

Abstract
Double crossover molecules are DNA structures containing two Holliday junctions connected by two double helical arms. There are several types of double crossover molecules, differentiated by the relative orientations of their helix axes, parallel or antiparallel, and by the number of double helical half-turns (even or odd) between the two crossovers. They are found as intermediates in meiosis and they have been used extensively in structural DNA nanotechnology for the construction of one-dimensional and two-dimensional arrays and in a DNA nanomechanical device. Whereas the parallel double helical molecules are usually not well behaved, we have focused on the antiparallel molecules; antiparallel molecules with an even number of half-turns between crossovers (termed DAE molecules) produce a reporter strand when ligated, facilitating their characterization in a ligation cyclization assay. Hence, we have estimated the flexibility of antiparallel DNA double crossover molecules by means of ligation-closure experiments. We are able to show that these molecules are approximately twice as rigid as linear duplex DNA.
INTRODUCTION
A key aim of biotechnology and structural DNA nanotechnology (Seeman, 1982) is a rational approach to the construction of new biomaterials, such as individual geometrical objects and nanomechanical devices, but including extended constructions, particularly periodic matter. In the last decade, DNA has been shown to be capable of fulfilling all of these roles in prototype systems (Seeman, 1999). A key element in this work has been the use of DNA double crossover (DX) molecules. DX molecules are model systems for structures proposed to be involved in genetic recombination initiated by double strand breaks (Thaler and Stahl, 1988; Sun et al., 1991), as well as in meiotic recombination (Schwacha and Kleckner, 1995). They contain two crossovers that connect two helical domains (more crossovers may connect two helical domains in artificial systems (Sha et al., 2000a)). DX molecules have been used to establish physical features of the Holliday junction (Fu et al., 1994a; Zhang and Seeman, 1994; Sun et al., 1998, 1999; Sha et al., 2000a), to characterize enzymatic cleavage of Holliday junctions (Fu et al. 1994b; Sha et al., 2000b), and as a system to explore DNA conductivity (Odom et al., 2000). They also have been proposed as a system wherein it would be possible to reduce cellular automata or Wang tiles to practice in a molecular context (Winfree, 1996).
There are five different isomers of DX molecules, shown in Fig. 1 (Fu and Seeman, 1993). The helical domains are parallel in three of the five isomers, and they are antiparallel in the other two. Parallel molecules have a dyad axis of backbone symmetry parallel to the helix axes; in antiparallel DX molecules, the dyad axis is perpendicular to the helix axes. Another way to think about the difference between parallel and antiparallel molecules is that the crossovers that lead to DX molecules occur between strands of the same polarity in parallel molecules and between strands of opposite polarity in antiparallel molecules (Seeman, 2001); consequently there is about a half-turn difference in phasing between the helical domains when parallel and antiparallel species are compared. In DX molecules with small separations between crossovers, those with parallel domains are not as well-behaved as those with antiparallel domains. Hence, most of the focus has been on antiparallel DX molecules, particularly in nanotechnological systems. The two antiparallel isomers differ by containing an even (DAE) or odd (DAO) number of double helical half turns between crossover points. Note that the DAE molecule is unique in that it contains a cyclic central strand.
FIGURE 1.
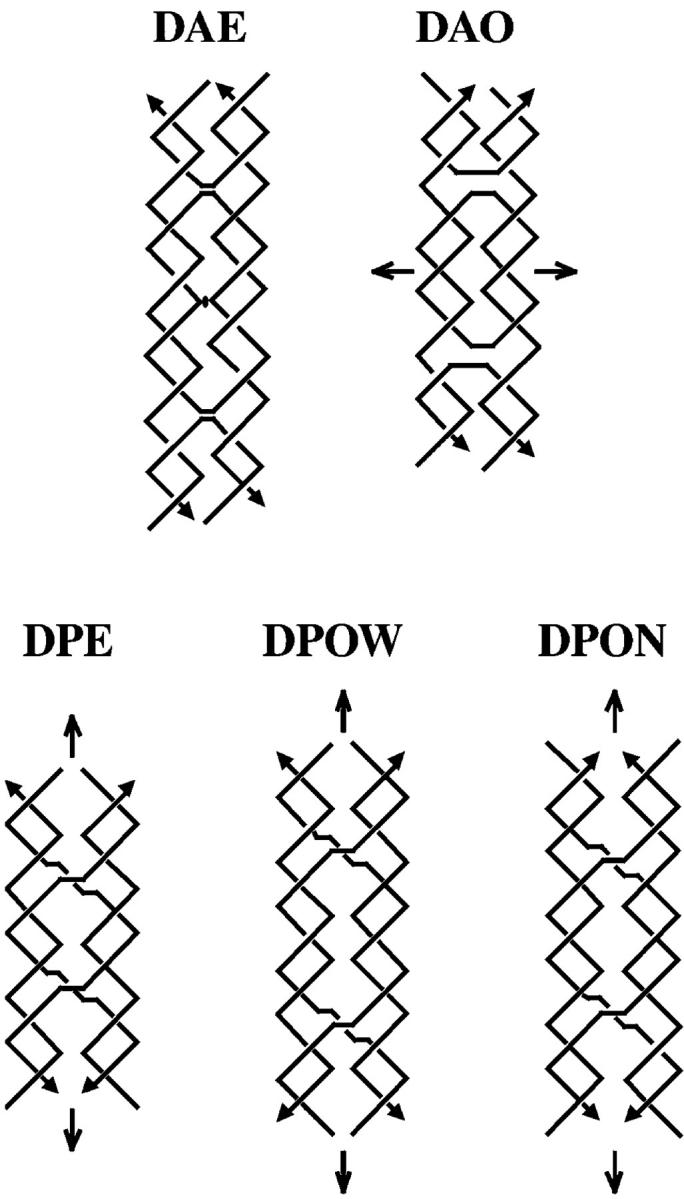
Schematic drawings of the five different structural arrangements of double crossover structures. The structures shown are named by the acronym describing their basic characteristics. All names begin with “D” for double crossover. The second character refers to the relative orientations of their two double helical domains, “A” for antiparallel and “P” for parallel. The third character refers to the number (modulus 2) of helical half-turns between crossovers, “E” for an even number and “O” for an odd number. A fourth character is needed to describe parallel double crossover molecules with an odd number of helical half-turns between crossovers. The extra half-turn can correspond to a major (wide) groove separation designated by “W”, or an extra minor (narrow) groove separation designated by “N”. The strands are drawn as zigzag helical structures, where two consecutive, perpendicular lines correspond to a full helical turn for a strand. The arrowheads at the ends of the strands designate their 3′ ends. The structures contain implicit symmetry, which is indicated by the conventional markings: a lens-shaped figure (DAE) indicating a potential dyad perpendicular to the plane of the page, and arrows indicating a twofold axis lying in the plane of the page. Note that the dyad in the DAE molecule shown is only approximate if the central strand contains a nick, which destroys the symmetry. The DAE molecule shown contains two turns between crossovers, like the molecules used in this study. An attempt has been made to portray the differences between the major and minor grooves. Note the differences between the central portions of DPOW and DPON. Also note that the symmetry brings symmetrically related portions of backbones into apposition along the center lines in parallel molecules in these projections. The same contacts are seen to be skewed in projection for the antiparallel molecules.
Several years ago, we demonstrated qualitatively that DNA DX molecules are relatively rigid molecules (Li et al., 1996). This was an important discovery for structural DNA nanotechnology, because flexible molecules are not appropriate components with which to build nanomechanical devices or periodic arrays. The earliest examples of one-dimensional DNA arrays (Yang et al., 1998), two-dimensional DNA arrays (Winfree et al., 1998; Liu et al., 1999), and DNA nanomechanical devices (Mao et al., 1999) all entailed the use of DX molecules. Our early analysis used DAE molecules, because they are easier to characterize; their ligation leads to a reporter strand through which it is possible to characterize the products conveniently.
Recently Podtelezhnikov et al. (2000) developed a method to estimate the structural features of units that can be assembled in multimers. The method derives estimates for the persistence length, the bend angle and helicity from the distributions of linear and circular multimers obtained during ligation of the monomeric units. This method was shown to be effective on a well-characterized linear duplex molecule. However, one of the key strengths of the method is that it can be applied to the ligation products of unusual DNA motifs, as well as to linear DNA. Here, we use this approach to characterize the flexibility of the DX molecules more quantitatively than in the previous study. We concentrate here on DAE molecules, although we no longer rely on reporter strands exclusively for characterization of the products. The generic DAE molecule we have studied is shown at the upper left of Fig. 1; it contains two turns of DNA between its crossover points. We have examined four different DAE molecules, three with sealed central circles of length 42 nucleotides; the molecules vary by their end-to-end lengths, 41, 42, or 43 nucleotide pairs (designated DAE-41, DAE-42, and DAE-43). A fourth molecule (designated DAE-42N) contains a nick in the central 42-nucleotide strand. We find that the DAE molecules we analyze have persistence lengths roughly twice the persistence length of the corresponding duplex molecule with the same sequence.
MATERIALS AND METHODS
Methods
Synthesis and purification of DNA
All DNA molecules in this study were designed by use of the program SEQUIN (Seeman, 1990). They were synthesized on an Applied Biosystems 380B automatic DNA synthesizer (Applied Biosystems, Foster City, CA), removed from the support, and deprotected, using routine phosphoramidite procedures (Caruthers, 1985). Biotin-Virtual Nucleotide phosphoramidites were purchased from Clontech (Palo Alto, CA). The DNA was purified by denaturing gel electrophoresis; ethidium bromide stained bands were excised from 12%–20% denaturing gels and eluted in a solution containing 500 mM ammonium acetate, 10 mM magnesium acetate, and 1 mM EDTA. The eluates were subjected to extraction with n-butanol to remove the ethidium, followed by ethanol precipitation. The amount of purified DNA strand was estimated by OD260.
Sealing of the central strand in DAE
Nonradioactive phosphorylation. 220–230 pmol of individual strand of DNA were dissolved in 20 μL solution of 1× kinase buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 10 mM 2-mercaptoethanol, US Biochemical (USB), Cleveland, OH) where additional ATP was added to a final concentration of 1 mM, and incubated with six units of polynucleotide kinase (USB) for 90 min at 37°C. The reaction was stopped by heating at 90°C for 15 min, followed by a denaturing gel purification.
Radioactive phosphorylation. 5 pmol of an individual strand of DNA were dissolved in 10 IL of 1× kinase buffer (USB) where γ-32P-ATP (10 mCi/mL, ICN Pharmaceuticals, Costa Mesa, CA) was added to the final concentration of 0.2 μM, and incubated with three units of polynucleotide kinase (USB) for 90 min at 37°C. The reaction was stopped by heating at 90°C for 15 min, followed by a denaturing gel purification.
Ligation-sealing of nick. Phosphorylated central strand (both radioactive and nonradioactive), which formed the nick in the DAE assembly, was mixed with excess amount of nonphosphorylated remaining strands to exclude the potential occurrence of radioactive middle strand not being incorporated into the final DAE assembly. 30 pmol of the nonradioactive phosphorylated strands and no more than 5 pmol of the radioactive counterpart were mixed with 38 pmol of other component strands in 15 μL ligation buffer (66 mM Tris-HCl, pH 7.6, 6.6 mM MgCl2, 10 mM DTT, 66 μM ATP, USB), heated to 90°C for 5 min and cooled to room temperature by the following protocol: 20 min at 65°C, 15 min at 50°C, 20 min at 37°C, 20 min at room temperature, and 20 min at 16°C. 10 units of T4 DNA ligase (USB) were added and the reaction was allowed to proceed at 16°C for up to 16 h.
Exonuclease III digestion. After 16 h incubation, half of the ligation mixture was withdrawn and incubated with 100 units of exonuclease III (USB) at 37°C for 1 h. Reaction was stopped by heating at 65°C for 20 min. The ligation product and exonuclease III-treated counterpart were then visualized on a 15% denaturing gel, and the amount of cyclized central strand was quantitated using a BioRad G5-525 Molecular Imager (BioRad, Hercules, CA).
Construction of experimental molecules
Radioactive phosphorylation.1.5 pmol of an individual strand of DNA was dissolved in 11 μL of a solution containing 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 10 mM DTT, 0.2 μM γ-32P-ATP (10 mCi/mL, NEN Life Science Products, Boston, MA) and incubated with three units of polynucleotide kinase (USB) for 60 min at 37°C. Radioactive labeling was followed by addition of 1 μl of 1× ligation buffer from New England Biolabs (NEB, Beverly, MA) containing 1 mM ATP in 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 10 mM DTT, 25 μg/mL bovine serum albumin; incubation proceeded for another 5 min. The reaction was stopped by heating the solution to 65°C for 20 min, and filtered through the G-25 micro-spin column (Amersham Pharmacia Biotech, Piscataway, NJ) to remove unincorporated γ-32P-ATP before denaturing gel purification.
Nonradioactive phosphorylation. Phosphorylations were performed in the ligation buffer (NEB). 90 pmol of individual strand of DNA were dissolved in 22.5 μL solution of 1× ligation buffer (NEB) and incubated with 3.6 units of polynucleotide kinase (USB) for 60 min at 37°C.
Ligations-construction of intermediates. All phosphorylated strands were mixed stoichiometrically in the presence of trace amount of labeled strand of DNA (no more than 0.7 pmol of labeled strand for 90 pmol of nonlabel strand counterpart), and the final reaction solution was adjusted to contain 128.6 μL ligation buffer. An aliquot containing 1.8 pmol of DNA was withdrawn and kept under −20°C until the specific activity was estimated on an SL30 Liquid Scintillation Spectrometer (Intertechnique, Dover, NJ); the specific activity is defined here as the ratio of counts per minute to pmol of DNA. The remaining ligation solution was then heated to 90°C for 5 min and cooled to room temperature by the following protocol: 20 min at 65°C, 15 min at 50°C, 20 min at 37°C, and 20 min at room temperature. 70 units of T4 DNA ligase (NEB) were added and the reaction was allowed to proceed at 16°C for up to 16 h. The reaction mixture was then subjected to purification on a denaturing gel. After electrophoresis, gels were exposed to Kodak X-OMAT AR films (Fisher Scientific, Springfield, NJ). Bands corresponding to the target topologically closed molecules were then excised, eluted in a solution containing 500 mM ammonium acetate, 10 mM magnesium acetate, and 1 mM EDTA, and the target molecules recovered by ethanol precipitation. The amount of recovered DNA was estimated by measuring the radioactivity and dividing the count by the specific activity established on the same occasion under identical conditions (normally, samples were measured in an Eppendorf tube, using high energy 3H analysis mode).
Restriction endonuclease digestions. 15–20 pmol of DNA samples that were to be used in the ligation multimerization assay were restricted with Bbs I (NEB) in NEB buffer 2 at 37°C for 2 h under the condition where 3.8 units of enzyme was used for the restriction of 1 pmol of DNA and the enzyme concentration was maintained at 1.4 units/μL. The reaction was further treated with streptavidin-coated magnetic beads to remove biotinylated material. When the concentration of the restriction product was below the desired concentration for the ligation-multimerization, the solution was filtered through a Microcon-10 column (Amicon, Beverly, MA) to concentrate the DNA without altering the buffer concentration.
Streptavidin bead treatment
Streptavidin beads were purchased from Promega (Madison, WI). They were supplied as 1 mg/ml magnetic particles in phosphate-buffered saline, 1 mg/ml bovine serum albumin, and 0.02% sodium azide. The binding capacity was 0.75–1.25 nmol/mg. 5 μL bead solutions were used for each pmol of DNA sample present at the beginning of the Bbs I restriction. An appropriate volume of bead solution was transferred into a siliconized Eppendorf tube and set on a magnetic stand for ∼30 s. The buffer was removed and the beads were washed three times with an equal volume of phosphate-buffered saline buffer and then three times with equal volume of NEB buffer 2. The Bbs I restriction solution was then added to the beads, mixed well, and allowed to incubate at 37°C for 5 min and then at room temperature for another 5 min. The tube was then set on the magnetic stand to separate the beads from the solution, which now contained no biotinylated material. Streptavidin bead purification was usually repeated twice. Homogeneity of ligatable materials obtained this way was checked by running aliquots of the materials on both denaturing and nondenaturing polyacrylamide gel electrophoreses. Denaturing gels contained 15% acrylamide (19:1 acrylamide:bisacrylamide) . All denaturing gels used in this study contained 8.3 M urea and were run at 55°C. The running buffer consisted of 89 mM Tris-HCl, pH 8.0, 89 mM boric acid, 2 mM EDTA (TBE). Nondenaturing gels contained 6% acrylamide (19:1 acrylamide:bisacrylamide) and were run at room temperature. The running buffer consisted of 40 mM Tris-HCl, pH 8.0, 20 mM acetic acid, 2 mM EDTA, and 12.5 mM magnesium acetate (TAEMg).
Experimental ligations
A streptavidin-bead treated solution containing monomers was brought to a final monomer concentration of 75 nM containing 1 mM ATP and 0.25 units/μL T4 DNA ligase (NEB). The appropriate incubation time was determined by preliminary time-course ligations that yielded 10%–20% monomer at the end of ligation reactions. The reactions were terminated by phenol/chloroform extraction followed by ethanol precipitation. Ligation products were then analyzed on a two-dimensional denaturing gel electrophoresis. The first dimension contained 4% acrylamide, and the second contained 6% acrylamide (19:1 acrylamide:bisacrylamide). These gels contained 8.3 M urea and were run at 55°C. After electrophoresis, the gels were dried onto Whatman 3MM paper (Whatman International, UK) and quantitated using a BioRad G5-525 Molecular Imager (BioRad) or a Storm 860 Gel and Blot Imaging System (Amersham Pharmacia Biotech).
Identification of cyclic ligation products
The sizing of a cyclic ligation product that appeared on the quantitative two-dimensional gel was aided by heat-induced random nicking of the cyclic molecule. An aliquot of the ligation multimerization products was run on a separate two-dimensional denaturing gel and a few representative bands corresponding to cyclic products of unknown sizes were excised from the gel, eluted, and recovered in an aqueous solution after ethanol precipitation. Half of the recovered material was kept as a control, whereas the other half was subjected to heating at 90°C for 15–20 min to induce moderate random single nicking in the molecule. A denaturing gel of this heat-induced nicked cyclic product revealed bands of relatively higher migration rate, in addition to the original band that was not subjected the treatment. These lower bands corresponded to species of reduced degrees of catenation. Depending on the nature of the catenated form of the original ligation product, bands corresponding to catenanes or single-stranded circles and their linearized versions were obtained. The size of the original circular products was inferred by comparing the mobility of the resulting linearized bands with the well-defined linear markers.
Analysis of flexibility
The procedure developed by Podtelezhnikov et al. (2000) was used to extract conformational parameters of DAE-42, DAE-41, DAE-43, and DAE-42N. First we found the values of j-factors for different multimers of each monomeric unit. This part of the procedure is based on fitting computed and measured distributions of linear and circular multimers obtained in the ligation reaction. Second, the set of j-factors was plotted in semilogarithmic scale as a function of multimer length and fitted by a second order polynomial. Two parameters of the fitting polynomial, the length corresponding to the maximum, Smax, and the width of the parabola at half-height, ΔS1/2, were used to evaluate conformational parameters by the empirical equations found by Podtelezhnikov et al.:
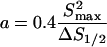 |
(1) |
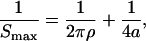 |
(2) |
where a is the persistence length of a polymer constituted from the monomeric units and ρ is the radius of curvature of the minimum energy conformation of the polymer. Equations 1and 2 were obtained for monomeric units modeled by homogeneous elastic rods with isotropic bending rigidity and uniformly distributed intrinsic curvature. This does not mean that the analysis cannot be applied to monomeric units with conformational features that are not homogeneous, although one should keep in mind that only effective, overall values of bending rigidity and intrinsic bend of the monomeric units can be obtained in this approach. For example, no differentiation can be made between the part of the monomeric unit between the crossover points (where two double helices are bound together), the parts just beyond the crossover points (where a single double helix is in close proximity to a second one), and the parts near the sticky ends (where there is a single double helix); the properties of these different segments are combined in our result.
The above analysis is applicable to monomeric units with an essential intrinsic curvature (Podtelezhnikov et al., 2000). To obtain the persistence length for an intrinsically straight linear duplex control molecule (designated LD-42), the set of j-factors was fitted by the theoretical dependence obtained by Shimada and Yamakawa (1984) for the wormlike chain model.
RESULTS
Helicity issues in the assembly of the experimental molecules
Before constructing the experimental molecules, one issue that needed to be resolved was the appropriate helicity for the central strand of the DAE molecule in those systems where it was to be sealed. Stacked DNA molecules in crystals typically display a helicity of 10 nucleotide pairs per turn (e.g., Qiu et al., 1997), but DNA in solution is usually characterized by a helicity of 10.5 (Wang, 1979; Rhodes and Klug, 1980). For a molecule whose stacking is as extensive as that of the DX molecule (Fu and Seeman, 1993), the correct number to choose is not obvious. We examined this issue by comparing ligation cyclization efficiencies between two DAE molecules, one with 20 nucleotide pairs between crossovers on each domain, leading to a cyclic 40-mer, and the other with 21 nucleotide pairs between crossovers, leading to a cyclic 42-mer. The results of this ligation are shown in Fig. 2. Shown left to right are the molecule with the nicked 40-mer circle, its ligation products, and the treatment of those products with exonucleases. The next three lanes contain analogous contents for the molecule with the nicked 42-mer circle. Quantitation of the ligation efficiencies shows that 13% of the 40-mer cyclized, but 82% of the 42-mer cyclized. We concluded that 10.5 nucleotide pairs per turn was the appropriate helicity to use, and we designed the molecules used in the experiments reported here accordingly. We also used a repeat distance of 42 nucleotides for the length of the DX molecules that were linked together, in a molecule called DAE-42. As a control, we varied this number from 41 to 43, in molecules called DAE-41 and DAE-43. In further controls, we compared these results to a molecule with a nicked 42-mer in the “cyclic” strand (DAE-42N), and to a linear duplex molecule containing 42 nucleotide pairs (LD-42).
FIGURE 2.

Comparison of the closure of the central cyclic 40-mer and 42-mer strands in DAE molecules. This is a denaturing gel autoradiogram that compares ligation efficiencies for two possible species of DAE molecules. Lane 1 contains linear markers (labeled No. L, where No. represents their lengths) and lanes 8 and 9 contain cyclic markers (labeled No. C, where No. again represents their lengths). Lanes 2–4 contain bands derived from the labeled 40-mer, and lanes 5–7 contain bands derived from the labeled 42-mer. Lanes 2 and 5 contain unligated material, lanes 3 and 6 contain the ligation products of these strands in the DAE context, and lanes 4 and 7 contain the results of treating the material in lanes 3 and 6, respectively, with exonuclease III. It is evident that under the conditions used, the 40-mer cyclizes only partially, whereas the 42-mer cyclizes almost completely.
Overall synthetic approach
The sequences of the molecules used in this work are shown in Fig. 3. The individual strands that are ligated to form the molecules are indicated: Two biotin-containing hairpins are ligated onto the ends of a DAE molecule with 42 nucleotide pairs in its cyclic central strand. In previous work (Podtelezhnikov et al., 2000), it was found that virtually complete phosphorylation was necessary for good agreement between experiment and theory. Here, we have adapted a method we developed previously for the ligation of DNA triangles (Yang et al., 1998); we solve the phosphorylation requirement problem by restricting to create phosphorylated sticky ends. The phosphate group has to be there for strand continuity, and only a contaminant with phosphatase activity could lead to its absence in the ligation mixture. The ultimate product of the ligations involved in producing the molecules we use is a cyclic molecule or a catenane. Consequently it is easy to purify the products on a denaturing gel, which readily separates failure products from target molecules. The restricted product can be purified readily from incompletely restricted molecules by streptavidin bead treatment.
FIGURE 3.

The sequences of the molecules used. The three molecules, DAE-41, DAE-42, and DAE-43, are shown. Small arrowheads indicated 3′ ends of strands. The central strand in each was sealed in the experiments, but the preligation 5′ and 3′ ends are indicated. Scission points by restriction enzymes are indicated by filled triangles. Recognition sites for restriction enzymes are indicated by hollow type. Biotin groups are indicated by bold, larger “B” characters. The molecules are prepared by ligating them together to form the structures indicated as topologically closed species. The final molecules to ligate are then prepared by restricting away the hairpin ends. A control linear duplex molecule, LD-42, is also shown in dumbbell form as DB-42. The nicked molecule DAE-42N is the same as the molecule DAE-42, except that its central strand is not sealed.
Characterization of the experimental molecules
The characterization of the experimental molecules is shown in a denaturing gel in Fig. 4 A. The species shown are DAE-42N, DAE-43, and DAE-42, left to right. It is clear that the starting species have been purified to homogeneity by denaturing gel electrophoresis. The point made via this gel is that Bbs I restriction of these molecules may be incomplete; nevertheless, after streptavidin bead treatment, only labeled species of the proper length are visible on the gel. Fig. 4 B shows the same species on a nondenaturing gel. Here, it is clear that the only species that remains after streptavidin bead treatment is the molecule that will be used in the ligation-closure study. There is some smearing in the target bands because of the complementarity of the sticky ends on the molecule. The smearing disappears if the gel is run at 37°C (data not shown).
FIGURE 4.
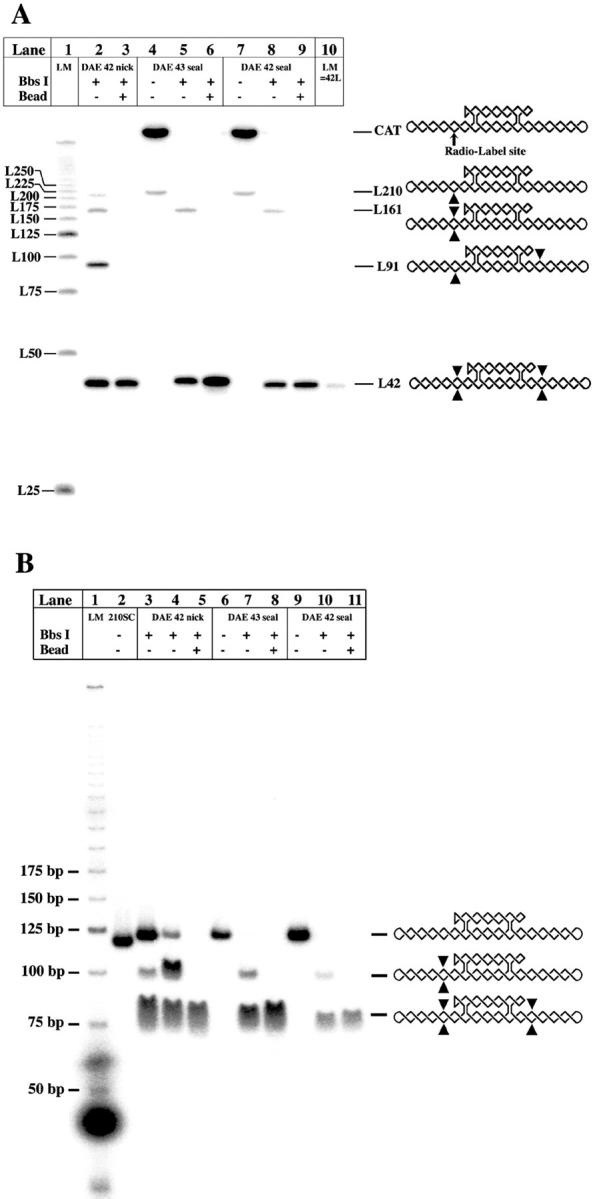
Characterization of the ligatable material. (A) Denaturing gel analysis of DAE preparation. This autoradiogram shows the preparation of DAE-42N, DAE-43, and DAE-42. Lane 1 contains a series of linear markers and lane 10 contains a 42-mer marker. To the right are the expected products reflecting various degrees of enzymatic degradation, with complete degradation indicated by the L42 species at the bottom. The radioactively labeled site is indicated in the undigested molecule. Sites where cleavage has occurred in a particular species are indicated by triangles; note that the linear molecule containing 210 nucleotides can be nicked at any of its restriction sites, not just the one indicated. Bbs I treatment is shown in lanes 2, 5, and 8, and streptavidin bead purification is shown in lanes 3, 6, and 9. It is clear that no impurities remain for any of the species in these lanes. (B) Nondenaturing gel characterization of the same species. Lanes 5, 8, and 11 contain the final materials after streptavidin bead purification. It is evident that they are uncontaminated by larger products.
Ligation-closure experiments
The nature of the ligation-closure experiment is illustrated in Fig. 5. The upper part of the figure shows a helical version of the molecules shown in Fig. 3; the biotin groups, included in the loop, are drawn outside the main chain for clarity. The molecules prepared as described above are restricted by Bbs I, and then purified by streptavidin bead treatment. The restricted molecule is shown below the original molecule. As shown in Fig. 4, these are pure species. These molecules are then ligated, and the products are characterized on denaturing two-dimensional gels. Two such gels are illustrated in Figs. 6 and 7. The gel in Fig. 6 contains the ligation products of DAE-42, and the gel in Fig. 7 contains the ligation products of DAE-42N. The products can be seen in three different arcs: an arc (virtually linear) corresponding to linear molecules (labeled L), a second arc corresponding to single-stranded circles (labeled C′), and an arc corresponding to double helical circles, which are cyclic catenanes (labeled C). Both the single-stranded circles and the double helical circles are catenated molecules. The single-stranded circles are catenanes of the inner strand with one cyclic 42-mer per repeat unit; there are likely single-stranded circle catenanes of the outer strand present as well, but they are unlabeled. The double helical circles are complex catenanes involving both the linking of the inner and outer strands, as well as the small cyclic strands. We have sought and failed to find evidence for the cleavage of the small cyclic strands in these systems; hence, the successive bands correspond to different numbers of molecules ligated into cyclic systems, rather than different extents of catenation to the small circles. The molecules electrophoresed in the gel shown in Fig. 7 are somewhat simpler. The nicking of the small cyclic strands results in their dissociation from the products; consequently the single-stranded circle bands correspond only to a single-stranded circle, and the double-stranded circle contains only two strands, although the linking between them is not expected to be that of comparable double helices of the same length. The individual bands have been identified and quantitated as described in Methods.
FIGURE 5.

The ligation-closure experiment. The initial purified topologically closed product is shown at the top of the figure. The biotin groups along the lower domain backbone helices are shown outside for clarity. The molecule is restricted, and its biotinylated hairpins are then removed (along with partial digestion products) by treatment with streptavidin beads. The product of this restriction is shown below the unrestricted molecule. The resulting molecules are then ligated to yield a collection of linear and cyclic molecules. A distribution of molecules is obtained, but both the linear and cyclic species are represented here only by hexamers. Note that the cyclic molecule is a catenane, but if the unlabeled strand were nicked, a single-stranded circle would result. Removal of one of the two continuous strands from the linear system would result in a rotaxane. Under denaturing conditions, the strands of the linear molecule are capable of dissociating.
FIGURE 6.

A two-dimensional denaturing gel showing the products of ligating DAE-42. The gel has been divided into two portions for clarity. The smaller (primarily linear) molecules are shown in a lower panel, and the rest in an upper panel. Linear products are indicated by the designation Ln, where the number n indicates the number of fundamental (42-mer) repeats are contained in the molecule. Single circles are indicated by the notation C′n, and cyclic catenated products are indicated as Cn. Note how successive species form arcs on the gel.
FIGURE 7.
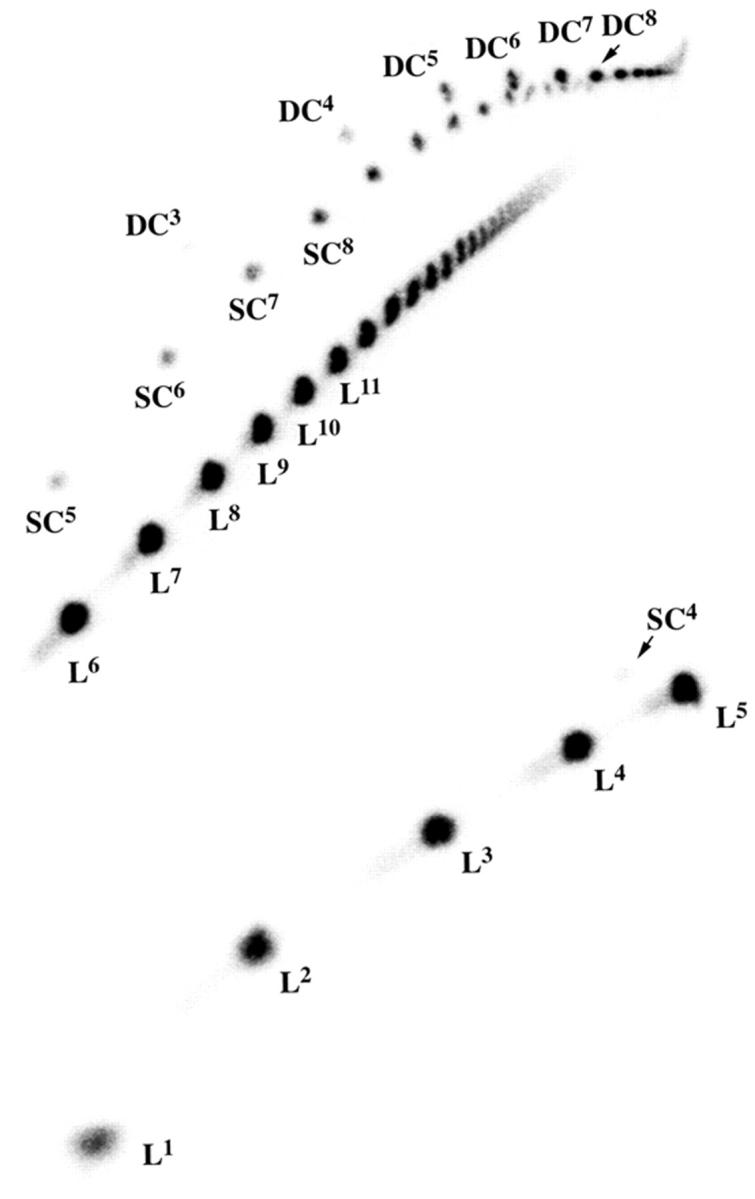
A two-dimensional denaturing gel showing the products of ligating DAE-42N. As in Fig. 6, the gel is shown in two portions. Linear molecules are indicated by “L” with a superscript indicating their size, single circles indicated by “SC” with a superscript, and double circles are indicated by “DC” with a superscript. The species are readily identified from the gel for purposes of quantitation.
Analysis of ligation experiments
The values of j-factors for different multimers of each monomeric unit were obtained by fitting computed and measured distributions of linear and circular multimers. Fig. 8 shows that a good fit between the measured and computed distributions of the ligation products was obtained in this procedure. The extracted sets of j-factors are shown in Fig. 9. Analysis of these sets of j-factors allowed us to obtain conformational parameters presented in Table 1. The persistence length estimated for the double helical fragment is 130 basepairs, near the values estimated by other investigators (Hagerman, 1988). Three sealed molecules were studied to control for the possibility that the twist of the molecules would have an effect on the results. Likewise, a molecule with a nicked circle was used, partly as another control, and partly because this is the molecule used most frequently in nanotechnology experiments (Yang et al., 1998; Winfree et al., 1998; Liu et al., 1999). The values of persistence length obtained for all four of these molecules, including nicked ones, are ∼2 times larger than the value for the double helix. Although the values of the persistence length and bend angles obtained for these molecules are slightly different, we believe that the difference reflects the accuracy of our analysis rather than the difference in the conformational properties. Thus, under the conditions assayed, DX molecules represent a substantial increase in rigidity over double helical DNA.
FIGURE 8.

Comparison of experiment and calculations for two systems. The top two panels indicate the fit between observed and calculated proportions of linear and cyclic products of various lengths for the DAE-42 system. The filled bars are experimental results, and the open bars are calculated based on the parameters derived from the analysis. The bottom two panels show the same extent of agreement for the LD-42 molecules.
FIGURE 9.
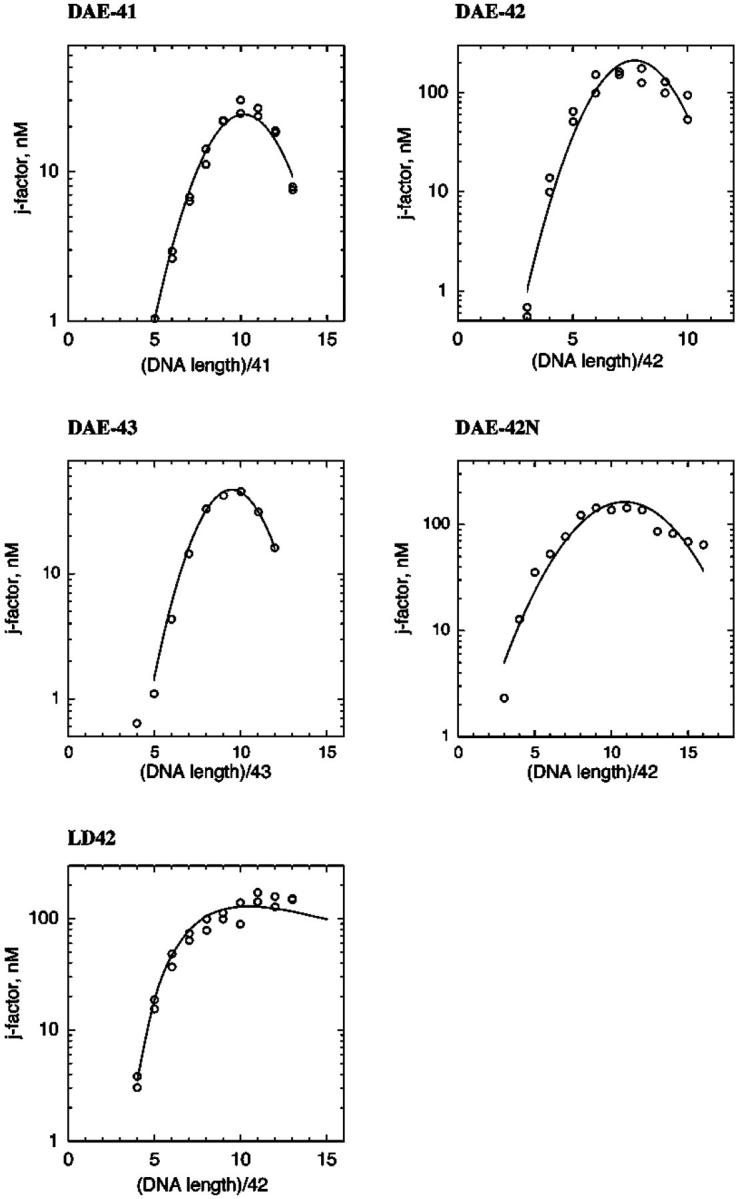
Extracted sets of j-factors for the species studied. The j-factors for multimers of different molecules are shown as a function of their length. The second order polynomial fits to the data (solid lines) are used to derive the parameters that characterize the molecules.
TABLE 1.
Parameters derived from the experiments performed here
| Monomer | Smax | ΔS1/2 | Persistence length, bp | Bend angle (per 10 bp) | Torsional fit |
|---|---|---|---|---|---|
| DAE-42 | 7.7 | 3.4 | 295 bp | 8° | Best |
| DAE-41 | 10.1 | 4.9 | 350 bp | 6° | |
| DAE-43 | 9.5 | 4.0 | 380 bp | 7° | |
| DAE-nicked | 10.9 | 7.0 | 285 bp | 5° | |
| Duplex-42 | – | – | 130 bp | 0° |
It should be noted that our analysis cannot address changes of DNA bending or the distribution of the intrinsic bend along a monomeric unit. Clearly, the rigidity is different for the regions consisting of one double helix or two double helices and the bends are likely to be localized at certain points within the unit. Also, the crossover sites might behave as points with reduced rigidity. What we have estimated in this analysis is the effective bending rigidity of the entire monomeric unit and its combined bend angle.
DISCUSSION
Rigidity of the DAE molecule
We have shown that the DX molecule represents a DNA molecule about twice as stiff as linear DNA molecules. We had demonstrated this result qualitatively several years ago (Li et al., 1996), but in those experiments no cyclic molecules were obtained, so no means of quantitating the stiffness was available. We do not know whether this result extends to other DX molecules, such as DAO molecules. Likewise, other systems with two double helical domains, such as paranemic crossover molecules or JX2 molecules (Yan et al., 2002) have not been characterized quantitatively, despite their use in nanomechanical devices. In addition, larger systems, such as triple crossover molecules, used to produce periodic arrays (LaBean et al., 2000) and in DNA-based computation (Mao et al., 2000) remain uncharacterized.
It is also important to emphasize that we have characterized the stiffness of the molecule in the direction of the helix axis, but we do not know about the flexibility around the helix axis. Thus, we have measured the effect on one DNA double helix of having a second double helix “fused” to it. However, we have not characterized the relative flexibility of the two helical domains. Thus, the two helices could flex about the axis that runs between them. The ability of arrays containing DX molecules to remain planar will be very much a function of this parameter.
Closure of the central strand
We have analyzed the relative closure probabilities of central strands, comparing a 40-mer to a 42-mer. This experiment was a test of the helicity of the DNA when confined within exactly two turns of double helix. Solution studies of DX molecules have variously assumed 10-fold (Li et al., 1996; Yang et al., 1998) or 10.5-fold (Sha et al., 2000b) helicity for DNA when designing these motifs. If we assume that ligation frequency reflects stability, these experiments suggest that DNA DX motifs to be used in solution should approximate the helicity as nearly as possible to 10.5 nucleotide pairs per turn. Our approach, however, does not allow us to determine the helical repeat of the molecules since we do not know their torsional rigidity (see Podtelezhnikov et al., 2000).
Nanotechnology: the generation of catenanes and rotaxanes
A byproduct of these experiments with sealed central strands is a collection of relatively long DNA catenanes. The molecules we have generated are complex catenanes, wherein the cyclic strands are linked to both of the continuous strands. In our heat-induced breakdown used for analysis, we have generated simple cyclic catenanes of DNA. These catenanes could be generated reliably if we included a restriction site on the hairpins of the DX molecule. Likewise, similar restriction of the linear molecules at ambient temperatures would lead to a precursor for a rotaxane, a linear species on which cyclic molecules have been threaded (e.g., Sauvage and Dietrich-Buchecker, 1999); removal of the restricted strands by the techniques of Yurke et al. (2000) would produce a long multiple rotaxane.
Acknowledgments
We thank Dr Lisa Wenzler Savin for guidance and discussions in the early stages of this project.
This work has been supported by grants GM-29554 from the National Institute of General Medical Sciences, N00014-98-1-0093 from the Office of Naval Research, DMI-0210844, EIA-0086015, DMR-01138790, and CTS-0103002 from the National Science Foundation, and F30602-01-2-0561 from the Defense Advanced Research Projects Agency/Air Force Office of Scientific Research to N.C.S.; and by grant GM54215 from the National Institute of General Medical Sciences to A.V.V.
References
- Caruthers, M. H. 1985. Gene synthesis machines: DNA chemistry and its uses. Science. 230:281–285. [DOI] [PubMed] [Google Scholar]
- Fu, T.-J., and N. C. Seeman. 1993. DNA double crossover molecules. Biochemistry. 32:3211–3220. [DOI] [PubMed] [Google Scholar]
- Fu, T.-J., Y.-C. Tse-Dinh, and N. C. Seeman. 1994a. Holliday junction crossover topology. J. Mol. Biol. 236:91–105. [DOI] [PubMed] [Google Scholar]
- Fu, T.-J., B. Kemper, and N. C. Seeman. 1994b. Endonuclease VII cleavage of DNA double crossover molecules. Biochemistry. 33:3896–3905. [DOI] [PubMed] [Google Scholar]
- Hagerman, P. J. 1988. Flexibility of DNA. Annu. Rev. Biophys. Biophys. Chem. 17:265–286. [DOI] [PubMed] [Google Scholar]
- LaBean, T., H. Yan, J. Kopatsch, F. Liu, E. Winfree, J. H. Reif, and N. C. Seeman. 2000. The construction, analysis, ligation and self-assembly of DNA triple crossover complexes. J. Am. Chem. Soc. 122:1848–1860. [Google Scholar]
- Li, X., X. Yang, J. Qi, and N. C. Seeman. 1996. Antiparallel DNA double crossover molecules as components for nanoconstruction. J. Am. Chem. Soc. 118:6131–6140. [Google Scholar]
- Liu, F., R. Sha, and N. C. Seeman. 1999. Modifying the surface features of two-dimensional DNA crystals. J. Am. Chem. Soc. 121:917–922. [Google Scholar]
- Mao, C., T. H. LaBean, J. H. Reif, and N. C. Seeman. 2000. Logical computation using algorithmic self-assembly of DNA triple crossover molecules. Nature. 407:493–496. [DOI] [PubMed] [Google Scholar]
- Mao, C., W. Sun, Z. Shen, and N. C. Seeman. 1999. A DNA nanomechanical device based on the B-Z transition. Nature. 397:144–146. [DOI] [PubMed] [Google Scholar]
- Odom, D. T., E. A. Dill, and J. K. Barton. 2000. Robust charge transport in DNA double crossover assemblies. Chem. Biol. 7:475–481. [DOI] [PubMed] [Google Scholar]
- Podtelezhnikov, A., C. Mao, N. C. Seeman, and A. Vologodskii. 2000. Multimerization-cyclization of DNA fragments as a method of conformational analysis. Biophys. J. 79:2692–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, H., J. C. Dewan, and N. C. Seeman. 1997. A DNA decamer with a sticky end: the crystal structure of d-CGACGATCGT. J. Mol. Biol. 267:881–898. [DOI] [PubMed] [Google Scholar]
- Rhodes, D., and A. Klug. 1980. Helical periodicity of DNA determined by enzyme digestion. Nature. 286:573–578. [DOI] [PubMed] [Google Scholar]
- Sauvage, J.-P., and C. Dietrich-Buchecker. 1999. Molecular Catenanes, Rotaxanes and Knots. Wiley-VCH, Weinheim, Germany.
- Schwacha, A., and N. Kleckner. 1995. Identification of double Holliday junctions as intermediates in meiotic recombination. Cell. 83:783–791. [DOI] [PubMed] [Google Scholar]
- Seeman, N. C. 1982. Nucleic acid junctions and lattices. J. Theor. Biol. 99:237–247. [DOI] [PubMed] [Google Scholar]
- Seeman, N. C. 1990. De novo design of sequences for nucleic acid structure engineering. J. Biomol. Struct. Dyn. 8:573–581. [DOI] [PubMed] [Google Scholar]
- Seeman, N. C. 1999. DNA engineering and its application to nanotechnology. Trends Biotechnol. 17:437–443. [DOI] [PubMed] [Google Scholar]
- Seeman, N. C. 2001. DNA nicks and nodes and nanotechnology. Nano Lett. 1:22–26. [Google Scholar]
- Sha, R., F. Liu, and N. C. Seeman. 2000a. Direct evidence for spontaneous branch migration in antiparallel DNA Holliday junctions. Biochemistry. 39:11514–11522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha, R., H. Iwasaki, F. Liu, H. Shinagawa, and N. C. Seeman. 2000b. Cleavage of symmetric immobile DNA junctions by Escherichia coli RuvC. Biochemistry. 39:11982–11988. [DOI] [PubMed] [Google Scholar]
- Shimada, J., and H. Yamakawa. 1984. Ring-closure probabilities for twisted wormlike chains. Application to DNA. Macromolecules. 17:689–698. [Google Scholar]
- Sun, W., C. Mao, H. Iwasaki, B. Kemper, and N. C. Seeman. 1999. No braiding of Holliday junctions in positively supercoiled DNA molecules. J. Mol. Biol. 294:683–699. [DOI] [PubMed] [Google Scholar]
- Sun, W., C. Mao, F. Liu, and N. C. Seeman. 1998. Sequence dependence of branch migratory minima. J. Mol. Biol. 282:59–70. [DOI] [PubMed] [Google Scholar]
- Sun, H., D. Treco, and J. W. Szostak. 1991. Extensive 3′-overhanging single-stranded DNA associated with the meiosis-specific double-strand breaks. Cell. 64:1155–1161. [DOI] [PubMed] [Google Scholar]
- Thaler, D. S., and F. W. Stahl. 1988. DNA double-chain breaks in recombination of phage λ and of yeast. Annu. Rev. Genet. 22:169–197. [DOI] [PubMed] [Google Scholar]
- Wang, J. C. 1979. Helical repeat of DNA in solution. Proc. Natl. Acad. Sci. USA. 76:200–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winfree, E. 1996. On the computational power of DNA annealing and ligation. In DNA Based Computers. R. J. Lipton and E. B. Baum, editors. American Mathematical Society, Providence, RI. 199–221.
- Winfree, E., F. Liu, L. A. Wenzler, and N. C. Seeman. 1998. Design and self-assembly of two-dimensional DNA crystals. Nature. 394:539–544. [DOI] [PubMed] [Google Scholar]
- Yan, H., X. Zhang, Z. Shen, and N. C. Seeman. 2002. A robust DNA mechanical device controlled by hybridization topology. Nature. 415:62–65. [DOI] [PubMed] [Google Scholar]
- Yang, X., L. A. Wenzler, J. Qi, X. Li, and N. C. Seeman. 1998. Ligation of DNA triangles containing double crossover molecules. J. Am. Chem. Soc. 120:9779–9786. [Google Scholar]
- Yurke, B., A. J. Turberfield, A. P. Mills, Jr., F. C. Simmel, and J. L. Neumann. 2000. A DNA-fueled molecular machine made of DNA. Nature. 406:605–608. [DOI] [PubMed] [Google Scholar]
- Zhang, S., and N. C. Seeman. 1994. Symmetric Holliday junction crossover isomers. J. Mol. Biol. 238:658–668. [DOI] [PubMed] [Google Scholar]