chromophore, the magnitude of the change in the dipole moment is diminished compared to the wild-type, while E46Q exhibits electronic properties similar to WT-PYP. In addition, the
which provide us with a direct measure of the degree of charge motion induced by photon absorption (Liptay, 1974). Therefore, one obtains information about the electronic charge distribution of the excited-state species in the Franck-Condon region of the potential energy surface (PES) on initiating the photocycle. In bacteriorhodopsin, which has a similar photocycle (Mathies et al., 1991), a difference dipole moment on the order of 10 Debye has been measured in the isolated retinal chromophore (Locknar and Peteanu, 1998). Similarly in PYP, the process that initiates the photocycle would be expected to involve a large change in dipole moment (charge separation) on excitation, because the major event in the PYP photocycle is a configurational change of the chromophore, i.e., isomerization. Here, in this direct measurement of the degree of charge separation in PYP, we have found that the Stark signal is dominated by a contribution due to a large change in dipole moment, which is somewhat surprising in such a small chromophore. In L-PYP, containing the locked chromophore, the magnitude of the change in the dipole moment is diminished compared to the wild-type, while E46Q exhibits electronic properties similar to WT-PYP. In addition, the 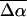 values indicate that the excited states of WT-PYP and E46Q are more extensively delocalized than in L-PYP. The similarity between the electrostatic properties of E46Q and WT-PYP highlights the importance of the intrinsic electronic properties of the cofactors in the PYP photocycle. We discuss the implications of these observations on the mechanism for the initiation of the PYP photocycle and provide additional support for our interpretations based on published electronic-structure calculations (Groenhof et al., 2002; Mollina and Merchán, 2001; Sergi et al., 2001).
values indicate that the excited states of WT-PYP and E46Q are more extensively delocalized than in L-PYP. The similarity between the electrostatic properties of E46Q and WT-PYP highlights the importance of the intrinsic electronic properties of the cofactors in the PYP photocycle. We discuss the implications of these observations on the mechanism for the initiation of the PYP photocycle and provide additional support for our interpretations based on published electronic-structure calculations (Groenhof et al., 2002; Mollina and Merchán, 2001; Sergi et al., 2001).
All the Stark experiments presented here are performed in glycerol-buffer glasses at 77 K. We note that, as in other low-temperature experiments (Cordfunke et al., 1998; Imamoto et al., 1996; Zhou et al., 2001), there is a light-induced blue-shift in the absorption maxima of WT-PYP and E46Q accompanied by the appearance of a weak absorption band at lower energy, although we use a much weaker light regime than the aforementioned authors. Previously, these changes in the absorption spectrum had been proposed to arise due to the formation of hypsochromic (PYPH or E46QH) and bathochromic (PYPB or E46QB) photoproducts (Imamoto et al., 1996; Zhou et al., 2001). Here, it is possible to differentiate between the properties of the dark-state species from that of the photoproducts in both the absorption and Stark spectra obtained at 77 K. We find that the red photoproduct, PYPB, which is considered to be comparable to the room-temperature intermediate I0 or pR510 (Imamoto et al., 1996; Imamoto et al., 2001a), exhibits different electronic properties than the dark-state species.
MATERIALS AND METHODS
Preparation of photoactive yellow protein and derivatives
ApoPYP and its site-directed mutant, E46Q, were produced and isolated as hexa-histidine tagged apo-proteins in Escherichia coli (Kort et al., 1996). The apoproteins were reconstituted with the 1,1-carbonyldiimidazole derivative of the respective chromophores, i.e., p-coumaric acid and 7-hydroxy-coumarin-3-carboxilic acid (Hendriks et al., 2002). Protein samples were used without removal of their hexahistidine containing N-terminal tag in 10 mM Tris/HCl at pH 8.
Sample apparatus
A sample cell similar to that used in other near-UV Stark experiments (Chowdhury et al., 1999) is also used here. The sample is embedded between a pair of semitransparent inconel-coated quartz slides using double-sided sticking tape as spacers. The metallic coating on the slides has an OD of 0.3 (Melles Griot, 0.3 ND filters). The sample consists of a 66–70% (w/v) solution of 99% glycerol in a pH 7.5 Tris buffer (10 mM) solution of PYP. To form a clear glass that is crack-free, the glycerol-buffer solution is cooled down slowly to 77 K. The thickness of each sample, individually determined from interference patterns generated in the near IR, is usually between 95 and 100 μm. The OD of the samples at the absorption maxima varied between ≈0.3 and 0.5/100 μm. The sample is immersed in LN2 in a dewar with quartz windows (HS Martin UV-Vis LN2 dewar) by means of a homemade sample holder. To set the angle between the electric field and the electric field vector of the light beam effectively at 54.7°, the external angle between the sample and the incoming light beam is set at 46° in 77 K. This accounts for the difference in refractive index between liquid nitrogen (n = 1.28) and the sample medium, which is approximated to be 1.5 for a glycerol-buffer glass at 77 K. Spectra obtained at the magic angle have been corrected for the above refractive index changes that occur in the path of the light beam. Variation in the external angle by ±5 degrees (accounting for a possible error of ±0.1 in the estimated refractive index of the glycerol-buffer glass) does not significantly change the size or shape of the Stark signal. The error, thereof, is less than 5% and smaller than the standard deviation, resulting from performing multiple trials.
Acquiring spectra
The broadband output of a (150-W) Xe-arc lamp was passed through a 0.4-m Oriel monochromator (1200 g/mm grating blazed at 350 nm) with 2-mm slit width. The output beam (<1 μW) was horizontally polarized using a Glan-Taylor polarizer. A homebuilt high-voltage AC power supply (Beekman et al., 1997) is used to apply a voltage across the sample on the order of 800–2000 V at ≈310 Hz. The field experienced by the molecule in the matrix cavity is determined by dividing the voltage applied across the sample by the sample thickness. The voltage output from the silicon photodiode (Hamamatsu), that measures the light transmitted by the sample, is sent to a lockin amplifier (EG&G Model 5210). The total transmission (I) and the signal due to the field-induced modulation (ΔIrms ≈ 1 × 10−4) are detected simultaneously via the analog and lockin ports, respectively, of the lockin detector. The locked signal is detected at the second harmonic (2 × 310 Hz) of the applied field. The monochromator and lockin detector are interfaced to a Unix machine using a homewritten program in C to automate data collection.
Each independent result is the average of at least three reproducible trials at a particular field strength. The electronic parameters reported in Table 2 are the average of three to five independent results. For both WT-PYP and E46Q, scans that are obtained following illumination with blue light (<440 nm for WT-PYP and <450 nm for E46Q) contain photoproduct(s). These products are immediately apparent in both the absorption and Stark signals at the red edge. In this manuscript, blue-light illumination refers to the exposure of the sample to light at lower wavelengths (≈1 μW <450 nm) in the process of acquiring the first scan (scan rates of 2 s/nm or 4 s/nm). All scans obtained subsequent to the first one across the entire absorption band, manifest the presence of photoproducts, primarily at the red edges of the absorption and Stark spectra (Fig. 2: 18,000–20,000 cm−1). However, after the initial diminution of the main peaks and troughs of the Stark signal (Fig. 2: 15,000–18,000 cm−1) and the appearance of a new Stark signal at the red edge of the absorption spectrum, all subsequent stepwise scans do not exhibit a further change in the Stark spectrum, nor in the overall transmission. The dominant part of the Stark signal in the region of absorption of the dark-state species remains relatively unaltered.
TABLE 2.
Electrostatic properties of WT-PYP, E46Q, and L-PYP
| Overall fit | Main transition | Red transition | ||||
|---|---|---|---|---|---|---|
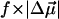 (D) (D) |
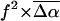 (Å3) (Å3) |
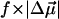 (D) (D) |
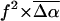 (Å3) (Å3) |
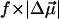 (D) (D) |
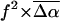 (Å3) (Å3) |
|
| WT-PYP | 25 ± 3 | 990 ± 180 | 26 ± 1 | 1020 ± 150 | 10 ± 4 | 2000 ± 500 |
| E46Q | 20 ± 2 | 950 ± 160 | 22 ± 4 | 1200 ± 260 | 12 ± 4 | 2400 ± 900 |
| L-PYP | 13 ± 1 | 150 ± 30 | – | – | – | – |
Values of 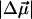 are in Debye and
are in Debye and 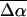 in Å3. These electrostatic properties include an enhancement due to an internal (reaction and cavity) field factor, f, that depends on the polarity of the environment. The
in Å3. These electrostatic properties include an enhancement due to an internal (reaction and cavity) field factor, f, that depends on the polarity of the environment. The 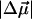 and
and 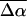 values obtained from the fit to the overall absorption lineshape are reported in the first two columns. The results from the fits obtained by separating out the absorption of the red photoproduct (PYPB for WT-PYP and E46QB for E46Q) from the main absorption band are shown in the respective columns labeled Red transition for PYPB and E46QB and Main transition for the dark-state species for WT-PYP (pG) and E46Q. The electronic properties for L-PYP are for the dark-state species corresponding to the main transition.
values obtained from the fit to the overall absorption lineshape are reported in the first two columns. The results from the fits obtained by separating out the absorption of the red photoproduct (PYPB for WT-PYP and E46QB for E46Q) from the main absorption band are shown in the respective columns labeled Red transition for PYPB and E46QB and Main transition for the dark-state species for WT-PYP (pG) and E46Q. The electronic properties for L-PYP are for the dark-state species corresponding to the main transition.
FIGURE 2.

(a) WT-PYP and (b) E46Q. Before illumination with blue light (<440 nm for WT-PYP and <450 nm for E46Q) the first Stark and absorption spectra that are acquired are shown in the light and dark dot-dashed lines, respectively (scaled for easier comparison). Absorption (light solid lines) and Stark (dark solid lines) spectra acquired following the initial illumination with blue light. The presence of photoproducts (see <20,000 cm−1) in these spectra are marked with the symbol * in a and b.
Theory of stark effect and fitting data
The field-induced change of the main absorption band of the chromophore in the protein pocket, which gives rise to the Stark signal, is analyzed using the Liptay formalism (Liptay, 1974). The effect of the electric field on the absorption spectrum, 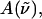 of an ensemble of molecules is measured by determining the difference in the intensity of the light transmitted through the sample in the presence of an applied field. This change in the absorbance (
of an ensemble of molecules is measured by determining the difference in the intensity of the light transmitted through the sample in the presence of an applied field. This change in the absorbance (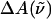 ), or molar extinction coefficient, averaged over all orientations, is given by
), or molar extinction coefficient, averaged over all orientations, is given by
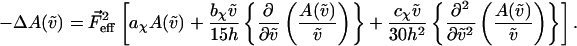 |
(1) |
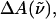 expressed as a function of the wavenumber,
expressed as a function of the wavenumber, 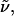 is proportional to the square of the effective field at the site of the solute,
is proportional to the square of the effective field at the site of the solute, 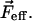 It includes the enhancement of the applied electric field due to the cavity field factor (Bottcher, 1952).
It includes the enhancement of the applied electric field due to the cavity field factor (Bottcher, 1952). 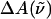 is proportional to the weighted sum of the 0th, first, and second derivatives of the unperturbed (field-free) absorption spectrum as a function of the angle (χ) between the applied AC electric-field vector and the electric-field vector of the polarized light. At the magic angle, 54.7°, the coefficients of the derivatives, aχ, bχ, and cχ are related to the molecular parameters of interest as shown in the expressions below.
is proportional to the weighted sum of the 0th, first, and second derivatives of the unperturbed (field-free) absorption spectrum as a function of the angle (χ) between the applied AC electric-field vector and the electric-field vector of the polarized light. At the magic angle, 54.7°, the coefficients of the derivatives, aχ, bχ, and cχ are related to the molecular parameters of interest as shown in the expressions below.
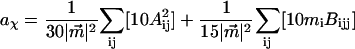 |
(2) |
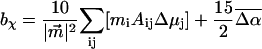 |
(3) |
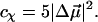 |
(4) |
The transition dipole moment, 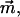 the change in polarizability,
the change in polarizability, 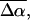 and the change in dipole moment,
and the change in dipole moment, 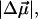 are related to the electric-field induced change in the absorption spectrum as shown in Eqs. 2, 3, and 4, respectively. The tensor components of the transition moment polarizability, A, and transition moment hyperpolarizability, B, present in Eqs. 2 and 3, are presumed to be small for strongly allowed transitions, such as the ones studied here. The subscripts i and j of these parameters refer to the axes in the Cartesian-coordinate system that are fixed in space. From b54.7, e.g., Eq. 3, we can determine the average change in polarizability of the system,
are related to the electric-field induced change in the absorption spectrum as shown in Eqs. 2, 3, and 4, respectively. The tensor components of the transition moment polarizability, A, and transition moment hyperpolarizability, B, present in Eqs. 2 and 3, are presumed to be small for strongly allowed transitions, such as the ones studied here. The subscripts i and j of these parameters refer to the axes in the Cartesian-coordinate system that are fixed in space. From b54.7, e.g., Eq. 3, we can determine the average change in polarizability of the system, 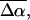 which is a measure of the induced dipole moment in the presence of a weak external field. This scalar quantity,
which is a measure of the induced dipole moment in the presence of a weak external field. This scalar quantity, 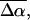 is one third the trace of the change in the second-rank polarizability tensor, Δα, (1/3Tr(Δα) = 1/3(Δαxx + Δαyy + Δαzz)). The other electrostatic property that can be determined at the magic angle is the magnitude of the change in permanent dipole moment,
is one third the trace of the change in the second-rank polarizability tensor, Δα, (1/3Tr(Δα) = 1/3(Δαxx + Δαyy + Δαzz)). The other electrostatic property that can be determined at the magic angle is the magnitude of the change in permanent dipole moment, 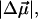 which can be derived from c54.7, e.g., Eq. 4. These electronic quantities reported in Table 2 have not been corrected for the internal field factor, f, which includes a reaction field due to the dielectric properties of the environment, and a cavity field (Bottcher, 1952).
which can be derived from c54.7, e.g., Eq. 4. These electronic quantities reported in Table 2 have not been corrected for the internal field factor, f, which includes a reaction field due to the dielectric properties of the environment, and a cavity field (Bottcher, 1952).
The coefficients, aχ, bχ, and cχ, are extracted by means of a linear least-squares fit of the electroabsorption signal to the sum of derivatives of 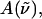 using a program written in MATLAB. All the Stark spectra have been smoothed using a combination of polynomial filtering and a sliding-average mean method. The absorption spectra are fit to a sum of three to five Gaussians using a simplex algorithm to generate smooth derivatives. The absorption spectra of WT-PYP have also been modeled using only two Gaussians (Hendriks et al., 2002). However, due to the presence of photoproducts, and to model the vibronic features that are present in the spectra, we require four to five Gaussians to model the absorption spectra here.
using a program written in MATLAB. All the Stark spectra have been smoothed using a combination of polynomial filtering and a sliding-average mean method. The absorption spectra are fit to a sum of three to five Gaussians using a simplex algorithm to generate smooth derivatives. The absorption spectra of WT-PYP have also been modeled using only two Gaussians (Hendriks et al., 2002). However, due to the presence of photoproducts, and to model the vibronic features that are present in the spectra, we require four to five Gaussians to model the absorption spectra here.
RESULTS
Absorption spectra
In Fig. 1, the absorption spectra of WT-PYP, L-PYP and E46Q in standard Tris-HCl buffer solutions at pH 7.5 are shown along with their respective absorption spectra in glycerol/buffer solutions at room temperature and in the corresponding glycerol/ buffer glasses that are formed at 77 K (see Table 1 for absorption maxima). In WT-PYP, the absorption maximum in Tris HCl buffer at pH 7.5, shown in the solid line in Fig. 1 a (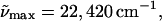 λmax = 446 nm), red-shifts by 100 cm−1 (2 nm) on the addition of two equivalents of glycerol (by weight) to the buffer solution at room temperature (Fig. 1 a, dotted line). On lowering the temperature to 77 K, the absorption maximum, now blue shifts by 100 cm−1 (Fig. 1 a, dot-dash line). Furthermore, at low temperature the absorption spectrum sharpens to reveal a shoulder at 23,920 cm−1 (418 nm) corresponding to a vibronic progression of 1500 cm−1. This mode is associated with the CC stretch around which trans-cis isomerization occurs (Brudler et al., 2001; Unno et al., 2002). The electronic transition energy of E46Q, where the glutamate residue present in WT-PYP is replaced by a glutamine, is much lower than that of WT-PYP at room temperature in the Tris-HCl buffer (
λmax = 446 nm), red-shifts by 100 cm−1 (2 nm) on the addition of two equivalents of glycerol (by weight) to the buffer solution at room temperature (Fig. 1 a, dotted line). On lowering the temperature to 77 K, the absorption maximum, now blue shifts by 100 cm−1 (Fig. 1 a, dot-dash line). Furthermore, at low temperature the absorption spectrum sharpens to reveal a shoulder at 23,920 cm−1 (418 nm) corresponding to a vibronic progression of 1500 cm−1. This mode is associated with the CC stretch around which trans-cis isomerization occurs (Brudler et al., 2001; Unno et al., 2002). The electronic transition energy of E46Q, where the glutamate residue present in WT-PYP is replaced by a glutamine, is much lower than that of WT-PYP at room temperature in the Tris-HCl buffer (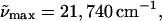 λmax = 460 nm: solid line in Fig. 1 b). On the addition of glycerol to the buffer solution there is a similar 90 cm−1 red-shift of the absorption maximum to 21,650 cm−1 as in WT-PYP (Fig. 1 b, dotted line), which on cooling to 77 K is blue shifted by 140 cm−1 to 21,790 cm−1 (459 nm) (Fig. 1 b, dot-dash line).
λmax = 460 nm: solid line in Fig. 1 b). On the addition of glycerol to the buffer solution there is a similar 90 cm−1 red-shift of the absorption maximum to 21,650 cm−1 as in WT-PYP (Fig. 1 b, dotted line), which on cooling to 77 K is blue shifted by 140 cm−1 to 21,790 cm−1 (459 nm) (Fig. 1 b, dot-dash line).
FIGURE 1.

Absorption spectra of (a) WT-PYP, (b) L-PYP, and (c) E46Q, in Tris-HCl buffer (pH 7.5) at 298 K (solid line), in glycerol-buffer (55:45 v:v) solution at 298 K (dotted line), glycerol-buffer glass at 77 K of dark-state sample (dot-dash line).
TABLE 1.
Electronic transition energies of WT-PYP, E46Q, and L-PYP
(a) 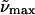 (λmax) buffer 298K (λmax) buffer 298K |
(b) 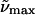 (λmax) gly/buffer 298K (λmax) gly/buffer 298K |
(c) 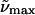 (λmax) gly/buffer 77K (λmax) gly/buffer 77K |
(d) 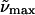 (λmax) gly/buffer 77K (λmax) gly/buffer 77K |
(e) 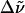 (a)–(b) (a)–(b) |
(f) 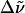 (a)–(c) [(a)–(d)] (a)–(c) [(a)–(d)] |
|
|---|---|---|---|---|---|---|
| WT-PYP | 22420 (446) | 22320 (448) | 22420 (446) | 22520 (444) | +100 | 0 [−100] |
| E46Q | 21740 (460) | 21650 (462) | 21790 (459) | 21980 (455) | +90 | 50 [−240] |
| L-PYP | 22520 (444) | 22420 (446) | 22520 (444) | 22520 (444) | +100 | 0 [0] |
All absorption maxima are reported in wavenumbers. The values in nanometers are given in parentheses. Experimental conditions: (a) Room temperature in Tris-HCl buffer at pH 7.5. (b) Room temperature in Tris-HCl buffer and 66% w:v glycerol solution. (c) 77 K glycerol-buffer glass before illumination with blue light (see Experimental). (d) 77 K glycerol-buffer glass following illumination with blue light. (e) Difference in energy between the absorption maxima in Tris-HCl buffer versus glycerol-buffer solution at room temperature. (f) Difference in energy between the absorption maxima in the glycerol-buffer glasses at 77 K versus in buffer solution at room temperature, before and after illumination with blue light.
The cofactor in L-PYP is generated by modifying the chromophore in WT-PYP to prevent it from undergoing trans-cis isomerization (Scheme 2 c). However, in contrast to E46Q the protein environment is unaltered from that of WT-PYP. Therefore the narrowing of the absorption spectrum of L-PYP by nearly 1800 cm−1, compared to WT-PYP and E46Q, can be attributed to the alteration of the chromophore (Fig. 1 c, dark solid line). On addition of glycerol to the buffer solution, the maximum of the electronic transition energy of L-PYP (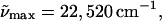 λmax = 444 nm: solid line in Fig. 1 c) red-shifts by 100 cm−1, similar to WT-PYP at room temperature (Fig. 1 c, solid line). On cooling to 77 K, the absorption maximum of L-PYP in the glycerol-buffer glass blue-shifts by 100 cm−1 (dot-dash line). In addition, the absorption spectrum of L-PYP, also shows greater structure than that observed in WT-PYP and it is possible to discern a distinct shoulder at 23,980 cm−1 (417 nm). Note that the addition of glycerol at room temperature produces a similar shift of the absorption maxima of all three species, while there is only a 100 cm−1 difference between the shift in the absorption maximum between WT-PYP and L-PYP when the temperature is lowered. This may lead one to conclude that the protein environment is not very polar, given the large difference in the electronic properties between WT-PYP and L-PYP (see below). However, to draw such a conclusion, it is necessary to know the gas-phase electronic transition energies of both species.
λmax = 444 nm: solid line in Fig. 1 c) red-shifts by 100 cm−1, similar to WT-PYP at room temperature (Fig. 1 c, solid line). On cooling to 77 K, the absorption maximum of L-PYP in the glycerol-buffer glass blue-shifts by 100 cm−1 (dot-dash line). In addition, the absorption spectrum of L-PYP, also shows greater structure than that observed in WT-PYP and it is possible to discern a distinct shoulder at 23,980 cm−1 (417 nm). Note that the addition of glycerol at room temperature produces a similar shift of the absorption maxima of all three species, while there is only a 100 cm−1 difference between the shift in the absorption maximum between WT-PYP and L-PYP when the temperature is lowered. This may lead one to conclude that the protein environment is not very polar, given the large difference in the electronic properties between WT-PYP and L-PYP (see below). However, to draw such a conclusion, it is necessary to know the gas-phase electronic transition energies of both species.
Photoproduct formation
In Fig. 2 we compare the effects on the absorption and Stark spectra of WT-PYP (a) and E46Q (b) in the glycerol/buffer glasses before and following blue-light illumination at 77 K. In both WT-PYP and in E46Q, before illumination with blue-light, not only the absorbances, but the Stark signals are also close to zero, below 20,000 cm−1 (Fig. 2, dot-dashed lines). However, it is not possible to prevent photoproduct generation in WT-PYP, nor in E46Q, in the process of obtaining multiple reproducible scans in the wavelength region between 370 and 570 nm (see Materials and Methods section). Exposure of these protein samples to blue light (<440 nm for WT-PYP and <450 nm for E46Q at 1 to 2 μW) results in two changes that are evident in the absorption spectra of WT-PYP and E46Q (Fig. 2 a and b, light solid lines): the absorption maxima blue-shift and there is a simultaneous appearance of red absorbing photoproducts. In WT-PYP, the additional blue-shift of the absorption maximum is on the order of 100 cm−1 (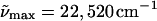 ) while it is more than a factor-of-two larger in E46Q (
) while it is more than a factor-of-two larger in E46Q (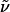 max = 21,980 cm−1) (see Table 1). The red photoproduct that is formed in the WT-PYP sample with an absorption maximum at ≈20,400 cm−1 (490 nm) is somewhat narrower than that formed in E46Q and more strongly overlapped with the absorbance of the dark-state species (Fig. 2: compare light solid lines between 18,000 and 21,000 cm−1 in a and b). The appearance of the red absorbing species is accompanied by a corresponding decrease in the absorbance of the dark-state species (≈20% in WT-PYP and ≈35% in E46Q).
max = 21,980 cm−1) (see Table 1). The red photoproduct that is formed in the WT-PYP sample with an absorption maximum at ≈20,400 cm−1 (490 nm) is somewhat narrower than that formed in E46Q and more strongly overlapped with the absorbance of the dark-state species (Fig. 2: compare light solid lines between 18,000 and 21,000 cm−1 in a and b). The appearance of the red absorbing species is accompanied by a corresponding decrease in the absorbance of the dark-state species (≈20% in WT-PYP and ≈35% in E46Q).
In previous low-temperature studies, the hypsochromic shifts in the absorption maxima of WT-PYP and E46Q have been attributed to the formation of the intermediates, PYPH and E46QH, while the red absorption is ascribed to the formation of PYPB and E46QB, respectively (Imamoto et al., 1996; Zhou et al., 2001). While we do not observe A440, the photoproduct previously observed by Hoff and co-workers (Hoff et al., 1992), it is possible that this species could be similar to PYPH. We note that PYPM (pB355), the protonated intermediate with a strongly blue-shifted absorption (λmax = 355 nm), is not formed because the large structural changes required of the protein (partial unfolding) are restricted at 77 K. In contrast to WT-PYP and E46Q, in L-PYP at low temperature, red photoproducts are not generated on excitation with blue light and nor is there an additional blue-shift of the absorption maximum, in agreement with the observations of Cordfunke and co-workers (Cordfunke et al., 1998). The diminution in the intensity of the absorption spectrum previously observed (Cordfunke et al., 1998) is not evident in our current measurements because a much weaker light regime is used.
The Stark signals of WT-PYP and E46Q, that are dominated by the field-dependent change of the main absorption band, also exhibit changes that coincide with those observed in the absorption spectra on blue-light illumination (dark solid lines in Fig. 2). After the first scan acquired across the entire absorption band, the intensities of these Stark signals decrease by ≈10–15% in WT-PYP and 25–30% in E46Q. Concurrently, a new but weak Stark signal is detected in the region of the red photoproduct absorbance for both samples (Fig. 2, dark solid lines. 18,000–21,000 cm−1). This loss in intensity of the main Stark signal corresponds to the overall decrease in absorbance of the dark state species. And although the decrease in the intensity of the absorption spectra of WT-PYP and E46Q is comparatively smaller than that of the corresponding Stark signals, an estimation of the electronic parameters obtained from the fits to these initial spectra indicate that they do not differ significantly from those extracted following photoproduct generation (results not reported here). In addition, we tried to mimic the results of Imamoto et. al, where we used a strong light regime (500 μW at 500 nm for 15 min) to bleach the red photoproduct absorption for both WT-PYP and E46Q (Imamoto et al., 1996; Mataga et al., 2000). We were able to successfully bleach the absorption in this region and also recover most of the absorbance and the Stark signal of the dark-state species (results not shown). Slight differences are observed with respect to the spectra obtained before bleaching the sample with high light intensity. Whether these changes in the Stark signal are indicative of changes in the electronic properties of these systems under these treatment conditions, is currently under investigation. Here, we only present results that have been obtained from Stark experiments on samples that have been subjected to a weak light regime. Note that during the acquisition of successive stepwise scans, following the generation of photoproducts, the intensity of the red absorption band in both WT-PYP and E46Q, relative to the main absorption, does not change under continuing illumination, ensuring that reproducible absorption and electroabsorption spectra are obtained under our experimental conditions.
Stark/electroabsorption spectra
The absorption spectra at 77 K and the corresponding Stark spectra and their fits are shown in Figs. 3–5 for WT-PYP, E46Q, and L-PYP, respectively. The absorption spectra obtained at 77 K in a glycerol/buffer glass are shown in a of each figure. The Stark spectra, shown in b (dark solid lines in Figs. 3 b, 4 b, and 5 b), have been normalized to the square of the applied electric field (V/cm) because the change in absorption, 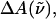 is proportional to this quantity, e.g., Eq. 1. These electroabsorption spectra are fit to the (wavenumber-normalized) derivatives of the absorption spectra to obtain the electronic properties of interest,
is proportional to this quantity, e.g., Eq. 1. These electroabsorption spectra are fit to the (wavenumber-normalized) derivatives of the absorption spectra to obtain the electronic properties of interest, 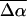 and
and 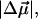 from the coefficients of
from the coefficients of 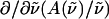 and
and 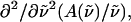 respectively. The fits obtained from the linear least-squares fits to the absorption lineshapes are shown in the dark-dashed line in Figs. 3 b, 4 b, and 5 b. The absorption spectra are fit to a sum of (four or five) Gaussians to yield smooth lineshapes to generate smooth derivatives. Shown along with the Stark spectra and the fits, are the zeroth (light dotted), first (light dashed) and second (light dot-dashed) derivative components of the fit, scaled by their respective coefficients. These scaled derivatives are the first, second, and third terms, in Eq. 1. The average change in polarizability,
respectively. The fits obtained from the linear least-squares fits to the absorption lineshapes are shown in the dark-dashed line in Figs. 3 b, 4 b, and 5 b. The absorption spectra are fit to a sum of (four or five) Gaussians to yield smooth lineshapes to generate smooth derivatives. Shown along with the Stark spectra and the fits, are the zeroth (light dotted), first (light dashed) and second (light dot-dashed) derivative components of the fit, scaled by their respective coefficients. These scaled derivatives are the first, second, and third terms, in Eq. 1. The average change in polarizability, 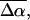 and the magnitude of the change in dipole moment,
and the magnitude of the change in dipole moment, 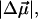 obtained from the coefficients of the first- and second-derivatives of the fit (bχ and cχ) to the overall absorption band are reported in Table 2.
obtained from the coefficients of the first- and second-derivatives of the fit (bχ and cχ) to the overall absorption band are reported in Table 2.
FIGURE 3.

(a) Absorption spectra of WT-PYP in Tris-HCl buffer: glycerol (55:45 v:v) at 77 K (dark solid line). The absorption lineshape is deconvolved into the absorption due to pG and the red photoproduct, PYPB (light solid lines; the original absorption spectrum is shown in the light dashed line). (b) The electroabsorption spectra (dark solid line) and fit (dark dot-dashed line) to the overall absorption lineshape (dark solid line in a) are shown along with the individual contributions of the zeroth (light dotted), first derivative (light dashed), and second derivative (light dot-dashed) components of the absorption spectrum (see Eq. 1). (c) Here the fit to the derivatives of the absorption bands of pG and PYPB (light solid lines in a) is shown. The legend key is the same as in b.
FIGURE 5.

(a) Absorption spectra of L-PYP in Tris-HCl buffer: glycerol (54:46 v:v) at 77 K (dark solid line). (b) The electroabsorption spectra (dark solid line) and fit (dark dot-dashed line) to the absorption spectrum (dark solid line in a) are shown along with the individual contributions of the zeroth (light dotted), first derivative (light dashed), and second derivative (light dot-dashed) components of the absorption spectrum.
FIGURE 4.

(a) Absorption spectra of E46Q in Tris-HCl buffer: glycerol (55:45 v:v) at 77 K (dark solid line). The absorption lineshape is deconvolved into the absorption due to E46Q and the red photoproduct, E46QB (light solid lines). (b) The electroabsorption spectra (dark solid line) and fit (dark dot-dashed line) to the overall absorption lineshape (dark solid line in a) are shown along with the individual contributions of the zeroth (light dotted), first derivative (light dashed), and second derivative (light dot-dashed) components of the absorption spectrum (see Eq. 1). (c) Here the fit to the derivatives of the absorption bands of E46Q and E46QB (light solid lines in a) is shown. The legend key is the same as in b.
In WT-PYP, the largest contribution to the field-dependent change in the absorbance arises due to the second-derivative component (Fig. 3 b: light dot-dash line) from which the change in dipole moment, 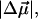 is calculated to be 25 D. The difference between the second- derivative component and the Stark signal is compensated for by the much smaller first- derivative component. This corresponds to an average change in polarizability,
is calculated to be 25 D. The difference between the second- derivative component and the Stark signal is compensated for by the much smaller first- derivative component. This corresponds to an average change in polarizability, 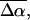 on the order of 1000 Å3. A relatively good fit to the dominant part of the electroabsorption signal is obtained from the weighted sum of the derivatives of the overall absorption spectrum. As it is possible to reproduce the Stark signal quite well using a single set of electro-optical parameters, the values of
on the order of 1000 Å3. A relatively good fit to the dominant part of the electroabsorption signal is obtained from the weighted sum of the derivatives of the overall absorption spectrum. As it is possible to reproduce the Stark signal quite well using a single set of electro-optical parameters, the values of 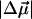 and
and 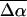 obtained from this fit provide a good estimation of the change in electronic properties for the main electronic transition. However, the fit is poor at the red edge of the absorption (<21,000 cm−1). Although the intensity of the electroabsorption signal is reproduced here, the fit to the overall shape is poor, particularly in comparison to the rest of the fit (residuals not shown). This strongly suggests that underlying this part of the absorption band there is either an electronic or a vibronic transition with electronic properties that are different from that of the dark-state species. Indeed, this wavelength region corresponds to the absorption of PYPB and therefore it is not surprising that this species, which is considered to be structurally different from pG, exhibits different electronic properties. In contrast, the good fit to the main absorption band, where both pG and PYPH absorb, indicates that PYPH is electronically quite similar to pG and therefore perhaps structurally similar as well.
obtained from this fit provide a good estimation of the change in electronic properties for the main electronic transition. However, the fit is poor at the red edge of the absorption (<21,000 cm−1). Although the intensity of the electroabsorption signal is reproduced here, the fit to the overall shape is poor, particularly in comparison to the rest of the fit (residuals not shown). This strongly suggests that underlying this part of the absorption band there is either an electronic or a vibronic transition with electronic properties that are different from that of the dark-state species. Indeed, this wavelength region corresponds to the absorption of PYPB and therefore it is not surprising that this species, which is considered to be structurally different from pG, exhibits different electronic properties. In contrast, the good fit to the main absorption band, where both pG and PYPH absorb, indicates that PYPH is electronically quite similar to pG and therefore perhaps structurally similar as well.
To distinguish between the properties of these two species, the absorption spectrum is modeled as two separate bands corresponding to pG (plus PYPH) and the red photoproduct, PYPB (Fig. 3 a, see bands labeled as pG and PYPB). The electroabsorption signal is then fit to the weighted sum of the derivatives of both these bands, i.e., six components. Indeed the quality of this fit (Fig. 3 c, dark dot-dashed line) is distinctly superior to the previous fit, particularly at the red edge (compare to Fig. 3 b). Because the absorption spectrum is separated into the absorption arising from pG and PYPB, it is possible to obtain the electronic properties for each of these species independently. The corresponding 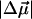 and
and 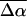 values for pG and PYPB obtained from this fit are also reported in Table 2 (see columns labeled Main transition and Red transition, respectively). We find that the electronic properties of the main absorption band (pG) obtained from this fit,
values for pG and PYPB obtained from this fit are also reported in Table 2 (see columns labeled Main transition and Red transition, respectively). We find that the electronic properties of the main absorption band (pG) obtained from this fit, 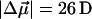 and
and 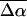 = 1000 Å3, are quite similar to the values obtained from the fit to the overall absorption spectrum. Although it is now possible to determine the change in electronic properties of the red photoproduct independently, because the signal is very small and the standard deviation of these properties is quite large, we caution that the results from this fit should be regarded primarily as an estimate for the electronic properties of PYPB. Overall, we find that the change in dipole moment (≈10 D) associated with this latter transition is smaller than that of pG while the change in polarizability (≈2000 Å3) is twice as large as that of pG.
= 1000 Å3, are quite similar to the values obtained from the fit to the overall absorption spectrum. Although it is now possible to determine the change in electronic properties of the red photoproduct independently, because the signal is very small and the standard deviation of these properties is quite large, we caution that the results from this fit should be regarded primarily as an estimate for the electronic properties of PYPB. Overall, we find that the change in dipole moment (≈10 D) associated with this latter transition is smaller than that of pG while the change in polarizability (≈2000 Å3) is twice as large as that of pG.
The fits and results obtained for E46Q are similar to WT-PYP. The fit to the weighted sum of the derivatives of the overall absorption lineshape (dark dot-dash line in Fig. 4 b) also comprises a dominant second-derivative component (light dot-dash line in Fig. 4 b). The corresponding change in dipole moment, 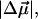 of 20 D although somewhat smaller than in WT-PYP, is also indicative of extensive charge motion. The fit to the overall absorption lineshape in the region of the red photoproduct (<20,000 cm−1) and at the red edge of the main absorption band (20,000–21,000 cm−1 in Fig. 4 b) is of even lower quality than in WT-PYP. Therefore, here too the absorption band is deconvolved into two separate bands (Fig. 4 a) corresponding to E46QB to the red (18,000–20,000 cm−1), and the dark-state species E46Q (and E46QH) underlying the main absorption band (20,000–25,000 cm−1). The distinctly different change in electronic properties that the red-shifted species undergoes on excitation is the cause of the poor fit to the overall absorption lineshape. Here too, the fit to the derivatives of two bands is markedly improved from that to a single electronic transition (compare dark dot-dash line in Fig. 4 b to that in c). From this fit, the values of
of 20 D although somewhat smaller than in WT-PYP, is also indicative of extensive charge motion. The fit to the overall absorption lineshape in the region of the red photoproduct (<20,000 cm−1) and at the red edge of the main absorption band (20,000–21,000 cm−1 in Fig. 4 b) is of even lower quality than in WT-PYP. Therefore, here too the absorption band is deconvolved into two separate bands (Fig. 4 a) corresponding to E46QB to the red (18,000–20,000 cm−1), and the dark-state species E46Q (and E46QH) underlying the main absorption band (20,000–25,000 cm−1). The distinctly different change in electronic properties that the red-shifted species undergoes on excitation is the cause of the poor fit to the overall absorption lineshape. Here too, the fit to the derivatives of two bands is markedly improved from that to a single electronic transition (compare dark dot-dash line in Fig. 4 b to that in c). From this fit, the values of 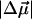 and
and 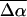 for the dark-state species are found to be 22 D and 1200 Å3, respectively. The red photoproduct, E46QB, exhibits a larger change in polarizability (2400 Å3) and a much smaller change in dipole moment (12 D) than the dark-state species. For the same reasons as outlined for WT-PYP, here too the electronic properties of the red band reported in Table 2 must be regarded as an estimate.
for the dark-state species are found to be 22 D and 1200 Å3, respectively. The red photoproduct, E46QB, exhibits a larger change in polarizability (2400 Å3) and a much smaller change in dipole moment (12 D) than the dark-state species. For the same reasons as outlined for WT-PYP, here too the electronic properties of the red band reported in Table 2 must be regarded as an estimate.
We note that the fit to the blue edge of the absorption band of both WT-PYP (>24,000 cm−1) and E46Q (>23,500 cm−1) are not good compared to the main absorption band (residuals not shown). We did not however attempt to determine the electrostatic properties of the species that gives rise to a field-dependent signal in this region that differs from that of the main absorption at lower energy, for two reasons. First, it appears that only a small amount of this species is present in the sample as evident from the much smaller magnitude of the Stark signal. Secondly, the Gaussian lineshape that would be used to model the absorption of these species would significantly overlap with the absorption of the dark-state species thereby sharply reducing the reliability of the electro-optical parameters extracted for this species from such fits.
In contrast to WT-PYP and E46Q, the absorption spectrum of L-PYP in a glycerol-buffer glass at 77 K does not show evidence for the presence of any red photoproducts, even after 2–3 h of obtaining repetitive scans down to 370 nm (Fig. 5 a). Previously, Cordfunke and co-workers (Cordfunke et al., 1998) have shown that following illumination with a strong light regime, there are minor changes in the intensity of the low-temperature absorption spectrum of L-PYP. The bleaching of the absorption spectrum was then attributed to the formation of a pR-like intermediate in L-PYP. We do not observe such a bleaching in the absorption spectrum and more importantly, neither do we observe a change in the Stark spectrum in the process of acquiring multiple scans. We are able to avoid bleaching of our L-PYP sample probably because of the comparatively lower light intensity (1 μW) than that used by Cordfunke and co-workers (Cordfunke et al., 1998).
The electroabsorption spectrum (dark solid line) and fit (dark dashed line) to the absorption band of L-PYP are shown in Fig. 5 b. The fit to the derivatives of the absorption band is indeed very good, and unlike WT-PYP and E46Q, at the red and blue edges as well. From this fit we find that 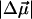 in L-PYP is 13 D. Although it is only about half the magnitude of that measured in WT-PYP, it is nonetheless quite large and probably also arises due to translocation of the negative charge localized on the phenolate oxygen in the ground state (Scheme 2 c). Here too, the major contribution to the field-dependent response arises due to the change in dipole moment (second-derivative term) with a much smaller contribution from the first derivative component.
in L-PYP is 13 D. Although it is only about half the magnitude of that measured in WT-PYP, it is nonetheless quite large and probably also arises due to translocation of the negative charge localized on the phenolate oxygen in the ground state (Scheme 2 c). Here too, the major contribution to the field-dependent response arises due to the change in dipole moment (second-derivative term) with a much smaller contribution from the first derivative component. 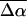 is ≈ 150 Å3, indicating that a smaller change in the induced dipole moment or a relatively smaller difference in the degree of delocalization in the excited versus the ground state occurs in L-PYP compared to WT-PYP. Although the magnitude of the field-dependent signal of L-PYP is similar to that of WT-PYP, the values of
is ≈ 150 Å3, indicating that a smaller change in the induced dipole moment or a relatively smaller difference in the degree of delocalization in the excited versus the ground state occurs in L-PYP compared to WT-PYP. Although the magnitude of the field-dependent signal of L-PYP is similar to that of WT-PYP, the values of 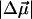 and
and 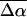 of L-PYP are smaller than that of WT-PYP. This is because the absorption band of L-PYP is much narrower than WT- PYP (compare Fig. 1 a to c) and therefore the magnitudes of the derivatives of the more steeply sloping absorption band of L-PYP are a factor of 2–3 larger. Consequently the coefficients of these derivatives, that are related to
of L-PYP are smaller than that of WT-PYP. This is because the absorption band of L-PYP is much narrower than WT- PYP (compare Fig. 1 a to c) and therefore the magnitudes of the derivatives of the more steeply sloping absorption band of L-PYP are a factor of 2–3 larger. Consequently the coefficients of these derivatives, that are related to 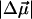 and
and 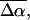 are a factor-of-two smaller than in WT-PYP.
are a factor-of-two smaller than in WT-PYP.
DISCUSSION
Electronic properties of WT-PYP
The electrostatic properties, 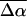 and
and 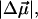 of the dark state species of WT-PYP (20,000–26,000 cm−1) in a glycerol-buffer glass at 77 K are obtained from the first- and second-derivative components of the fits to the absorption lineshape in two different ways, as described in the previous section. The change in the electrostatic properties in this electronic region shows that photo-excitation results in a significant change in the degree of charge separation in the excited state compared to the ground state. This is evident from the large
of the dark state species of WT-PYP (20,000–26,000 cm−1) in a glycerol-buffer glass at 77 K are obtained from the first- and second-derivative components of the fits to the absorption lineshape in two different ways, as described in the previous section. The change in the electrostatic properties in this electronic region shows that photo-excitation results in a significant change in the degree of charge separation in the excited state compared to the ground state. This is evident from the large 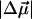 of 26 D (Table 2). The dipole moment which is quite large in such a small chromophore, suggests that it would be very sensitive to changes in the dielectric properties of its environment and even a small increase in the polarity of the protein pocket could produce large spectral shifts. However, not only is the magnitude of the shift in the absorption maximum small (100 cm−1), but we also find that at 77 K the electronic transition energy of WT-PYP shifts to lower energy in the glycerol-buffer environment. Typically, when the temperature is lowered the polarity (static dielectric) of the environment is expected to increase, which should result in a red-shift of the absorption maximum if μe > μg, resulting from greater stabilization of the excited state dipole. Indeed the opposite seems to be the case, assuming that the polarity of the environment does increase on lowering the temperature. DFT calculations which yield a value of 25 D for the ground state dipole moment in the gas phase (F. Buda, Leiden University, personal communication, 2002), might then imply that μe is actually close to zero, given that the measured value of
of 26 D (Table 2). The dipole moment which is quite large in such a small chromophore, suggests that it would be very sensitive to changes in the dielectric properties of its environment and even a small increase in the polarity of the protein pocket could produce large spectral shifts. However, not only is the magnitude of the shift in the absorption maximum small (100 cm−1), but we also find that at 77 K the electronic transition energy of WT-PYP shifts to lower energy in the glycerol-buffer environment. Typically, when the temperature is lowered the polarity (static dielectric) of the environment is expected to increase, which should result in a red-shift of the absorption maximum if μe > μg, resulting from greater stabilization of the excited state dipole. Indeed the opposite seems to be the case, assuming that the polarity of the environment does increase on lowering the temperature. DFT calculations which yield a value of 25 D for the ground state dipole moment in the gas phase (F. Buda, Leiden University, personal communication, 2002), might then imply that μe is actually close to zero, given that the measured value of 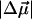 is 26 D. (The difference in charge distributions of the excited versus the ground state (Mollina and Merchán, 2001) however indicates that the excited state dipole moment is unlikely to be zero, but may be very small). We note that the values of
is 26 D. (The difference in charge distributions of the excited versus the ground state (Mollina and Merchán, 2001) however indicates that the excited state dipole moment is unlikely to be zero, but may be very small). We note that the values of 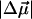 and
and 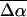 include an additional enhancement factor in the protein cavity due to the local reaction field of the environment in addition to the cavity field (Bottcher, 1952). We can estimate this factor to be on the order of 10% assuming a static dielectric constant of 5–10 (Demchuk et al., 2000) in the protein cavity and a molecular volume of 60 Å3 (see Chowdhury et al., 1999) for obtaining an estimate of this enhancement factor). Note that the small shifts produced in the absorption maxima on the addition of glycerol and on lowering the temperature suggest that there is only a small change in the polarity in the protein cavity due to these events.
include an additional enhancement factor in the protein cavity due to the local reaction field of the environment in addition to the cavity field (Bottcher, 1952). We can estimate this factor to be on the order of 10% assuming a static dielectric constant of 5–10 (Demchuk et al., 2000) in the protein cavity and a molecular volume of 60 Å3 (see Chowdhury et al., 1999) for obtaining an estimate of this enhancement factor). Note that the small shifts produced in the absorption maxima on the addition of glycerol and on lowering the temperature suggest that there is only a small change in the polarity in the protein cavity due to these events.
The value of the dipole moment change gives an estimate of the extent of charge separation between positive and negative moieties in the molecules, where one unit of electronic charge separated by 1 Å corresponds to ≈4.8 D in vacuo. This implies that on excitation there is a change in the charge separation of one unit of electronic charge over a distance of ≈5 Å in WT-PYP. In charged molecules, such as the ones studied here, it is difficult to precisely define the separation of unique positive and negative moieties in the ground or excited state of the absorbing species. However, the change in dipole moment may be defined with respect to a fixed origin, such that 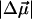 is a measure of the relative difference in the charge distribution of the excited versus the ground state. It is then not necessary to uniquely identify such an origin because it would be the same in the ground state and in the Frank-Condon region. Interestingly, the
is a measure of the relative difference in the charge distribution of the excited versus the ground state. It is then not necessary to uniquely identify such an origin because it would be the same in the ground state and in the Frank-Condon region. Interestingly, the 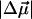 measured in WT-PYP is much larger than that measured in some other coumarins. Changes in dipole moment on the order of 5–7 D have been measured in coumarin 153 (Chowdhury et al., 1999), while initial experiments on protonated thio-methyl pCA, the chromophore in WT-PYP, in glycerol/buffer glasses yield
measured in WT-PYP is much larger than that measured in some other coumarins. Changes in dipole moment on the order of 5–7 D have been measured in coumarin 153 (Chowdhury et al., 1999), while initial experiments on protonated thio-methyl pCA, the chromophore in WT-PYP, in glycerol/buffer glasses yield 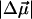 's of a similarly smaller magnitude (work in progress). The crucial difference in these aforementioned coumarins, that have been studied in (organic) solvent environments, is that they are neutral in the ground state. It appears therefore, that in WT-PYP the large
's of a similarly smaller magnitude (work in progress). The crucial difference in these aforementioned coumarins, that have been studied in (organic) solvent environments, is that they are neutral in the ground state. It appears therefore, that in WT-PYP the large 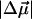 is a direct consequence of the charged nature of the chromophore in the ground state. The estimated charge-transfer distance of 5 Å implies that the negative charge localized on the phenolate oxygen atom, O1, in the ground state, is displaced toward the thio-ester moiety (Scheme 3). This proposal is further substantiated by calculations on model pCA systems (Groenhof et al., 2002; Mollina and Merchán, 2001; Sergi et al., 2001).
is a direct consequence of the charged nature of the chromophore in the ground state. The estimated charge-transfer distance of 5 Å implies that the negative charge localized on the phenolate oxygen atom, O1, in the ground state, is displaced toward the thio-ester moiety (Scheme 3). This proposal is further substantiated by calculations on model pCA systems (Groenhof et al., 2002; Mollina and Merchán, 2001; Sergi et al., 2001).
SCHEME 3.

Model for charge redistribution on excitation.
In Scheme 3 we outline a model for the propagation of the negative charge in the excited state. Charge can be shifted toward the electrophilic carbon double bond, C7C8 (Scheme 3, IA–IIA), or to the electrophilic carbonyl oxygen, O2 (Scheme 3, IB–IIB). The latter proposal is also suggested by CASSCF calculations on the transisomer of an isolated thio-methyl pCA molecule (Mollina and Merchán, 2001). These calculations show that overall electronic charge shifts from O1 to O2. The molecular orbital charge difference picture clearly shows a hole on O1 with increased electronic charge density on not only O2, but C7 as well. Furthermore, DFT calculations show that the electronic charge localized on the phenolate anion in the ground state is mostly shifted to C7 (Sergi et al., 2001). What we suggest, and which is also evident in electronic structure calculations, is that the negative electronic charge shifted from O1, is actually delocalized over the C CCO unit (Scheme 3, IIC). The crux of the proposal is that the single-bond character of the C7C8 bond increases due to electronic charge displacement. The greater flexibility in the C7C8 bond resulting from this charge shift would thereby facilitate the twisting of this moiety in the excited state. This could explain why the bathochromic intermediates, which are considered to be in twisted conformations, are so easily generated even at 77 K despite the isomerization process having an activation barrier of 25 kcal/mol (van Brederode et al., 1996; Sergi et al., 2001). Clearly a weakening of the CC double bond would significantly decrease the activation barrier for the trans-cis isomerization in the excited state. Furthermore, this proposal is supported by DFT calculations where the decrease in the double-bond character of the C7C8 bond in the excited state is clearly followed by a lengthening of this bond in the transition state, which, these authors also conclude, lowers the barrier to isomerization (Sergi et al., 2001).
CCO unit (Scheme 3, IIC). The crux of the proposal is that the single-bond character of the C7C8 bond increases due to electronic charge displacement. The greater flexibility in the C7C8 bond resulting from this charge shift would thereby facilitate the twisting of this moiety in the excited state. This could explain why the bathochromic intermediates, which are considered to be in twisted conformations, are so easily generated even at 77 K despite the isomerization process having an activation barrier of 25 kcal/mol (van Brederode et al., 1996; Sergi et al., 2001). Clearly a weakening of the CC double bond would significantly decrease the activation barrier for the trans-cis isomerization in the excited state. Furthermore, this proposal is supported by DFT calculations where the decrease in the double-bond character of the C7C8 bond in the excited state is clearly followed by a lengthening of this bond in the transition state, which, these authors also conclude, lowers the barrier to isomerization (Sergi et al., 2001).
The other effect of such a charge transfer is the conversion of O1 from a single-bonded negative phenolate oxygen to a double-bonded neutral oxygen. Such a change would affect the nature of the hydrogen bonding interaction with the Glu46/Gln46 and Tyr42 residues. This might be expected to result in weakening of the hydrogen bonding with the adjacent protein residues due to a loss of negative charge from the phenolate oxygen (Scheme 3, IA or IB). However, a carbonyl oxygen is itself very electrophilic and it may be argued that the conversion of O1 from a single bonded negative oxygen to a neutral double-bonded oxygen (O− toO0) would provide better electronic overlap with the adjacent hydrogen atom of either Tyr42 or Glu46, thereby strengthening the hydrogen bonds with these residues. Therefore the hydrogen bonding interactions within the protein pocket and, by extension, the whole protein is preserved on excitation. Low-temperature FTIR measurements on PYPH, which is proposed to be a product state generated on excitation of pG, exhibits a downshift in the phenolate CO stretching mode, indicating that the hydrogen bond strength with the Glu46 residue is stronger than in the ground state of pG (Imamoto et al., 2001a; Xie et al., 1996). Therefore, it is possible that the excited state electronic structure of pG would be similar to ground-state PYPH, where a change in the hybridization of the phenolate oxygen, O1, results in the conversion of O− toO0. The strengthening of the hydrogen bond with Glu46 is also evident in the well-characterized I1 intermediate from time-resolved FTIR spectra (Brudler et al., 2001; Xie et al., 2001). Such a strengthening of the hydrogen bond to Glu46 in WT-PYP, which would be induced by photon absorption, could be visualized as a tether which supports the phenyl group and facilitates the flipping of the thio-ester moiety. In contrast, the consequence of charge shift toward the carbonyl oxygen, O2, would have the opposite effect to that on O1. In this case, increased electronic charge on O2 (Scheme 3, IIB) would result in poorer π-orbital overlap with the adjacent hydrogen atoms and consequently weaken the hydrogen bonding with the cysteine residue. This would provide additional flexibility to this oxygen atom whereby the whole thio-ester moiety will also be in a position to twist more easily. Within such a model, it appears that excitation of the dark-state species of WT-PYP and the consequent charge motion prepares the species for trans-cis isomerization by facilitating the rotation of the thio-ester moiety. Furthermore, the increase in hydrogen bonding strength with O1 would also be expected to facilitate proton transfer, however, this would probably have to be preceded by a back electron transfer to the phenolate oxygen.
In addition to the change in dipole moment, we find that the average change in polarizability, 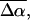 for WT-PYP is extremely large, on the order of 1000 Å3. The fit to the Stark spectra of WT-PYP is primarily comprised of second-derivative components with much smaller contributions from the first-derivative components (Fig. 3, b and c) and therefore the values of
for WT-PYP is extremely large, on the order of 1000 Å3. The fit to the Stark spectra of WT-PYP is primarily comprised of second-derivative components with much smaller contributions from the first-derivative components (Fig. 3, b and c) and therefore the values of 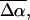 are less reliable than the values of
are less reliable than the values of 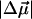 (Table 2). However, it is not surprising that the induced dipole moments (that are directly related to
(Table 2). However, it is not surprising that the induced dipole moments (that are directly related to 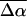 ) in these systems are large, given that the coupling between the negatively charged ground- and excited-state species and the external field will be strong. Initial experiments (work in progress) on the isolated chromophore of WT-PYP, thio-methyl pCA, indicate that
) in these systems are large, given that the coupling between the negatively charged ground- and excited-state species and the external field will be strong. Initial experiments (work in progress) on the isolated chromophore of WT-PYP, thio-methyl pCA, indicate that 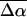 in the protein is significantly larger than in the isolated chromophore and the enhancement of this electrostatic property may be unique to the protein environment. Calculations on a species that mimics the chromophore in the protein pocket show that the excited state is delocalized beyond the chromophore and over the arginine residue as well (Groenhof et al., 2002) while there is greater localization in the ground state.
in the protein is significantly larger than in the isolated chromophore and the enhancement of this electrostatic property may be unique to the protein environment. Calculations on a species that mimics the chromophore in the protein pocket show that the excited state is delocalized beyond the chromophore and over the arginine residue as well (Groenhof et al., 2002) while there is greater localization in the ground state.
Electronic properties of photoproducts (PYPH and PYPB)
In WT-PYP the main absorption band is considered to be composed of the dark-state species, pG. The blue shift in the absorption maximum observed in WT-PYP upon cooling, before blue-light illumination, would arise due to solvation effects. An alteration in the interaction between the chromophore and the surrounding protein residues would generally be expected to stabilize both the ground and excited-state energies on lowering the temperature, thereby producing a red-shift. The initial blue-shift to 446 nm before illumination with blue light, in the absence of photoproducts, also suggests that μe < μg for pG. Nevertheless, given the large value of 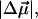 the mere 100 cm−1 blue-shift of the electronic transition energy indicates that there is only a small perturbation in the chromophore-protein interactions on lowering the temperature.
the mere 100 cm−1 blue-shift of the electronic transition energy indicates that there is only a small perturbation in the chromophore-protein interactions on lowering the temperature.
The additional 100 cm−1 blue-shift of the absorption maximum that results from blue-light exposure could be (partially) accounted for by the generation of the hypsochromically shifted species, PYPH, which has been shown to be a precursor to the cis-isomer PYPL (I1 or pR465) and also undergoes the PYP photocycle (Imamoto et al., 2001b). However, because we use a much weaker light regime, we probably generate a much smaller proportion of the photoproducts and we do not believe that more than half of pG is converted to PYPH and PYPB, as the previous authors concluded (Imamoto et al., 2001a). Therefore, the main Stark signal of WT-PYP (21,000–26,000 cm−1 in Fig. 3 b) could be considered to arise from the combined field-dependent response of two structurally disparate species: pG (λmax = 446 nm) and PYPH (λmax = 442 nm). However, we find that the Stark signal, after photoproduct generation, is similar in shape and only somewhat diminished in magnitude compared to the signal measured before illumination with blue light (Fig. 2 a). Furthermore, we obtain a good fit to the Stark signal from the derivatives of the overall absorption lineshape (Fig. 3, b and c) indicating that the dominant component in this wavelength region is likely to be composed of the dark-state species, pG. Alternatively, this could be due to the fact that PYPH is electronically similar to pG, arguing against the suggestion that it is in a cis configuration, which was assigned based on the downshift of the 1294 cm−1 mode of the C7C8 double bond (Imamoto et al., 2001a). And although its blue-shifted absorbance is an indication that PYPH is likely to be in a more strained conformation than pG, the relatively small increase in the electronic transition energy of 200 cm−1 (λmax = 442 nm) (Imamoto et al., 1996) suggests that it is likely to be a small change in geometry (a partial twist) as opposed to a complete rotation of the thioester moiety.
The red photoproduct, PYPB, however clearly exhibits electronic properties that differ from that of the dark-state species. The red-shifted absorbance probably arises due to a change in the geometry of the chromophore, which, based on low-temperature FTIR spectra, is also proposed to be in a cis configuration (Imamoto et al., 2001a). Such a change in the geometry would probably be manifested in a field-dependent response that is distinct from that of pG, which contains the chromophore in a trans configuration. We find that PYPB exhibits a significantly larger change in polarizability and a much smaller change in the dipole moment than pG. The increase in polarizability can be explained by invoking the sum-over-states expression where strong coupling to the higher lying excited states would significantly increase the excited state polarizability of PYPB. A much smaller change in dipole moment would imply that the movement of charge in these intermediates is smaller than on the excitation of the dark-state species. Such a trend where the change in dipole moment decreases on increasing the twist angle of the chromophore has also been measured in the retinal chromophore which is present in bacteriorhodopsin (Locknar and Peteanu, 1998).
Electronic properties of E46Q
At room temperature, the absorption maximum of E46Q red-shifts by 775 cm−1(λmax = 460 nm), due to a change in the protein environment of the chromophore. While this effect can be correlated to the change in counterions (Kroon et al., 1996) it may also be understood in terms of solvation effects arising from the alteration in the hydrogen bonding interactions in the immediate vicinity of the chromophore. Although the hydrogen bonding within the protein pocket and the rest of the protein is preserved, the hydrogen bonding strength of the negative phenolate oxygen, O1, of E46Q with the glutamine residue is weaker than that with the glutamate residue present in WT-PYP (Scheme 2, a versus b). This would effectively decrease the strain on the chromophore in E46Q thereby lowering the electronic transition energy. Nevertheless, on lowering the temperature to 77 K, and inducing the formation of photoproducts (illumination <450 nm), the overall hydrogen bonding network is not disrupted in E46Q either (Imamoto et al., 2001b). Similar to WT-PYP, the Stark spectrum obtained for the dark species is very similar to that obtained after blue-light illumination. Therefore, using similar arguments as we did for WT-PYP above, we conclude that here too the Stark spectrum of the main absorption band reflects the change in electrostatic properties that occurs in the first step of the E46Q photocycle on excitation of the dark-state species.
We find that the large change in the dipole moment is not uniquely manifested in WT- PYP but is also present in E46Q (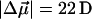 ). Charge motion in E46Q, where the chromophore is the same as in WT-PYP, would be expected to follow a similar pattern of translocation as shown in Scheme 3 for WT-PYP. The biochemical modification of E46 to Q, which affects the electronic transition energy, is also likely to be responsible for the 3 Debye difference in
). Charge motion in E46Q, where the chromophore is the same as in WT-PYP, would be expected to follow a similar pattern of translocation as shown in Scheme 3 for WT-PYP. The biochemical modification of E46 to Q, which affects the electronic transition energy, is also likely to be responsible for the 3 Debye difference in 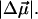 Because the amide hydrogen in glutamine is not as strong an electrophile as the hydroxyl hydrogen in glutamate, the hydrogen bond strength between the phenolate oxygen and Gln46 in E46Q is weaker (Zhou et al., 2001). This may, in turn, modulate the extent of charge separation in the ground and/or excited states. Nevertheless, E46Q which also undergoes a photocycle similar to WT-PYP (Borucki et al., 2002.; Devanathan et al., 2000; Genick et al., 1997; Zhou et al., 2001) exhibits signification charge motion as a result of photon absorption, despite the alteration of a glutamate to a glutamine. The similarity in the excited-state dynamics of E46Q to that of WT-PYP, particularly in the initial stages of the photocycle (Devanathan et al., 2000), may therefore not be surprising in light of the fact that photon absorption by E46Q produces a similar effect on the chromophore as in WT-PYP. Therefore, it would appear that it is the charge motion induced by excitation of the chromophore that provides the driving force for initiating the photocycle, where, the protein itself is effectively a spectator during this excitation process while nevertheless ensuring the execution of this process by stabilizing the negative charge on the phenolate anion in the ground state.
Because the amide hydrogen in glutamine is not as strong an electrophile as the hydroxyl hydrogen in glutamate, the hydrogen bond strength between the phenolate oxygen and Gln46 in E46Q is weaker (Zhou et al., 2001). This may, in turn, modulate the extent of charge separation in the ground and/or excited states. Nevertheless, E46Q which also undergoes a photocycle similar to WT-PYP (Borucki et al., 2002.; Devanathan et al., 2000; Genick et al., 1997; Zhou et al., 2001) exhibits signification charge motion as a result of photon absorption, despite the alteration of a glutamate to a glutamine. The similarity in the excited-state dynamics of E46Q to that of WT-PYP, particularly in the initial stages of the photocycle (Devanathan et al., 2000), may therefore not be surprising in light of the fact that photon absorption by E46Q produces a similar effect on the chromophore as in WT-PYP. Therefore, it would appear that it is the charge motion induced by excitation of the chromophore that provides the driving force for initiating the photocycle, where, the protein itself is effectively a spectator during this excitation process while nevertheless ensuring the execution of this process by stabilizing the negative charge on the phenolate anion in the ground state.
In E46Q, low-temperature intermediates E46QB and E46QH have been detected, similar to PYPB and PYPH (Devanathan et al., 2000; Imamoto et al., 2001b) and are also considered to be geometrically distorted with respect to the dark-state species. Indeed the bathochromic photoproduct, E46QB also exhibits a much larger 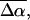 while
while 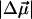 is much smaller than the dark-state species. This disparity in the electrostatic properties of E46QB versus E46Q suggests that the red photoproduct has a different geometry, and consequently exhibits different electronic properties than the dark state, just as in WT-PYP. Once again, while it is difficult to make a precise conclusion about the electronic properties of the hypsochromic photoproduct, E46QH, it is also likely to be similar to the dark-state species.
is much smaller than the dark-state species. This disparity in the electrostatic properties of E46QB versus E46Q suggests that the red photoproduct has a different geometry, and consequently exhibits different electronic properties than the dark state, just as in WT-PYP. Once again, while it is difficult to make a precise conclusion about the electronic properties of the hypsochromic photoproduct, E46QH, it is also likely to be similar to the dark-state species.
Electronic properties of L-PYP
In L-PYP, the 4-hydroxycinnamyl cysteine thioester chromophore present in WT-PYP and E46Q is replaced by 7-hydroxy-coumarin3-carboxylic acid (Scheme 2 c). L-PYP initially red-shifts by 100 cm−1 (2 nm) to 22,420 cm−1 (446 nm) on the addition of glycerol to the Tris-HCl buffer solution at room temperature and then blue-shifts back to 22,520 cm−1 (444 nm) at 77 K. In the absence of evidence for structural changes in the chromophore on photoexcitation at 77 K, the change in the spectroscopic properties of the chromophore are likely to arise due to environmental/solvation effects. Because the protein environment in L-PYP is the same as in WT-PYP, the solvating effect on the two different chromophores in both systems would be expected to be similar. This also supports the assertion that the 200 cm−1 blue-shift of the absorption maximum in WT-PYP to 22,520 cm−1 (444 nm) can be (partially) accounted for by temperature-dependent solvation effects of the protein environment. Note that the chromophore in WT-PYP would be expected to produce a larger blue-shift, given similar solvation effects, because the change in dipole moment is a factor-of-two larger than that of L-PYP.
The change in dipole moment of 13 D in L-PYP, where the chromophore is chemically modified to prevent it from undergoing a trans-cis isomerization step, although quite large, is however, a factor-of-two smaller than that of WT-PYP. Charge motion in this system, which is also probably initiated from the negative charge isolated on the phenolate oxygen, would be expected to move toward the electrophilic carbonyl oxygens, O3 and O4, or the somewhat less electrophilic oxygen atom, O2 (see Scheme 2 b). Here, the value of 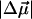 indicates that on photon absorption there is a charge separation of one unit of electronic charge over 2–3 Å. In L-PYP, the potential electron acceptors which include the carbonyl oxygens, O3 and O4, are 7 Å and 8.5 Å away from the negatively charged oxygen atom, O1, respectively. As O3 and O4 are quite far from O1, a transfer of partial electronic charge over this distance can also account for the smaller
indicates that on photon absorption there is a charge separation of one unit of electronic charge over 2–3 Å. In L-PYP, the potential electron acceptors which include the carbonyl oxygens, O3 and O4, are 7 Å and 8.5 Å away from the negatively charged oxygen atom, O1, respectively. As O3 and O4 are quite far from O1, a transfer of partial electronic charge over this distance can also account for the smaller 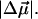 Additionally, in L-PYP it is the structure of the chromophore and not the protein environment, which is identical to that in WT-PYP, that accounts for the smaller change in the dipole moment. This is also manifested in the measured changes in polarizability. In contrast to the 1000 Å3
Additionally, in L-PYP it is the structure of the chromophore and not the protein environment, which is identical to that in WT-PYP, that accounts for the smaller change in the dipole moment. This is also manifested in the measured changes in polarizability. In contrast to the 1000 Å3 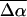 measured in WT-PYP and E46Q, a much smaller change in polarizability of 150 Å3 is measured in L-PYP. Although these
measured in WT-PYP and E46Q, a much smaller change in polarizability of 150 Å3 is measured in L-PYP. Although these 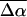 values are not very reliably estimated in WT-PYP and E46Q, it certainly appears that the
values are not very reliably estimated in WT-PYP and E46Q, it certainly appears that the 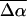 of L-PYP is smaller. Indeed, the smaller
of L-PYP is smaller. Indeed, the smaller 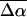 in L-PYP is consistent with the smaller change in permanent dipole moment as well. Qualitatively, the
in L-PYP is consistent with the smaller change in permanent dipole moment as well. Qualitatively, the 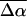 values indicate that the increase in delocalization of the excited states of WT-PYP and E46Q with respect to the ground state are much greater than in L-PYP. Essentially, the ‘volume’ of the excited state is 1000 Å3 larger in WT-PYP and E46Q and 150 Å3 larger in L-PYP. Depending on the cavity size used to estimate this volume change, the increase in the radius of the excited state is between 0.5 Å and 1.5 Å in these systems. The difference in
values indicate that the increase in delocalization of the excited states of WT-PYP and E46Q with respect to the ground state are much greater than in L-PYP. Essentially, the ‘volume’ of the excited state is 1000 Å3 larger in WT-PYP and E46Q and 150 Å3 larger in L-PYP. Depending on the cavity size used to estimate this volume change, the increase in the radius of the excited state is between 0.5 Å and 1.5 Å in these systems. The difference in  between these systems could alternatively, be due to a comparatively greater localization of the ground states of WT-PYP and E46Q compared to L-PYP. It is also possible that unlike WT-PYP, the excited state is localized on the chromophore in L-PYP, and does not extend over the surrounding amino acid residues—thus explaining the smaller
between these systems could alternatively, be due to a comparatively greater localization of the ground states of WT-PYP and E46Q compared to L-PYP. It is also possible that unlike WT-PYP, the excited state is localized on the chromophore in L-PYP, and does not extend over the surrounding amino acid residues—thus explaining the smaller 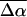 value.
value.
CONCLUSION
Stark effect studies show that excitation of the chromophore in PYP, 4-hydroxycinnamyl thioester, could provide the driving force for initiating the photocycle due to the large change in charge distribution (dipole moment) that occurs instantaneously on photon absorption. The resultant charge motion from the negatively charged phenolate oxygen in the ground state toward the thio-ester moiety are proposed to facilitate the rotation (or twisting) of this unit by inducing an increase in the single bond character of the C7C8 double bond, and by weakening the hydrogen bonding of the carbonyl oxygen with the backbone nitrogen of the cysteine residue. Furthermore, E46Q exhibits similar electro-optical properties to WT-PYP in contrast to L-PYP, where charge-transfer is significantly diminished, suggesting that the change in electrostatic properties undergone by the chromophore on photon absorption plays a crucial role in initiating the PYP photocycle.
Acknowledgments
We thank R. Cordfunke for help with sample preparations, and L.L.P. thanks Francesco Buda for discussions and additional information about the DFT calculations.
This project was supported by the Foundation for Fundamental Research on Matter of The Netherlands (L.L.P. and R.v.G.).
References
- Beekman, L. M. P., R. N. Frese, G. J. S. Fowler, R. Picorel, R. J. Cogdell, I. H. M. van Stokkum, C. N. Hunter, and R. van Grondelle. 1997. Characterization of the light harvesting antennas of photosynthetic purple bacteria by Stark spectroscopy. 2. LH2 complexes: influence of the protein environment. J. Phys. Chem. B. 101:7293–7301. [Google Scholar]
- Birge, R. H., and L. Hubbard. 1980. Molecular dynamics of cis-trans isomerization in rhodopsins. J. Am. Chem. Soc. 102:2195–2205. [Google Scholar]
- Borucki, B., S. Devanathan, H. Otto, M. A. Cusanovich, G. Tollin, and M. P. Heyn. 2002. Kinetics of proton uptake and dye binding by photoactive yellow protein in wild type and in the E46Q and E46A mutants. Biochemistry. 41:10026–10037. [DOI] [PubMed] [Google Scholar]
- Bottcher, C. J. F. 1952. Theory of Electric Polarisation. Elsevier Publishing Company, Amsterdam, The Netherlands.
- Boxer, S. G., R. A. Goldstein, D. J. Lockhart, T. R. Middendorf, and L. Takiff. 1989. Excited states, electron- transfer reactions, and intermediates in bacterial photosynthetic reaction centers. J. Phys. Chem. 93:8280–8294. [Google Scholar]
- Brudler, R., R. Rammelsberg, T. T. Woo, E. D. Getzoff, and K. Gerwert. 2001. Structure of the I1 early intermediate of photoactive yellow protein by FTIR spectroscopy. Nature. 8:265–270. [DOI] [PubMed] [Google Scholar]
- Changenet, P., H. Zhang, M. J. van der Meer, K. J. Hellingwerf, and M. Glasbeek. 1998. Subpicosecond fluorescence upconversion measurements of primary events in the yellow proteins. Chem. Phys. Lett. 282:276–282. [Google Scholar]
- Chowdhury, A., S. Locknar, L. Premvardhan, and L. A. Peteanu. 1999. Effects of matrix temperature and rigidity on the electronic properties of solvatochromic molecules: electroabsorption of coumarin 153. J. Phys. Chem. A. 103:9614–9625. [Google Scholar]
- Cordfunke, R., R. Kort, A. Pierik, B. Gobets, G.-J. Koomen, J. W. Verhoeven, and K. J. Hellingwerf. 1998. Trans/cis (Z/E) photoisomerization of the chromophore of photoactive yellow protein is not a prerequisite for the initiation of the photocycle of this photoreceptor protein. Proc. Natl. Acad. Sci. USA. 90:7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demchuk, E., U. K. Genick, T. T. Woo, E. D. Getzoff, and D. Bashford. 2000. Protonation states and pH titration in the photocycle of photoactive yellow protein. Biochemistry. 39:1100–1113. [DOI] [PubMed] [Google Scholar]
- Devanathan, S., S. Linm, M. A. Cusanovich, N. Woodbury, and G. Tollin. 2000. Early intermediates in the photocycle of the Glu46Gln mutant of photoactive yellow protein: femtosecond spectroscopy. Biophys. J. 79:2132–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genick, U. K., S. Devanthan, T. E. Meyer, I. L. Canestrelli, E. Williams, M. A. Cusanovich, G. Tollin, and E. D. Getzoff. 1997. Active site mutants implicate key residues for control of color and light cycle kinetics of photoactive yellow protein. Biochemistry. 36:8–14. [DOI] [PubMed] [Google Scholar]
- Gensch, T., C. C. Gardinaru, I. H. M. van Stokkum, J. Hendriks, K. J. Hellingwerf, and R. van Grondelle. 2002. The primary photoreaction of photoactive yellow protein (PYP): anisotropy changes and excitation wavelength dependence. Chem. Phys. Lett. 356:347–354. [Google Scholar]
- Groenhof, G., M. F. Lensink, H. J. C. Berendsen, J. G. Snijders, and A. E. Mark. 2002. Signal transduction in the photoactive yellow protein. I. Photon absorption and the isomerization of the chromophore. Prot. Struct. Funct. Gen. 48:202–211. [DOI] [PubMed] [Google Scholar]
- Gottfried, D. S., J. W. Stocker, and S. G. Boxer. 1991. Stark-effect spectroscopy of bacteriochlorophyll in light-harvesting complexes from photosynthetic bacteria. Biochim. Biophys. Acta. 1059:63–75. [Google Scholar]
- Hendriks, J., T. Gensch, L. Hviid, M. A. van der Horst, K. J. Hellingwerf, and J. J. van Thor. 2002. Transient exposure of hydrophobic surface in the photoactive yellow protein monitored with Nile red. Biophys. J. 82:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff, W. D., S. L. S. Kwa, R. van Grondelle, and K. J. Hellingwerf. 1992. Low temperature absorbance and fluorescence spectroscopy of the photoactive yellow protein from Ectothiorhodospira halophila. Photochem. Photobiol. 56:529–539. [Google Scholar]
- Hoff, W. D., P. Dux, K. Hard, B. Devreese, I. M. Nugteren-Roodzant, W. Crielaard, R. Boelens, and R. Kaptein. 1994. Thiol-ester linked p-coumaric acid as a new photoactive prosthetic group in a protein with rhodopsin-like photochemistry. Biochemistry. 33:13959–13962. [DOI] [PubMed] [Google Scholar]
- Imamoto, Y., M. Kataoka, and F. Tokunaga. 1996. Photoreaction cycle of photoactive yellow protein from Ectothiorhodospira halophila studied by low-temperature spectroscopy. Biochemistry. 35:14047–14053. [DOI] [PubMed] [Google Scholar]
- Imamoto, Y., Y. Shirahige, F. Tokunaga, T. Kinoshita, K. Yoshihara, and M. Kataoka. 2001a. Low-temperature Fourier transform infrared spectroscopy of photoactive yellow protein. Biochemistry. 40:8997–9004. [DOI] [PubMed] [Google Scholar]
- Imamoto, Y., K. Mihara, F. Tokunaga, and M. Kataoka. 2001b. Spectroscopic characterization of the photocycle intermediates of photoactive yellow protein. Biochemistry. 40:14336–14343. [DOI] [PubMed] [Google Scholar]
- Kim, M., R. A. Mathies, W. D. Hoff, and K. J. Hellingwerf. 1995. Resonance Raman evidence that the thioester- linked 4-hydroxycinnamyl chromophore of photoactive yellow protein is deprotonated. Biochemistry. 34:12669–12672. [DOI] [PubMed] [Google Scholar]
- Kort, R., H. Vonk, X. Xu, W. D. Hoff, W. Crielaard, and K. J. Hellingwerf. 1996. Evidence for trans-cis isomerization of the p-coumaric acid chromophore as the photochemical basis of the photocycle of photoactive yellow protein. FEBS Lett. 382:73–78. [DOI] [PubMed] [Google Scholar]
- Kroon, A. R., W. D. Hoff, H. P. M. Fennema, J. Gijzen, G.-J. Koomen, J. W. Verhoeven, W. Crielaard, and K. J. Hellingwerf. 1996. Spectral tuning, fluorescence, and photoactivity in hybrids of photoactive yellow protein, reconstituted with native or modified chromophores. J. Biol. Chem. 271:31949–31956. [DOI] [PubMed] [Google Scholar]
- Liptay, W. 1974. Dipole Moments and Polarizabilities of Molecules in Excited States. E. C. Lim, editor. Academic Press, New York. pp. 129–229.
- Locknar, S. A., and L. A. Peteanu. 1998. Investigation of the relationship between dipolar properties and cis-trans configuration in retinal polyenes: a comparative study using stark spectroscopy and semiempirical calculations. J. Phys. Chem. B. 102:4240–4246. [Google Scholar]
- Mataga, H., Y. Chosrowjan, Y. Shibata, F. Imamoto, and N. Tokunaga. 2000. Effects of modification of protein nanospace structure and change of temperature on the femtosecond to picosecond fluorescence dynamics of photoactive yellow protein. J. Phys. Chem. B. 104:5191–5199. [Google Scholar]
- Mathies, R. A., S. W. Lin, J. B. Ames, and W. T. Pollard. 1991. From femtoseconds to biology: mechanism of bacteriorhodopsin's light-driven proton pump. Annu. Rev. Biphys. Biochem. 20:491–518. [DOI] [PubMed] [Google Scholar]
- Meyer, T. E., E. Yakali, M. Cusanovich, and G. Tollin. 1987. Properties of a water-soluble, yellow protein isolated from a halophilic phototrophic bacterium that has photochemical activity analogous to sensory rhodopsin. Biochemistry. 26:418–423. [DOI] [PubMed] [Google Scholar]
- Meyer, T. E., M. A. Cusanovich, and G. Tollin. 1993. Transient proton uptake and release is associated with the photocycle of the photoactive yellow protein from the purple phototrophic bacterium Ectothiorhodospira halophila. Arch. Biochem. Biophys. 306:515–517. [DOI] [PubMed] [Google Scholar]
- Mollina, V., and M. Merchán. 2001. On the absorbance changes in the photocycle of the photoactive yellow protein: a quantum-chemical analysis. Proc. Natl. Acad. Sci. USA. 98:4299–4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergi, A., M. Grüning, M. Ferrario, and F. Buda. 2001. Density functional study of the photoactive yellow protein's chromophore. J. Phys. Chem. B. 105:4386–4391. [Google Scholar]
- Sprenger, W. W., W. D. Hoff, J. P. Armitage, and K. J. Hellingwerf. 1993. The eubacterium Ectothiorhodospira halophila is negatively phototactic, with a wavelength dependence that fits the absorption spectrum of the photoactive yellow protein. J. Bacteriol. 175:3096–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujj, L., S. Devanathan, T. E. Meyer, M. A. Cusanovich, G. Tollin, and G. H. Atkinson. 1998. New photocycle intermediates in the photoactive yellow protein from Ectothiorhodospira halophila: picosecond transient absorption spectroscopy. Biophys. J. 75:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno, M., M. Kamauchi, J. Sasaki, F. Tokunaga, and S. Yamauchi. 2002. Resonance Raman spectroscopy and quantum calculations reveal structural changes in the active site of photoactive yellow protein. Biochemistry. 41:5668–5674. [DOI] [PubMed] [Google Scholar]
- van Brederode, M. E., W. D. Hoff, I. H. M. van Stokkum, M.-L. Groot, and K. J. Hellingwerf. 1996. Protein folding thermodynamics applied to the photocycle of photoactive yellow protein. Biophys. J. 71:365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, A., W. D. Hoff, A. R. Kroon, and K. J. Hellingwerf. 1996. Glu46 donates a proton to the 4-hydroxycinnamate anion chromophore during the photocycle of photoactive yellow protein. Biochemistry. 35:14671–14678. [DOI] [PubMed] [Google Scholar]
- Xie, A., L. Kelemen, J. Hendriks, B. J. White, K. J. Hellingwerf, and W. D. Hoff. 2001. Formation of a new buried charge drives a large-amplitude protein quake in photoreceptor activation. Biochemistry. 40:1510–1517. [DOI] [PubMed] [Google Scholar]
- Zadok, U., A. Khatchatouriants, A. Lewis, M. Ottolenghi, and M. Shreves. 2002. Light-induced charge redistribution in the retinal chromophore is required for initiating the Bacteriorhodopsin photocycle. J. Am. Chem. Soc. 124:11844–11845. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., L. Ujj, T. E. Meyer, M. A. Cusanovich, and G. H. Atkinson. 2001. Photocycle dynamics and vibrational spectroscopy of the E46Q mutant of photoactive yellow protein. J. Phys. Chem. A. 105:5719–5726. [Google Scholar]