

Abstract
Membrane proteins are generally classified into the following five types: 1), type I membrane protein; 2), type II membrane protein; 3), multipass transmembrane proteins; 4), lipid chain-anchored membrane proteins; and 5), GPI-anchored membrane proteins. In this article, based on the concept of using the functional domain composition to define a protein, the Support Vector Machine algorithm is developed for predicting the membrane protein type. High success rates are obtained by both the self-consistency and jackknife tests. The current approach, complemented with the powerful covariant discriminant algorithm based on the pseudo-amino acid composition that has incorporated quasi-sequence-order effect as recently proposed by K. C. Chou (2001), may become a very useful high-throughput tool in the area of bioinformatics and proteomics.
INTRODUCTION
A cell is enclosed by the plasma membrane (cell envelope). Inside the cell there are various organelles such as the endoplasmic reticulum, Golgi apparatus, mitochondria, and other membrane-bound organelles. Although the basic structure of biological membranes is provided by the lipid bilayer, most of the specific functions are carried out by the membrane proteins. Membrane proteins consist of transmembrane proteins and anchored membrane proteins. The transmembrane proteins contain one or more transmembrane segments with one or more hydrophobic segments to ensure stable association with the hydrophobic interior of the membrane, and hence are relatively easily discriminated from nonmembrane proteins (Rost et al., 1995). The anchored membrane proteins do not have the hydrophobic membrane spanning portions, but they have a consensus sequence motif at either the N- or C-terminus (Casey, 1995; Resh, 1994). Accordingly, membrane proteins can be reliably distinguished by using existing methods, as elaborated by many previous investigators (Chou and Elrod, 1999a; Reinhardt and Hubbard, 1998; Rost et al., 1995). Membrane proteins are generally classified into the following five types: 1), type I membrane protein; 2), type II membrane protein; 3), multipass transmembrane proteins; 4), lipid chain-anchored membrane proteins; and 5), GPI-anchored membrane proteins (Fig. 1). The way that a membrane-bound protein is associated with the lipid bilayer usually reflects the function of the protein. The transmembrane proteins, for example, can function on both sides of membrane or transport molecules across it, whereas proteins that function on only one side of the lipid bilayer are often associated exclusively with either the lipid monolayer or a protein domain on that side.
FIGURE 1.

Schematic drawing showing the following five types of membrane proteins: (a) type I transmembrane, (b) type II transmembrane, (c) multipass transmembrane, (d) lipid-chain anchored membrane, and (e) GPI-anchored membrane. As shown from the figure, although both type I and type II membrane proteins are of single-pass transmembrane, type I has a cytoplasmic C-terminus and an extracellular or luminal N-terminus for plasma membrane or organelle membrane, respectively, while the arrangement of N- and C-termini in type II membrane proteins is the reverse. No such distinction was drawn between the extracellular (or luminal) and cytoplasmic sides for the other three types in the current classification scheme. Reproduced from Chou (2001) with permission.
Accordingly, it will certainly expedite the function determination for new membrane proteins if a fast and effective algorithm is available to predict their types. In a pioneer work, Chou and Elrod introduced the covariant discriminant algorithm (Chou and Elrod, 1999a) to predict the types of membrane proteins based on the amino acid composition. According to the conventional definition, the amino acid composition of a protein consists of 20 components, representing the occurrence frequency of each of its 20 native amino acids (Chou, 1989; Nakashima et al., 1986). Obviously, if using the conventional amino acid composition as the representation for a protein, all the sequence-order and sequence-length effects would be missed. To improve this situation, a novel concept, the so-called pseudo-amino acid composition, was proposed recently by Chou (2001). Based on the concept, an elegant formulation was given that can incorporate part of sequence effects or the quasi-sequence order effect (Chou, 2000), remarkably improving the prediction quality. Stimulated by the concept of pseudo-amino acid composition, the present study was initiated in an attempt to incorporate the sequence-order effects by a different approach, the so-called functional domain composition.
PSEUDO-AMINO ACID COMPOSITION AND FUNCTIONAL DOMAIN COMPOSITION
First of all, let us give a brief introduction about the pseudo-amino acid composition. Instead of 20 discrete numbers as defined in the conventional amino acid composition (Chou, 1989; Nakashima et al., 1986), the pseudo-amino acid composition consists of 20 + λ discrete numbers (Chou, 2001), and hence a protein can be expressed as a vector in a (20 + λ)-D space, as given by
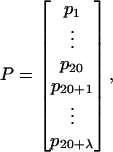 |
(1) |
where the first 20 components are the same as those in the conventional amino acid composition and the components p20+1, …, p20+λ are related to λ different ranks (Fig. 2) of sequence-order correlation factors as formulated by the following equation (Chou, 2002):
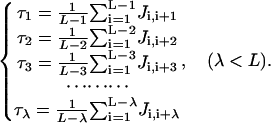 |
(2) |
In the above equation, L is the chain length of the protein concerned, τ1 is called the first-rank coupling factor that harbors the sequence-order correlation between all the most contiguous residues along a protein chain (Fig. 2 a), τ2 the second-rank coupling factor that harbors the sequence-order correlation between all the second most contiguous residues (Fig. 2 b), τ3 the third-rank coupling factor that harbors the sequence-order correlation between all the third most contiguous residues (Fig. 2 c), and so forth. The coupling factor Ji,j in Eq. 2 is a function of amino acids Ri and Rj, such as the physicochemical distance (Schneider and Wrede, 1994) from Ri to Rj (Chou, 2000) or some combination of several biochemical quantities related to Ri and Rj (Chou, 2001, 2002). As we can see from Fig. 2, the sequence-order effect of a protein can be, to some extent, reflected through a set of discrete numbers τ1, τ2, τ3, …, tλ, as defined by Eq. 2. Accordingly, the first 20 components of Eq. 1 reflect the effect of the amino acid composition, while the components from 20 + 1 to 20 + λ reflect the effect of sequence order. A set of such 20 + λ components as formulated by Eqs. 1–2 is called the pseudo-amino acid composition for protein P. Using such a name is because it still has the main feature of amino acid composition, but on the other hand, it contains the information beyond the conventional amino acid composition. The pseudo-amino acid composition thus defined has the following advantage: compared with the 210-D pair-coupled amino acid composition (Chou, 1999) and the 400-D first-order coupled amino acid composition (Liu and Chou, 1999) that contain the sequence-order effect only for a very short range (i.e., within two adjacent amino acid residues along a chain), the pseudo-amino acid composition incorporate much more sequence effects, i.e., those not only for the short range but for the medium range and long range as well, as reflected by a series of sequence-coupling factors with different tiers of correlation (see Fig. 2 and Eqs. 1–2). Therefore, the prediction quality can be significantly improved by using the pseudo-amino acid composition to represent a protein. The detailed formulation and application of the pseudo-amino acid composition are given in two recent articles (Chou, 2001, 2002).
FIGURE 2.

A schematic drawing to show (a) the first rank, (b) the second rank, and (c) the third-rank sequence-order correlation mode along a protein sequence. (a) Reflects the correlation mode between all the most contiguous residues, (b) that between all the secondmost contiguous residues, and (c) that between all the thirdmost contiguous residues.
Now, let us use a different approach to incorporate the sequence-order effects. By searching 139,765 annotated protein sequences, Murvai and co-workers (Murvai et al., 2001) have constructed a database called SBASE-A that contains 2005 sequences with well-known structural and functional domain types. Based on the 2005 functional domains, a protein can be defined in a 2005-D space according to the following procedures.
Use BLASTP to compare a protein with each of the 2005 domain sequences in SBASE-A to find the high-scoring segment pairs and the smallest sum probability. A detailed description about this operation can be found in Altschul (Altschul et al., 1990).
If the high-scoring segment pairs score ≫75 and smallest sum probability <0.8 in comparing the protein sequence with the ith domain sequence, then the ith component of the protein in the 2005-D space is assigned 1; otherwise, 0.
- The protein can thus be explicitly formulated as
where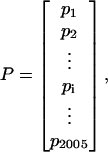
(3) 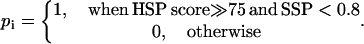
(4)
Thus, a protein is corresponding to a 2005-D vector by using each of the 2005 functional domain sequences as a base; i.e., rather than the 20-D space (Chou, 1995; Nakashima et al., 1986) in terms of the amino acid composition or the (20 + λ)-D space of the pseudo-amino acid composition (Chou, 2001), a protein is defined in terms of the functional domain composition. By using such a representation, not only some sequence-order effects but also some functional information are included. In other words, the representation thus obtained for a protein would bear some sequence-order mark as well as the structural and functional type mark. Since the function of a membrane protein is closely related to its type, the prediction algorithm established based on the new representation would naturally incorporate those factors that might be directly correlated with the membrane protein type.
SUPPORT VECTOR MACHINES
Support vector machines (SVMs) are a kind of learning machine based on statistical learning theory. The most remarkable characteristics of SVMs are the absence of local minima, the sparseness of the solution, and the use of the kernel-induced feature spaces. The basic idea of applying SVMs to pattern classification can be outlined as follows. First, map the input vectors into a feature space (possible with a higher dimension), either linearly or nonlinearly, which is relevant to the selection of the kernel function. Then, within the feature space, seek an optimized linear division; i.e., construct a hyper-plane which can separate two classes (this can be extended to multiclasses) with the least errors and maximal margin. The SVMs training process always seeks a global optimized solution and avoids over-fitting, so it has the ability to deal with a large number of features. A complete description to the theory of SVMs for pattern recognition is given in the book by Vapnik (1998).
SVMs have been used to deal with protein fold recognition (Ding and Dubchak, 2001), protein-protein interactions prediction (Bock and Gough, 2001), and protein secondary structure prediction (Hua and Sun, 2001).
In this article, the Vapnik's Support Vector Machine (Vapnik 1995) was introduced to predict the types of membrane proteins. Specifically, the SVMlight, which is an implementation (in C Language) of SVM for the problems of pattern recognition, was used for computations. The optimization algorithm used in SVMlight can be found in Joachims (1999). The relevant mathematical principles can be briefly formulated as follows. Given a set of N samples, i.e., a series of input vectors
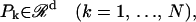 |
(5) |
where 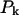 can be regarded as the kth protein or vector defined in the 2005-D space according to the functional domain composition, and
can be regarded as the kth protein or vector defined in the 2005-D space according to the functional domain composition, and 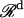 is a Euclidean space with d dimensions. Since the multiclass identification problem can always be converted into a two-class identification problem, without loss of the generality the formulation below is given for the two-class case only. Suppose the output derived from the learning machine is expressed by hk∈{;+1,−1} (k = 1, …, N), where the indexes −1 and +1 are used to stand for the two classes concerned, respectively. The goal here is to construct one binary classifier or derive one decision function from the available samples that has a small probability of misclassifying a future sample. Here, both the basic linear separable case, and the most useful linear nonseparable case for most real life problems, are taken into consideration.
is a Euclidean space with d dimensions. Since the multiclass identification problem can always be converted into a two-class identification problem, without loss of the generality the formulation below is given for the two-class case only. Suppose the output derived from the learning machine is expressed by hk∈{;+1,−1} (k = 1, …, N), where the indexes −1 and +1 are used to stand for the two classes concerned, respectively. The goal here is to construct one binary classifier or derive one decision function from the available samples that has a small probability of misclassifying a future sample. Here, both the basic linear separable case, and the most useful linear nonseparable case for most real life problems, are taken into consideration.
The linear separable case
In this case, there exists a separating hyper-plane whose function is 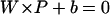 , which implies:
, which implies:
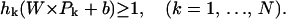 |
(6) |
By minimizing 1/2‖W‖2 subject to the above constraint, the SVM approach will find a unique separating hyper-plane. Here ‖W‖2 is the Euclidean norm of W, which maximizes the distance between the hyper-plane, or the optimal separating hyper-plane (Cortes and Vapnik, 1995), and the nearest data points of each class. The classifier thus obtained is called the maximal margin classifier. By introducing Lagrange multipliers 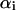 , and using the Karush-Kuhn-Tucker conditions (Cristianini and Shawe-Taylor, 2000; Karush, 1939) as well as the Wolfe dual theorem of optimization theory (Wolfe, 1961), the SVM training procedure amounts to solving the following convex quadratic programming problem,
, and using the Karush-Kuhn-Tucker conditions (Cristianini and Shawe-Taylor, 2000; Karush, 1939) as well as the Wolfe dual theorem of optimization theory (Wolfe, 1961), the SVM training procedure amounts to solving the following convex quadratic programming problem,
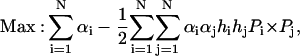 |
(7) |
subject to the following two conditions:
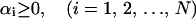 |
(8) |
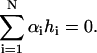 |
(9) |
The solution is a unique globally optimized result, which can be expressed with the following expansion:
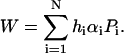 |
(10) |
Only if the corresponding 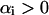 are these
are these  called the support vectors. Now suppose P is a query protein defined in the same 2005-D space based on the functional domain composition. After the SVM has been trained, the decision function for identifying which class the query protein belongs to can be formulated as:
called the support vectors. Now suppose P is a query protein defined in the same 2005-D space based on the functional domain composition. After the SVM has been trained, the decision function for identifying which class the query protein belongs to can be formulated as:
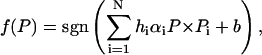 |
(11) |
where sgn( ) in the above equation is a sign function, which equals to +1 or −1 when its argument is ≥0 or ≤0, respectively.
The linear nonseparable case
For this case, two important techniques are needed that are given below respectively.
The “soft margin” technique
To allow for training errors, Cortes and Vapnik (1995) introduced the slack variables
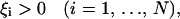 |
(12) |
and the relaxed separation constraint given by
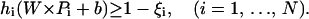 |
(13) |
The optimal separating hyper-plane can be found by minimizing
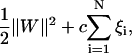 |
(14) |
where c is a regularization parameter used to decide a tradeoff between the training error and the margin.
The kernel substitution technique
The SVM performs a nonlinear mapping of the input vectors from the Euclidean space  into a higher dimensional Hilbert space H, where the mapping is determined by the kernel function. Then like in the linear separable case, it finds the optimal separating hyper-plane in the Hilbert space H that would correspond to a nonlinear boundary in the original Euclidean space. Two typical kernel functions are listed below:
into a higher dimensional Hilbert space H, where the mapping is determined by the kernel function. Then like in the linear separable case, it finds the optimal separating hyper-plane in the Hilbert space H that would correspond to a nonlinear boundary in the original Euclidean space. Two typical kernel functions are listed below:
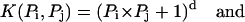 |
(15) |
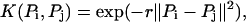 |
(16) |
where the first one is called the polynomial kernel function of degree d which will eventually revert to the linear function when d = 1, and the second one is called the Radial Basic Function kernel. Finally, for the selected kernel function, the learning task amounts to solving the following quadratic programming problem:
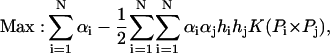 |
(17) |
subject to:
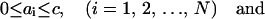 |
(18) |
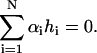 |
(19) |
Accordingly, the form of the decision function is given by
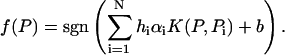 |
(20) |
For a given data set, only the kernel function and the regularity parameter c must be selected to specify the SVM.
RESULTS AND DISCUSSION
The same data set constructed by Chou and Elrod (1999a) was used to demonstrate the current method. The data set contains 2059 membrane protein sequences, of which 435 are type I transmembrane proteins, 152 type II transmembrane proteins, 1311 multipass transmembrane proteins, 51 lipid-chain anchored membrane proteins, and 110 GPI anchored membrane proteins (Fig. 1). The names of the 2059 membrane proteins, classified into five groups, were given in Table 1 of Chou and Elrod (1999a).
TABLE 1.
Overall rates of correct prediction for the five membrane protein types by different algorithms and test methods
| Algorithm | Input form | Test method self-consistency* | Jackknife* | Independent data set† |
|---|---|---|---|---|
| Least Hamming distance (Chou, 1980) | Amino acid composition | 1293/2059 = 62.8% | 1279/2059 = 62.1% | 1751/2625 = 66.7% |
| Least Euclidean distance (Nakashima et al., 1986) | Amino acid composition | 1307/2059 = 63.5% | 1293/2059 = 62.8% | 1816/2625 = 69.2% |
| ProtLock (Cedano et al., 1997) | Amino acid composition | 1372/2059 = 66.6% | 1348/2059 = 65.5% | 1674/2625 = 63.8% |
| Covariant discriminant (Chou and Elrod, 1999a) | Amino acid composition | 1670/2059 = 81.1% | 1573/2059 = 76.4% | 2085/2625 = 79.4% |
| Augmented covariant discriminant (Chou, 2000) | Pseudo-amino acid composition (Chou, 2001) | 1872/2059 = 90.9% | 1665/2059 = 80.0% | 2298/2625 = 87.5% |
| Support vector machines | Functional domain composition | 1934/2059 = 93.9% | 1776/2059 = 86.3% | 1773/2625 = 67.5% |
Conducted for the 2059 membrane proteins classified into five different types as described in the text and Fig. 1.
Conducted based on the rule parameters derived from the 2059 membrane proteins for the 2625 independent membrane proteins (see text).
During the operation, the width of the Gaussian Radial Basic Functions was selected for minimizing the estimation of the VC-dimension (Vapnik-Chervonenkis-dimension (1998). The parameter C that controlled the error-margin tradeoff was set at 1000. After being trained, the hyper-plane output by the SVM was obtained. This indicates that the trained model, i.e., hyper-plane output which is including the important information, has the function to identify the membrane protein types.
The demonstration was conducted by three different approaches, the resubstitution test, jackknife test, and independent data set test, as reported below.
Resubstitution test
The so-called resubstitution test is an examination for the self-consistency of an identification method. When the resubstitution test is performed for the current study, the type of each membrane protein in a data set is, in turn, identified using the rule parameters derived from the same data set, the so-called training data set. The success rate thus obtained for the 2059 membrane proteins is summarized in Table 1, from which we can see that the overall success rate is 93.9%, indicating that after being trained, the SVMs model has grasped the complicated relationship between the functional domain composition and the types of membrane proteins. However, during the process of the resubstitution test, the rule parameters derived from the training data set include the information of the query protein later plugged back in the test. This will certainly underestimate the error and enhance the success rate because the same proteins are used to derive the rule parameters and to test themselves. Accordingly, the success rate thus obtained represents some sort of optimistic estimation (Cai, 2001; Chou, 1995; Chou and Elrod, 1999b; Zhou and Assa-Munt, 2001). Nevertheless, the resubstitution test is absolutely necessary because it reflects the self-consistency of an identification method, especially for its algorithm part. An identification algorithm certainly cannot be deemed as a good one if its self-consistency is poor. In other words, the resubstitution test is necessary but not sufficient for evaluating an identification method. As a complement, a cross-validation test for an independent testing data set is needed because it can reflect the effectiveness of an identification method in practical application. This is especially important for checking the validity of a training database to determine whether it contains sufficient information to reflect all the important features concerned so as to yield a high success rate in application.
Jackknife test
As is well known, the independent data set test, subsampling test, and jackknife test are the three methods often used for cross-validation in statistical prediction. Among these three, however, the jackknife test is deemed as the most effective and objective one; see, for example, Chou and Zhang (1995) for a comprehensive discussion about this, and Mardia et al. (1979) for the mathematical principle. During jackknifing, each membrane protein in the data set is in turn singled out as a tested protein and all the rule parameters are calculated based on the remaining proteins. In other words, the type of each membrane protein is identified by the rule parameters derived using all the other membrane proteins except the one which is being identified. During the process of jackknifing both the training data set and testing data set are actually open, and a protein will in turn move from one to the other. The results of jackknife test thus obtained for the 2059 membrane proteins are also given in Table 1.
Independent data set test
Moreover, as a demonstration of practical application, predictions were also conducted for the 2625 independent membrane proteins based on the rule parameters derived from the 2059 proteins in the training data set. The 2625 independent proteins were also taken from Chou and Elrod (1999a), of which 478 are type I transmembrane proteins, 180 type II transmembrane proteins, 1867 multipass transmembrane proteins, 14 lipid-chain anchored membrane proteins, and 86 GPI anchored membrane proteins. The predicted results thus obtained are also given in Table 1.
From Table 1 the following can be observed. 1), The success prediction rates, by both the functional domain composition approach and the pseudo-amino acid composition approach, are significantly than those by the other approaches. This is fully consistent with what is expected because both these two approaches bear some sequence-order effects, although by means of different avenues. 2), A comparison between the functional domain composition approach and the pseudo-amino acid composition approach indicates that the success rates by the former are ∼3–6% higher than those by the latter in the self-consistency test and jackknife test, indicating the current functional domain composition approach is very promising with a high potential for further development. However, it had a remarkable setback in predicting the 2065 independent proteins: the success rate is 20% lower that that by the pseudo-amino acid composition approach. The setback might be due to the reason that the functional domain database used in the current study is far from a complete one yet. Accordingly, many of the 2065 independent proteins cannot be effectively defined based on the current limited functional domain database. It is anticipated that with the continuous improvement of the functional domain database, the setback would be naturally overcome. 3), The goal of this study is not to determine the possible upper limit of the success rate for membrane protein type predictions, but to propose a novel and different approach to incorporate the sequence-order effect because this is both vitally important and a notoriously difficult task in this area, and so far only the pseudo-amino acid composition approach (Chou, 2001) has been proved really useful, widely applied in various sequence-based (both protein and DNA) prediction projects. Also, it is too premature to construct a complete or quasi-complete training data set based on the protein sequences available so far. Without a complete or quasi-complete training data set, any attempt to determine such an upper limit would be unjustified, and the result thus obtained might be misleading no matter how powerful the prediction algorithm is.
CONCLUSION
The above results, together with those obtained by the covariant discriminant prediction algorithm (Chou, 2001; Chou and Elrod, 1999a), have indicated that the types of membrane proteins are predictable with a considerable accuracy. The development in statistical prediction of protein attributes generally consists of two aspects: constructing a training data set and formulating a prediction algorithm. The latter also consists of two aspects; i.e., how to define a protein and how to operate the prediction. The process in expressing a protein from the 20-D amino acid composition space (Chou, 1995, 1980, 1989; Nakashima et al., 1986), to the (20 + λ)-D pseudo-amino acid composition space (Chou, 2001), and to the current 2005-D functional domain composition space reflects the development in defining a protein. The process in introducing the simple geometry distance algorithm (Chou, 1980; 1989; Nakashima et al., 1986), the Mahalanobis distance algorithm (Cedano et al., 1997; Chou, 1995; Chou and Zhang, 1994), the covariant discriminant algorithm (Chou and Elrod 1999a,b; Chou et al., 1998; Liu and Chou, 1998; Zhou, 1998), and the current SVM algorithm reflects the development in operating algorithms. The pseudo-amino acid composition and the functional domain composition each have their own advantages. For some cases, the functional domain composition yields better results than the pseudo-amino acid composition; but for some other cases, the outcome may be the reverse. This is exactly the same in the comparison of the covariant discriminant algorithm with the SVM algorithm. Therefore, when we are still in the situation of lacking a complete training data set and functional domain database, it would be wise to complement the covariant discriminant algorithm, based on the pseudo-amino acid composition, with the SVM algorithm, based on the functional domain composition, for conducting practical predictions.
References
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Bock, J. R., and D. A. Gough. 2001. Predicting protein-protein interactions from primary structure. Bioinformatics. 17:455–460. [DOI] [PubMed] [Google Scholar]
- Cai, Y. D. 2001. Is it a paradox or misinterpretation? Protein Struct. Funct. Genet. 43:336–338. [DOI] [PubMed] [Google Scholar]
- Casey, P. J. 1995. Protein lipidation in cell signalling. Science. 268:221–225. [DOI] [PubMed] [Google Scholar]
- Cedano, J., P. Aloy, J. A. P'erez-pons, and E. Querol. 1997. Relation between amino acid composition and cellular location of proteins. J. Mol. Biol. 266:594–600. [DOI] [PubMed] [Google Scholar]
- Chou, K. C. 1995. A novel approach to predicting protein structural classes in a (20–1)-D amino acid composition space. Protein Struct. Funct. Genet. 21:319–344. [DOI] [PubMed] [Google Scholar]
- Chou, K. C. 1999. Using pair-coupled amino acid composition to predict protein secondary structure content. J. Protein Chem. 18:473–480. [DOI] [PubMed] [Google Scholar]
- Chou, K. C. 2000. Prediction of protein subcellular locations by incorporating quasi-sequence-order effect. Biochem. Biophys. Res. Commun. 278:477–483. [DOI] [PubMed] [Google Scholar]
- Chou, K. C. 2001. Prediction of protein cellular attributes using pseudo-amino-acid-composition. Protein Struct. Funct. Genet. 43:246–255. [DOI] [PubMed] [Google Scholar]
- Chou, K. C. 2002. A new branch of proteomics: prediction of protein cellular attributes. In Gene Cloning and Expression Technologies. P. W. Weinrer, and Q. Lu, editors. Eaton Publishing, Westborough, MA. pp. 57–70.
- Chou, K. C., and D. W. Elrod. 1999a. Prediction of membrane protein types and subcellular locations. Protein Struct. Funct. Genet. 34:137–153. [PubMed] [Google Scholar]
- Chou, K. C., and D. W. Elrod. 1999b. Protein subcellular location prediction. Protein Eng. 12:107–118. [DOI] [PubMed] [Google Scholar]
- Chou, K. C., W. Liu, G. M. Maggiora, and C. T. Zhang. 1998. Prediction and classification of domain structural classes. Protein Struct. Funct. Genet. 31:97–103. [PubMed] [Google Scholar]
- Chou, K. C., and C. T. Zhang. 1994. Predicting protein folding types by distance functions that make allowances for amino acid interactions. J. Biol. Chem. 269:22014–22020. [PubMed] [Google Scholar]
- Chou, K. C., and C. T. Zhang. 1995. Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol. 30:275–349. (Review.) [DOI] [PubMed] [Google Scholar]
- Chou, P. Y. 1980. Amino acid composition of four classes of proteins. Abstracts of Papers, Part I, Second Chemical Congress of the North American Continent, Las Vegas.
- Chou, P. Y. 1989. Prediction of protein structural classes from amino acid composition. In Prediction of Protein Structure and The Principles of Protein Conformation. G. D. Fasman, editor. Plenum Press, New York. pp. 549–586.
- Cortes, C., and V. Vapnik. 1995. Support vector networks. Machine Learning. 20:273–293. [Google Scholar]
- Cristianini, N., and J. Shawe-Taylor. 2000. Support Vector Machines. Cambridge University Press, Cambridge.
- Ding, C. H., and I. Dubchak. 2001. Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics. 17:349–358. [DOI] [PubMed] [Google Scholar]
- Hua, S. J., and Z. R. Sun. 2001. A novel method of protein secondary structure prediction with high segment overlap measure: support vector machine approach. J. Mol. Biol. 308:397–407. [DOI] [PubMed] [Google Scholar]
- Joachims, T. 1999. Making large-scale SVM learning practical. In Advances in Kernel Methods—Support Vector Learning. B. Schölkopf, C. J. C. Burges, and A. J. Smola, editors. MIT Press. pp. 169–184.
- Karush, W. 1939. Minima of functions of several variables with inequalities as side constraints. University of Chicago, Chicago, IL. (M.Sc. thesis.)
- Liu, W., and K. C. Chou. 1998. Prediction of protein structural classes by modified Mahalanobis discriminant algorithm. J. Protein Chem. 17:209–217. [DOI] [PubMed] [Google Scholar]
- Liu, W., and K. C. Chou. 1999. Protein secondary structural content prediction. Protein Eng. 12:1041–1050. [DOI] [PubMed] [Google Scholar]
- Mardia, K. V., J. T. Kent, and J. M. Bibby. 1979. Multivariate Analysis. Academic Press, London. pp. 322, 381.
- Murvai, J., K. Vlahovicek, E. Barta, and S. Pongor. 2001. The SBASE protein domain library, release 8.0: a collection of annotated protein sequence segments. Nucleic Acids Res. 29:58–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima, H., K. Nishikawa, and T. Ooi. 1986. The folding type of a protein is relevant to the amino acid composition. J. Biochem. 99:152–162. [DOI] [PubMed] [Google Scholar]
- Reinhardt, A., and T. Hubbard. 1998. Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res. 26:2230–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resh, M. D. 1994. Myristylation and palmitylation of Src family members: the fats of the matter. Cell. 76:411–413. [DOI] [PubMed] [Google Scholar]
- Rost, B., R. Casadio, P. Fariselli, and C. Sander. 1995. Transmembrane helices predicted at 95% accuracy. Protein Sci. 4:521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, G., and P. Wrede. 1994. The rational design of amino acid sequences by artificial neural networks and simulated molecular evolution: de novo design of an idealized leader peptidase cleavage site. Biophys. J. 66:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vapnik, V. 1998. Statistical Learning Theory. Wiley-Interscience, New York.
- Vapnik, V. N. 1995. The Nature of Statistical Learning Theory. Springer-Verlag.
- Wolfe, P. 1961. A duality theorem for nonlinear programming. Quart. Applied Math. 19:239–244. [Google Scholar]
- Zhou, G. P. 1998. An intriguing controversy over protein structural class prediction. J. Protein Chem. 17:729–738. [DOI] [PubMed] [Google Scholar]
- Zhou, G. P., and N. Assa-Munt. 2001. Some insights into protein structural class prediction. Protein Struct. Funct. Genet. 44:57–59. [DOI] [PubMed] [Google Scholar]