

Abstract
The molecular mechanism by which DNA-binding proteins find their specific binding sites is still unclear. To gain insights into structural and energetic elements of this mechanism, we used the crystal structure of the nonspecific BamHI-DNA complex as a template to study the dominant electrostatic interaction in the nonspecific association of protein with DNA, and the possible sliding pathways that could be sustained by such an interaction. Based on calculations using the nonlinear Poisson-Boltzmann method and Brownian dynamics, a model is proposed for the initial nonspecific binding of BamHI to B-form DNA that differs from that seen in the crystal structure of the nonspecific complex. The model is electrostatically favorable, and the salt dependence as well as other thermodynamic parameters calculated for this model are in good agreement with experimental results. Several residues in BamHI are identified for their important contribution to the energy in the nonspecific binding model, and specific mutagenesis experiments are proposed to test the model on this basis. We show that a favorable sliding pathway of the protein along DNA is helical.
INTRODUCTION
Specific protein-DNA interactions are essential for normal biological function of the cell, including transcription, replication and recombination. All these processes relating to cellular growth require the regulatory proteins to bind to specific DNA sites. Information about the molecular mechanism of specific protein-DNA recognition has accumulated from a variety of studies that provided information about the structural characteristics (Bewley et al., 1998; Harrison and Aggarwal, 1990; Pabo and Sauer, 1992), thermodynamics (Hard and Lundback, 1996; Jen-Jacobson, 1997; Record et al., 1991), and kinetics (Oda and Nakamura, 2000; Pingoud and Jeltsch, 1997; Record et al., 1991) of the protein-DNA complexes. Because specific DNA-binding proteins also have a finite affinity for nonspecific DNA sequences (Jen-Jacobson, 1997; von Hippel, 1994), it is necessary to study both specific and nonspecific protein-DNA interaction in order to fully understand the mechanism of specific recognition. In fact, nonspecific protein-DNA interactions have been shown to be important elements of the process of searching for, and binding to the cognate specific sites on DNA (Jen-Jacobson, 1997; Pingoud and Jeltsch, 1997; Record et al., 1991).
Because only four crystal structures are currently available for protein-DNA complexes considered to represent nonspecific interactions (Albright et al., 1998; Luisi et al., 1991; Viadiu and Aggarwal, 2000; Winkler et al., 1993), our understanding of such interactions derives mainly from thermodynamic and kinetic studies. Nonspecific binding is shown to be accompanied by negligible heat capacity change (Engler, 1998; Frank et al., 1997; Ladbury et al., 1994; Merabet and Ackers, 1995; Takeda et al., 1992) and is dominated by electrostatic interactions showing stronger salt-concentration dependence than the corresponding specific binding (Engler, 1998; Frank et al., 1997; Record et al., 1991). The process is considered to be enthalpy driven with a small positive entropy change (Jen-Jacobson, 1997). The protein and DNA are thought to be more hydrated at the protein-DNA interface than that in the specific complex (Garner and Rau, 1995; Sidorova and Rau, 1996) with long-range Coulombic interaction dominating the loose association between protein and DNA. There is no intimate protein-DNA interface as both the protein and DNA are hydrated in the complex (Breyer and Matthews, 2001; Hard and Lundback, 1996; Jen-Jacobson, 1997; Oda and Nakamura, 2000; Record et al., 1991). This is exemplified well in the recently reported structure of BamHI bound to noncognate DNA (Viadiu and Aggarwal, 2000).
The electrostatic interaction in protein-DNA complexes has long been the focus of theoretical study (Anderson and Record, 1995; Manning, 1978; Record et al., 1978). The development of nonlinear Poisson-Boltzmann (NLPB) theory has made it possible to quantify computationally all the electrostatic free energy components in the protein-DNA system based on the detailed atomic structures (Sharp and Honig, 1990), and NLPB has been successfully applied to several protein-DNA systems (Fogolari et al., 1997; Misra et al., 1994; Misra et al., 1998; Zacharias et al., 1992). In these studies, the total electrostatic free energy of binding, as well as its dependence on salt concentration was calculated for the specific protein-DNA interaction based on the known structures of the complexes. Nonspecific interaction was studied in a similar manner based on a model of the nonspecific complex that was built from the specific complex structure (Fogolari et al., 1997; Zacharias et al., 1992). The nonspecific binding of Cro repressor to a long stretch of DNA was also studied with Brownian dynamics (BD) simulation (Thomasson et al., 1997). In this context, the crystal structure for the nonspecific complex of BamHI with DNA (Viadiu and Aggarwal, 2000) makes it possible now to review the inferences obtained from these models of nonspecific complexes in a structural context of an authentic system (see also Breyer and Matthews, 2001).
BamHI is a homodimeric enzyme belonging to the type II family of restriction endonucleases that requires only Mg2+ as a cofactor for double-stranded cleavage of its cognate site (GGATCC), after the first guanine in the sequence (Pingoud and Jeltsch, 1997). A study based on BamHI complexes is advantageous because of the availability of crystal structures for the unbound (Newman et al., 1994), specifically bound forms of the enzyme (Newman et al., 1995), as well as the recently determined structure of the nonspecific complex (Viadiu and Aggarwal, 2000). In the latter complex, the second Guanine in the cognate site was mutated to Adenine (GAATCC). In addition, crystal structures of metal bound pre- and post-reactive intermediates provide a comprehensive picture of the BamHI reaction pathway. Together, these structures offer a unique opportunity to study the structural adaptations from the free protein to nonspecific DNA binding, and finally to the rearrangements to specific binding and catalysis.
In order to relate the structural insights to the thermodynamic properties of the nonspecific complex, we studied the electrostatic interaction in the nonspecific BamHI-DNA complex using NLPB theory. Analysis of the results reveals an unexpected orientation of the molecules in the complex, in which the enzyme is rotated ∼60° relative to that seen in the nonspecific crystal structure. This new orientation positions the BamHI dimer along a DNA sliding pathway that is favored electrostatically—unlike that discussed for the crystal structure (Viadiu and Aggarwal, 2000), and agrees with measurements of salt dependence and thermodynamic data. We show that the structure of this nonspecific complex provides a basis for understanding the mechanism of protein sliding along DNA (Breyer and Matthews, 2001; Pingoud and Jeltsch, 1997; Viadiu and Aggarwal, 2000).
METHODS
Formulation of the energy terms in calculations with NLPB theory
The electrostatic potential of a macromolecular system with a monovalent salt solution is given by the NLPB equation:
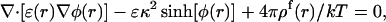 |
(1) |
where φ is the dimensionless electrostatic potential in units of kT/e (k is Boltzmann's constant, T is the absolute temperature, and e is the proton charge). In Eq. 1, ɛ is the dielectric constant, ρf is the fixed charge density and 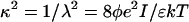 (where λ is the Debye length and I is the ionic strength of the bulk solution). The total electrostatic energy of the system can be expressed as (Sharp and Honig, 1990):
(where λ is the Debye length and I is the ionic strength of the bulk solution). The total electrostatic energy of the system can be expressed as (Sharp and Honig, 1990):
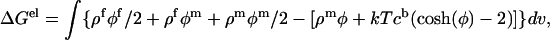 |
(2) |
where the potential φ and charge density ρ have been split up into contribution from the fixed (f) and mobile (m) charges. Based on the thermodynamic pathway, the total electrostatic energy can be decomposed into a salt-independent contribution 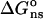 , and a salt-dependent contribution
, and a salt-dependent contribution 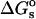 . As derived by Sharp and Honig (Sharp and Honig, 1990) ((see also Fogolari et al., 1997) for alternative expressions), the salt-dependent term can be further divided into three contributions: the entropic work of organizing the ionic atmosphere around solute,
. As derived by Sharp and Honig (Sharp and Honig, 1990) ((see also Fogolari et al., 1997) for alternative expressions), the salt-dependent term can be further divided into three contributions: the entropic work of organizing the ionic atmosphere around solute, 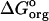 ; the self energy of charging the ionic atmosphere,
; the self energy of charging the ionic atmosphere, 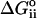 ; the interaction between solute and salt,
; the interaction between solute and salt, 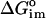 .
.
For the binding reaction: 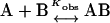 if the salt dependence of the reaction is assumed to be entirely in the electrostatic part of the binding free energy, then:
if the salt dependence of the reaction is assumed to be entirely in the electrostatic part of the binding free energy, then:
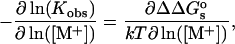 |
(3) |
where 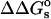 is the change in the salt-dependent contribution to the total electrostatic energy upon binding:
is the change in the salt-dependent contribution to the total electrostatic energy upon binding:
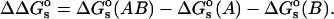 |
(4) |
Computational protocols for NLPB calculations
The program UHBD (Madura et al., 1995) was used for all the NLPB calculations. Atom charges and radii were adapted from the CHARMM27 parameter set (Foloppe and Mackerell, 2000), with polar hydrogen atom radius set as 0.8 Å. The dielectric constant of DNA and protein was set to 4, and the solvent to 78. The solute-solvent boundary was taken as defined either by the van der Waals surface, or as the molecular surface calculated with probes of radius 1.0 Å and 1.4 Å. The radius of the ion-exclusion region was set to 2 Å. A monovalent salt concentration of 0.1 M was used in all calculations, except for salt-dependence calculation, where the concentration was varied from 0.1 M to 0.4 M.
A 189 × 189 × 189 grid was used in the calculation, with grid size set to 1.2 Å in the first calculation, then focused at 0.6 Å. A 0.4-Å final grid and two steps of focusing were also tested and showed ∼1% difference in binding energy calculation.
Molecular models
The nonspecific BamHI-DNA complex (PDB code 1esg) includes 11 basepairs of DNA. To enable comparisons with experimental studies on the same system, a 28-mer DNA was built with the program JUMNA (Lavery et al., 1995) based on the available 8-mer DNA in the crystal structure; details of this method were described in a recent paper (Lebrun et al., 2001). Briefly, the backbone atoms of DNA in the crystal structure were used as the NOE constraints, then these constraints were used in the energy minimization of a canonical B form DNA with FLEX force field (Lavery et al., 1995). The final structure, termed N-DNA28, was used in the calculations. When the sequence of the central hexamer was mutated back to the BamHI cognate site (GAATCC → GGATCC), the model was named C-DNA28, was built based on N-DNA28. The canonical B-form DNA was named BDNA28 and BMUT28 with DNA sequence corresponding to N-DNA28 and C-DNA28, respectively. A short DNA model, DNA11, was obtained directly from N-DNA28.
The last residue in monomer A of BamHI and the last four residues in monomer B were disordered in the crystal structure, and these residues were built in extended conformations with INSIGHTII (Biosym Technologies). This model was named BAM_X. In the model of the complex N-DNA28-BAM_X, there were some steric clashes between N-DNA28 and flexible segments of BamHI around residues 79–84 and 194–196. Residues 79–91 are disordered in free BamHI (Newman et al., 1994) but fold in the presence of DNA, albeit to a different conformation in the specific and the nonspecific complexes, respectively. Energy minimization with CHARMM (Brooks et al., 1983) was done with all heavy atoms in N-DNA28 and BAM_X fixed, except for residues 79–84 and 194–196 in each BamHI monomer (ɛ = R; 1000 steps of steepest descent). The minimized structure of BAM_X was named BAM_XM. A symmetric model of BamHI, BAM_SYM, was constructed by rebuilding the last eight residues in monomer B as α-helix in INSIGHTII (Biosym Technologies). The electrostatic contribution to the nonspecific interaction was calculated as for the rigid body association of DNA and BamHI, in which these loop regions remain folded as in the nonspecific complex. All solvent accessible surface area (SASA) calculations were done in CHARMM using a probe radius of 1.4 Å.
RESULTS AND DISCUSSION
The nonspecific BamHI-DNA complex (PDB code 1esg) includes 11 base pairs of DNA. To enable comparisons with experimental studies on the same system, a 28-mer DNA was built with the program JUMNA (Lavery et al., 1995) based on the available 8-mer DNA in the crystal structure; details of this method were described in a recent paper (Lebrun et al., 2001) and are briefly reviewed in the Methods section. The energy terms were calculated with NLPB theory as described in Methods. The values were used to calculate the electrostatic part of the binding free energy and its salt dependence (see Methods).
Electrostatic interaction in the crystal model (ns-crys/28)
NLPB calculations of the crystallographically determined nonspecific complex, translated to a 28-mer DNA stretch (Fig. 1, left) (see Methods), showed that the electrostatic interaction contributed about +16.9 kcal/mol to the nonspecific DNA binding at 0.1 M monovalent salt concentration. The large positive value of the electrostatic free energy of binding, discussed qualitatively by Viadiu and Aggarwal (Viadiu and Aggarwal, 2000), challenges the common view of the nonspecific protein-DNA interaction as electrostatically favorable (Jen-Jacobson, 1997; Manning, 1978; Record et al., 1978; Record et al., 1991). Because the exact binding value calculated with the NLPB method depends strongly on the parameters used in the calculation, whereas the calculated salt dependence of electrostatic interaction between protein and DNA (the “polyelectrolyte signature” of protein-DNA interaction) is less sensitive to the parameter sets (Fogolari et al., 1997; Misra et al., 1994), we proceeded to calculate the salt-dependent property of the electrostatic interaction in the crystal complex model. The electrostatic interaction was calculated for a range of monovalent salt concentrations from 0.1 M to 0.4 M, for which the experimental data are available for the nonspecific BamHI-DNA interaction (Engler, 1998; Engler et al., 2001). Surprisingly, the plot of electrostatic free energy of binding ΔΔGe against ln[S] (where [S] represents the salt concentration), showed little dependence of binding on salt concentration (Fig. 2, top) for this complex. Moreover, the slope of the fitted line is only 0.38 (Fig. 2, top), much smaller than the experimental value of 5∼6 (Engler, 1998).
FIGURE 1.


Models of nonspecific BamHI-DNA complex. Models of 28-mer DNA (N-DNA28) and BamHI dimer (BAM_XM) were built as described in Methods, based on the crystal structure of the nonspecific complex. The depicted models of the nonspecific complex were constructed from these components: (Left) The ns-crys/28 model was built directly from the crystal structure (PDB code 1esg); (Right) The ns-xt15r/28 model was constructed by positioning BAM_XM 34 Å away from the DNA axis, with its dimer axis parallel to the DNA axis, to model the configuration found in the NLPB calculations to have optimal interaction energy (see text). This positions BamHI ∼15 Å away from its position in the crystal structure.
FIGURE 2.



Calculated salt dependence of electrostatic interaction in the models of nonspecific BamHI-DNA complex. NLPB calculations were conducted at salt concentration from 0.1 M to 0.4 M, van der Waals surface was used to define solute-solvent boundary. (Top) ns-crys/28 with N-DNA28-BAM_XM; (middle) ns-xt15r/28 with N-DNA28-BAM_XM; (bottom) ns-xt15r/28 with BDNA28 and symmetrical BamHI model BAM_SYM in the optimal configuration (see text).
Another feature of the protein-DNA interaction that can indicate the nature of the binding interaction is the change of heat capacity (ΔCp) associated with the binding process, the so-called thermodynamic signature (Jen-Jacobson, 1997; Spolar and Record, 1994). Like other specific binding proteins, binding of BamHI to its cognate DNA site was shown to be associated with a large negative heat capacity change (ΔCp = −1.3 kcal/mol/K), while there is no detectable ΔCp in nonspecific binding (ΔCp ∼ 0.0 kcal/mol/K) (Engler, 1998; Engler et al., 2001). There are several sources for the ΔCp in protein-DNA association including the hydrophobic contribution from the release of surface water during association, the effect of association on the internal vibrations (Sturtevant, 1977), binding-coupled protein folding (Spolar and Record, 1994), water trapped at the protein-DNA interface (Engler, 1998), and hydration of the released ions from DNA (Oda et al., 1998). Among them, the hydrophobic effect has been considered as the major contribution to ΔCp (Sturtevant, 1977). We estimated the possible ΔCp for nonspecific binding based on the crystal model by considering the hydrophobic effect. Neglecting the refolding of the 79–91 loop region, and treating nonspecific binding as rigid body association, the lower limit of buried solvent accessible surface area (SASA) for nonspecific BamHI-DNA binding was calculated to be ∼2500 Å2, composed of 1300 Å2 of nonpolar surface, and 1200 Å2 of polar surface. According to the empirical formula for ΔCp from SASA (Spolar and Record, 1994):
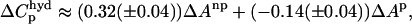 |
(5) |
the estimation of ΔCp from the hydrophobic effect is about −0.25 ± 0.10 kcal/mol/K, significantly different from experimental data showing almost no heat capacity change upon nonspecific BamHI-DNA binding (Engler, 1998).
Electrostatically favorable model (ns-xt15r/28)
The divergence between salt-dependence experimental results and the results from NLPB calculations motivated the search for an alternative form of the nonspecific BamHI-DNA complex. BD simulations were used to study the association of BamHI with a 90-mer nonspecific B-form DNA using the program SDA (Gabdoulline and Wade, 1998) with the effective charge method (Gabdoulline and Wade, 1996), and taking into consideration the desolvation term (Elcock et al., 1999). Similar BD simulations had been performed for the Cro repressor-DNA system (Thomasson et al., 1997). From the BD simulation (data not shown), we find that the energy minimum is located about ∼34 Å away from the DNA axis, and when BamHI approaches this energy minimum, the dimer axis of BamHI is nearly parallel to the helix axis of DNA, as shown in Fig. 1, right. The energetics of this configuration of a putative nonspecific BamHI-DNA complex was analyzed with NLPB calculations. The electrostatic interaction between BamHI and the 28-mer DNA was calculated at several distances while keeping BamHI parallel to the DNA axis. The most favorable configuration is shown in Fig. 1, right, where the distance between BamHI and DNA axis is ∼34 Å, 15 Å larger than in the crystal structure. This configuration was therefore used throughout to explore the properties of the nonspecific BamHI-DNA complex. In this alternative model of the nonspecific complex, referred to as ns-xt15r/28, the calculated electrostatic free energy of binding was favorable (−3.55 kcal/mol), and exhibited a strong salt dependence with a slope of the ΔΔGe versus ln[S] of ∼3.0. The buried SASA was only ∼120 Å2, with negligible ΔCp according to Eq. 5. Thus, the results for this alternative model agree with all the elements of the experimental findings about the nonspecific complex in solution.
The mechanism of stabilization of the complex in the ns-xt15r/28 model is best appreciated from the calculated electrostatic potential surface of BamHI displayed with GRASP program (Nicholls et al., 1991) (Fig. 3). The BamHI dimer generates a negative potential on one side of the surface, whereas the opposite side of the surface is mostly positive. Not surprisingly, the DNA binding cleft is located in the positive side. Viadiu and Aggarwal (Viadiu and Aggarwal, 2000) compared the electrostatic surface of BamHI in the nonspecific and specific complexes, and showed that the protein-DNA interface in the nonspecific complex is located at the bottom of the DNA binding cleft, which has a mixed positive and negative electrostatic potential surface. Because of the strong negative potential of the DNA surface, the electrostatic interaction calculated with NLPB for the crystal model was inherently unfavorable. However, the most positive potential in the DNA binding side is a region located at the C-terminal of each monomer, which includes the C-terminal helix (helix 7) and the loop preceding it. This region of BamHI, comprising ∼20 residues, includes seven Lys and one Arg, but only two Asp and one Glu in each monomer. Thus, it is reasonable that when the BamHI dimer approaches DNA, these two regions with the most positive potential will point toward DNA to form favorable electrostatic interactions, as in the alternative model (Fig. 1, right).
FIGURE 3.

Electrostatic surface potential and contours of BamHI model BAM_SYM. Electrostatic potential was calculated with program UHBD, displayed with GRASP. Solvent accessible surface with probe radius 1.4 Å was used to define solute-solvent boundary.
The mutual orientation of the interacting molecules and implications for sliding
The orientation of BamHI relative to the axis of DNA in the various complexes reflects the different modes of interaction. In the computed model, the BamHI dimer is parallel to the DNA axis, and in the nonspecific crystal structure it is tilted by ∼60° from the DNA axis. In the specific complex, the axis through the BamHI dimer is nearly perpendicular (∼80°) to the DNA axis (Newman et al., 1994; Viadiu and Aggarwal, 2000). To better understand the role of electrostatic interaction in determining the preferred orientation of BamHI relative to DNA, we performed a series of NLPB calculations around the theoretical new model, in which BamHI was rotated in steps of 15° along an axis (x) that is perpendicular to DNA helix axis (z axis) and passes through the center of BamHI. Similar calculations have been done in the Homeodomain-DNA system to study the orientation effect of binding (Fogolari et al., 1997). In that system, NLPB calculation showed that rotation along x axis did not affect the electrostatic interaction in the complex, while rotation along the z axis dramatically changed the interaction, leading to the conclusion that electrostatic forces might ensure the approach of the homeodomain to DNA with correct orientation regarding to z axis, but not along the x axis. Unlike the homeodomain-DNA system, our calculations demonstrated that in the BamHI-DNA system the electrostatic force could also steer the BamHI to approach DNA with a “correct” orientation with respect to the x axis (refer to the orientation in the ns-xt15r/28 nonspecific complex model), as shown in Fig. 4. The difference between the optimal orientation (parallel to DNA helix axis) and the energetically least favorable orientation (45°) was calculated to be ∼7 kcal/mol, while relative to the optimal configuration, this difference can be as high as ∼9 kcal/mol (see below). Thus, in the nonspecific BamHI-DNA system, the electrostatic interaction itself seems capable of steering BamHI to a unique orientation relative to the DNA axis.
FIGURE 4.

Calculated mutual orientation effect on the electrostatic interaction in ns-xt15r/28 model. Electrostatic binding energy was calculated with UHBD. Different relative orientations between BamHI and DNA were generated through rotation of BamHI along the axis defined by the centers of DNA and BamHI. Line with circle, BDNA28 with the center of BAM_SYM facing DNA major groove; line with square, BDNA28 with the center of BAM_SYM facing DNA minor groove; dashed line with triangle, BDNA28 with the center of BAM_SYM facing DNA minor groove, and boundary between solute and solvent defined by solvent accessible surface with probe radius 1.4 Å.
To understand how the enzyme searches its cognate site along a stretch of DNA, we undertook a study of the electrostatic interaction between BamHI and DNA along the possible sliding paths indicated by the profile of the electrostatic interaction energy. A “sliding” model has been proposed and tested experimentally in several type II endonuclease restriction enzymes (Pingoud and Jeltsch, 1997). According to the sliding model, the protein would perform one-dimensional random walks along DNA driven by thermal energy. In the kinetic study on EcoRI and EcoRV system, these experiments indirectly suggested that the protein would follow a helical path along the DNA (Jeltsch et al., 1994). However, the detailed sliding path of the protein along DNA was not determined, and it is not clear whether the protein follows one face of the DNA along the axis or slides along the helical path. With the assumption that electrostatic interaction is the major contribution to nonspecific binding (Jen-Jacobson, 1997; Manning, 1978; Record et al., 1978; Record et al., 1991), the electrostatic interaction may determine the preferred sliding pathway.
If BamHI slides along one face of DNA, in a “straight path”, it will face alternating major groove and minor groove sections during the sliding process. Since this is sequence independent, the sliding process can be simulated in our theoretical model by rotating BamHI around the DNA axis so as to face the different grooves on DNA. In our model (Fig. 1, right), the N-termini of helix 3 (residues 58–70) and helix 7 (residues 200–211) of BamHI are facing the minor groove while the center of the dimer is close to the DNA major groove. Fig. 5 shows that the initial configuration with ΔΔGe of about −3.6 kcal/mol is not optimal; in the optimal configuration the N-termini of two helices (helix 3 and 7) approach the major groove (ΔΔGe −5.9 kcal/mol). This configuration also yields a salt dependence of the interaction in the model that is in better agreement with experiment (the slope of plot ΔΔGe versus ln[S] is 3.9 (Fig. 2, bottom)). With the symmetrical BamHI model and B form DNA, the binding energy varies in a range between −4.2 kcal/mol and −6.8 kcal/mol, indicating that while sliding along a B-form like DNA in a straight path, BamHI would encounter energy barriers of 2.6 kcal/mol when the center of the dimer moves from the minor groove to the next major groove. On the other hand, if BamHI slides along the helical path, it can maintain throughout the optimal interaction configuration. In considering the calculated values of the barriers, it is noteworthy that the continuum solvent model, unlike an explicit solvent model, cannot accurately represent the role of the interface water molecules, such as the water-bridged hydrogen bonds between the protein and the DNA. However, we expect the role of the interface water molecules would be similar in the minor and major grooves, so that the overall conclusion regarding the relative probabilities of the alternative paths should not be affected by this approximation.
FIGURE 5.

Calculated energy profile of simulated sliding along a straight path. DNA in ns-xt15r/28 model was rotated along its axis in steps of 15°, started from Fig. 1. Line with circle, N-DNA28 with BAM_X; line with square: BDNA28 with BAM_SYM; dashed line with triangle, BDNA28 with BAM_SYM, and boundary between solute and solvent defined by solvent accessible surface with probe radius 1.4 Å.
Effects of mutations on the nonspecific complex: a test of the molecular model
Mutations in BamHI were used to identify the possible catalytic residues before the crystal structures of free BamHI and specific complex were solved (Dorner and Schildkraut, 1994; Xu and Schildkraut, 1991). In this early analysis of the BamHI, two mutants—E77K and E113K—were reported to have enhanced nonspecific DNA binding. These two residues are located at the bottom of the DNA binding cleft and have been suggested to be involved in catalysis (Dorner and Schildkraut, 1994; Viadiu and Aggarwal, 1998). We calculated the effect of the mutations on the electrostatic binding interactions to nonspecific DNA based on the optimal configuration in the theoretical model. In agreement with the experimental results, the calculated ΔΔGe showed that both mutants had increased binding affinity to nonspecific DNA, by ∼0.4 kcal/mol (E77K) and 1.2 kcal/mol (E113K). The closest distance of Glu-113 to DNA backbone is ∼22 Å, whereas for Glu-77 it is ∼27 Å, with the corresponding screened Coulombic interactions with DNA yielding +5.1 kcal/mol and +7.1 kcal/mol, respectively. The results support the computed model of the nonspecific complex, as the differential increase in binding affinities correlates well with the corresponding distance to DNA in the theoretical model, as well as with the solvent-screened Coulombic interaction with DNA.
Table 1 lists all the charged residues with strong solvent-screened Coulombic interaction with DNA, together with their distance to the DNA backbone. The salt-independent electrostatic contribution to the total binding free energy has been decomposed into two parts: the desolvation from the low dielectric cavity, and the solvent-screened Coulombic interaction (Gilson and Honig, 1988; Misra et al., 1998). Since the desolvation part is always unfavorable, the favorable interaction could only come from the solvent-screened Coulombic interaction. The charged residues located at the protein-DNA interface (N-terminal of helix 3 and helix 7, and their preceding loop regions) should have a strong impact on the protein-DNA interaction. Two negatively charged residues, Glu-51 and Asp-196, are located in the two loop regions closest to the DNA backbone, at 10 Å and 9 Å, respectively. From the theoretical complex model, the replacement of any one of these two residues by a neutral or positively charged residue would greatly enhance the nonspecific binding affinity. NLPB calculations indicate the increased nonspecific binding affinity for neutral mutation to be 2.2 kcal/mol (E51Q) and 2.4 kcal/mol (D196N), while mutations to the charged residues should lead to an increase of ∼4.4 kcal/mol (E51K) and 5.3 kcal/mol (D196K). Since Asp-196 is also involved in specific recognition (Newman et al., 1995), mutation of this residue would affect the specific interaction as well. The role of Glu-51 in the specific interaction may not be as important as that of Asp-196 according to the crystal structure of the specific complex (Newman et al., 1995). Therefore we expect that a E51Q or E51K mutant would increase only the nonspecific interaction, and according to the sliding model, would affect the sliding ability of BamHI along DNA, as suggested earlier in the study of EcoRV mutants (Jeltsch et al., 1996).
TABLE 1.
Calculated solvent-screened Coulombic interactions of charged residues with DNA
| Residue | ΔGsc (kcal mol−1) | Distance (Å) |
|---|---|---|
| Glu-51 | 9.8 | 10.1 |
| Lys-52 | −7.3 | 15.3 |
| Lys-61 | −8.3 | 18.5 |
| Glu-62 | 7.4 | 17.0 |
| Arg-76 | −7.2 | 19.3 |
| Glu-86 | 7.2 | 22.5 |
| Glu-13 | 7.1 | 21.8 |
| Arg-122 | −7.9 | 19.6 |
| Lys-126 | −7.2 | 24.0 |
| Asp-154 | 9.2 | 9.9 |
| Arg-155 | −9.2 | 9.9 |
| Glu-161 | 10.6 | 10.5 |
| Lys-193 | −10.6 | 10.5 |
| Glu-196 | 10.0 | 9.1 |
| Lys-200 | −7.2 | 17.1 |
| Lys-205 | −10.3 | 7.0 |
| Lys-209 | −8.0 | 9.6 |
The van der Waals surface was used to define the solute-solvent boundary. Salt concentration is 0 M.
CONCLUSIONS
In the association of protein with double-stranded DNA, the long-range Coulombic interaction has been considered the dominant contribution to nonspecific protein-DNA binding that brings proteins to the proximity of DNA to form a loose, dynamic complex (Jen-Jacobson, 1997; Oda and Nakamura, 2000; Record et al., 1991). After this nonspecific binding, the specific binding occurs at the cognate site where specific hydrogen bonds and van der Waals interactions as well as hydrophobic interaction play a more important role than in the initial nonspecific binding (Hard and Lundback, 1996). From the analysis of the electrostatic interaction in the nonspecific binding of BamHI to DNA, using the NLPB method and the crystal structure of the nonspecific BamHI-DNA complex, we found an electrostatically favorable configuration in which BamHI is driven to an orientation parallel to the DNA helix axis, that better recapitulates the experimental data. This new model for the nonspecific complex made possible an energy-based analysis of the structural basis for protein sliding along the DNA in search for the cognate site. The results suggest that electrostatic interaction alone constrain BamHI to slide along a preferred DNA helix path. According to our model of nonspecific binding, residues Glu-51 and Asp-196 in BamHI would strongly affect the nonspecific interaction to DNA and therefore determine the protein's sliding ability along the helical path.
DNA sliding requires not only moderate affinity, but also equal affinity for different binding sites along DNA (Breyer and Matthews, 2001; Jen-Jacobson, 1997). However, due to the intrinsic periodic double helix structure of DNA with alternative major and minor grooves, the electrostatic surface potential of DNA is not uniform, with the minor groove more negative than the major groove (Jayaram et al., 1989). This feature of DNA produces an energy barrier if BamHI were to slide from the minor groove region to the next major groove (Fig. 5). Here, the energy barrier was estimated from NLPB calculations to be ∼2.5 kcal/mol. To avoid this barrier during sliding, BamHI could slide smoothly along the DNA helical pitch in the orientation described here, determined solely by the electrostatic interaction. Such a configuration could contribute to the quick scan of the nonspecific DNA by the protein, and could be a common feature of type II endonucleases where the biological function is predominantly kinetically controlled (Jeltsch et al., 1996).
A comparison of the free BamHI and BamHI-DNA complex structures (Breyer and Matthews, 2001; Viadiu and Aggarwal, 2000) had suggested that the DNA would be positioned at the concave binding site formed by the BamHI dimer. However, the quantitative analysis of the electrostatic interaction undertaken here suggested that binding in the concave surface could not achieve a favorable electrostatic interaction for sliding. Although there is a fairly large interaction interface in the nonspecific crystal structure, most of it is surrounded by electrostatic potential that is usually negative at pH 7 and would therefore contribute unfavorably to the complex formation. We suggest that the structure of the complex with DNA bound in the concave surface may represent an intermediate state in the transition from the nonspecific complex to the specific complex. The initial nonspecific binding may be better represented by the alternative model proposed here, which has a smaller interaction surface but is in better agreement with the experimental measurements, including salt dependence, favorable electrostatic interaction and negligible heat capacity change (Engler, 1998). Thus, during a search for its cognate site, BamHI may slide along this electrostatically favorable configuration, punctuated by a transition to the orientation seen in the nonspecific crystal structure (Viadiu and Aggarwal, 2000), and facilitated by local structural features. Such local features could modify the protonation states of selective residues in the protein, and hence the electrostatic potential in the interacting surface (data not shown). The larger interaction interface observed crystallographically, may be critical in sensing the target sequence and triggering the conformational changes that lead to specific hydrogen bonding, van der Waals and hydrophobic interactions in the specific complex.
Acknowledgments
The SDA program was kindly provided by Dr. Wade. Computations were performed on the National Science Foundation Terascale Computing System at the Pittsburgh Supercomputing Center.
The work is supported by National Institutes of Health grants DA-00060 (to H.W.) and GM44006 (to A.A.).
References
- Albright, R. A., M. C. Mossing, and B. W. Matthews. 1998. Crystal structure of an engineered Cro monomer bound nonspecifically to DNA: possible implications for nonspecific binding by the wild-type protein. Protein Sci. 7:1485–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, C. F., and M. T. Record, Jr. 1995. Salt-nucleic acid interactions. Annu. Rev. Phys. Chem. 46:657–700. [DOI] [PubMed] [Google Scholar]
- Bewley, C. A., A. M. Gronenborn, and G. M. Clore. 1998. Minor groove-binding architectural proteins: structure, function, and DNA recognition. Annu. Rev. Biophys. Biomol. Struct. 27:105–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyer, W. A., and B. W. Matthews. 2001. A structural basis for processivity. Protein Sci. 10:1699–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Dorner, L. F., and I. Schildkraut. 1994. Direct selection of binding proficient/catalytic deficient variants of BamHI endonuclease. Nucleic Acids Res. 22:1068–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcock, A. H., R. R. Gabdoulline, R. C. Wade, and J. A. McCammon. 1999. Computer simulation of protein-protein association kinetics: acetylcholinesterase-fasciculin. J. Mol. Biol. 291:149–162. [DOI] [PubMed] [Google Scholar]
- Engler, L. E. 1998. Specificity determinants in the BamHI endonulease-DNA interaction. Ph.D. thesis. University of Pittsburgh. 255 pp.
- Engler, L. E., P. Sapienza, L. F. Dorner, R. Kucera, I. Schildkraut, and L. Jen-Jacobson. 2001. The energetics of the interaction of BamHI endonuclease with its recognition site GGATCC. J. Mol. Biol. 307:619–636. [DOI] [PubMed] [Google Scholar]
- Fogolari, F., A. H. Elcock, G. Esposito, P. Viglino, J. M. Briggs, and J. A. McCammon. 1997. Electrostatic effects in homeodomain-DNA interactions. J. Mol. Biol. 267:368–381. [DOI] [PubMed] [Google Scholar]
- Foloppe, N., and A. D. Mackerell, Jr. 2000. All-atom Empirical force field for nucleic acids: I. parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 21:86–104. [Google Scholar]
- Frank, D. E., R. M. Saecker, J. P. Bond, M. W. Capp, O. V. Tsodikov, S. E. Melcher, M. M. Levandoski, and M. T. Record, Jr. 1997. Thermodynamics of the interactions of lac repressor with variants of the symmetric lac operator: effects of converting a consensus site to a non-specific site. J. Mol. Biol. 267:1186–1206. [DOI] [PubMed] [Google Scholar]
- Gabdoulline, R. R., and R. C. Wade. 1996. Effective charges for macromolecules in solvent. J. Phys. Chem. 100:3868–3878. [Google Scholar]
- Gabdoulline, R. R., and R. C. Wade. 1998. Brownian dynamics simulation of protein-protein diffusional encounter. Methods. 14:329–341. [DOI] [PubMed] [Google Scholar]
- Garner, M. M., and D. C. Rau. 1995. Water release associated with specific binding of gal repressor. EMBO J. 14:1257–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson, M. K., and B. Honig. 1988. Calculation of the total electrostatic energy of a macromolecular system: solvation energies, binding energies, and conformational analysis. Proteins. 4:7–18. [DOI] [PubMed] [Google Scholar]
- Hard, T., and T. Lundback. 1996. Thermodynamics of sequence-specific protein-DNA interactions. Biophys. Chem. 62:121–139. [DOI] [PubMed] [Google Scholar]
- Harrison, S. C., and A. K. Aggarwal. 1990. DNA recognition by proteins with the helix-turn-helix motif. Annu. Rev. Biochem. 59:933–969. [DOI] [PubMed] [Google Scholar]
- Jayaram, B., K. A. Sharp, and B. Honig. 1989. The electrostatic potential of B-DNA. Biopolymers. 28:975–993. [DOI] [PubMed] [Google Scholar]
- Jeltsch, A., J. Alves, H. Wolfes, G. Maass, and A. Pingoud. 1994. Pausing of the restriction endonuclease EcoRI during linear diffusion on DNA. Biochemistry. 33:10215–10219. [DOI] [PubMed] [Google Scholar]
- Jeltsch, A., C. Wenz, F. Stahl, and A. Pingoud. 1996. Linear diffusion of the restriction endonuclease EcoRV on DNA is essential for the in vivo function of the enzyme. EMBO J. 15:5104–5111. [PMC free article] [PubMed] [Google Scholar]
- Jen-Jacobson, L. 1997. Protein-DNA recognition complexes: conservation of structure and binding energy in the transition state. Biopolymers. 44:153–180. [DOI] [PubMed] [Google Scholar]
- Ladbury, J. E., J. G. Wright, J. M. Sturtevant, and P. B. Sigler. 1994. A thermodynamic study of the trp repressor-operator interaction. J. Mol. Biol. 238:669–681. [DOI] [PubMed] [Google Scholar]
- Lavery, R., K. Zakrzewska, and H. Sklenar. 1995. Jumna (Junction Minimization of Nucleic-Acids). Comput. Phys. Commun. 91:135–158. [Google Scholar]
- Lebrun, A., R. Lavery, and H. Weinstein. 2001. Modeling multi-component protein-DNA complexes: the role of bending and dimerization in the complex of p53 dimers with DNA. Protein Eng. 14:233–243. [DOI] [PubMed] [Google Scholar]
- Luisi, B. F., W. X. Xu, Z. Otwinowski, L. P. Freedman, K. R. Yamamoto, and P. B. Sigler. 1991. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature. 352:497–505 [see comments]. [DOI] [PubMed] [Google Scholar]
- Madura, J. D., J. M. Briggs, R. Wade, M. E. Davis, B. A. Luty, A. Ilin, A. Antosiewicz, M. K. Gilson, B. Bagheri, S. L. Ridgway, and J. A. McCammon. 1995. Electrostatic and diffusion of molecules in solution: simulation with the University of Houston Brownian Dynamics program. Comput. Phys. Commun. 91:57–95. [Google Scholar]
- Manning, G. S. 1978. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q. Rev. Biophys. 11:179–246. [DOI] [PubMed] [Google Scholar]
- Merabet, E., and G. K. Ackers. 1995. Calorimetric analysis of lambda cI repressor binding to DNA operator sites. Biochemistry. 34:8554–8563. [DOI] [PubMed] [Google Scholar]
- Misra, V. K., J. L. Hecht, K. A. Sharp, R. A. Friedman, and B. Honig. 1994. Salt effects on protein-DNA interactions. The lambda cI repressor and EcoRI endonuclease. J. Mol. Biol. 238:264–280. [DOI] [PubMed] [Google Scholar]
- Misra, V. K., J. L. Hecht, A. S. Yang, and B. Honig. 1998. Electrostatic contributions to the binding free energy of the lambdacI repressor to DNA. Biophys. J. 75:2262–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, M., T. Strzelecka, L. F. Dorner, I. Schildkraut, and A. K. Aggarwal. 1994. Structure of restriction endonuclease BamHI and its relationship to EcoRI. Nature. 368:660–664. [DOI] [PubMed] [Google Scholar]
- Newman, M., T. Strzelecka, L. F. Dorner, I. Schildkraut, and A. K. Aggarwal. 1995. Structure of Bam HI endonuclease bound to DNA: partial folding and unfolding on DNA binding. Science. 269:656–663. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., K. A. Sharp, and B. Honig. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 11:281–296. [DOI] [PubMed] [Google Scholar]
- Oda, M., K. Furukawa, K. Ogata, A. Sarai, and H. Nakamura. 1998. Thermodynamics of specific and non-specific DNA binding by the c-Myb DNA-binding domain. J. Mol. Biol. 276:571–590. [DOI] [PubMed] [Google Scholar]
- Oda, M., and H. Nakamura. 2000. Thermodynamic and kinetic analyses for understanding sequence-specific DNA recognition. Genes Cells. 5:319–326. [DOI] [PubMed] [Google Scholar]
- Pabo, C. O., and R. T. Sauer. 1992. Transcription factors: structural families and principles of DNA recognition. Annu. Rev. Biochem. 61:1053–1095. [DOI] [PubMed] [Google Scholar]
- Pingoud, A., and A. Jeltsch. 1997. Recognition and cleavage of DNA by type-II restriction endonucleases. Eur. J. Biochem. 246:1–22. [DOI] [PubMed] [Google Scholar]
- Record, M. T., Jr., C. F. Anderson, and T. M. Lohman. 1978. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: the roles of ion association or release, screening, and ion effects on water activity. Q. Rev. Biophys. 11:103–178. [DOI] [PubMed] [Google Scholar]
- Record, M. T., Jr., J. H. Ha, and M. A. Fisher. 1991. Analysis of equilibrium and kinetic measurements to determine thermodynamic origins of stability and specificity and mechanism of formation of site-specific complexes between proteins and helical DNA. Methods Enzymol. 208:291–343. [DOI] [PubMed] [Google Scholar]
- Sharp, K. A., and B. Honig. 1990. Calculation total electrostatic energies with the nonlinear Poisson-Boltzmann equation. J. Phys. Chem. 94:7684–7692. [Google Scholar]
- Sidorova, N. Y., and D. C. Rau. 1996. Differences in water release for the binding of EcoRI to specific and nonspecific DNA sequences. Proc. Natl. Acad. Sci. USA. 93:12272–12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spolar, R. S., and M. T. Record, Jr. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science. 263:777–784 [see comments]. [DOI] [PubMed] [Google Scholar]
- Sturtevant, J. M. 1977. Heat capacity and entropy changes in processes involving proteins. Proc. Natl. Acad. Sci. USA. 74:2236–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda, Y., P. D. Ross, and C. P. Mudd. 1992. Thermodynamics of Cro protein-DNA interactions. Proc. Natl. Acad. Sci. USA. 89:8180–8184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomasson, K. A., I. V. Ouporov, T. Baumgartner, J. Czlapinski, T. Kaldor, and S. H. Northrup. 1997. Free energy of nonspecific binding of Cro repressor protein to DNA. J. Phys. Chem. 101:9127–9136. [Google Scholar]
- Viadiu, H., and A. K. Aggarwal. 1998. The role of metals in catalysis by the restriction endonuclease BamHI. Nat. Struct. Biol. 5:910–916. [DOI] [PubMed] [Google Scholar]
- Viadiu, H., and A. K. Aggarwal. 2000. Structure of BamHI bound to nonspecific DNA: a model for DNA sliding. Mol. Cell. 5:889–895. [DOI] [PubMed] [Google Scholar]
- von Hippel, P. H. 1994. Protein-DNA recognition: new perspectives and underlying themes. Science. 263:769–770. [DOI] [PubMed] [Google Scholar]
- Winkler, F. K., D. W. Banner, C. Oefner, D. Tsernoglou, R. S. Brown, S. P. Heathman, R. K. Bryan, P. D. Martin, K. Petratos, and K. S. Wilson. 1993. The crystal structure of EcoRV endonuclease and of its complexes with cognate and non-cognate DNA fragments. EMBO J. 12:1781–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S. Y., and I. Schildkraut. 1991. Isolation of BamHI variants with reduced cleavage activities. J. Biol. Chem. 266:4425–4429. [PubMed] [Google Scholar]
- Zacharias, M., B. A. Luty, M. E. Davis, and J. A. McCammon. 1992. Poisson-Boltzmann analysis of the lambda repressor-operator interaction. Biophys. J. 63:1280–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]