

Abstract
The topological model proposed for the Kir2.1 inward rectifier predicts that seven of the channel 13 cysteine residues are distributed along the N- and C-terminus regions, with some of the residues comprised within highly conserved domains involved in channel gating. To determine if cytosolic cysteine residues contribute to the gating properties of Kir2.1, each of the N- and C-terminus cysteines was mutated into either a polar (S, D, N), an aliphatic (A,V, L), or an aromatic (W) residue. Our patch-clamp measurements show that with the exception of C76 and C311, the mutation of individual cytosolic cysteine to serine (S) did not significantly affect the single-channel conductance nor the channel open probability. However, mutating C76 to a charged or polar residue resulted either in an absence of channel activity or a decrease in open probability. In turn, the mutations C311S (polar), C311R (charged), and to a lesser degree C311A (aliphatic) led to an increase of the channel mean closed time due to the appearance of long closed time intervals (Tc ≥ 500 ms) and to a reduction of the reactivation by ATP of rundown Kir2.1 channels. These changes could be correlated with a weakening of the interaction between Kir2.1 and PIP2, with C311R and C311S being more potent at modulating the Kir2.1-PIP2 interaction than C311A. The present work supports, therefore, molecular models whereby the gating properties of Kir2.1 depend on the presence of nonpolar or neutral residues at positions 76 and 311, with C311 modulating the interaction between Kir2.1 and PIP2.
INTRODUCTION
Inward rectifying potassium channels are membrane-bound proteins formed of four subunits each containing two transmembrane segments flanking a well-conserved pore region. Members of the Kir2 family exhibit a strong inward rectification produced by a voltage- and external K+-dependent channel block involving intracellular Mg2+ and polyamines (Fakier et al., 1995; Lu and MacKinnon, 1994; Silver and DeCoursey, 1990; Wible et al., 1994). These channels have been found to play a prominent role in a number of cellular processes including regulation of resting potential, cell proliferation, and control of vascular tone.
Cysteine residues contribute to the structural stability of proteins through the formation of disulfide bonds while being key target sites for redox related processes. The primary structure of the human Kir2.1 channel comprises a total of 13 cysteine residues, with two external cysteines at positions 122 and 154 highly conserved among Kir channels. It was suggested that these specific residues form either intra- or intersubunit disulfide bonds or both (Leyland et al., 1999; Cho et al., 2000). Similar conclusions were reported for the cysteines at position 113 and 145 in Kir2.3 (Bannister et al., 1999). The current topological model proposed for Kir2.1 also predicts that the cysteines at positions 89 and 101 should be located in the channel TM1 transmembrane segment whereas the pore and TM2 regions should contain the cysteines C149 and C169, respectively. The remaining seven cysteine residues are expected in turn to be distributed along the channel N- and C-terminus regions, with several residues comprised within conserved domains documented to contribute to channel gating (Lopes et al., 2002; Plaster et al., 2001; Shyng et al., 2000; Schulte et al., 1999). Fig. 1 presents amino acid alignments highlighting several N- and C-terminus domains common to a large variety of channels within the Kir superfamily. This analysis reveals a common highly conserved motif TTxxDxxWR, located in the N-terminus in close proximity to TM1. This mildly hydrophobic area, termed Q region, has already been reported to be involved in the gating and conduction properties of Kir channels (Choe et al., 1997). Interestingly, the Kir2.1 channel contains a cysteine at position 76 within the Q region, suggesting that this residue may be important for proper channel functioning. Another feature of the Kir channel family is the presence in the C-terminus region of a highly conserved motif QxRxSY (see Fig. 1). Amino acids within this motif have been found to be of critical importance to channel gating mediated by the interaction between PIP2 and Kir (Lopes et al., 2002; Liou et al., 1999). The Kir2.1 channel contains a cysteine at position 311 within the 310-QCRSSY-315 sequence. Hence, modification of this residue may potentially affect the channel-gating properties.
FIGURE 1.

Amino acid sequence alignments of representative members of the Kir superfamily showing sequence similarities in conserved C- and N-terminus regions. Alignments were performed with Clustalw using the BLOSUM equivalence matrix. Sequence similarities of 100% are shaded in black. The numbers refer to the location of the cysteine residues along the Kir2.1 sequence. ROMK and ROMK2 channels are referred to as Kir1.1 and Kir1.1b, respectively.
Although most of the cysteines in Kir2.1 appeared to be cytosolic, their functional role has not so far been extensively investigated. Experiments carried out using hydrophilic thiol modifying agents have already provided evidence that the cysteines at positions 54 and 76 are modifiable by internal MTSET and contribute to the MTSET-dependent inhibition of wild-type Kir2.1 (Lu et al., 1999a). It was proposed that these two residues could participate to the formation of an inner vestibule facing the channel central pore (Lu et al., 1999b), but their contribution to channel gating was never established. A patch-clamp study was thus undertaken in which we examined the effects of substituting each of the cysteines in the N- and C-termini of Kir2.1 by either a polar (S, D, N), an aliphatic (A, V, L), or an aromatic (W) residue. Our results indicate that the substitution of the N-terminus cysteine C76 or the C-terminus cysteine C311 by polar residues strongly modifies the channel intrinsic kinetic properties with the mutations at position 311 introducing long-lasting closed time intervals through a destabilization of the channel PIP2-Kir2.1 interaction.
MATERIALS AND METHODS
Site-directed mutagenesis of the Kir2.1 channel
The Kir2.1 channel used in these experiments was cloned from HeLa cells as described elsewhere (Klein et al., 1999). Site-directed mutagenesis of Kir2.1 channels were carried out using the QuikChange Site-Directed Mutagenesis kit (Stratagene, La Jolla. CA, USA). To obtain the 23 C/X consecutive point mutations, amino acid changes were introduced by using 25 mer oligonucleotides (C43S, C54/S/V, C76/S/L/V/F/R/N/W, C89/S/G, C122S, C101S, C149S, C154S, C169S, C209S, C311/S/A/R, C356S, and C375S) and wild-type Kir2.1 as template. All mutations were confirmed by sequencing the entire coding region in both directions on both strands.
Oocytes
Mature oocytes (stage V or VI) were obtained from Xenopus laevis frogs anesthetized with 3-aminobenzoic acid ethyl ester. The follicular layer was removed by incubating the oocytes in a Ca2+-free Barth's solution containing 1.6 mg/ml collagenase (Sigma, Oakville, Ontario, Canada) for 45 min. The composition of the Barth's solution was (in mM): 88 NaCl, 3 KCl, 0.82 MgSO4, 0.41 CaCl2, 0.33 Ca(NO3)2, and 5 HEPES (pH 7.6). Defolliculated oocytes were stored at 18°C in a Barth's solution supplemented with 5% horse serum, 2.5 mM Na pyruvate, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. Oocytes were studied 4–6 days after coinjection (0.92 ng–9.2 ng) of the cDNA coding for Kir2.1 channels and 1.38 ng of cDNA coding for a green fluorescent protein that was used as a marker for nuclear injection (Klein et al., 1999).
Before patch-clamping, defolliculated oocytes were shrunk in a hypertonic solution containing (in mM) 250 KCl, 1 MgSO4, 1 EGTA, 50 sucrose, and 10 HEPES buffered at pH 7.4 with KOH. The vitelline membrane was then peeled off using forceps, and the oocyte was transferred to a superfusion chamber for patch-clamp measurements.
Patch-clamp recording
Patch-clamp recordings were carried out in the inside-out or cell-attached patch-clamp configuration using an Axopatch 200A amplifier (Axon Instruments, Union City, CA). Patch pipettes were pulled from borosilicate capillaries using a Narishige pipette puller (Model PP-83, Narishige, Tokyo, Japan) and used uncoated. The resistance of the patch electrodes ranged from 4 to 10 MΩ. Unless specified otherwise, the membrane potential is expressed as −Vp, where Vp is the pipette applied potential. Data acquisition was performed using a Digidata 1320A acquisition system (Axon Instruments, Union City, CA) at a sampling rate of 3.0 kHz with filtering at 500 Hz. Unless specified otherwise, the change of bath solution in inside-out recordings was carried out with a RSC-160 rapid solution changer system (BioLogic, Grenoble, France). When required, the open channel probability, Po, was estimated from current amplitude histograms on the basis of a binomial distribution as described elsewhere (Morier and Sauvé, 1994). Po stationarity as a function of time was tested according to the criteria defined in a previous work (Denicourt et al., 1996). The voltage dependence of the channel open probability Po was fitted to a Boltzmann equation defined as
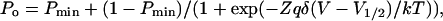 |
(1) |
where V1/2 is the applied voltage for half activation, Pmin the minimal Po, δ the electrical fractional distance, Z the valence of the interacting particle, q the electronic charge, and k and T the Boltzmann constant and temperature, respectively. Because most of our patches contained multiple channels, a detailed analysis of the dwell time distribution for the open and closed states could not be systematically performed. The channel mean open (To) and closed (Tc) times could nevertheless be estimated from current recordings with multiple channels using
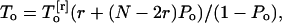 |
(2) |
where To[r] is the mean dwell time interval when r channels among N are open (see Appendix). As a rule, To[r] was measured using the SKM subroutine provided by the QuB single channel analysis package with r corresponding to the current level having the highest probability (Qin et al., 1996, 1997).
Time course of the poly-lysine induced current inhibition was estimated by fitting a single exponential function to the slow current decay that followed the instantaneous current reduction observed after poly-lysine application (Lopes et al., 2002).
In experiments involving the ATP-induced reactivation of rundown Kir2.1 channels (Fig. 7 B), the percent of reactivation was estimated from the ratio 〈I〉after/〈I〉before, where 〈I〉before is the average Kir2.1 current measured over a 5 s period after patch excision, and 〈I〉after the resulting Kir2.1 current obtained after ATP addition averaged over the same period of time. Experiments were performed at room temperature (22°C).
FIGURE 7.

Contribution of the C311 residue to the interaction Kir2.1-PIP2. (A) Poly-Lys inhibition of the Kir2.1wild-type and C311S, C311R, and C311A mutants. Normalized currents measured in inside-out patches in 200 mM K2SO4 conditions at an applied voltage (−Vp) of −60 mV. As seen, the poly-Lys inhibition constituted of two components: an initial very fast channel block followed by a slow inhibition reflecting a weakening of the interaction Kir2.1-PIP2. The slow component was affected by mutating the C311 residue with a potency rank C311A < C311S < C311R, indicating an increasingly more important weakening of the Kir2.1-PIP2 interaction. (B) Typical patch-clamp recording showing the absence of ATP regeneration after rundown of the wild-type C311A, C311R, and C311S mutant channels. The internal medium consisted in a 200 mM KCl solution with or without ATP added (see Materials and Methods). The applied potential (−Vp) was equal to −60 mV throughout. In contrast to the results obtained with the wild-type Kir2.1 channel, ATP failed to restore channel activity with the C311R/S/A mutant.
Solutions
The solution referred to as 200 K2SO4 had a composition as follows (in mM): 200 K2SO4, 1.8 MgCl2, 10 Hepes buffered with KOH at pH 7.4. Sulfate salts were used to minimize the contribution of endogenous calcium-activated chloride channels while ensuring that the levels of contaminant divalent cations never exceeded 0.5 nM for Ba2+ and 90 nM for Pb2+, the solubility constants for BaSO4 and PbSO4 being equal to 1.1 × 10−10 M and 1.8 × 10−8 M, respectively. In experiments performed in the presence of 2 mM K2ATP, the Mg2+ concentration was increased to 3.6 mM to keep the free Mg2+ concentration to a constant value of 1.8 mM. In some experiments, patch-clamp recordings were performed in the cell-attached configuration in a bath containing the 200 K2SO4 solution. The extrapolated zero current potential obtained under these conditions revealed a maximum voltage error of 7 ± 3 mV (n = 15) for an external K+ activity of 150 mM. For experiments performed in KCl conditions, we used a solution containing (in mM): 200 KCl, 1.8 MgCl2, 10 HEPES buffered with KOH at pH 7.4. Poly-l-lysine (MW 4000–15000) and DTT were purchased from Sigma.
Secondary structure predictions
DSSP, STRIDE, and STR secondary structure predictions were performed using the SAM-T02 package for sequence alignment and modeling (Karplus et al., 1998). Helical wheel projections were carried out with the Antheprot 2000 V5.2 software.
RESULTS
Kinetic properties of the wild-type Kir2.1 channel
Fig. 2 A illustrates examples of inside-out recordings performed in symmetrical K2SO4 solutions for potentials ranging from −60 mV to −150 mV. Whereas the channel open probability, Po, remained high (≈1) for potentials positive to −80 mV, long closed time intervals were systematically present at more negative potential leading to a reduction in the Po value. The voltage dependence of the channel open probability is illustrated in Fig. 2 B. As seen, the data points could be fitted to a Boltzmann equation with a half-activation potential, V1/2, and a fractional electrical distance, δ, equal to −143 ± 15 mV (n = 3) and 0.86 ± 0.15 (n = 3), respectively. These observations agree with the results reported previously for the Kir2.1 channel expressed in Xenopus oocytes (Choe et al., 1999) and for the native inward rectifier currents measured in bovine aortic endothelial cells (Elam and Lansman, 1995). In the latter case it was proposed that the decrease in Po at hyperpolarizing potentials resulted from the binding of external Mg2+ to a site (Kd = 300 μM) located at an electrical distance of 0.38. Such a conclusion is compatible with the present findings as experiments performed in zero external Mg2+ led to a Po voltage dependence characterized by a Pmin (Eq. 1) of 0.65 ± 0.07 (n = 2) as compared with Pmin < 0.1 in 1.8 mM Mg2+ conditions (data not shown). Fig. 2 C presents a plot of the channel mean open (To) and closed (Tc) times as a function of voltage. The results obtained indicate that the voltage-dependence of the channel Po results from an 11-fold increase in Tc over the voltage range from −60 mV to −180 mV, coupled to a 1.8-fold decrease in To for voltages ranging from −60 mV to −180 mV. This analysis also reveals that the channel fluctuation pattern under physiologically relevant voltage conditions consists essentially in channel openings of 250 ms mean duration interrupted by brief closed intervals of <15 ms leading to an open probability nearly equal to one.
FIGURE 2.

Gating properties of Kir2.1 channels in 200 mM K2SO4 + 1.8 mM Mg2+ external conditions. (A) Examples of inside-out recordings performed with pipettes containing 200 mM K2SO4 + 1.8 mM Mg2+ for membrane potentials (−Vp) ranging from −60 mV to −150 mV. The label c refers to the current level for the closed channel conformation. (B) Kir2.1 channel open probability, Po, plotted as a function of applied voltage (−Vp). The data points were fitted to a Boltzmann equation with a half-activation potential, V1/2, and a fractional electrical distance, δ, equal to −143 ± 15 mV (n = 3) and 0.86 ± 0.15 (n = 3) respectively. (C) Variations as a function of voltage of the channel mean open (To, open squares) and closed (Tc, filled squares) times measured in 200 mM K2SO4 + 1.8 mM Mg2+ conditions. Data obtained from three different experiments. These measurements indicate that the voltage dependence of Po arises from an 11-fold increase in Tc over the voltage range from −60 mV to −180 mV, coupled to a 1.8 decrease in To over the same voltage range.
Mutations of cytosolic cysteine affect Kir2.1 gating and permeation properties
The structural role of cysteine residues was next investigated by substituting each of the channel cysteines to a serine with the exception of C122 and C154, which were reported to be essential for proper channel folding (Cho et al., 2000). The cysteine to serine mutation consists essentially in replacing a sulfur by an oxygen with an overall decrease in the bond length of ∼0.7 Å while maintaining hydrogen bonding capability. Hence, changes in the channel-gating properties under these conditions could argue for stringent requirements for a residue with cysteinelike physicochemical properties at a particular location. Table 1 summarizes the effects of cysteine to serine mutations on the Po voltage dependence and single channel conductance for the nine mutants tested in this study. Because of the nonohmic behavior of the current/voltage relationship at negative potentials, the single channel conductance was estimated 1), for voltages ranging from −20 mV to −120 mV (Λ low) and 2), for voltages more negative than −120 mV (Λ high). This analysis first reveals that most substitutions neither affected the channel conductance nor its rectifying properties. An inhibition of 20% of the channel conductance was, however, observed with the C89S mutant, suggesting that this residue is involved in the ion permeation process. Changes were also observed in the parameters describing the Po voltage dependence as computed according to Eq. 1 (see Materials and Methods). A significant decrease in voltage sensitivity (δ) was apparent with the C209S and to a lesser extent with C43S and C375S mutants. For instance, the average Po for C209S was estimated at 0.91 at −40 mV and 0.60 at −200 mV. These values contrast with the results obtained under the same conditions for the wild-type Kir2.1, where Po remained lower than 0.35 at potentials more negative than −170 mV (Fig. 2 B). Globally, the C209S channel behaved like the wild-type Kir2.1 channel in the absence of external Mg2+. Moreover, patch-clamp experiments performed with the mutations of the conserved C54 residue (C54S, C54V) failed to provide evidence for significant changes in gating properties relative to wild-type Kir2.1 (data not shown). The most drastic effects on channel gating were observed with the C76S and C311S channels.
TABLE 1.
Voltage gating and unitary conductance for the wild-type and serine mutant Kir2.1 channels
| V1/2 (mV) | Zδ | Λlow(pS) | Λhigh(pS) | |
|---|---|---|---|---|
| Wild-type | −143 ± 15 (3) | 0.86 ± 0.15 (3) | 34 ± 2 (3) | 40 ± 3 (3) |
| C43S | −174 ± 9 (3) | 0.56 ± 0.02 (3) | 34 ± 1 (3) | 44 ± 1 (3) |
| C54S | −162 ± 5 (2) | 0.96 ± 0.24 (2) | 33 ± 0.5 (2) | 42 ± 3 (2) |
| C76S | – | – | 28 ± 4 (4) | 41 ± 3 (4) |
| C89S | −146 ± 16 (3) | 0.66 ± 0.06 (3) | 24 ± 3 (6) | 32 ± 1 (6) |
| C89G | −121 ± 6 (2) | 0.94 ± 0.26 (2) | ||
| C101S | −130 ± 10 (2) | 0.96 ± 0.14 (2) | 36 ± 2 (3) | 43 ± 1 (3) |
| C209 | −146 ± 50 (3) | 0.44 ± 0.18 (3) | 32 ± 2 (6) | 36 ± 3 (6) |
| C311S | – | – | 29 ± 2 (8) | – |
| C356S | −145 ± 2 (2) | 0.66 ± 0.04 (2) | 36 ± 1 (2) | 41 ± 1 (2) |
| C375S | −156 (3) | 0.54 (3) | 35 ± 1(3) | 39 ± 4 (3) |
The C123S and C154S mutants are not included, as no currents could be recorded in both cases. Because of the nonohmic properties of the channel current/voltage relationships, the single channel conductance was estimated separately for voltages ranging from −20 mV to −120 mV (Λ low) and for voltages more negative than −120 mV (Λ high). With the exception of the C89S mutant, none of the mutations tested caused a significant change in unitary conductance relative to wild-type. The voltage dependence of the channel open probability was estimated according to the parameters of the Boltzmann equation (Eq. 1). A decrease in voltage sensitivity was observed with the C209S and to a lesser extent C43S and C375S channels as these mutants showed reduced δ values. The most important modifications in channel gating without significant changes in single-channel conductance were recorded with the C76S and C311S mutants.
Contribution of the N-terminal C76 residue to channel gating
As mentioned previously, the cysteine residue at position 76 in Kir2 channels constitutes an integral part of a highly conserved N-terminus motif TTxxDxxWR located in the mildly hydrophobic Q region. Inside-out experiments were thus conducted in which the C76 residue in Kir2.1 was replaced by residues of increasing polarity as to investigate the contribution of C76 to channel gating. Notably, no currents could be detected with the C76D/N (polar) and C76W (aromatic) mutants. Fig. 3 illustrates representative patch-clamp recordings obtained with the C76S, C76V, and C76L channels. As seen, the C76S channel experienced a drastic change in channel gating as compared with wild-type, with a current fluctuation pattern characterized by brief channel openings for voltages ranging from −60 mV to −140 mV. Internal addition of ATP and/or DTT has been reported to reverse the rundown of Kir2.1 channels expressed in Xenopus oocytes (Ruppersberg and Fakler, 1996). Inside-out experiments were thus performed to determine if the decrease in channel activity observed with the C76S mutant could be due to a facilitation of the channel rundown process. Internal applications of either ATP (2 mM) (n = 4) or DTT (5 mM) (n = 4) failed to activate the C76S mutant channel, suggesting that the state of the C76S channel is distinct from rundown Kir2.1.
FIGURE 3.

Effects of mutating the C76 residue on channel gating. Patch-clamp recordings performed in the cell-attached (C76S, C76V) or inside-out configuration (C76L) with pipettes containing 200 mM K2SO4 + 1.8 mM Mg2+. The potential (−Vp) was varied from −60 mV to −140 mV. The label c refers to the current level for the closed channel conformation. The C76S mutation led to a drastic change in channel gating characterized by a destabilization of the channel open state. Such behavior was not observed with the C76F, C76V mutants, which showed current fluctuation features equivalent to wild-type Kir2.1. Current traces were filtered at 1kHz and sampled at 3kHz.
Fig. 4 A presents a plot of Po as a function of voltage for the C76S, C76V, and C76L mutants. The average Po value for C76S was estimated at 0.05 ± 0.02 (n = 10) for −Vp ranging from −100 mV to 0 mV, in contrast to wild-type Kir2.1 where Po remained very high (≈1) for voltages positive to −100 mV. Fig. 4 B illustrates the variations in To and Tc computed for the C76S and C76V channels as a function of voltage. The results of this analysis indicated that the C76S mutation caused a drastic inhibition of channel activity with To and Tc values equal to 7.0 ± 0.4 ms (n = 8) and 135 ± 15 ms (n = 8), respectively (open triangles). In contrast, both To and Tc varied as a function of voltage for the C76V mutant (filled squares) with Tc increasing from 34 ± 2 ms (n = 3) at −40 mV to 247 ± 31 ms (n = 3) at −200 mV and To decreasing from 275 ± 10 ms (n = 3) at −40 mV to 97 ± 12 ms (n = 3) at −200 mV. Globally, these results support a model whereby the C76 residue within the N-terminal K64-V93 domain plays a prominent role in the channel-gating process, with the mutation of the cysteine at 76 to the more polar amino acid serine leading to a destabilization of the channel open state.
FIGURE 4.

Voltage-dependent gating of the C76 channel mutants. (A) Po voltage dependence measured for the C76L (open circles) and C76V (filled squares) mutants. Data points were fitted to a Boltzmann equation with V1/2 = −147 ± 7 mV and δ = 0.46 ± 0.09 (n = 3) for C76V and V1/2 = −158 ± 5 mV and δ = 0.60 ± 0.05 (n = 2) for C76L. The average Po value for C76S (open triangles) was equal to 0.05 ± 0.02 (n = 10). (B) Voltage dependence of the mean open (To) and closed (Tc) times for the C76S and C76V mutants. The C76S mutation caused a drastic inhibition of channel activity with To and Tc values equal to 7.0 ± 0.4 ms (n = 8) and 135 ± 15 ms (n = 8) respectively (open triangles). In contrast, both To and Tc varied as a function of voltage for the C76V mutant (filled squares) with Tc increasing from 34 ± 2 ms (n = 3) at −40 mV to 247 ± 31 ms (n = 3) at −200 mV and To decreasing from 275 ± 10 ms (n = 3) at −40 mV to 97 ± 12 ms (n = 3) at −200 mV.
Contribution of the C-terminal C311 residue to channel gating
The Kir2.1 channel contains a cysteine at position 311 that lies within the highly conserved C-terminus motif QxRxSY. Recent studies have in this regard provided evidence that the 310-QCRSSY-315 region in Kir2.1 participates in the interaction between Kir2.1 and PIP2 and as such is likely to be involved in channel gating (Lopes et al., 2002). Patch-clamp recordings of the C311S, C311A, and C311R mutant channels are shown in Fig. 5. As seen, the current recordings obtained with the C311R and C311S mutants contained long closed time intervals absent in C311A. At more negative potentials, these silent periods generated a typical burstlike fluctuation pattern with long interburst intervals separating periods of channel activity with multiple closed intervals. These results are summarized in Fig. 6 A where the channel Po is plotted as a function of −Vp. The main effect of C311R/S substitutions consisted in decreasing the channel Po for voltages ranging from −100 mV to −20 mV, resulting in a partial loss of the Po voltage dependence. Such a behavior was not observed with the C311A channel. The variation of Tc and To as a function of voltage for the C311S, C311A, and C311R channels is plotted in Fig. 6 B. As seen, there was a strong increase in Tc for the C311S and C311R mutants with values ranging from 500 ms to 1 s for voltages positive to −100 mV, whereas C311A showed wild-type characteristics with Tc decreasing from 57 ms to 7 ms over the same voltage range. In contrast, the C311S and C311R mutations led to wild-type features for To with values increasing twofold over the voltage range from −200 mV to −60 mV, whereas the C311A substitution resulted in voltage insensitive To with values averaging 125 ms (Fig. 6 B). Hence C311 differs from C76 where the cysteine to serine substitution caused an important destabilization of the channel open state. These observations support therefore complementary roles for the C76 and C311 residues in channel gating.
FIGURE 5.

Effects of mutating the C311 residue on channel gating. Patch-clamp recordings performed in the cell-attached configuration with pipettes containing 200 mM K2SO4 + 1.8 mM Mg2+. The potential (−Vp) was varied from −60 mV to −160 mV. The label c refers to the current level for the closed channel conformation. The presence of a polar (S) or charged (R) residues at 311 led to the appearance for −Vp ranging from −100 mV to −40 mV of long closed time intervals absent in wild-type Kir2.1 and C311A recordings.
FIGURE 6.

Voltage-dependent gating of C311 channel mutants. (A) Po voltage dependence measured for the C311A (open circles), C311S (filled triangles), and C311R (open triangles) channel mutants. The data points for C311A were fitted to a Boltzmann equation with V1/2 = −127 ± 9 mV and δ = 0.63 ± 0.2 (n = 2). The mutation C311S led to an important decrease in Po for voltages positive to −120 mV as compared with C311A or wild-type Kir2.1. Similarly, the mutation of C311R caused a larger decrease in Po relative to C311A, with Po values never exceeding 0.3 over the entire voltage range from −180 mV to −40 mV. (B) Mean open and closed times plotted as a function of voltage for the C311A (open circles), C311R (open triangles), and C311S (filled triangles) channel mutants. Whereas the C311A channel showed wild-type characteristics for Tc with values decreasing from 57 ms to 7 ms for potential positive to −100 mV, the mutations C311S and C311R led to Tc values ranging from 500 ms to 1 s over the same voltage range. In contrast, the C311S and C311R mutants showed wild-type features for To with a twofold increase over the voltage range from −200 mV to −60 mV, whereas the C311A mutation resulted in a voltage insensitive To with values averaging 125 ms. These results support a model whereby the presence of a polar or charged residue at position 311 significantly modifies the gating properties of the Kir2.1 channel by favoring the appearance of long-lasting closed intervals.
Contribution of the C-terminal C311 residue to the interaction Kir2.1-PIP2
The C311 residue is adjacent to the R312 that is documented to play a prominent role in the interaction of Kir2.1 with PIP2 (Lopes et al., 2002). Experiments were thus performed to test the hypothesis that mutating the C311 residue modifies the 310-QCRSSY-315 C-terminus domain as to weaken the interactions Kir2.1-PIP2. Fig. 7 A presents a series of inside-out recordings where poly-Lys (300 μg/ml) was applied internally onto the Kir2.1 wild-type channel and to the C311S, C311R, and C311A channel mutants. The positively charged poly-Lys has been documented to compete with the Kir2.1 channel for the available PIP2, thus initiating an inhibition of channel activity (Lopes et al., 2002). As already reported, we observed two components to the Kir2.1 inhibition by poly-Lys: first a very fast initial block that was followed by a slow channel inhibition due to a weakening of the interaction Kir2.1-PIP2 (Lopes et al., 2002). Our results clearly show that the C311S and C311R mutations led to a faster poly-Lys-induced inhibition of channel activity with time constants of 9 ± 4 s (n = 3) for C311S and 8 ± 4 s (n = 4) for C311R as compared with 55 ± 12 s (n = 3) for wild-type, suggesting in both cases a weakening of the Kir2.1-PIP2 interaction. The mutation C311A appeared, however, less potent in modulating the inhibition by poly-Lys with a time constant of 34 ± 2 s (n = 4). This observation indicates that the cysteine to alanine substitution at 311 did not alter the interaction of Kir2.1 with PIP2 to the same extent as that observed when C311 was replaced by a charged (R) or polar (S) residue.
Kir2.1 channels have also been reported to undergo rapid rundown in inside-out patch-clamp experiments carried out in the absence of internal ATP. This effect was attributed to the absence of PI- and/or PIP-kinase activity in the patch membranes as both kinases require hydrolysable ATP to maintain a proper concentration of PIP2. Conditions that result in a weakening of the interaction PIP2-Kir2.1 are thus expected to impair the ATP-mediated recovery of channel activity after rundown. Fig. 7 B shows inside-out current recordings of the wild-type Kir2.1 channel performed in symmetrical KCl in the absence of ATP. As expected, a rapid decrease in single-channel activity was observed in KCl after patch excision with the channel mean current value decaying by 94 ± 2% (n = 12) within 2 min. Such a behavior was not observed in K2SO4 conditions, suggesting that the presence of the SO42− anions prevented somehow channel rundown. Channel activity could be recovered by the internal addition of ATP (2 mM K2ATP with 3.6 mM MgCl2). The effect of ATP was, however, variable, with the percentage of reactivation ranging from 35% to 100% (mean 49%; n = 8). As seen in Fig. 7 B, ATP failed to restore channel activity to a similar degree in inside-out experiments performed with the C311S/R/A mutants. For instance, the percent of reactivation was estimated to 15 ± 11% for C311S (n = 6) and to <3% for the C311A (n = 4) and C311R (n = 3) mutants, respectively. These results suggest, therefore, that the integrity of the C311 is important for the ATP-Mg reactivation effect of Kir2.1 after rundown probably by stabilizing the interaction PIP2-Kir2.1.
DISCUSSION
The inward rectifying Kir2.1 channel contains seven cytosolic cysteines with the C76 and C311 residues critically located in highly conserved domains of the channel N- and C-terminus regions. Mutation of either one of these cysteines by polar residues caused an important change in the channel-gating properties without significant modifications of the channel unitary conductance. Furthermore, substituting the C-terminus cysteine C311 by a polar or charged residue impaired the interaction Kir2.1-PIP2 and prevented the reactivation by ATP-Mg of rundown channels. These results constitute the first evidence that cytosolic cysteine residues play a role in the gating properties of Kir2.1, with C311 modulating the interaction between Kir2.1 and PIP2.
The N-terminus C54 residue
The N-terminus end of Kir2.1 contains four cysteine residues, respectively located at positions 43, 54, 76, and 89. The amino acid alignment presented in Fig. 1 shows that the cysteine residue at position 54 is highly conserved among members of the Kir channel family, with C54 corresponding to C49 in Kir1.1. Notably, the C49 residue in Kir1.1 has been reported to react with the thiol-modifying agent DTNB in the closed channel configuration only. It was suggested that C49 participates to the N- and C-terminus interactions underlying the pH-dependent gating of Kir1.1 (Ruppersberg, 2000). The C54 residue in Kir2.1 has also been found to be modifiable by thiol-specific reagents and to contribute to the MTSET-dependent inhibition of Kir2.1 (Lu et al., 1999a). Our results failed, however, to demonstrate important changes in channel gating with the C54S/V mutants. This observation does not support therefore a model whereby Kir2.1 functioning would critically depend on the integrity of the C54 residue but suggest that C54 in Kir2.1 is not functionally equivalent to C49 in Kir1.1.
Contribution of the N-terminus C76 residue to channel gating
Members of the Kir superfamily also contain a highly conserved motif, TTxxDxxWR, located in a mildly hydrophobic area, termed Q region. This region has already been reported to be involved in the gating and conduction properties of Kir channels (Choe et al., 1997). For instance, the Kir1.1 V72E mutant (equivalent to C76 in Kir2.1) detected in a subset of patients with the antenatal Bartter syndrome (Derst et al., 1997) has been documented to cause a decrease in channel activity. Similarly we found that mutating C76 to either a charged (D) or polar (N) amino acid results in an absence of detectable channel activity. These data confirm the functional role attributed to the 74-TTCVDIRWR-82 Q domain for proper Kir2.1 channel gating while providing evidence for a significant contribution of C76 residue.
The mechanism by which the mutation of C76 to polar residues causes a destabilization of the channel open state still remains unclear. Addition of ATP and/DTT failed to restore channel activity, arguing for a mechanism not related to the regulation of Kir2.1 by kinase-phosphatase-dependent processes. Chemical modification of the C76 residue by the hydrophilic sulphydryl reagents MTSET has already been reported to cause a 35% inhibition in whole-cell current (Lu et al., 1999a). These findings argue for C76 being water accessible while contributing to the formation of a long inner vestibule along the channel pore (Lu et al., 1999a). A more detailed analysis of the Kir2.1 channel secondary structure in the Q region based on the DSSP, STRIDE, and STR algorithms is presented in Fig. 8. Clearly each of these approaches strongly suggests the presence of a helical structure starting at residue K64. Notably, the STRIDE representation also predicts a β-sheet-turn-β-sheet structure for the F46-K49 and H53A-I59 regions, in qualitative agreement with the x-ray structures reported by Nishida and MacKinnon for the GIRK1 channel (Nishida and MacKinnon, 2002). A helical wheel projection for the region extending from K64 to M84 is illustrated in Fig. 8 D. According to this representation, the α-helix that contains the Kir2.1 Q domain is organized such that hydrophilic and hydrophobic residues are located on opposite sides with C76 centered on the helix hydrophobic face (Fig. 8 D). This spatial organization could optimize hydrophobic interactions and suggests a specific organization of the hydrophobic residues. This proposal is strongly supported by the findings presented in this work where the substitution of C76 to hydrophilic residues led either to nonfunctional channels (C76D, C76N) or channels characterized by an unstable open state configuration (C76S), whereas the substitutions to nonpolar residues such as L or V resulted in mutant channels with wild-type properties. It is thus possible that the introduction of a hydrophobic residue at C76 changes the orientation of the helical structure of the Q region leading to a destabilization of the channel open state.
FIGURE 8.

Secondary structure predictions for the Kir2.1 N-terminus Q domain. Secondary structure predictions for the N-terminus region extending from S45 to V93 computed according to the STRIDE (A), DSSP (B), and STR (C) algorithms for the wild-type Kir2.1. In panels A and B, the probability of a β-strand (E), short β-bridge (B), 310 helix (G), α-helix (H), turn (T), and random coil (C) is plotted as a function of the amino acid position number. In panel C, the STR β-strand (E) is subdivided into β-strand surrounded by two antiparalleled partners (A), two paralleled partners (P), one paralleled partner and one antiparalleled partner (M), one paralleled partner (Q), and one antiparalleled partner (Z), whereas I represented π-helix and S bends. This analysis clearly supports the presence of an α-helix starting at K64. (D) Helical wheel representation of the region K64–M84 for which a high probability of an α-helix structure was obtained. This representation suggests that the helix hydrophobic (open circles) and hydrophilic (filled circles) residues are located on opposite sides with C76 centered on the helix hydrophobic face. Squares represent cysteine residues or residues not strictly considered hydrophobic or hydrophilic.
Contribution of the C-terminus C311 residue to channel gating
Members of the Kir superfamily contain several highly conserved domains in C-terminus (Fig. 1). In this regard, the most conserved amino acids in the QxRxSY motif (310-QCRSSY-315 in Kir2.1) were shown to play a prominent role in Kir channel gating by participating to the functional interactions between the channel and PIP2 (Jones et al., 2001; Lopes et al., 2002). Recent reports have demonstrated, for instance, that the C-terminus region in Kir6.2 extending from 301 to 314 (312–325 in Kir2.1) is important for the maintenance of channel function and interactions with PIP2 (Shyng et al., 2000; Cukras et al., 2002). Similarly, the Kir1.1 mutations R311Q/W (equivalent to R312 in Kir2.1) associated with the antenatal variant of Bartter syndrome were found to severely affect the strength of the channel-PIP2 interactions (Schulte et al., 1999; Lopes et al., 2002). These observations suggest by analogy, that the 311–325 region in Kir2.1 may be involved in the channel interactions with PIP2. In support of this proposal is the finding that the Kir2.1-PIP2 interaction is drastically weakened by the R312Q mutation (Lopes et al., 2002). Altogether, these data support a model where the arginine residue in the Kir2.1, Kir1.1, and Kir6.2 QxRxSY motif participates to the electrostatic-based interactions between the channel and PIP2 (Lopes et al., 2002). Notably, the results presented in Fig. 7 A show that the C311 residue could also contribute to interaction channel-PIP2 in Kir2.1. As seen, mutating C311 to either a polar (S) or charged (R) amino acid caused a faster poly-Lys-induced inhibition of channel activity indicating a weaker Kir2.1-PIP2 interaction. Moreover, the physicochemical properties of the residue at this position were found to be of critical importance in this case with polar residues (C311R and C311S) being more potent in interfering the poly-Lys-dependent channel inhibition than the nonpolar one (C311A). These findings are correlated with the changes in channel gating recorded at the single-channel level where the most prominent variations in mean closed time were observed with the C311R and C311S channel mutants. The overall results obtained with the C311 mutant channels are thus in agreement with the previous reports indicating that decreasing the interaction between Kir and PIP2 induces an increase in the channel mean closed time (Lopes et al., 2002; Enkvetchakul et al., 2000).
Although the results in Fig. 7 A clearly support a modulatory action of C311 on the Kir2.1-PIP2 interaction, the exact molecular mechanism by which C311 exerts its action remains undetermined. A model whereby the C311 residue would directly interact with the negatively charged PIP2 through electrostatic interactions is unlikely, as the C311R mutation weakens interactions between Kir2.1 and PIP2. Because the C311 residue is adjacent to the R312 residue, it is possible, however, that the mutation C311R allosterically modifies the 310-QCRSSY-315 C-terminus domain, causing a weakening of the interactions of R312 with PIP2. These allosteric-induced changes in 3D-structure would need to be less prominent with hydrophobic (A) than charged (R) or polar (S) residues to account for our poly-Lys results presented in Fig. 7. Globally our results suggest that the residues close to R312 may contribute indirectly to the interaction PIP2-Kir2.1 via a structural change involving the R312 residue.
C311 and ATP reactivation effects
Our results also indicate that modifications of the cysteine residue at 311 affect the ATP-dependent recovery of channel activity after rundown. It has been proposed that the ATP-induced reactivation of most Kir was related to the presence of PI and/or PIP kinases in the patch membranes that require hydrolysable ATP to maintain a proper concentration of PIP2 (Derst et al., 1997). Furthermore, evidence was presented suggesting that the activation of Kir1.1 by PKA results from an enhancement of the Kir1.1-PIP2 interaction (Liou et al., 1999). In support of this proposal is the observation that the S313A mutation (equivalent to S314A in Kir2.1) in the Kir1.1 C-terminal phosphorylation site 309-QVRTSY-314 weakened the interaction PIP2-channel (Liou et al., 1999). A reduction of the ATP-mediated reactivation of rundown C311R/S/A mutant channels would thus be compatible with the weakening in Kir2.1-PIP2 interaction we observed in poly-Lys experiments (Fig. 7). Furthermore, as the C311 in Kir2.1 residue is adjacent to a putative PKA phosphorylation consensus motif (312-RSS-314), we cannot rule out that the C311S/R/A mutations initiate a series of allosteric effects susceptible to prevent a proper regulation by PKA-phosphorylation of the interaction PIP2-Kir2.1 at the level of the 310-QCRSSY-315 domain.
CONCLUSIONS
Our results demonstrate that two cytosolic cysteine residues respectively located in conserved N-terminus and C-terminus regions of Kir2.1 affect channel gating. These residues thus represent potential targets for a regulation by redox processes of the Kir2.1 channel activity. Such a regulation may be of particular importance in pathological conditions such as those prevailing during oxidative stress.
Acknowledgments
We thank Ms. Julie Verner for expert oocyte preparation. L.P. is a senior scholar from the Fonds de la Recherche en Santé du Québec.
This work was performed with a grant from the Canadian Institutes of Health Research (MOP 7769) to R.S. and with a joint grant from the FCAR Équipe to L.P. and R.S.
APPENDIX
Let us consider a channel represented by No open states and Nc closed states with Ki,j the rate of transition from state i to j and LO, LC the current levels for the open and closed conformations respectively. Let 〈KOC〉 and 〈KCO〉 correspond to the average numbers of observable transition per s from LO to LC and LC to LO respectively. The overall kinetic scheme can now be reduced to
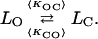 |
The average rate of transition 〈KOC〉 and 〈KCO〉 can be formally expressed as
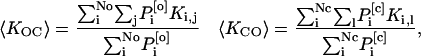 |
where {P1[o],P2[o],…,PNo[o]} represents the set of all the open states with the summation on j limited to the subset of states for which the transition i to j is observable (Roux and Sauvé, 1985). Similarly let {P1[c],P2[c],…,PNc[c]} represents the set of all the closed states with the summation on l being limited to the subset of states for which the transition i to l is observable. For a population of N identical channels the number of transitions between the current levels L0, L1, L2,…,Lr,…,LN, where r represents the number of open channels is given by
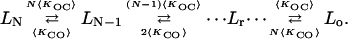 |
It follows that the mean dwell time for the current level corresponding to r open channels among N is given by To[r] = 1/((N − r)〈KCO〉 + r 〈KOC〉). Eq. 2 can now be obtained directly using Po = 〈KCO〉/(〈KOC〉 + 〈KCO〉) and To = 1/〈KOC〉.
Abbreviations used: MTSET, [2-(Trimethylammonium)ethyl] methanethiosulfonate bromide; PKA, cAMP-dependent protein kinase; PIP2, phosphatidylinositole 4,5-bisphosphate; DDT, dithiothreitol; DTNB, 5,5′-Dithio-bis(2-nitrobenzioc acid).
References
- Bannister, J. P., B. A. Young, A. Sivaprasadarao, and D. Wray. 1999. Conserved extracellular cysteine residues in the inwardly rectifying potassium channel Kir2.3 are required for function but not expression in the membrane. FEBS Lett. 458:393–399. [DOI] [PubMed] [Google Scholar]
- Cho, H. C., R. G. Tsushima, T. T. Nguyen, H. R. Guy, and P. H. Backx. 2000. Two critical cysteine residues implicated in disulfide bond formation and proper folding of Kir2.1. Biochemistry. 39:4649–4657. [DOI] [PubMed] [Google Scholar]
- Choe, H., L. G. Palmer, and H. Sackin. 1999. Structural determinants of gating in inward-rectifier K+ channels. Biophys. J. 76:1988–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe, H., H. Zhou, L. G. Palmer, and H. Sackin. 1997. A conserved cytoplasmic region of ROMK modulates pH sensitivity, conductance, and gating. Am. J. Physiol. 273:F516–F529. [DOI] [PubMed] [Google Scholar]
- Cukras, C. A., I. Jeliazkova, and C. G. Nichols. 2002. Structural and functional determinants of conserved lipid interaction domains of inward rectifying kir6.2 channels. J. Gen. Physiol. 119:581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denicourt, N., S. Cai, L. Garneau, M. Gagnan-Brunette, and R. Sauvé. 1996. Evidence from incorporation experiments for an anionic channel of small conductance at the apical membrane of the rabbit distal tubule. Biochim. Biophys. Acta. 1285:155–166. [DOI] [PubMed] [Google Scholar]
- Derst, C., M. Konrad, A. Kockerling, L. Karolyi, G. Deschenes, J. Daut, A. Karschin, and H. W. Seyberth. 1997. Mutations in the ROMK gene in antenatal Bartter syndrome are associated with impaired K+ channel function. Biochem. Biophys. Res. Commun. 230:641–645. [DOI] [PubMed] [Google Scholar]
- Elam, T. R., and J. B. Lansman. 1995. The role of Mg2+ in the inactivation of inwardly rectifying channels in aortic endothelial cells. J. Gen. Physiol. 105:463–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul, D., G. Loussouarn, E. Makhina, S. L. Shyng, and C. G. Nichols. 2000. The kinetic and physical basis of K(ATP) channel gating: toward a unified molecular understanding. Biophys. J. 78:2334–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakier, B., U. Brändle, E. Glowatzki, S. Weldemann, H.-P. Zenner, and J. P. Ruppersberg. 1995. Strong voltage-dependent inward rectification on inward rectifier K+ channels is caused by intracellular spermine. Cell. 80:149–154. [DOI] [PubMed] [Google Scholar]
- Jones, P. A., S. J. Tucker, and F. M. Ashcroft. 2001. Multiple sites of interaction between the intracellular domains of an inwardly rectifying potassium channel, Kir6.2. FEBS Lett. 508:85–89. [DOI] [PubMed] [Google Scholar]
- Karplus, K., C. Barrett, and R. Hughey. 1998. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 14:846–856. [DOI] [PubMed] [Google Scholar]
- Klein, H., L. Garneau, M. Coady, G. Lemay, J. Y. Lapointe, and R. Sauvé. 1999. Molecular characterization of an inwardly rectifying K+ channel from HeLa cells. J. Membr. Biol. 167:43–52. [DOI] [PubMed] [Google Scholar]
- Leyland, M. L., C. Dart, P. J. Spencer, M. J. Sutcliffe, and P. R. Stanfield. 1999. The possible role of a disulphide bond in forming functional Kir2.1 potassium channels. Pflugers Arch. 438:778–781. [DOI] [PubMed] [Google Scholar]
- Liou, H. H., S. S. Zhou, and C. L. Huang. 1999. Regulation of ROMK1 channel by protein kinase A via a phosphatidylinositol 4,5-bisphosphate-dependent mechanism. Proc. Natl. Acad. Sci. USA. 96:5820–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes, C. M., H. Zhang, T. Rohacs, T. Jin, J. Yang, and D. E. Logothetis. 2002. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 34:933–944. [DOI] [PubMed] [Google Scholar]
- Lu, T., Y. G. Zhu, and J. Yang. 1999a. Cytoplasmic amino and carboxyl domains form a wide intracellular vestibule in an inwardly rectifying potassium channel. Proc. Natl. Acad. Sci. USA. 96:9926–9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, T., B. Nguyen, X. Zhang, and J. Yang. 1999b. Architecture of a K+ channel inner pore revealed by stoichiometric covalent modification. Neuron. 22:571–580. [DOI] [PubMed] [Google Scholar]
- Lu, Z., and R. MacKinnon. 1994. Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature. 371:243–246. [DOI] [PubMed] [Google Scholar]
- Morier, N., and R. Sauvé. 1994. Analysis of a novel double-barreled anion channel from rat liver rough endoplasmic reticulum. Biophys. J. 67:590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida, M., and R. MacKinnon. 2002. Structural basis of inward rectification. Cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell. 111:957–965. [DOI] [PubMed] [Google Scholar]
- Plaster, N. M., R. Tawil, M. Tristani-Firouzi, S. Canun, S. Bendahhou, A. Tsunoda, M. R. Donaldson, S. T. Iannaccone, E. Brunt, R. Barohn, J. Clark, F. Deymeer, A. L. George, Jr., F. A. Fish, A. Hahn, A. Nitu, C. Ozdemir, P. Serdaroglu, S. H. Subramony, G. Wolfe, Y. H. Fu, and L. J. Ptacek. 2001. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell. 105:511–519. [DOI] [PubMed] [Google Scholar]
- Qin, F., A. Auerbach, and F. Sachs. 1996. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys. J. 70:264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, F., A. Auerbach, and F. Sachs. 1997. Maximum likelihood estimation of aggregated Markov processes. Proc. R. Soc. Lond. B Biol. Sci. 264:375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, B., and R. Sauvé. 1985. A general solution to the time interval omission problem applied to single channel analysis. Biophys. J. 48:149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppersberg, J. P. 2000. Intracellular regulation of inward rectifier K+ channels. Pflugers Arch. 441:1–11. [DOI] [PubMed] [Google Scholar]
- Ruppersberg, J. P., and B. Fakler. 1996. Complexity of the regulation of Kir2.1 K+ channels. Neuropharmacology. 35:887–893. [DOI] [PubMed] [Google Scholar]
- Schulte, U., H. Hahn, M. Konrad, N. Jeck, C. Derst, K. Wild, S. Weidemann, J. P. Ruppersberg, B. Fakler, and J. Ludwig. 1999. pH gating of ROMK (K(ir)1.1) channels: control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome. Proc. Natl. Acad. Sci. USA. 96:15298–15303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S. L., C. A. Cukras, J. Harwood, and C. G. Nichols. 2000. Structural determinants of PIP(2) regulation of inward rectifier K(ATP) channels. J. Gen. Physiol. 116:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver, M. R., and T. E. DeCoursey. 1990. Intrinsic gating of inward rectifier in bovine pulmonary artery endothelial cells in the presence or absence of internal Mg2+. J. Gen. Physiol. 96:109–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wible, B. A., M. Taglialatela, E. Ficker, and A. M. Brown. 1994. Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature. 371:246–249. [DOI] [PubMed] [Google Scholar]