

Abstract
HIV-1 reverse transcriptase (RT) is the primary target for anti-AIDS chemotherapy. Nonnucleoside RT inhibitors (NNRTIs) are very potent and most promising anti-AIDS drugs that specifically inhibit HIV-1 RT. The binding and unbinding processes of α-APA, an NNRTI, have been studied using nanosecond conventional molecular dynamics and steered molecular dynamics simulations. The simulation results show that the unbinding process of α-APA consists of three phases based on the position of α-APA in relation to the entrance of the binding pocket. When α-APA is bound in the binding pocket, the hydrophobic interactions between HIV-1 RT and α-APA dominate the binding; however, the hydrophilic interactions (both direct and water-bridged hydrogen bonds) also contribute to the stabilizing forces. Whereas Tyr-181 makes significant hydrophobic interactions with α-APA, Tyr-188 forms a strong hydrogen bond with the acylamino group (N14) of α-APA. These two residues have very flexible side chains and appear to act as two “flexible clamps” discouraging α-APA to dissociate from the binding pocket. At the pocket entrance, two relatively inflexible residues, Val-179 and Leu-100, gauge the openness of the entrance and form the bottleneck of the inhibitor-unbinding pathway. Two special water molecules at the pocket entrance appear to play important roles in inhibitor recognition of binding and unbinding. These water molecules form water bridges between the polar groups of the inhibitor and the residues around the entrance, and between the polar groups of the inhibitor themselves. The water-bridged interactions not only induce the inhibitor to adopt an energetically favorable conformation so the inhibitor can pass through the pocket entrance, but also stabilize the binding of the inhibitor in the pocket to prevent the inhibitor's dissociation. The complementary steered molecular dynamics and conventional molecular dynamics simulation results strongly support the hypothesis that NNRTIs inhibit HIV-1 RT polymerization activity by enlarging the DNA-binding cleft and restricting the flexibility and mobility of the p66 thumb subdomain that are believed to be essential during DNA translocation and polymerization.
INTRODUCTION
Human immunodeficiency virus type 1 reverse transcriptase (HIV-1 RT) is one of the most important antiretroviral targets in the chemotherapy of acquired immune deficiency syndrome (AIDS) patients. The drug discovery researches targeting on HIV-1 RT have been extensively enhanced in the past two decades (De Clercq, 1996, 1998, 1999, 2001, 2002; Sala and Vartanian, 1998; Turner and Summers, 1999; Chen et al., 2000; Buckheit, 2001; Gupta, 2002; Campiani et al., 2002). Currently approved drugs targeting on HIV-1 RT to treat HIV infection fall mainly into two classes: nucleoside-analogous RT inhibitors (NRTIs) and nonnucleoside RT inhibitors (NNRTIs). NRTIs, such as 3′-azido-3′-deoxythymidine (AZT), 2′,3′-dideoxyinosine (ddI) and 2′,3′-dideoxycytidine (ddC), are competitive inhibitors that bind at the deoxynucleoside triphosphate (dNTP) binding pocket and function as chain terminator in DNA elongation. These inhibitors are effective in inhibiting HIV-1 RT; however, their application is limited due to the high cytotoxicity (inhibition to other normal polymerization process in vivo) and the emergence of drug-resistant mutants (reviewed in Larder, 1993; Tantillo et al., 1994; De Clercq, 1994, 2001; Ding et al., 1997). NNRTIs are a chemically diverse set of compounds with high specificity to HIV-1 RT and, therefore, have the potential to become the most promising anti-AIDS drugs. Three NNRTIs, Nevirapine (a dipyridodiazepinone derivative), Delavirdine (BHAP U-90152), and Efavirenz, have been approved by the U.S. Food and Drug Administration (FDA) and are used in combination with NRTIs and PIs (protease inhibitors) to treat AIDS patients (De Clercq, 1998, 1999, 2001, 2002; Buckheit, 2001; Gupta, 2002). However, similar to NRTIs, NNRTIs' Achilles heel is the emergence of drug resistant mutations in the enzyme, and there is no final treatment solution to AIDS yet (Larder, 1993; Richman, 1993; Campiani et al., 2002; De Clercq, 2002).
A number of high-resolution crystal structures of HIV-1 RT in complexing with NNRTIs in both wild-type and mutant-type have been reported in the past decade (Kohlstaedt et al., 1992; Ding et al., 1995a,b; Ren et al., 1995a,b, 2001; Das et al., 1996; Hsiou et al., 1998, 2001; Lindberg et al., 2002). Crystal structures demonstrate that HIV-1 RT is composed by two subunits, p66 and p51, and that NNRTIs bind to the p66 subunit. The three-dimensional (3D) model of the p66 subunit is schematically represented in Fig. 1. In general, the structure of p66 looks like a human hand, comprising fingers subdomain, thumb subdomain, palm subdomain, and connection subdomain linking with Rnase H subdomain (not shown in Fig. 1). The apoenzyme adopts a “close” conformation in which the p66 thumb subdomain folds down into the DNA binding cleft and makes contacts with the tip of fingers (Jager et al., 1994; Esnouf et al., 1997; Rodgers et al., 1995; Hsiou et al., 1996). Binding with template-primer of DNA or inhibitors, the p66 thumb subdomain takes an upright or “open” conformation and, together with the p66 fingers and palm subdomains, forms a large cleft to accommodate the template-primer substrate (Jacobo-Molina et al., 1993; Ding et al., 1998). Interestingly, NNRTIs can burrow a naturally inexistent hydrophobic pocket to accommodate themselves. The NNRTI binding pocket (NNIBP) is located ∼10 Å away from the polymerase active site and is primarily consisted of Leu-100, Lys-101, Lys-103, Val-106, Thr-107, Val-108, Val-179, Tyr-181, Tyr-188, Val-189, Gly-190, Phe-227, Trp-229, Leu-234, and Tyr-318 in p66 (Kohlstaedt et al., 1992; Ding et al., 1995a). The putative entrance of the pocket is proposed to be located at the interface of the p66/p51 heterodimer and is primarily formed by Pro-95, Leu-100, Lys-101, Lys-103, Val-179, and Tyr-181 of p66 (Ding et al., 1995a; Ren et al., 1995a; Esnouf, et al., 1997). The side chains of several residues, especially those of Tyr-181 and Tyr-188, assume significant conformational changes on the binding of NNRTIs. The secondary structural elements around the NNIBP rearrange as well in comparison with the structure of the apoenzyme (Ding et al., 1995a).
FIGURE 1.

A schematic representation of the 3D structure feature of p66 subunit of HIV-1 RT and the diagram of the SMD simulation system. The p66 subunit is shown in solid ribbon and α-APA in ball-and-stick model. The NNRTI binding pocket is represented with the VDW surface. During the SMD simulation, the bound α-APA is pulled away from the binding pocket using a harmonic potential symbolized by an artificial spring that is connected to the C12 atom of α-APA (labeled in green color). This pulling potential moves with a constant velocity of 0.02 Å/ps in the direction indicated by the arrow. Three snapshots of α-APA in the unbinding pathway are shown, representing the initial state, the state passing through the entrance of the binding pocket, and the final state in the SMD trajectory, respectively. This picture was generated using WebLab ViewerPro program (MSI, 1998) and rendered using the POV-Ray program (Pov-Ray-Team, 1999). The numbering of α-APA is shown in the schematic diagram of α-APA.
This unique ligand-enzyme interaction phenomenon raises interesting questions: how do NNRTIs induce the formation of the NNIBP? How do NNRTIs inhibit the polymerization activity of HIV-1 RT? So far, no convincing data have been reported concerning these two questions. To investigate the microscopic dynamic processes of NNRTIs binding and the formation of NNIBP, large scale conventional molecular dynamics (CMD) and steered molecular dynamics (SMD) simulations were performed. For the CMD simulation, the inhibitor was harmonically constrained in the NNIBP; and for the SMD simulation, a harmonic potential was applied to the inhibitor to pull the inhibitor out from the binding pocket artificially. Through simulations and comparisons with various crystal structures of HIV-1 RT bound with and without NNRTIs, we intend to explore the detailed protein-inhibitor interactions related to inhibitor binding and unbinding and gain insights into the inhibition mechanism of NNRTIs to HIV-1 RT.
SMD is an extended MD simulation method mimicking the principle of the atomic force microscopy (AFM) (Binning et al., 1986; Block and Svoboda, 1994; Evans et al., 1995). SMD has been widely used to explore the binding and unbinding properties of biomolecules and their responses to external mechanical manipulations at the atomic level (Izrailev et al., 1998). SMD has been successfully applied to identify several ligand binding pathways (Grubmüller et al., 1996; Isralewitz et al., 1997; Stepaniants et al., 1997; Marrink et al., 1998; Wriggers and Schulten, 1997) and to explain the elastic properties of some proteins (Lu et al., 1998; Lu and Schulten, 1999; Heymann and Grubmüller, 1999a; Krammer et al., 1999; Heymann and Grubmüller, 2001). In SMD simulations, time-dependent external forces are applied to ligand to facilitate its unbinding with a protein as shown in Fig. 1. From the accelerated dissociation process of the ligand, SMD simulation can reveal information about the enzyme's flexibility and its response to the dissociation of ligand. Analyses of interactions between the dissociating ligand and the protein (especially residues forming the binding pocket) and the relationship between the applied forces and the ligand position could yield important information about the structure-function relationships of the protein-ligand complex, the binding and unbinding pathway(s), and possible mechanisms of ligand recognition and inhibition.
In this work, we present the molecular dynamics studies of the unbinding process of α-APA, an NNRTI, to HIV-1 RT. The simulation results show that the polar groups of α-APA play important roles in inhibitor recognition and binding. Water-bridged hydrogen bonds appear to not only mediate the protein-inhibitor interactions but also induce conformational change of the inhibitor and, thus, facilitate the inhibitor binding and unbinding. Comparison of the complementary SMD and CMD simulation results strongly supports the hypothesis that NNRTIs inhibit HIV-1 RT polymerization activity by enlarging the DNA-binding cleft and restricting the mobility of the p66 thumb subdomain.
MATERIAL AND METHODS
Molecular model
As a pregeneration of Loviride, α-APA is a typical NNRTI with high activity (IC50 of 5 nM) and high selectivity (selectivity index of 84,000) (Pauwels and Janssen, 1993). It is selected to explore the binding and unbinding properties of NNRTIs in the present molecular dynamics simulations. The simulation model was constructed based on the x-ray crystal structure of HIV-1 RT/α-APA complex (PDB entry 1HNI) (Ding et al., 1995a). The side chains with missing coordinates in the crystal structure were reconstructed using the fragment library of the Biopolymer module in Sybyl version 6.7 (Tripos Inc., 1999). The modified structure was subjected to energy minimization using steepest descent method up to the gradient tolerance of 0.05 kcal/(mol·Å) to relieve possible steric clashes and overlaps of side chains.
The entire structure of the HIV-1 RT/α-APA complex is too large to be simulated by our current computational facilities. Structural and biochemical data indicate that the NNIBP is mainly composed of structural elements of the p66 subunit (Kohlstaedt et al., 1992; Ding et al., 1995a; Ren et al., 1995a) and that the RNase H subdomain and the p51 subunit are not directly involved in NNRTI binding and play a minor role in the sensitivity to NNRTIs (Boyer et al., 1992; Auwerx et al., 2002). Therefore, in the current simulation studies, both the RNase H subdomain and the p51 subunit were omitted to facilitate the simulation and save the computation time.
The simulation model was solvated into a TIP3P (Jorgensen et al., 1983; Neria et al., 1996) water sphere droplet with a radius of 52 Å using the program SOLVATE (Grubmüller, 1996). The large solvent radius ensures the whole protein surface to be covered by water molecules at least 12 Å thick, which is large enough for the conformational change of HIV-1 RT model within the water sphere. Moreover, a layer of 4 Å thick in the brim of the droplet was subjected to SBOUND forces (Brooks and Karplus, 1983) to ensure that the water molecules at the edge are restrained in a given volume. In total, the simulation system contains 3927 protein atoms, 26 inhibitor atoms, and 52,000 water molecule atoms.
MD simulations
The MD simulations were performed with the parallelized MD program EGO (Eichinger and Grubmüller, 1996) running on an SW-I supercomputer at the Shanghai Supercomputer Center. CHARMM19 force field (Brooks et al., 1983) was adopted in the program. The force field of α-APA was generated using program XPLO2D (Kleywegt and Jones, 1997) and the partial atomic charges of α-APA were determined using the RESP fitting method (Bayly et al., 1993) implemented in the NWChem software package version 3.3.1 (Anchell et al., 1998). All simulations were carried out with an integration step of 1 fs and coordinates of the simulation model were recorded per 1 ps. Translation and rotation corrections were enabled during MD simulations to ensure that structures in trajectory were well superimposed, which is convenient for the structure analysis. The chemical bond lengths involving hydrogen atoms were fixed with SHAKE algorithm (Brooks and Karplus, 1983). The application of EGO program in large-scale molecular dynamics simulations has been reviewed by Eichinger et al. (2000).
The solvated model was first minimized using the minimization routine of EGO to reduce steric conflicts between water molecules and the protein. During minimization, the velocities of atoms were rescaled by a friction factor τ of 0.1 at the end of each integration step, and the maximal positional movement of atoms per integration step was set to be no more than 0.08 Å. The solvent molecules in the minimized model were then heated up to 300 K and equilibrated with the protein coordinates being constrained until the total energy of the system reaches a stable state. Finally, the whole solvated model was heated up to 300 K by a coupling constant of 0.1 ps and then subjected to a free equilibrium simulation for 900 ps with α-APA being harmonically constrained in the binding pocket. By the time of 900 ps, the simulation system reached an equilibrium state (see Results and Discussion); thus, the system was subjected to two MD simulations simultaneously: one is an SMD simulation and the other is a conventional MD (CMD) simulation for comparison.
In the SMD simulation, the bound α-APA was pulled out from the binding pocket of HIV-1 RT by employing an artificial harmonic potential on α-APA as shown by the symbolic “spring” in Fig. 1. Based on the averaged conformation between 800–900 ps, the moving direction of α-APA was determined using the criterion that the inhibitor can pass through the putative entrance with the smallest collision with the protein residues (mainly considering the collisions with Lys-101, Lys-103, and Val-179; see Results and Discussion). Translation and rotation corrections of the protein were switched on for the convenience of the structure analysis; the translation and rotation corrections of α-APA were switched off, and thus it can adjust its orientation during moving. Because of switching on the rotation correction for the protein, if the potential is applied to the end atom of the ligand as previous SMD did, it is possible that the ligand may not adjust its orientation once the moiety around the potential assigned atom rams the protein. However, assigning the pulling potential to an atom close to the center of mass (CM) of the ligand may overcome this problem because CM has low possibility to clash the pocket wall of the protein and, moreover, the two ends of the ligand divided by the CM have flexibility to avoid the clash with the protein. Therefore, the pulling potential was assigned to atom C12 of α-APA that is close to the center of mass of the inhibitor molecule. To mimic the unbinding process of NNRTIs in the real biological system, the pulling speed of the spring potential was fixed at 0.02 Å/ps and the spring constant was set to be 4 kcal/(mol·Å2). The SMD simulation ran for 2 ns. After the inhibitor was completely pulled out of the binding pocket and entered the solvent region, the SMD simulation system was continued with a CMD simulation for additional 1 ns. For comparison, the parallel CMD simulation was performed for a similar time period in which α-APA was harmonically constrained in the binding pocket.
Quantum mechanics calculation
Substantial conformational changes of α-APA have been observed to occur during the SMD simulation, and water-bridged interactions appear to play important role(s) in inhibitor binding and unbinding (see Results and Discussion). To examine the energetic properties of different α-APA conformations and the effects of water-bridged interactions in the inhibitor unbinding process, models of typical α-APA conformations with and without water molecules bound at different time of the SMD trajectory were recorded, and their relative energies were computed using the electronic correlation quantum mechanics method Möller-Plesset second order perturbation (MP2) (Pople and Head-Gordon, 1988; Saebo and Almlof, 1989; Perdew and Wang, 1992) at the 6-31G** basis set level. All quantum mechanics calculations were carried out using Gaussian 98 software (Frisch et al., 1998) on a Power Challenger R-10000 supercomputer.
RESULTS and DISCUSSION
MD simulations
The testing CMD simulations indicated that α-APA might depart from the equilibrious position in the binding pocket during the MD simulation. To keep α-APA moving only around its equilibrious position derived from the x-ray structure, harmonic constraint was assigned to the heavy atoms of α-APA with a coefficient of 0.1 kcal/(mol·Å2) in the CMD simulations, including the MD simulation for system equilibration.
System temperature and total energy of the simulation model versus simulation time are shown in Fig. 2, A and B, which indicates that the system temperature is coupled at 300 K and the total energy fluctuates within less than 0.7% after short time equilibration. The main chain root mean square deviation (RMSD) from the starting structure is an important criterion for the convergence of the protein system. The RMSDs of the main chains of the whole model and residues composing the binding pocket are also shown in Fig. 2, C and D, which indicate that both the whole simulation system and the binding pocket appear to have been stabilized after 750 ps equilibration. Hence, at the time of 900 ps, the system was subjected to two different MD simulations: a CMD simulation in which α-APA was harmonically constrained in the binding pocket, and an SMD simulation in which α-APA was pulled out from the binding pocket by an artificial external potential. The SMD simulation started from 900 ps and ended at 1873 ps. At this time, α-APA had been completely pulled out of the binding pocket and entered into the solvent region. Therefore, the SMD simulation was terminated and the simulation system was continued with an additional 1 ns CMD simulation (from 1873 to 3000 ns). The parallel CMD simulation started from 900 ps and ended at 2500 ps.
FIGURE 2.

The temperature (A) and total energy (B) fluctuations and the RMSDs of the main chain of the whole model (C) and the residues composing the binding pocket (Pro-95–Thr-108, Val-179–Tyr-181, Tyr-188–Gly-191, Phe-227–Trp-229, Val-234–Pro-236, and Tyr-318) (D) of the simulation system in the free equilibrium period.
Three phases of the inhibitor unbinding process
Before the SMD simulation, α-APA is tightly bound in the NNIBP and the pocket entrance is narrowly open. In the SMD simulation, the bound α-APA dissociates from the binding pocket, passes through the pocket entrance, and finally enters the solvent region. Analyses of the SMD simulation trajectory and the protein-inhibitor interactions suggest that the unbinding process of α-APA can be divided into three phases based on the position of α-APA in relation to the inhibitor-binding pocket (Figs. 3–5, see discussions later). In phase I (from 900 to 1150 ps), α-APA dissociates from the binding pocket and moves toward the pocket entrance. In phase II (from 1150 to 1300 ps), α-APA overcomes the steric conflicts with the residues at the entrance and passes through the entrance of the pocket. After crossing over the entrance and exiting the binding pocket, the unbinding process enters phase III. During this phase (from 1300 to 1600 ps), α-APA moves further away from the binding pocket and enters the solvent region.
FIGURE 3.

Rupture force exerted on α-APA during inhibitor unbinding versus simulation time at a pulling velocity of 0.02 Å/ps. The pulling force was smoothened with a Gaussian filter kernel of 10 ps width.
FIGURE 5.

Variations of the numbers of direct hydrogen-bonding interactions (A), water-bridged interactions (B), and hydrophobic interactions (C) between α-APA and HIV-1 RT in the SMD simulation. Regions I, II, and III denote the three phases in the unbinding process of α-APA from the inhibitor-binding pocket. The calculation of interactions is recorded every 10 ps.
Correlation of the rupture force with the protein-inhibitor interactions
The rupture force profile derived from the SMD simulation shows multiple force peaks and troughs, implying that the unbinding process of the inhibitor might be complicated (Fig. 3). To probe the relationship between the calculated pulling forces and the protein-inhibitor interactions, 12 specific snapshots of the simulation model along the unbinding pathway were taken, corresponding to five force peaks (925, 997, 1095, 1205, and 1250 ps) and seven force troughs (935, 1010, 1150, 1240, 1280, 1345, and 1460 ps) in Fig. 4. For each snapshot, three types of interactions between HIV-1 RT and α-APA, the direct hydrogen-bonding interaction (DHBI), water-bridged interaction (inhibitor-protein interaction via water molecule, WBI), and hydrophobic interaction (HI), were analyzed using LIGPLOT program (Wallace et al., 1995) (Fig. 5). LIGPOT expresses the hydrophobic interaction as van der Waals contacts involving nonpolar atoms between a ligand and a protein. We use the number of van der Waals contacts of nonpolar atoms (NPAVDWs) to represent the hydrophobic interaction strength throughout this paper.
FIGURE 4.
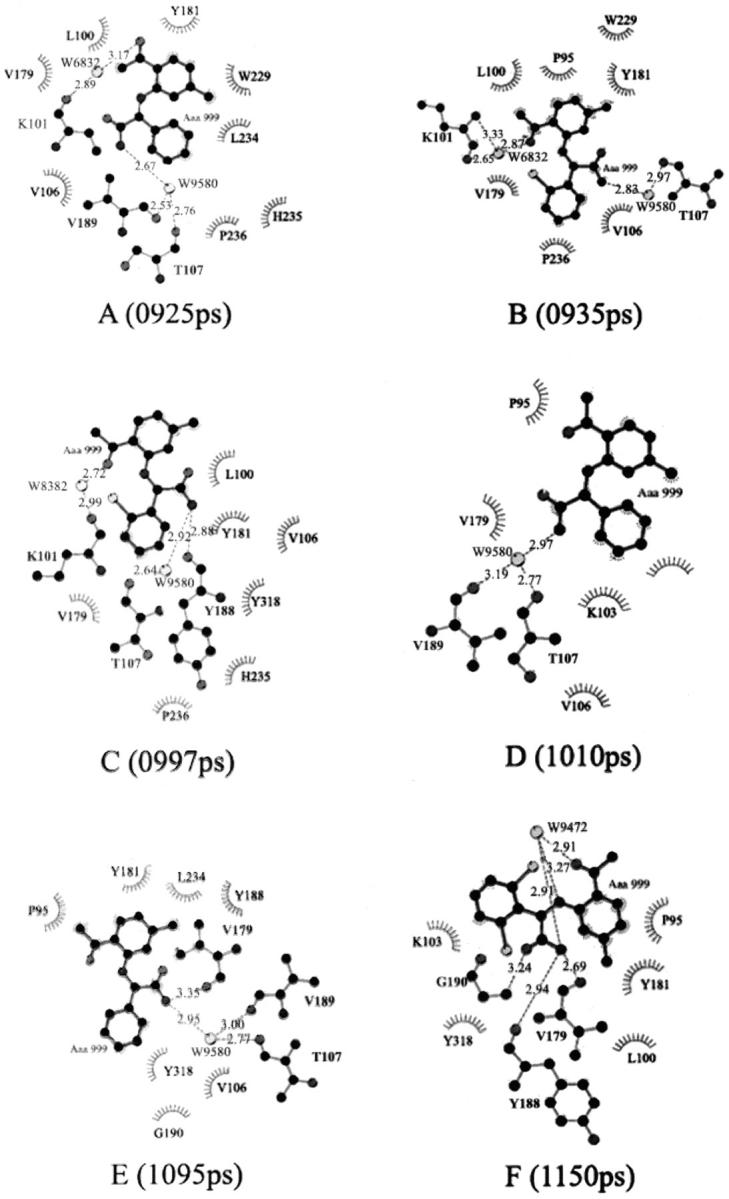
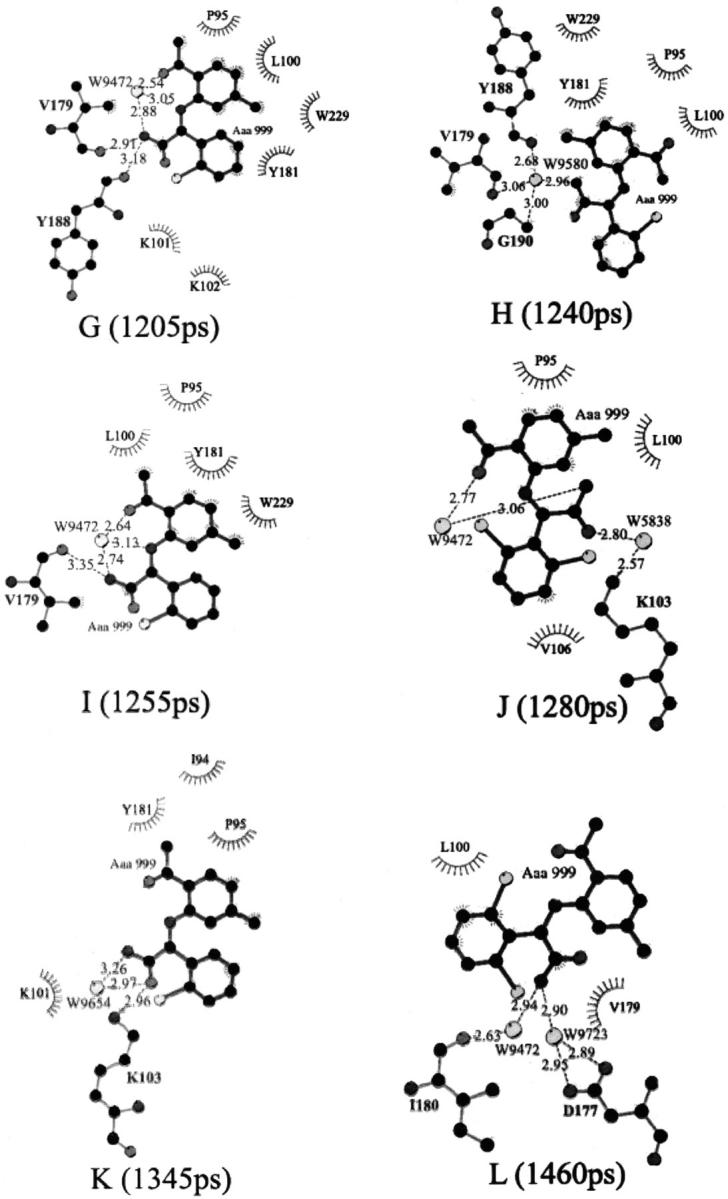
Schematic diagrams showing hydrogen-bonding, water-bridged, and hydrophobic interactions between α-APA and HIV-1 RT at different stages of the unbinding process. Amino acid residues of HIV-1 RT that have hydrophobic interactions with α-APA are shown as spiked spheres (hydrophobic interactions with distances of less than 3.5 Å). Direct and water-bridged hydrogen-bonding interactions are indicated by dashed lines.
Along the inhibitor-unbinding pathway, the interactions between α-APA and HIV-1 RT change from time to time (Figs. 4 and 5). Overall, the hydrophobic interactions decrease gradually from phases I to III; the hydrogen-bonding interactions increase from phases I to II and decrease from phases II to III; and the water-bridged interactions decrease slightly from phases I to II and increase moderately from phases II to III (Figs. 4 and 5). The DHBIs and WBIs in phase II are particularly different from the other two phases, which is apparently related to the passage of α-APA through the entrance of the pocket (see discussions later).
In phase I, owing to the hydrophobic nature of both the NNIBP and the inhibitor, the interactions between α-APA and HIV-1 RT are dominated by the hydrophobic interactions. The number of the van der Waals contacts involving nonpolar atoms (NPAVDWs) reaches as high as 25 (Fig. 5 C). The hydrophilic interactions (including both DHBIs and WBIs) are less significant (always less than 3 each), but appear to play important roles in stabilizing the inhibitor binding (Fig. 5, A and B). The acylamino group (N14) of α-APA forms two direct hydrogen bonds with the carbonyl O atoms of Tyr-188 and Val-179, respectively, and two water-bridged interactions with Thr-107 and Val-189 (Fig. 4, A–E). Similar hydrogen-bonding interactions have also been observed in the crystal structure of HIV-1 RT/α-APA complex (Ding et al., 1995a). The carbonyl group (O4) of α-APA can also form one WBI with Lys-101 at the beginning of the SMD simulation (Fig. 4 A).
In phase II, α-APA penetrates through the entrance of the pocket. The hydrophobic interactions between α-APA and HIV-1 RT become weak, but NPAVDWs are still more than 10 (Fig. 5 C). The DHBIs between the acylamino group (N14) of α-APA and Tyr-188 and Val-179 still exist (Fig. 4, F–J). In addition, α-APA forms a DHBI via its carbonyl O15 atom with the amide N atom of Gly-190. In this region, the WBIs between α-APA and HIV-1 RT almost disappear. Instead, α-APA forms several intramolecular interactions via two special water molecules (Fig. 4, I and J). These water-bridged intramolecular interactions assist α-APA to adopt a proper conformation in a way that it can pass through the entrance of the pocket (see discussion later).
After α-APA exited the binding pocket (phase III), the hydrophobic interactions between α-APA and HIV-1 RT become very weak (Fig. 5 C), and α-APA forms one DHBI with the side chain Nζ atom of Lys-103 and a few WBIs with the residues at the outer rim of the entrance (Fig. 4, K and L). As α-APA moves further away from the binding pocket (after 1600 ps), it enters into the water solvent region and all of its interactions with HIV-1 RT diminish gradually (Fig. 5). During this period of time, α-APA forms hydrogen bonds with the surrounding water molecules and the rupture force comes primarily from the frictional force of water.
Comparison of Figs. 3 and 5 indicates that the rupture force profile correlates well with the interactions between α-APA and HIV-1 RT. The stronger rupture force appears to correspond to stronger interactions between α-APA and HIV-1 RT. At the first major force peak (at 997 ps), α-APA forms ∼20 pairs of NPAVDWs, 1 DHBI, and 2 WBIs with HIV-1 RT. The largest force peak occurs at around 1095 ps, where α-APA forms more than 20 pairs of NPAVDWs, 2 DHBI, and 2 WBIs with HIV-1 RT. At the third force peak (at 1205 ps), α-APA may form 10 pairs of NPAVDWs, 1 DHBI, and 1 WBI with HIV-1 RT (Figs. 4 and 5).
A bottleneck of the inhibitor unbinding pathway
The putative entrance of NNIBP is proposed to be formed by Pro-95, Leu-100, Lys-101, Lys-103, Val-179, and Tyr-181 of the p66 subdomain (Ding et al., 1995a; Ren et al., 1995a; Esnouf, et al., 1997). Several important distances between these residues were monitored during the SMD simulation. Two of them that are of particular interest are the distance between Nζ atoms of Lys-101 and Lys-103 and the distance between Cα atom of Leu-100 and Cγ atom of Val-179 (Fig. 6). Lys-101 and Lys-103 are located at the rim of the entrance (Figs. 1 and 7). Structural data show that these residues are involved in the formation of a network of hydrogen bonds at the entrance and might act as gatekeeper to discriminate whether an NNRTI can enter into the binding pocket (Hsiou et al., 2001). Biochemical data indicate that HIV-1 RT containing mutations at residues 101 and 103 (especially Lys-103-Asn mutation) confers significant drug resistance to most NNRTIs (Byrnes et al., 1993; Richman, 1993; De Clercq, 1998; Rubsamen-Waigmann et al., 1999; Kleim et al., 1999). The SMD simulation results show that the side chains of Lys-101 and Lys-103 are very flexible. In phase I, the distance between these two residues varies significantly between 6 and 10 Å (Fig. 6 A). In phase II, this distance increases gradually to ∼9 Å. In phase III, this distance fluctuates between 8 and 10 Å. After that, it drops to 5 Å and vibrates around 6 Å. The high flexibility of these residues might play some roles in inhibitors binding and/or unbinding processes.
FIGURE 6.

Time dependence of (A) the distance between the Nζ atoms of Lys-101 and Lys-103 that are located at the rim of the putative entrance, and (B) the distance between the Cβ atoms of Leu-100 and Val-179 which are located at the opposite edges of the putative entrance (see also Figs. 1 and 7). Regions I to III denote the three phases in the unbinding process of α-APA from the binding pocket.
FIGURE 7.
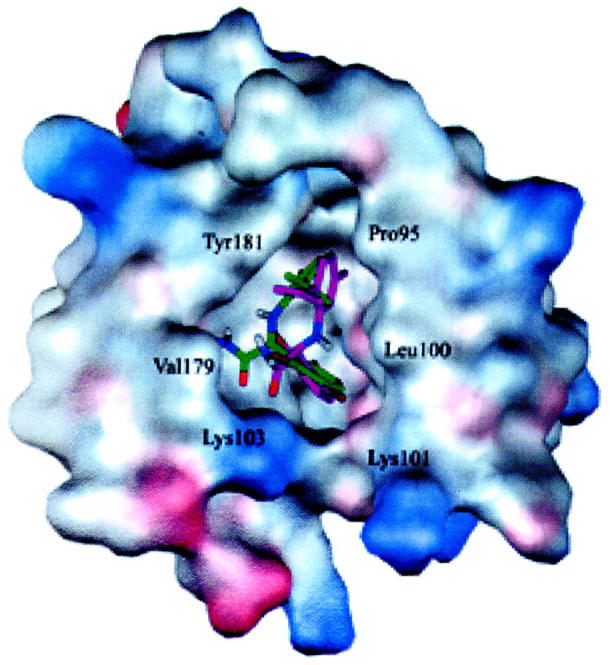
Structures of the HIV-1 RT/α-APA complex before α-APA passes through the bottleneck of the entrance (at 1250 ps, green) and right after α-APA crosses over the bottleneck of the entrance (at 1280 ps, pink). The inhibitor-binding pocket is represented by the electrostatic potential surface. This picture was generated using WebLab ViewerPro program (MSI, 1998) and then rendered using the POV-Ray program (Pov-Ray-Team, 1999).
Residues Val-179 and Leu-100 are located at the opposite edges of the entrance. The simulation results show that the side chains of Leu-100 and Val-179 are more rigid (Fig. 6 B). The distance between Leu-100 and Val-179 fluctuates only slightly around 9 Å in phase I. As α-APA passes through the entrance from inside of the pocket to the outside (phase II), the distance between them increases gradually from 9 to 12 Å. In phase III, as α-APA leaves further away from the pocket the distance between Leu-100 and Val-179 decreases a little bit to ∼11 Å and keeps relatively stable at 11 Å with a slight fluctuation (Fig. 6 B).
To examine the effect of Val-179 on inhibitor binding and unbinding, two particular snapshots of the HIV-1 RT/α-APA complex structure in the SMD trajectory were recorded at 1250 ps (right before α-APA passes through the pocket entrance) and at 1280 ps (right after α-APA crossed over the entrance) (Figs. 4 and 7). Analysis of the detailed interactions between α-APA and HIV-1 RT shows that when α-APA moves toward the narrow entrance of the binding pocket, the side chain of Val-179 apparently produces steric clash with the acylamino group (N14) of α-APA, discouraging the inhibitor to escape from the binding pocket. α-APA has to bump the Val-179 side chain away to enlarge the entrance, so it could pass through. It is conceivable that the increase of the distance between Leu-100 and Val-179 in phase II is caused by the steric clash between α-APA and the side chain of Val-179. Hence, the spatial distance between Leu-100 and Val-179 measures the openness of the pocket entrance and can be regarded as the bottleneck of the unbinding pathway, which prevents the inhibitors from escape. Inversely, we could infer that Val-179 might also act as a guard to discriminate NNRTIs to enter into the binding pocket during the inhibitor binding process. This would imply that mutation of Val-179 to any residues with larger side chains would cause more serious steric conflict with the inhibitor during the inhibitor entering process and, therefore, would discourage the inhibitor to bind. This suggestion seems in agreement with the biochemical data that mutations of Val-179 to Asp or Glu increase side-chain bulk and lead to a 2- to 3-fold decrease of the binding affinity (Byrnes et al., 1993; Boyer et al., 1993).
Possible functions of Tyr-181 and Tyr-188
Site-directed mutagenesis studies show that mutations at residues 181 and 188 affect the binding activity of NNRTIs and confer resistance to many NNRTIs (Nunberg et al., 1991; Richman et al., 1991; Balzarini et al., 1993). Biochemical and structural studies indicate that the hydrophobic interactions between Tyr-181/Tyr-188 and NNRTIs play an important role in the inhibitor binding (Shih et al., 1991; Condra et al., 1992; Das et al., 1996; Hsiou et al., 1998; Ren et al., 2001). In addition, structural studies of HIV-1 RT indicate that binding of NNRTIs to HIV-1 RT induces substantial conformational changes of the side chains of Tyr-181 and Tyr-188. In the unliganded and DNA-bound HIV-1 RT structures, the side chains of Tyr-181 and Tyr-188 in p66 both point away from the polymerase active and fill up the hydrophobic core (Jacobo-Molina et al., 1993; Rodgers et al., 1995; Hsiou et al., 1996; Ding et al., 1995a; Huang et al., 1998). In all NNRTI-bound HIV-1 RT structures, these two residues rotate their side chains away from the hydrophobic core and point toward the polymerase active side, creating space to accommodate the inhibitor (Kohlstaedt et al., 1992; Ding et al., 1995a,b; Ren et al., 1995a,b, 2001; Das et al., 1996; Hsiou et al., 1998, 2001; Lindberg et al., 2002). Our SMD simulation results indicate that Tyr-181 makes significant hydrophobic interactions with α-APA during phases I and II of the unbinding process of inhibitor. The hydrophobic interactions between Tyr-181 and α-APA vary with the simulation time, reaching 16 pairs of NPAVDWs at maximum in phase II (Fig. 8). Though Tyr-188 has less contribution to the hydrophobic interactions (Fig. 5), its carbonyl group forms a strong hydrogen bond with the acylamino group (N14) of α-APA. The hydrogen bond length (O···N14) varies between 2.6 and 3.3 Å and persists for nearly 300 ps (Fig. 9).
FIGURE 8.

Number of van der Waals contacts involving nonpolar atoms (NPAVWs) between α-APA and Tyr-181 versus SMD simulation time (distance of less than 3.5 Å).
FIGURE 9.

Hydrogen bond distance (Å) between the main chain oxygen atom of Tyr-188 and the N14 atom of α-APA versus SMD simulation time.
It is suggested that the hydrophobic interactions between the aromatic rings of NNRTIs and the side chains of Tyr-181 and Tyr-188 are important for NNRTIs binding (Ding et al., 1995a). Analysis of the protein-inhibitor interactions shows that the aromatic side chains of Tyr-181 and Tyr-188 are involved in interactions with the aromatic ring A of α-APA, but to a different extent (Fig. 10). In phases I and II (from 900 to 1300 ps), the center-to-center distance between the aromatic ring of the side chain of Tyr-181 and aromatic ring A of α-APA is relatively stable at 4 Å and the corresponding ring-ring distance between Tyr-188 and α-APA is longer with moderate fluctuation (between 6 to 8 Å). This is consistent with the observation that Tyr-181 makes more hydrophobic interactions with α-APA than Tyr-188 (see discussion above). After the inhibitor exited the pocket (phase III), the ring-ring distances between α-APA and Tyr-181 and Tyr-188 increase gradually to 12 and 16 Å, respectively, suggesting that the hydrophobic interactions between α-APA and the two tyrosines have been disrupted (Fig. 10).
FIGURE 10.

Center-to-center distances (Å) between the aromatic ring A of α-APA (see Fig. 1 for definition) and the side-chain aromatic rings of Tyr-188 (A) and Tyr-181 (B), respectively. Regions I to III denote the three phases in the unbinding process of α-APA from the binding pocket.
The relatively stable ring-ring distances between ring A of α-APA and the side chain of Tyr-181 in phases I and II is very interesting. It might suggest that the side chain of Tyr-181 follows α-APA moving toward the pocket entrance. Analysis of the structure models from the SMD simulation trajectory reveals that after the inhibitor left the binding pocket the side chain of Tyr-181 has changed its conformation substantially and points toward the pocket entrance that is similar to that observed in the unliganded HIV-1 RT structure. In other words, as α-APA dissociates from the binding pocket and moves toward the entrance of the pocket, the side chain of Tyr-181 changes its conformation and tends to occupy the empty space left over by the departing inhibitor. On the other hand, though the side chain of Tyr-188 appears also relatively flexible, it does not change back to the conformation as seen in the apoenzyme structure during the SMD simulation. The reason(s) for why the side chains of Tyr-181 and Tyr-188 behave differently after the inhibitor exited the binding pocket is not clear yet. This might be due, in part, to the time limitation of the simulation.
Taking together, it appears that Tyr-181 and Tyr-188 may act as two flexible clamps to interact with α-APA. The hydrophobic and hydrogen-bonding interactions of Tyr-181 and Tyr-188 with α-APA not only stabilize the inhibitor binding, but also prevent the inhibitor from leaving the binding pocket. It is possible that during the inhibitor binding process, these two residues might also facilitate NNRTIs to enter the binding pocket and adopt an appropriate conformation that could have the optimal interactions with the surrounding residues.
Interactions between α-APA and water molecules and conformational change of α-APA at the entrance
Analysis of the protein-inhibitor interactions shows that there are two special water molecules in the NNIBP that appear to play important roles in the inhibitor binding and unbinding. One is water molecule 9580 (Wat 1), which plays an important role for the binding of α-APA with HIV-1 RT in phase I; the other one is water molecule 9472 (Wat 2), which is essential for the conformation change of α-APA passing the entrance of the HIV-1 RT binding pocket. At the beginning of the SMD simulation, Wat 1 forms a stable water bridge, linking atom N14 of α-APA with the carbonyl O atom of Thr-107 (Fig. 4 A). As α-APA moves toward the pocket entrance, Wat 1 moves along with α-APA and forms WBI(s) with residues Thr-107, Val-189, Val-179, and Gly-190 respectively in the binding pocket along the disassociation pathway (Fig. 5 B). The strong hydrogen-bonding interaction between Wat 1 and atom N14 of α-APA was kept till to the entrance of the pocket (the Wat 1···N14 distance varies between 2.6 and 3.15 Å) (Fig. 11). In reverse, it is quiet possible that, after α-APA enters into the binding pocket, there is a water molecule that can regulate the dynamic WBIs between atom N14 of α-APA and the residues of HIV-1 RT mentioned above, orienting α-APA to adopt a conformation having proper interaction with the surrounding residues during the process of α-APA binding.
FIGURE 11.

Variation of the hydrogen bond distance between atom N14 of α-APA and Wat 1 in the SMD simulation trajectory.
Wat 2 is close to Val-179 at the beginning of the SMD simulation. When α-APA approaches to the bottleneck of the entrance, Wat 2 competes with Wat 1 by forming water-bridged intramolecular interactions with atoms N14, N11, and O4 of α-APA (Fig. 4, F, I, and J). Figs. 4 and 12 clearly show how Wat 2 squeezes out Wat 1 from atom N14 by forming water-bridged interactions with α-APA in phase II. At this time, the side chain of Val-179 clashes with the acylamino group (N14) of α-APA. To avoid steric conflict, the Val-179 side chain moves away to expand the pocket entrance, and α-APA forms a folded conformation by the acylamino group rotating toward the carbonyl group. The WBIs between Wat 2 and α-APA facilitate the conformational folding (Figs. 7 and 13) (see following discussion). After α-APA crossed over the bottleneck, the water-bridged interactions diminish and α-APA gradually resumes its unfolded conformation.
FIGURE 12.

Number of the hydrogen-bonding interactions between α-APA and Wat 1 and Wat 2 verses SMD simulation time. (A)–(D) represent the hydrogen bonding interactions between atoms N11, O4, N14, and O15 of α-APA with Wat 1 and Wat 2, respectively. The hydrogen-bonding interactions of α-APA with Wat 1 and Wat 2 are highlighted in gray and light gray, respectively. Before 1150 ps, Wat 1 forms hydrogen bond with N14 atom of α-APA. Between 1150 and 1260 ps, when α-APA passes through the entrance, Wat 2 replaces Wat 1 to form several hydrogen bonds with O4, N14, and N11 atoms of α-APA.
FIGURE 13.

Variations of the geometric parameters of α-APA versus simulation time in the SMD trajectory: (A) distance between N14 and O4, (B) dihedral angle C1-C10-N11-C12, (C) dihedral angle C10-N11-C12-C13, (D) dihedral angle N11-C12-C16-C22, (E) dihedral angle of N11-C12-C13-N14, and (F) dihedral angle of C10-C1-C2-O4. The atomic numbering of α-APA is shown in Fig. 1. Regions I to III denote the three phases in the unbinding process of α-APA from the binding pocket.
As discussed above, as α-APA passes through the bottleneck region of the pocket entrance, α-APA bumps the Val-179 side chain away, expanding the entrance wider. Meanwhile, conformation of α-APA also makes substantial change (Figs. 7 and 13). The distance between N14 and O4 atoms of α-APA decreases markedly from 5.5 to 3 Å and the dihedral angle C1-C10-N11-C12 flips from 25° to 125°. The dihedral angle between the two aromatic rings of α-APA is compressed and α-APA adopts a folded conformation. By doing so, α-APA can reduce the steric conflict with the side chain of Val-179, making it easier to pass through the entrance.
A quantum chemistry calculation was performed to investigate the intrinsic relationship between the formation of intramolecule interaction mediated by Wat 2 and the conformational changes of α-APA. The quantum mechanics calculations indicate the importance of Wat 2 bridged intramolecular interactions to the conformation folding of α-APA. Fig. 14 shows the profile of the relative energy of several representative α-APA conformations (1200, 1250, 1280, and 1310 ps) as α-APA passes through the bottleneck of the binding pocket. The quantum mechanics calculations indicate that the isolated α-APA conformation seen in the crystal structure of HIV-1 RT/α-APA complex does not have the lowest energy. The α-APA conformation of the lowest energy is observed at 1200 ps in the SMD trajectory, which is ∼3 kcal/mol energy lower than that seen in the crystal structure. Around the bottleneck, α-APA adopts a conformation that is ∼23 kcal/mol higher than that in the crystal structure, or ∼26 kcal/mol higher than that of the lowest energy. Such a high-energy barrier would be too costly for α-APA to leave the binding pocket. However, the formation of the water-bridged intramolecular hydrogen bonds induces the conformational change of the inhibitor and/or stabilizes the unstable conformation of α-APA of the high energy. That significantly lowers the energy barriers by ∼6.5 kcal/mol for α-APA unbinding and 2 kcal/mol for α-APA binding, respectively (Fig. 14).
FIGURE 14.

Profiles of the relative energies of α-APA conformations along the unbinding pathway of the inhibitor. A and B lines represent the profiles of α-APA in the isolated state and in complex with a water molecule, respectively. All conformations of α-APA and their complexes with water molecule were isolated from the SMD trajectory at times of 0, 1200, 1250, 1280, and 1310 ps, respectively. Dashed lines represent hydrogen-bonding interactions between α-APA and water molecule. The relative energy of each α-APA conformer was computed using the MP2/6-31G** method by setting that of α-APA in the crystal structure of HIV-1 RT/α-APA complex as 0.0 kcal/mol.
Water molecules have been observed at the entrance of the binding pocket and inside the binding pocket in the crystal structures of HIV-1 RT bound with and without NNRTIs (Ding et al., 1995b; Ren et al., 1995a,b; Hsiou, et al., 2001). These water molecules are involved in hydrogen-bonding interactions with several important residues at the entrance, including Lys-101, Lys-103, Tyr-181, and Tyr-188. These interactions are suggested to stabilize the structure of NNIBP and may play important roles in steering the NNRTI entering the pocket. Indeed, our SMD simulation results show that water molecules do play important roles in the inhibitor unbinding process. Some water molecules (such as Wat 1) may be involved in stabilizing the binding of inhibitor in the NNIBP and preventing the inhibitor's dissociation. Other molecules (such as Wat 2) may be involved in inducing the inhibitor to adopt a geometric favorable conformation that can pass through the pocket entrance during the unbinding process. In the inhibitor binding process, water molecules may stimulate NNRTIs to enter the binding pocket via their interactions with the polar groups of NNRTIs.
Conformational change of the p66 subdomain: implication for the NNRTI inhibition mechanism to HIV-1 RT
Based on structural and biochemical data, three possible inhibition mechanisms of NNRTIs have been postulated (Kohlstaedt et al., 1992; Ding et al., 1995a; Das et al., 1996; Ren et al., 1995a). The binding of NNRTIs and the formation of NNIBP upon the inhibitor binding may 1), distort the precise geometry and mobility of the nearby polymerase catalytic site; 2), alter the DNA-binding cleft and restrict the mobility of the p66 thumb subdomain which is believed to play an important role in the translocation of template-primer during DNA polymerization; and 3), deform the “primer grip” (β12-β13 hairpin of the p66 palm subdomain) and consequently affect the precise positioning of the primer strand relative to the polymerase active site. All of those hypotheses were proposed indirectly and may not be mutually exclusive to each other.
To explore the relationship between NNRTIs binding and the inhibition mechanism of HIV-1 RT, the SMD simulation was followed by an additional 1 ns CMD simulation after α-APA was completely pulled out from the binding pocket. For comparison, a 2.3 ns CMD simulation was carried out on the HIV-1 RT/α-APA complex with α-APA being constrained in the binding pocket. Structural studies of HIV-1 RT indicated that the p66 thumb subdomain closes down into the DNA-binding cleft (composed of the p66 fingers, palm, and thumb subdomains) in the absence of template-primer (Rodgers et al., 1995; Hsiou et al., 1996), and opens up to take an upright position when template-primer is bound (Jacobo-Molina et al., 1993; Ding et al., 1998; Huang et al., 1998). The conformational flexibility and mobility of the p66 thumb are suggested to be crucial during DNA translocation and polymerization (Jacobo-Molina et al., 1993; Ding et al., 1998; Huang et al., 1998).
To examine the conformational change of the p66 thumb subdomain and the change of the width of the DNA-binding cleft, the domain mass center distances (DMCD) between the p66 fingers subdomain (residues 27–82 and 120–150) and the p66 thumb subdomain (residues 241–314) were measured along the simulation trajectories for the above two MD simulations (Fig. 15). (Hereinafter, if not noted specifically, DMCD will refer to the domain mass center distance between the p66 fingers and thumb subdomains as defined above.) Fig.15 gave the DMCD values of the three different types of HIV-1 RT structures: the unliganded HIV-1 RT structure (1DLO, Hsiou et al., 1996), structures of HIV-1 RT in complexes of the template-primers (2HMI, Ding et al., 1998; 1RTD, Huang et al., 1998), and structures of HIV-1 RT bound with NNRTIs (1HNI, Ding et al., 1995a; 1HNV, Ding et al., 1995b; 1KLM, Esnouf et al., 1997). The DMCD range for HIV-1 RT/NNRTIs complexes is from 45.6 to 47 Å, that for HIV-1 RT/DNA complexes is from 39.6 to 42.7 Å, and that for unliganded HIV-1 RT is near 26.9 Å. It is evident that the three types of HIV-1 RT structures (unliganded, DNA bound, NNRTIs bound) can be clearly distinguished by DMCD. Therefore, DMCD can be used as a useful indicator to investigate the conformation change of the p66 thumb subdomain (and HIV-1 RT) along the simulation trajectory in the inhibitor unbinding process.
FIGURE 15.

The domain mass center distance (DMCD) between the p66 fingers subdomain (residues 12–85 and 120–150) and the p66 thumb subdomain (residue 241–314) versus simulation time: (A) the CMD simulation and (B) the SMD simulation. The SMD simulation was followed by a CMD simulation for 1 ns after α-APA was completely pulled out from the inhibitor-binding pocket. The typical variation ranges of DMCDs in different types of HIV-1 RT structures. The structures of HIV-1 RT are denoted with their PDB entry codes as follows: structure in unliganded state, 1DLO (Hsiou et al., 1996), structures of HIV-1 RT in complexes of the template-primers, 2HMI (Ding et al., 1998) and 1RTD (Huang et al., 1998), and structures of HIV-1 RT bound with NNRTIs, 1HNI (Ding et al., 1995a), 1HNV (Ding et al., 1995b), and 1KLM (Esnouf et al., 1997).
In the CMD simulation, the DMCD fluctuates between 45.5 and 47 Å throughout the whole simulation that falls in the range of DMCDs measured from various HIV-1 RT/NNRTIs complex structures (Fig. 15 A). This suggests that the p66 thumb subdomain as well as the DNA-binding cleft do not change significantly when α-APA is constrained in the NNIBP. On the other hand, in the SMD simulation (Fig. 15 B), the DMCD decreases slightly from 46 to 45 Å at the beginning of the simulation and then reaches a stable state at ∼45 Å when α-APA is bound in the NNIBP (phase I, from 900 to 1150 ps). As α-APA crosses over the entrance of the binding pocket and moves from inside of the binding pocket to the outside (phase II, from 1150 to 1300 ps), the DMCD decreases dramatically (Fig.15 B), which indicates that the p66 thumb subdomain starts to close accompanied by the shrinkage of the DNA-binding cleft. The decreasing of the DMCD continues after α-APA exited the binding pocket (phase III, from 1300 to 1600 ps). At ∼1600 ps, the DMCD reaches ∼40 Å that is similar to that observed in the structures of HIV-1 RT/DNA complexes (Jacobo-Molina et al., 1993; Huang et al., 1998; Ding et al., 1998). The p66 thumb keeps this conformation for a relatively long period of time (from 1600 to 2250 ps) with the variation of DMCD between 39 and 42 Å. This suggests that the p66 thumb closes down partially and adopts a relatively stable conformation similar to that seen in the structures of HIV-1 RT in complex with template-primer after the inhibitor dissociated from the binding pocket. At ∼2250 ps, the DMCD sharply decreases to ∼37 Å and keeps stable at this value to the end of the simulation (Fig. 15 B). This means that the p66 thumb continues to collapse and the DNA-binding cleft shrinks further, but does not reach the close conformation of the p66 thumb as observed in the unliganded HIV-1 RT structure (Hsiou et al., 1996).
The simulation results indicate that the p66 thumb subdomain indeed has the conformational flexibility to open and fold down. The binding of α-APA in the NNIBP enlarges the DNA-binding cleft and constrains the p66 thumb in the ultra upright conformation and, therefore, restricts the flexibility and mobility of the p66 thumb. Since the DNA binding cleft is expanded in the presence of the inhibitor, it is probable that the binding of α-APA to HIV-1 RT should not prohibit the binding of DNA template-primer, but may, to some extent, destabilize the DNA binding. This suggestion seems in agreement with the biochemical data that show that binding of some NNRTIs to HIV-1 RT does not markedly affect the binding of either template-primer or dNTP substrates (Debyser et al., 1991; Wu et al., 1991; Maga et al., 2000).
Taken together, the simulation results provide support for the hypothesis of NNRTI inhibition mechanism to HIV-1 RT that the inhibitor-binding site acts as a hinge between the p66 thumb and palm subdomains and the binding of NNRTIs expands the DNA-binding cleft and restricts the flexibility and mobility of the p66 thumb subdomain. The constrained p66 thumb might not be able to interact properly with the template-primer during the recognition and translocation of the template-primer, and consequently, impairs the polymerization activity of HIV-1 RT (Kohlstaedt et al., 1992; Ding et al., 1995a).
CONCLUSIONS
This work presents two nanosecond molecular dynamics (MD) simulation studies of the unbinding process of α-APA, an NNRTI, to HIV-1 RT. The first was a CMD simulation in which the inhibitor was harmonically constrained in the NNIBP. The other was an SMD simulation in which a harmonic potential was applied to the inhibitor to pull the inhibitor out from the binding pocket artificially. The pulling velocity (Vpull) is an important parameter in the SMD simulations. The simulations at higher pulling velocity may lead to remarkable nonequilibrium effects, which may introduce obvious errors to the simulation results (Isralewitz et al., 2001). The low-velocity SMD simulations that are carried out on a millisecond time scale can overcome these disadvantages and may reproduce actual atomic force microscopy experiments; however, the corresponding computational cost will be very expensive. However, the aim of this study is to address the dynamical process of the inhibitor/HIV-1 RT binding and the possible inhibition mechanism of the NNRTIs with HIV-1 RT rather than reproduce the accurate binding force or binding free energy. Heymann and Grubmüller (1999b) demonstrated that the global shapes of the force profiles are similar under different pulling velocities in the dissociation of AN02/DNP-hapten complex. We had performed an SMD simulation for the unbinding process of α-APA/HIV-1 RT complex under a velocity of 0.05 Å/ps (result is not shown in this paper). The force profile feature of this simulation is similar to that of the SMD simulation at a velocity of 0.02 Å/ps (Fig. 3). Therefore it can be concluded that the unbinding process of α-APA/HIV-1 RT complex may not be significantly affected by the pulling velocity, and that the SMD simulation result described in this paper can be used to illustrate the inhibitor/HIV-1 RT binding (unbinding) mechanism.
The simulation results have revealed many dynamic features of the inhibitor unbinding process and provided insights into the inhibition mechanism of NNRTIs. Analyses of the force profile of the SMD simulation and the interactions between HIV-1 RT and α-APA show that the force profile in general correlates well with the protein-inhibitor interactions. The peaks and troughs in the force profile can be associated with the formation and rupture of specific direct and water-bridged hydrogen-bonding interactions and hydrophobic interactions. The unbinding process of α-APA can be divided into three phases based on the position of α-APA in relation to the entrance of the binding pocket. When α-APA dissociates from the binding pocket and moves toward the pocket entrance (phase I), the interactions between HIV-1 RT and α-APA are dominated by hydrophobic interactions. However, the hydrophilic interactions (both direct and water-bridged hydrogen bonds) also contribute to the stabilizing forces. Whereas Tyr-181 makes significant hydrophobic interactions with α-APA, Tyr-188 forms strong hydrogen bond with the acylamino group (N14) of α-APA. Moreover, during the unbinding process of α-APA, the side chains of these two residues change their conformations. In particular, the side chain of Tyr-181 moves along with the inhibitor toward the pocket entrance and tends to occupy the space left by the departing inhibitor. These two residues appear to act as two flexible clamps discouraging α-APA to dissociate from the binding pocket. In phase II, α-APA penetrates through the entrance of the pocket. At the pocket entrance, two relatively inflexible residues Val-179 and Leu-100 gauge the openness of the entrance and, therefore, the spatial distance between them appears to be the bottleneck of the inhibitor-unbinding pathway. To escape from the binding pocket, α-APA not only bumps the side chain of Val-179 away to expand the size of the pocket entrance, but also folds its conformation to reduce the steric conflicts with Val-179. Two water molecules (Wat 1 and Wat 2) form water bridges between the polar groups of the inhibitor and the residues at the pocket entrance and between the polar groups of the inhibitors themselves. Wat 1 seems involved in stabilizing the binding of the inhibitor in the binding pocket and preventing the inhibitor's dissociation. Wat 2 seems involved in inducing the inhibitor to adopt a geometric favorable conformation so that the inhibitor can pass through the pocket entrance. During the inhibitor binding process, these water molecules might play important roles in inhibitor recognition and steering it into the binding pocket. After α-APA exits the binding pocket, the interactions between α-APA and the protein become weaker and eventually diminish. When the inhibitor enters the solvent region, α-APA forms hydrogen bonds with the surrounding water molecules and the rupture force comes primarily from the friction force of water.
Comparison of the CMD and the SMD simulation results shows that the binding of α-APA in the inhibitor-binding pocket enlarges the DNA-binding cleft and constrains the p66 thumb in an ultra upright conformation. The p66 thumb subdomain has a great conformational flexibility and tends to fold down to adopt a closed conformation after the bound inhibitor dissociated from the binding pocket. These results strongly support the hypothetic mechanism of NNRTI inhibition that the binding of NNRTIs alters the DNA-binding cleft and restricts the flexibility and mobility of the p66 thumb subdomain that are believed to play an important role in the translocation of the template-primer during DNA polymerization.
Acknowledgments
The authors thank Dr. Helmut Grubmüller for his kindness in offering the EGO program.
The research in H.J.'s laboratory was supported by grants from the National Natural Science Foundation of China (grants 29725203 and 20072042), the State Key Program of Basic Research of China (grant 1998051115), the Life Science Foundation for Young Scientists of CAS (grant STZ-00-06), and the Qi Ming Xing Foundation of Shanghai Ministry of Science and Technology (grant 00QB14034). The research in J.D.'s laboratory was supported by the National Science Foundation for Distinguished Young Scholars (grant 30125011) and the 863 Hi-Tech Program (grant 2001AA235071). The MD calculations were performed on the SW-I supercomputer at the Shanghai Supercomputer Center.
References
- Anchell, J. A., E. Apra, D. Bernholdt, P. Borowski, T. Clark, D. Clerc, H. Dachsel, M. Deegan, M. Dupuis, K. Dyall, G. Fann, H. Fruchtl, M. Gutowski, R. Harrison, A. Hess, J. Jaffe, R. Kendall, R. Kobayashi, R. Kutteh, Z. Lin, R. Littlefield, X. Long, B. Meng, J. Nichols, J. Nieplocha, A. Rendall, M. Stave, T. P. Straatsma, H. Taylor, G. Thomas, K. Wolinski, and A. Wong. 1998. NWChem, Version 3.2. High Performance Computational Chemistry Group, Pacific Northwestern National Laboratory, Richland, WA.
- Auwerx, J., T. W. North, B. D. Preston, G. J. Klarmann, E. De Clercq, and J. Balzarini. 2002. Chimeric human immunodeficiency virus type 1 and feline immuno-deficiency virus reverse transcriptases: role of the subunits in resistance/sensitivity to non-nucleoside reverse transcriptase inhibitors. Mol. Pharmacol. 61:400–406. [DOI] [PubMed] [Google Scholar]
- Balzarini, J., A. Karlsson, M. J. Perez-Perez, M. J. Camarasa, W. G. Tarpley, and E. De Clercq. 1993. Treatment of human immunodeficiency virus type 1 (HIV-1)-infected cells with combinations of HIV-1-specific inhibitors results in a different resistance pattern than does treatment with single-drug therapy. J. Virol. 67:5353–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly, C., P. Cieplak, W. D. Cornell, and P. A. Kollman. 1993. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—the RESP model. J. Phys. Chem. 97:10269–10280. [Google Scholar]
- Binning, G., C. F. Quate, and G. Gerber. 1986. Atomic force microscope. Phys. Rev. Lett. 56:930–933. [DOI] [PubMed] [Google Scholar]
- Block, S., and K. Svoboda. 1994. Biological applications of optical forces. Annu. Rev. Biophys. Biomol. Struct. 23:247–285. [DOI] [PubMed] [Google Scholar]
- Boyer, P. L., M. J. Currens, J. B. McMahon, M. R. Boyd, and S. H. Hughes. 1993. Analysis of nonnucleoside drug-resistant variants of human immuno-deficiency virus type 1 reverse transcriptase. J. Virol. 67:2412–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer, P. L., A. L. Ferris, and S. H. Hughes. 1992. Mutational analysis of the fingers domain of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 66:7533–7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B. R., and M. Karplus. 1983. Deformable stochastic boundaries in molecular dynamics. J. Chem. Phys. 79:6312–6325. [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Buckheit, R. W. 2001. Non-nucleoside reverse transcriptase inhibitors: perspectives on novel therapeutic compounds and strategies for the treatment of HIV infection. Expert Opin. Investig. Drugs. 10:1423–1442. [DOI] [PubMed] [Google Scholar]
- Byrnes, V. W., V. V. Sardana, W. A. Schleif, J. H. Condra, J. A. Waterbury, J. A. Wolfgang, W. J. Long, C. L. Schneider, and B. S. Wolanski. 1993. Comprehensive mutant enzyme and viral variant assessment of human immunodeficiency virus type 1 reverse transcriptase resistance to non-nucleoside inhibitors. Antimicrob. Agents Chemother. 37:1576–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campiani, G., A. Ramunno, G. Maga, V. Nacci, C. Fattorusso, B. Catalanotti, E. Morelli, and E. Novellino. 2002. Non-nucleoside HIV-1 reverse transcriptase (RT) inhibitors: past, present, and future perspectives. Curr. Pharm. Des. 8:615–657. [DOI] [PubMed] [Google Scholar]
- Chen, M., A. Elise, T. Sudbeck, K. Venkatachalam, and F. M. Uckun. 2000. Structure-based drug design for NNRTIs for wild-type and drug-resistant HIV-1 RT. Biochem. Pharmacol. 60:1251–1265. [DOI] [PubMed] [Google Scholar]
- Condra, J. H., E. A. Emini, L. Gotlib, D. J. Graham, A. J. Schlabach, J. A. Wolfgang, R. J. Colonno, and V. V. Sardana. 1992. Identification of the human immuno-deficiency virus reverse transcriptase residues that contribute to the activity of diverse nonnucleoside inhibitors. Antimicrob. Agents Chemother. 36:1441–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das, K., J. Ding, J. Hsiou, A. D. Clark, Jr., H. Moereels, L. Koyman, K. Andries, R. Pauwels, P. A. Janssen, P. L. Boyer, P. Clark, R. H. Smith, Jr., S. M. B. Kroeger, C. J. Michehda, S. H. Hughes, and E. Arnold. 1996. Crystla structure of 8-cl and 9-cl TIBO complexed with wild-type HIV-1 RT and 8-cl TIBO complexed with the Tyr 181 Cys HIV-1 RT drug-resistant mutant. J. Mol. Biol. 264:1085–1100. [DOI] [PubMed] [Google Scholar]
- Debyser, A., R. Pauwels, K. Andries, J. Desmyter, M. Kukla, P. Janssen and E. De Clercq. 1991. An antiviral target on reverse transcriptase of human immunodeficiency virus type 1 revealed by tetrahydroimidazo-[4,5,1-jk] [1,4]benzodiazepin-2(1H-one and -thione derivatives. Proc. Natl. Acad. Sci. USA. 88:1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq, E. 1994. HIV resistance to reverse transcriptase inhibitors. Biochem. Pharmacol. 47:155–169. [DOI] [PubMed] [Google Scholar]
- De Clercq, E. 1996. Non-nucleoside reverse transcriptase inhibitors (NNRTIs) for the treatment of human immunodeficiency virus type 1 (HIV-1) infections: strategies to overcome drug resistance development. Med. Res. Rev. 16:125–157. [DOI] [PubMed] [Google Scholar]
- De Clercq, E. 1998. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Res. 38:153–179. [DOI] [PubMed] [Google Scholar]
- De Clercq, E. 1999. Perspectives of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Farmaco. 54:26–45. [DOI] [PubMed] [Google Scholar]
- De Clercq, E. 2001. New developments in anti-HIV chemotherapy. Curr. Med. Chem. 8:1543–1572. [DOI] [PubMed] [Google Scholar]
- De Clercq, E. 2002. Strategies in the design of antiviral drugs. Nat. Rev. Drug. Discov. 1:13–25. [DOI] [PubMed] [Google Scholar]
- Ding, J., K. Das, C. Tantillo, W. Zhang, A. D. Clark, Jr., S. Jessen, X. Lu, Y. Hsiou, A. Jacobo-Monlina, K. Andries, R. Pauwels, H. Moereels, L. Koymans, P. A. J. Janssen, R. Smith, M. K. Koepke, C. Michejda, S. H. Hughes, and E. Arnold. 1995a. Structure of HIV-1 RT in a complex with NNRTI a-APA R 95845 at 2.8 Å. Structure. 3:365–379. [DOI] [PubMed] [Google Scholar]
- Ding, J., K. Das, H. Moereels, L. Koymans, K. Andries, P. A. Jassen, S. H. Hughes, and E. Arnold. 1995b. Structure of HIV-1 RT/TIBO R 86183 reveals similarity in the binding of diverse NNRTI. Nat. Struct. Biol. 2:407–415. [DOI] [PubMed] [Google Scholar]
- Ding, J., K. Das, P. N. S. Yadav, Y. Hsiou, W. Zhang, S. H. Hughes, and E. Arnold. 1997. A review of HIV-1 RT structural studies and implications for drug design. In Structure Based Drug Design. P. Veerapandian, editor. Marcel Dekker Inc., New York. 41–82.
- Ding, J., K. Das, H. Yu, S. G. Sarafianos, A. D. Clark, A. Jacobo-Molina, C. Tantillo, S. H. Hughes, and E. Arnold. 1998. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 Å resolution. J. Mol. Biol. 284:1095–1111. [DOI] [PubMed] [Google Scholar]
- Eichinger, M., and H. Grubmüller. 1996. EGOVIII. Version 2.0. Electronic access: http://www.mpibpc.gwdg.de/abteilungen/071/ego.html.
- Eichinger, M., H. Heller, and H. Grubmüller. 2000. EGO—An efficient molecular dynamics program and its application to protein dynamics simulations. In Workshop on Molecular Dynamics on Parallel Computers. R. Esser, P. Grassberger, J. Grotendorst, and M. Lewerenz, editors. John von Neumann Institute for Computing (NIC) Research Centre, Jüich, Germany. 397–402.
- Esnouf, R. M., J. Ren, A. L. Hopkins, C. K. Ross, E. Y. Jones, D. K. Stammers, and D. I. Stuart. 1997. Unique features in the structure of the complex between HIV-1 reverse transcriptase and the bis(heteroaryl)piperazine (BHAP) U-90152 explain resistance mutations for this nonnucleoside inhibitor. Proc. Natl. Acad. Sci. USA. 94:3984–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, E., K. Ritchie, and R. Merkel. 1995. Sensitive force technique to probe molecular adhesion and structural linkages at biological interfaces. Biophys. J. 68:2580–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle, and J. A. Pople. 1998. Gaussian 98. Gaussian, Inc., Pittsburgh, PA.
- Grubmüller, H. 1996. Solvate: a program to create atomic solvent models. Electronic publication: http://www.mpibpc.gwdg.de/abteilungen/071/hgrub/solvate/docu.html.
- Grubmüller, H., B. Heymann, and P. Tavan. 1996. Ligand binding: molecular mechanics calculation of the Streptavidin-Biotin rupture force. Science. 271:997–999. [DOI] [PubMed] [Google Scholar]
- Gupta, S. P. 2002. Advances in QSAR studies of HIV-1 reverse transcriptase inhibitors. Prog. Drug Res. 58:223–264. [DOI] [PubMed] [Google Scholar]
- Heymann, B., and H. Grubmüller. 1999a. Elastic properties of poly(ethylene-glycol) studied by molecular dynamics stretching simulations. Chem. Phys. Lett. 307:425–432. [Google Scholar]
- Heymann, B., and H. Grubmüller. 1999b. AN02-DNP-hapten unbinding forces studied by molecular dynamics atomic force microscopy simulations. Chem. Phys. Lett. 303:1–9. [Google Scholar]
- Heymann, B., and H. Grubmüller. 2001. Molecular dynamics force probe simulations of antibody/antigen unbinding: entropic control and non-additivity of unbinding forces. Biophys. J. 81:1295–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiou, Y., J. Ding, K. Das, A. D. Clark, Jr., S. H. Hughes, and E. Arnold. 1996. Structure of unliganded HIV-1 at 2.7 Å resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure. 4:853–860. [DOI] [PubMed] [Google Scholar]
- Hsiou, Y., K. Das, J. Ding, A. D. Clark, Jr., J. P. Kleim, M. Rosner, I. Winker, G. Riess, S. H. Hughes, and E. Arnold. 1998. Structures of Tyr188Leu mutant and wild-type HIV-1 reverse transcriptase complexed with the non-nucleoside inhibitor HBY 097: inhibitor flexibility is a useful design feature for reducing drug resistance. J. Mol. Biol. 284:313–323. [DOI] [PubMed] [Google Scholar]
- Hsiou, Y., J. Ding, K. Das, A. D. Clark, P. L. Boyer, P. Lewi, P. A. J. Janssen, J. P. Kleim, M. Roesner, S. H. Hughes, and E. Arnold. 2001. The Lys103Asn mutation of HIV-1 RT: a novel mechanism of drug resistance. J. Mol. Biol. 309:437–445. [DOI] [PubMed] [Google Scholar]
- Huang, H., R. Chopra, G. L. Verdine, and S. C. Harrison. 1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 282:1669–1675. [DOI] [PubMed] [Google Scholar]
- Isralewitz, B., J. Baudry, J. Gullingsrud, D. Kosztin, and K. Schulten. 2001. Steered molecular dynamics investigations of protein function. J. Mol. Graph. Model. 19:13–25. [DOI] [PubMed] [Google Scholar]
- Isralewitz, B., S. Izrailev, and K. Schulten. 1997. Binding pathway of retinal to bacterioopsin: a prediction by molecular dynamics simulations. Biophys. J. 73:2972–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izrailev, S., S. Stepaniants, B. Isralewitz, D. Kosztin, H. Lu, F. Molnar, W. Wrigers, and K. Schulten. 1998. Steered molecular dynamics. In Computational Molecular Dynamics: Challenges, Methods and Ideas, Lecture Notes in Computational Science and Engineering, Vol. 4. P. Denflhard, J. Hermans, B. Leimkuhler, A. Mark, R. Skeel, and S. Reich, editors. Springer-Verlag, Berlin. 39–65.
- Jacobo-Molina, A., J. Ding, R. G. Nanni, A. D. Clark, X. Lu, C. Tantillo, R. L. Willicams, G. Kamer, A. L. Ferris, P. Clark, A. Hiti, S. H. Hughes, and E. Arnold. 1993. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complex with double-stranded DNA at 3.0 Å resolution show bent DNA. Proc. Natl. Acad. Sci. USA. 90:6320–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager, J., S. J. Smerdon, J. Wang, D. C. Boisvert, and T. A. Steitz. 1994. Comparison of three different crystal forms shows HIV-1 reverse transcriptase displays an internal swivel motion. Structure. 2:869–876. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Kleim, J. P., M. Winters, A. Dunkler, J. R. Suarez, G. Riess, I. Winkler, J. Balzarini, D. Oette, and T. C. Merigan. 1999. Antiviral activity of the human immunodeficiency virus type 1-specific nonnucleoside reverse transcriptase inhibitor HBY 097 alone and in combination with zidovudine in a phase II study. HBY 097/2001 study group. J. Infect. Dis. 179:709–713. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G. J., and T. A. Jones. 1997. Model-building and refinement practice. Methods Enzymol. 277:208–230. [DOI] [PubMed] [Google Scholar]
- Kohlstaedt, L. A., J. Wang, J. M. Friedman, P. A. Rice, and T. A. Steitz. 1992. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 256:1783–1790. [DOI] [PubMed] [Google Scholar]
- Krammer, A., H. Lu, B. Isralewitz, K. Schulten, and V. Vogel. 1999. Forced unfolding of the fibronectin type III module reveals a tensile molecular recognition switch. Proc. Natl. Acad. Sci. USA. 96:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larder, B. A. 1993. Inhibitors of HIV-1 RT as antiviral agents and drug resistance. In Reverse Transcriptase. A. M. Skalka and S. P. Goff, editors. Cold Spring Harbor Laroratory Press, Plainview, NY. 205–222.
- Lindberg, J., S. Sigurdsson, S. Lowgren, H. O. Andersson, C. Sahlberg, R. Noreen, K. Fridborg, H. Zhang, and T. Unge. 2002. Structural basis for the inhibitory efficacy of efavirenz (Dmp-266), Msc194 and Pnu142721 towards the HIV-1 RT K103N mutant. Eur. J. Biochem. 269:1670–1677. [DOI] [PubMed] [Google Scholar]
- Lu, H., B. Isralewitz, A. Krammer, V. Vogel, and K. Schulten. 1998. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulations. Biophys. J. 75:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H., and K. Schulten. 1999. Steered molecular dynamics simulation of force-induced protein domain unfolding. Proteins. 35:453–463. [PubMed] [Google Scholar]
- Maga, G., D. Ubiali, R. Salvetti, M. Pregnolato, and S. Spadari. 2000. Selective interaction of the human immunodeficiency virus type 1 reverse transcriptase nonnucleoside inhibitor efavirenz and its Thio-substituted analog with different enzyme-substrate complexes. Antimicrob. Agents Chemother. 44:1186–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink, S. J., O. Berger, P. Tieleman, and F. Jahnig. 1998. Adhesion forces of lipids in a phospholipid membrane studied by molecular dynamics simulations. Biophys. J. 74:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neria, E., S. Fischer, and M. Karplus. 1996. Simulation of activation free energies in molecular systems. J. Chem. Phys. 105:1902–1921. [Google Scholar]
- Nunberg, J. H., W. A. Schleif, E. J. Boots, J. A. O'Brien, J. C. Quintero, J. M. Hoffman, E. A. Emini, and M. E. Goldman. 1991. Viral resistance to human immunodeficiency virus type 1-specific pyridinone reverse transcriptase inhibitors. J. Virol. 65:4887–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels, R., and P. A. J. Janssen. 1993. Potent and highly selective human immunodeficiency virus type 1 inhibition by a series of α-anilinophenylacet-amide derivatives targeted at HIV-1 reverse transcriptase inhibitors. Proc. Natl. Acad. Sci. USA. 90:1711–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdew, J. P., and Y. Wang. 1992. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B. 45:13244–13249. [DOI] [PubMed] [Google Scholar]
- Pople, J. A., and M. Head-Gordon. 1988. MP2 energy evaluation of direct methods. Chem. Phys. Lett. 153:503–506. [Google Scholar]
- Ren, J., R. Esnouf, E. Garman, D. Somers, C. Ross, I. Kirby, J. Keeling, G. Darby, Y. Jones, D. Stuart, and D. Stammers. 1995a. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Biol. 2:293–302. [DOI] [PubMed] [Google Scholar]
- Ren, J., R. Esnouf, E. Garman, D. Sommers, C. Ross, I. Kirby, J. Keeling, G. Darby, Y. Jones, D. Stuart, and D. Stammers. 1995b. The structure of HIV-1 RT complexed with 9-cloro-TIBO: lessons for inhibitor design. Structure. 3:915–926. [DOI] [PubMed] [Google Scholar]
- Ren, J., C. Nichols, L. Bird, P. Chamberlain, K. Weaver, S. Short, D. I. Stuart, and D. K. Stammers. 2001. Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J. Mol. Biol. 312:795–805. [DOI] [PubMed] [Google Scholar]
- Richman, D. D. 1993. Resistance of clinical isolates of HIV to antiretroviral agents. Antimocrob. Agents Chemother. 37:1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman, D. D., C. K. Shih, I. Lowy, J. Rose, P. Prodanovich, S. Goff, and J. Griffin. 1991. Human immunodeficiency virus type 1 mutants resistant to nonnucleoside inhibitors of reverse transcriptase arise in tissue culture. Proc. Natl. Acad. Sci. USA. 88:11241–11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers, D. W., S. J. Gamblin, B. A. Harris, S. Ray, J. S. Culp, B. Hellmig, D. J. Woolf, C. Debouck, and S. C. Harrison. 1995. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA. 92:1222–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubsamen-Waigmann, H., E. Huguenel, A. Shah, A. Paessens, H. J. Ruoff, H. von Briesen, A. Immelmann, U. Dietrich, and M. A. Wainberg. 1999. Resistance mutations selected in vivo under therapy with anti-HIV drug HBY 097 differ from resistance pattern selected in vitro. Antiviral Res. 42:15–24. [DOI] [PubMed] [Google Scholar]
- Saebo, S., and J. Almlof. 1989. Avoiding the integral storage bottleneck in LCAO calculations of electron correlation. Chem. Phys. Lett. 154:83–89. [Google Scholar]
- Sala, M., and J. P. Vartanian. 1998. HIV1 reverse transcriptase: an out-of-the-ordinary enzyme. Bull. Inst. Pasteur. 96:49–63. [Google Scholar]
- Shih, C., J. M. Rose, G. L. Hansen, J. C. Wu, A. Bacolla, and J. A. Griffin. 1991. Chimeric human immunodeficiency virus type 1/type 2 reverse transcriptases display reversed sensitivity to nonnucleoside analog inhibitors. Proc. Natl. Acad. Sci. USA. 88:9878–9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepaniants, S., S. Izarailev, and K. Schulten. 1997. Extraction of lipids from phospholipid membranes by steered molecular dynamics. J. Mol. Model. 3:473–475. [Google Scholar]
- Tantillo, C., J. Ding, A. Jacobo-Molina, R. G. Nanni, P. L. Boyer, S. H. Hughes, R. Pauwels, K. Andries, P. A. Janssen, and E. Arnold. 1994. Locations of anti-AIDS drug binding sites and resistance mutations in the three-dimensional structure of HIV-1 RT: implications for mechanisms of drug inhibition and resistance. J. Mol. Biol. 243:369–387. [DOI] [PubMed] [Google Scholar]
- Tripos Inc. 1999. Sybyl. Version 6.5. St. Louis, MO.
- Turner, B. G., and M. F. Summers. 1999. Structural biology of HIV. J. Mol. Biol. 285:1–32. [DOI] [PubMed] [Google Scholar]
- Wallace, A. C., R. A. Laskowski, and J. M. Thornton. 1995. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8:127–134. [DOI] [PubMed] [Google Scholar]
- Wriggers, W., and K. Schulten. 1997. Stability and dynamics of G-action: back door water diffusion and behavior of a subdomain 3/4 loop. Biophys. J. 73:1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. C., T. C. Warren, J. Adams, J. Proudfoot, J. Skiles, P. Raghavan, C. Perry, I. Potocki, P. R. Farina, and P. M. Grob. 1991. A novel dipyridodiazepinone inhibitor of HIV-1 reverse transcriptase acts through a nonsubstrate binding site. Biochemistry. 30:2022–2026. [DOI] [PubMed] [Google Scholar]