

Abstract
Mechanical forces exerted by laser tweezers or atomic force microscopes can be used to drive rare transitions in single molecules, such as unfolding of a protein or dissociation of a ligand. The phenomenological description of pulling experiments based on Bell's expression for the force-induced rupture rate is found to be inadequate when tested against computer simulations of a simple microscopic model of the dynamics. We introduce a new approach of comparable complexity to extract more accurate kinetic information about the molecular events from pulling experiments. Our procedure is based on the analysis of a simple stochastic model of pulling with a harmonic spring and encompasses the phenomenological approach, reducing to it in the appropriate limit. Our approach is tested against computer simulations of a multimodule titin model with anharmonic linkers and then an illustrative application is made to the forced unfolding of I27 subunits of the protein titin. Our procedure to extract kinetic information from pulling experiments is simple to implement and should prove useful in the analysis of experiments on a variety of systems.
INTRODUCTION
Pulling experiments using atomic-force microscopes (AFMs) or laser tweezers are widely used to probe rare molecular events, such as protein unfolding and ligand dissociation. In such experiments, an anchored molecule or molecular assembly is attached to a pulling spring, possibly via a linker molecule, as shown in Fig. 1 A. The pulling spring is then moved away from the anchored molecule, typically at a constant velocity v, building up mechanical stress in the system. Eventually, this forces a molecular transition, such as the dissociation of a molecular complex (Florin et al., 1994; Merkel et al., 1999), unfolding of a protein (Kellermayer et al., 1997; Marszalek et al., 1999; Rief et al., 1997; Tskhovrebova et al., 1997), or unwrapping of a higher-order structure (Cui and Bustamante, 2000).
FIGURE 1.

(A) Schematic illustration of a pulling experiment. (B) Free energy surface of the model of rupture. (C) Determination of the effective pulling spring constant from force-extension curves for a multimodule protein consisting of independently unfolding domains, such as titin.
In this paper, we consider the problem of extracting kinetic information from such experiments. Specifically, we are interested in obtaining the intrinsic rate of a rare molecular event along the pulling coordinate. Most currently used procedures to analyze pulling experiments (see e.g., Evans and Ritchie, 1997; Evans et al., 1991; Rief et al., 1997, 1998) involve an extension of Bell's expression (Bell, 1978) for the rate coefficient for rupture in the presence of a time-dependent external force F(t):
 |
(1) |
where k0 is the intrinsic rate constant we would like to determine, x‡ is the distance from the free-energy minimum to the barrier, and β−1 = kBT with kB Boltzmann's constant and T the absolute temperature. The attractive feature of these phenomenological procedures is their apparent generality. No assumption is made concerning the nature of the dynamics (it is subsumed into k0), and the underlying free-energy surface is characterized by a single parameter (x‡). However, by analyzing simulated data obtained from arguably the simplest microscopic model of pulling experiments, it will be shown below that the phenomenological description is inadequate in the experimentally relevant pulling regime. If the phenomenological approach, with no adjustable parameters, does not work even in such a simple context, it can hardly be expected to yield reliable intrinsic rate constants when applied to actual experimental data.
The purpose of this paper is to introduce a new procedure for analyzing experimental pulling data that is more reliable than the phenomenological approach but has comparable ease of implementation. Our approach has just one more parameter (i.e., the activation free energy) and is based on a rigorous analysis of a microscopic model that is sufficiently simple to yield analytic results but nevertheless captures the key features of pulling experiments. In this model, the system diffuses on a harmonic free-energy surface with a single sharp barrier and is pulled by a harmonic spring moving at a constant velocity. We obtain analytic expressions for the average force at rupture and the probability distribution of rupture forces. These expressions are generalizations of analogous results obtained within the framework of the phenomenological approach. Based on these results we propose a procedure for analyzing experimental data. After testing it against computer simulations of a many-particle microscopic model of a multimodule protein with anharmonic linkers, an illustrative application is made to the forced unfolding of linked I27 modules of the protein titin (Carrion-Vazquez et al., 1999).
In this article, we have taken the first step in removing the most serious deficiencies of phenomenological descriptions of force-induced rupture that are based on Bell's formula (Bell, 1978) for the rate coefficient in the presence of a time-dependent force. More sophisticated approaches with fewer assumptions can be readily envisaged but are difficult to implement. For example, one could first obtain the free energy profile along the pulling coordinate using the formalism we recently developed (Hummer and Szabo, 2001) based on the seminal work of Jarzynski (1997). If it is assumed that the dynamics on this surface is diffusive, the diffusion coefficient could be obtained by fitting the numerically calculated rupture-force distributions to the experimental ones. Having determined both the diffusion coefficient and the free energy surface, the intrinsic rate could be found using Kramers' theory (Kramers, 1940). Unfortunately, many practical problems must be overcome before such a procedure would become viable.
The outline of this paper is as follows. After reviewing the phenomenological description of force-induced rupture, we introduce our microscopic model and obtain accurate analytic expressions for the rupture-force probability distribution and the average force at rupture. These and the corresponding results obtained within the framework of the phenomenological approach are compared with Brownian dynamics simulations of the model. Our approach is next tested against computer simulations of a multimodule titin model with anharmonic linkers and then applied to experimental data for the unfolding of titin. Finally, we present some concluding remarks.
THEORY
Phenomenological description of the kinetics of rupture
We consider a system that is pulled over a barrier by the application of a time-dependent force F(t). If the free-energy surface has a single well, as shown in Fig. 1 B, this process is irreversible. In this case, the phenomenological formalism based on an approximate expression for the rate constant in the presence of an external force (Bell, 1978) can be reformulated in a simple way as follows. The survival probability S(t) of the system (i.e., the probability that rupture has not yet occurred at time t) is assumed to satisfy the first-order rate equation with a time-dependent rate coefficient
 |
(2) |
and thus
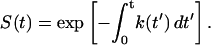 |
(3) |
The time-dependent rate coefficient k(t) is given by Bell's expression (Bell, 1978) generalized to time dependent forces, Eq. 1. The probability distribution of lifetimes t* is  , so that the mean lifetime is
, so that the mean lifetime is  . The probability distribution of forces F at rupture is related to the probability distribution of lifetimes by
. The probability distribution of forces F at rupture is related to the probability distribution of lifetimes by  . This formalism is generalized for multiple covalently linked subunits (such as titin) in Appendix A.
. This formalism is generalized for multiple covalently linked subunits (such as titin) in Appendix A.
Even for time-independent forces, Bell's expression is only valid for diffusive barrier crossing in the limit of small forces (Dembo et al., 1988). This is one of the reasons why the phenomenological approach has a limited range of validity. Another reason is that Bell's expression does not take into account the fact that the molecular coordinate fluctuates under the influence of the combined molecular and pulling potentials. In this article, we test the applicability of the phenomenological procedure for arguably the simplest microscopic model of the kinetics of rupture, and then propose a new procedure for analyzing experimental data that leads to more accurate estimates of the intrinsic rate constant k0. Before doing this, let us give a simple example of the phenomenological formalism when F(t) is a linear function of time: βF(t) = κsvt where v is the velocity of pulling, and κs is a harmonic force constant scaled by kBT = β−1. Using Eqs. 1 and 3, we have
 |
(4) |
The probability distribution of rupture forces  is
is
 |
(5) |
This result was first obtained in the pioneering work of Schulten and co-workers on the theory and simulation of pulling experiments (1997).
The mean force at rupture is
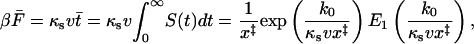 |
(6) |
where 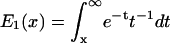 is the exponential integral. This result was obtained previously by Gergely et al. (2000). For low velocities,
is the exponential integral. This result was obtained previously by Gergely et al. (2000). For low velocities, 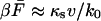 ; for high velocities, we obtain the following relation between the intrinsic rate constant and the average force at rupture
; for high velocities, we obtain the following relation between the intrinsic rate constant and the average force at rupture
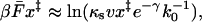 |
(7) |
where γ = 0.5772… is the Euler-Mascheroni constant.
The most probable force at rupture, βFmaxx‡ ≡ ksvtmax, satisfies this relation with γ set to zero, as previously found by Izrailev et al. (1997). Thus, plots of the mean and most probable force versus lnv will have the same slope but different intercepts. tmax is the most probable time at rupture which satisfies 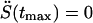 , and by using Eq. 3, it is found to be the solution of the equation
, and by using Eq. 3, it is found to be the solution of the equation 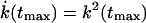 . In the limit where Eq. 7 is valid (i.e., for
. In the limit where Eq. 7 is valid (i.e., for 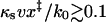 ), the difference between the maximum of the force distribution and the mean force is
), the difference between the maximum of the force distribution and the mean force is 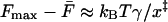 , independent of the pulling velocity. The asymmetry of the rupture-force distribution thus provides direct information about the distance to the barrier, x‡.
, independent of the pulling velocity. The asymmetry of the rupture-force distribution thus provides direct information about the distance to the barrier, x‡.
Equation 7 relates the intrinsic rate constant k0 to a measurable quantity, namely the force-loading rate κsv and the mean force at rupture 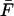 . Specifically, the phenomenological approach predicts that at high pulling speeds the average force grows linearly with the logarithm of the force-loading rate,
. Specifically, the phenomenological approach predicts that at high pulling speeds the average force grows linearly with the logarithm of the force-loading rate, 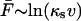 , as found by Evans and Ritchie (1997) and Izrailev et al. (1997). In the next section, we investigate the range of applicability of this expression by comparing it with simulations and the essentially exact solution for a simple microscopic model of rupture.
, as found by Evans and Ritchie (1997) and Izrailev et al. (1997). In the next section, we investigate the range of applicability of this expression by comparing it with simulations and the essentially exact solution for a simple microscopic model of rupture.
Microscopic theory of the kinetics of rupture
We assume that along the pulling coordinate x the potential of mean force is given by
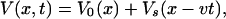 |
(8) |
where V0(x) is the molecular free energy surface of the type shown in Fig. 1 B. The molecular coordinate x is coupled to the pulling apparatus, which is moving at velocity v, through a time-dependent external force, −∂Vs(x − vt)/∂x. We further assume that x happens to be a good reaction coordinate so that the rate of escape along this coordinate in the absence of pulling gives the intrinsic dissociation rate of the system. This is rigorously valid only if the dynamics of all other degrees of freedom turn out to be sufficiently fast.
We assume that the pulling force is harmonic, 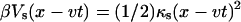 , where κs is an effective force constant (divided by β−1 = kBT) which is determined not only by the cantilever force constant of the pulling apparatus but also by the properties of the molecular linker. We further assume that the molecular free energy surface is given by
, where κs is an effective force constant (divided by β−1 = kBT) which is determined not only by the cantilever force constant of the pulling apparatus but also by the properties of the molecular linker. We further assume that the molecular free energy surface is given by
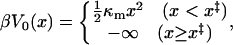 |
(9) |
where κm is the molecular spring constant. Assuming a cusplike barrier (see Fig. 1 B) at x = x‡ is not as limiting as it may appear at first sight. Because of the snapping motion at rupture, experiments contain limited information about the shape of the free energy surface near the transition state. Generalizations to more complex free energy surfaces are straightforward, but do not lead to analytic expressions for observables such as the distribution of forces at rupture.
Finally, we assume that the dynamics is diffusive in nature, i.e., the system undergoes Brownian motion on the free energy surface. Trajectories for our system can be generated by solving a stochastic differential equation, 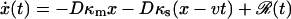 , where D is the diffusion coefficient, and ℛ(t) is a Gaussian random force with zero mean,
, where D is the diffusion coefficient, and ℛ(t) is a Gaussian random force with zero mean, 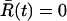 , and variance
, and variance 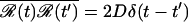 . In practice, this means that after a short time step Δt, the position is given by
. In practice, this means that after a short time step Δt, the position is given by 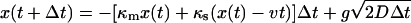 where g is a Gaussian random number with zero mean and unit variance (i.e., picked from the distribution
where g is a Gaussian random number with zero mean and unit variance (i.e., picked from the distribution 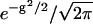 ). Equivalently, the probability distribution of finding the system at x at time t satisfies the Smoluchowski equation (Zwanzig, 2001).
). Equivalently, the probability distribution of finding the system at x at time t satisfies the Smoluchowski equation (Zwanzig, 2001).
The rate of rupture in the absence of pulling can be obtained from Kramers' theory (Kramers, 1940). For sufficiently high barriers, the kinetics is exponential with a rate constant given by
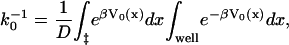 |
(10) |
where the first integral is over the barrier region, and the second integral is over the free-energy minimum. Applying this to our potential we find
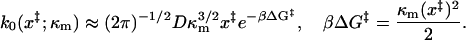 |
(11) |
Our goal is to extract this rate constant from pulling experiments. To do this we must calculate the statistics of the force at rupture
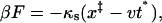 |
(12) |
where t* is the lifetime of our system defined as follows. The system diffuses on the time-dependent potential 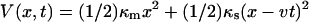 for x < x‡, and V(x, t) = −∞ for x ≥ x‡. At t = 0 we start from an equilibrium distribution for x < x‡ and are interested in the survival probability of the system S(t) in the region x < x‡. The distribution of lifetimes t* (and therefore the forces at rupture) is then given by
for x < x‡, and V(x, t) = −∞ for x ≥ x‡. At t = 0 we start from an equilibrium distribution for x < x‡ and are interested in the survival probability of the system S(t) in the region x < x‡. The distribution of lifetimes t* (and therefore the forces at rupture) is then given by  . Although there is no exact analytic solution to this simple problem, we shall now obtain an accurate solution for the entire range of parameters.
. Although there is no exact analytic solution to this simple problem, we shall now obtain an accurate solution for the entire range of parameters.
Before rupture, the pulling force will fluctuate about 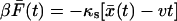 where
where 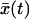 is the average position at time t. For our model,
is the average position at time t. For our model, 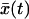 satisfies an ordinary differential equation since
satisfies an ordinary differential equation since 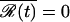 . For the initial condition
. For the initial condition 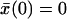 , we find
, we find
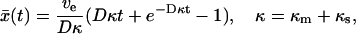 |
(13) |
with ve = vκs/κ. At short times (Dκt ≪ 1), 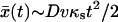 ; at long times,
; at long times, 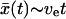 , so the molecular coordinate moves with a constant effective velocity ve.
, so the molecular coordinate moves with a constant effective velocity ve.
When the pulling velocity is sufficiently fast, the fluctuations about 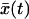 are irrelevant and the survival probability is simply given by S(t) = 1 for t < τ and zero otherwise, where τ is the time for
are irrelevant and the survival probability is simply given by S(t) = 1 for t < τ and zero otherwise, where τ is the time for 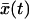 to reach x‡,
to reach x‡, 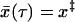 . Approximately solving
. Approximately solving 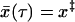 for τ, we find
for τ, we find
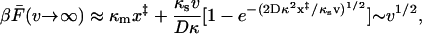 |
(14) |
so that the average force is proportional to the square root of the velocity for large velocities. This result is not general and would be affected by the inclusion of a linker. Moreover, as the pulling velocity increases, motions along degrees of freedom other than the pulling direction are given less time to relax, ultimately requiring inclusion of additional coordinates in the model.
If the pulling velocity is sufficiently slow so that the system ruptures when the activation energy is still large, then we can use an adiabatic approximation analogous to what is done in quantum mechanics. If we make the substitution 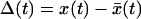 , our problem reduces to the dynamics of a harmonic oscillator with effective force constant κ = κs + κm in the presence of a time-dependent barrier at
, our problem reduces to the dynamics of a harmonic oscillator with effective force constant κ = κs + κm in the presence of a time-dependent barrier at 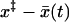 . If the motion is slow, the survival probability is given by
. If the motion is slow, the survival probability is given by
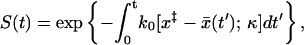 |
(15) |
where k0(x‡; κ) is the intrinsic rate constant for a fixed barrier at x‡ given in Eq. 11, with κm replaced by κ = κm + κs, the sum of pulling and molecular spring constants. If we use the long time limit 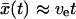 , Eq. 15 becomes:
, Eq. 15 becomes:
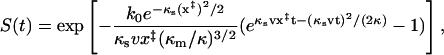 |
(16) |
where we used Eq. 11 for the intrinsic rate constant k0.
The probability distribution of forces 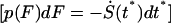 obtained analytically from Eqs. 12 and 16 is
obtained analytically from Eqs. 12 and 16 is
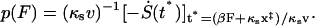 |
(17) |
The cumulative probability distribution of rupture forces is given by the survival probability,
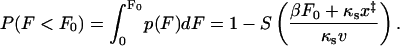 |
(18) |
To find an expression for S(t) valid over the entire velocity range, the simplest procedure (which turns out to be accurate when compared to computer simulations) is to use Eq. 16 only for times t < τ before the average trajectory reaches the barrier, 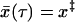 , and put S(t) = 0 for t > τ. Consequently, the average force at rupture is given by
, and put S(t) = 0 for t > τ. Consequently, the average force at rupture is given by
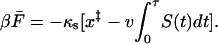 |
(19) |
The integral in Eq. 19 can be evaluated for very small velocities to give
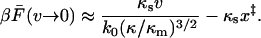 |
(20) |
If the molecular spring is stiff compared to the pulling spring, κm ≫ κs, the average force at rupture is zero approximately at v = k0x‡.
For intermediate velocities (i.e., before the v1/2 limit is reached) we find
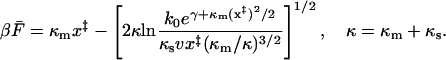 |
(21) |
This and Eq. 16 are the key relations used to analyze experimental data. These are generalizations of the corresponding phenomenological results given in Eqs. 4 and 7, to which they reduce in the limit of 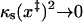 and
and 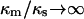 . Thus the phenomenological approach is a somewhat unphysical special case of ours.
. Thus the phenomenological approach is a somewhat unphysical special case of ours.
In summary, our theory predicts three pulling regimes: 1), In the limit of slow pulling speeds (v ≪ k0x‡), rupture is slowed down because the pulling spring holds back the molecular coordinate (Heymann and Grubmüller, 2000), and the mean force at rupture is negative, 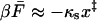 [see Eq. 20]. 2), In the limit of fast pulling (
[see Eq. 20]. 2), In the limit of fast pulling (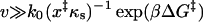 with βΔG‡ = κm(x‡)2/2 the height of the free energy barrier), stochastic motion is irrelevant, and the average force at rupture grows asymptotically as v1/2 with the pulling velocity (see Eq. 14), as seen in the simulations of Evans and Ritchie (1997). 3), For intermediate pulling speeds that are typical for most experiments
with βΔG‡ = κm(x‡)2/2 the height of the free energy barrier), stochastic motion is irrelevant, and the average force at rupture grows asymptotically as v1/2 with the pulling velocity (see Eq. 14), as seen in the simulations of Evans and Ritchie (1997). 3), For intermediate pulling speeds that are typical for most experiments 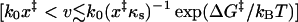 , both stochastic fluctuations and pulling are relevant. In this regime, the mean force at rupture is given by Eq. 21 and is a nonlinear function of the logarithm of the force-loading rate κsv. These regimes are related but not identical to the activation, drift, and diffusion regimes first discussed by Izrailev et al. (1997).
, both stochastic fluctuations and pulling are relevant. In this regime, the mean force at rupture is given by Eq. 21 and is a nonlinear function of the logarithm of the force-loading rate κsv. These regimes are related but not identical to the activation, drift, and diffusion regimes first discussed by Izrailev et al. (1997).
In typical experiments, the probe molecule and force measurement apparatus are connected by a linker molecule, affecting the behavior of the system under pulling (Evans and Ritchie, 1999). If the linker relaxes rapidly compared to rupturing and its force-extension curve is sufficiently harmonic then the microscopic theory is applicable when the pulling spring constant κs is replaced by an effective spring constant describing the softened combination of pulling spring and linker. If the spring describing the molecular process of interest is stiff compared to the combination of pulling and linker spring, κm ≫ κs, then the effective spring constant is given simply by the slope of the average force 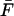 with respect to the extension x = vt before rupture, as illustrated in Fig. 1 C, and discussed in more detail in Appendix B.
with respect to the extension x = vt before rupture, as illustrated in Fig. 1 C, and discussed in more detail in Appendix B.
The above theory describes the irreversible rupture of a single subunit. Titin, however, consists of multiple folded modules connected by short linker peptides. In pulling experiments on natural and synthetic titin constructs, the modules are sequentially unfolded. If these unfolding events are uncorrelated, we can immediately apply the formalism of this section by simply multiplying k0 by the appropriate statistical factor. For a construct containing N subunits, the rate of unfolding of the first subunit in such construct is N times faster than it would be if the construct only contained a single subunit, and so on (Evans and Ritchie, 1999; Makarov et al., 2001; Zhang et al., 1999). Therefore, our theory can be applied to the i-th unfolding event by simply replacing k0 by (N − i + 1)k0 in Eqs. 16 and 21.
This implies that the average time between rupture events is shorter for a longer titin construct (Makarov et al., 2001; Zhang et al., 1999), resulting in smaller measured rupture forces (Evans and Ritchie, 1999) for the long titin constructs in laser-tweezer pulling experiments (Kellermayer et al., 1997) compared to shorter constructs in AFM experiments (Carrion-Vazquez et al., 1999). In addition, every unfolding event leads to softening of the effective spring constant. This reduces the average rupture force, as previously discussed by Evans and Ritchie (1999).
RESULTS AND DISCUSSION
Microscopic model
As a first illustration, we test the phenomenological formalism and the microscopic theory by comparing it with exact results obtained from Brownian dynamics simulations of a model system over a broad range of pulling velocities v. We choose κs = 1, κm = 10, D = 1, β = 1, and x‡ = 1, corresponding to a 5kBT barrier to rupture. As initial condition we use an equilibrium distribution on the interval −∞ < x < x‡. Initially, the time step is set to Δt = 10−4, but as the barrier is approached, Δt is reduced linearly in x‡ − x(t) to 10−6 (Pastor et al., 1996).
Fig. 2 shows the force at rupture as a function of the pulling speed. Qualitatively, we find the three pulling regimes predicted by our theory. In an intermediate range of pulling velocities (10−1 < v < 10), in which experiments will typically be conducted, the average force depends approximately linearly on the logarithm of the pulling velocity. Below, the average force at rupture becomes negative and linearly dependent on the pulling speed. In the deterministic limit above, the force at rupture grows as v1/2. Quantitatively, we find that the mean force estimated from Eq. 19 is in good agreement with the simulation data over the whole range of pulling velocities.
FIGURE 2.

Average force at rupture as a function of pulling velocity from simulation and theory. Symbols show reference results from Brownian dynamics simulations for κs = 1, κm = 10, D = 1, β = 1, and x‡ = 1, with statistical errors smaller than the symbol size. The solid line is obtained by integrating the approximate survival time distribution, Eq. 19. The long-dashed line shows the estimate of the mean force from the analytic approximation to the microscopic theory, Eq. 21. The short-dashed line is the phenomenological result for the average force at rupture, Eq. 7. A vertical arrow indicates a pulling speed of k0x‡ at the crossover from negative to positive rupture forces.
The phenomenological result based on an explicitly time-dependent force is in good agreement with the simulation data only for relatively small pulling speeds, 0.1 < v < 1. The analytic approximation to the microscopic theory, Eq. 21, covers a broader pulling regime (0.1 < v < 10). However, unlike the full microscopic theory, Eq. 19, it does not reproduce the rupture forces in the deterministic limit at the highest pulling speeds (v > 100) and the transitions to negative rupture forces at the lowest pulling speeds (v < 10−2). As expected from the theory, the rupture force curve crosses zero at a pulling velocity v ≈ k0x‡.
Pulling of a multimodule titin model with anharmonic linkers
The first step in our procedure to analyze experimental pulling data is to extract effective spring constants κs from the slope of force-extension curves just before rupture. The remaining model parameters (κm, x‡, and k0) are then fitted to the data for rupture forces as a function of pulling speed. This approach is correct when both the molecular coordinate and the pulling apparatus (including the linker) respond linearly to force. In many practical applications, in particular to biopolymers, this assumption may appear to be too restrictive. Here, we show that our procedure works well even for anharmonic linkers described by wormlike-chain models, and for systems with multiple sequentially rupturing subunits or bonds.
To test the applicability of the theory, we mimic unfolding of the protein titin by simulating an anharmonic model and comparing the results to the theory, Eq. 21. In this titin model, N = 10 independently unfolding modules are connected by linkers. In the folded state, the molecular coordinates move on a harmonic potential. Upon unfolding, each unit converts to a peptide coil described by an approximate wormlike-chain potential (Marko and Siggia, 1995). Details are given in Appendix C.
A simulated pulling trace is shown in Fig. 3. The force-extension curves are now nonlinear with 10 sharp peaks, each corresponding to rupturing of a module. From a straight-line fit to the force-extension curves just before each rupture event i, as illustrated in Fig. 1 C, we extract an effective force constant κs(v, i) at pulling velocity v using Eq. B3. The velocity-dependent κs(v, i) are obtained by averaging the slopes independently at each pulling velocity for the first (i = 1), second (i = 2), etc., rupture event. Our previous theory for N = 1 can be extended to analyze the i-th rupture event by using an intrinsic rate constant (N − i + 1)k0 instead of k0. To test whether our procedure for determining the effective force constant is applicable to this multimodule system with anharmonic linkers, we compare in Fig. 4 the observed forces at rupture to those calculated from Eq. 21. We find good agreement of the average forces from simulation and theory. In particular, the theory correctly predicts that the average force increases with the order of the unfolding event. However, that increase in the rupture force is significant only for the last four to five rupture events. We conclude from this that the simple microscopic theory, Eqs. 19 and 21, is applicable even for multimodule systems with anharmonic linkers if effective spring constants are used, and the appropriate statistical factor is used to multiply k0.
FIGURE 3.

(Top) Schematic representation of a titin model with N = 5 modules. Jagged lines indicate harmonic linkers connecting the C and N termini. Smooth lines represent the folded (units 1, 2, 3, and 5) and unfolded titin modules (unit 4), and a linker molecule connecting the titin construct to the AFM spring. In the folded state, the modules respond harmonically with a tight spring constant κm; in the unfolded state, they respond as wormlike chains. (Bottom) Force-extension curve from a pulling simulation at a pulling speed of 0.1 μm s−1 for a model with N = 10 modules. Peaks correspond to unfolding of single titin molecules.
FIGURE 4.

Force at rupture as a function of pulling velocity for a construct of 10 independently unfolding modules. The construct is connected to the pulling spring via a wormlike-chain linker. After unfolding, each module converts irreversibly into a wormlike-chain polymer. Forces are shown for the first, fifth, and tenth rupture event (symbols), and compared to the theory (lines), Eq. 21, modified for the multimodule structure of the system. Error bars correspond to one estimated standard deviation. The effective pulling spring constants in Eq. 21 were determined from straight-line fits to the force-extension curves before rupture, and averaged independently for each pulling velocity and the first, second, etc., rupture event. The inset shows a scatter plot of average forces from simulation and Eq. 21 for all velocities and rank order of rupture events.
Ideally, experiments should be analyzed by fitting the average force (and, even better, the distribution of forces) for the i-th rupture event using our theory with 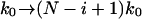 . However, with limited data, it is necessary to average rupture forces over all sequential events. For a model system with 10 titin modules with an average effective spring constant of kBTκs = 5.3 pN/nm, we fit the microscopic theory Eq. 21 to the average force as a function of pulling velocity, and obtain an apparent rate
. However, with limited data, it is necessary to average rupture forces over all sequential events. For a model system with 10 titin modules with an average effective spring constant of kBTκs = 5.3 pN/nm, we fit the microscopic theory Eq. 21 to the average force as a function of pulling velocity, and obtain an apparent rate 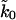 that is about ten times faster than the actual rate k0. In general, we expect for the apparent rate that
that is about ten times faster than the actual rate k0. In general, we expect for the apparent rate that 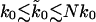 for a system with N identical and independently rupturing subunits.
for a system with N identical and independently rupturing subunits.
Analyzing an experiment: Unfolding of titin
A classic application of single-molecule force pulling experiments is the mechanical unfolding of proteins. One of the most widely studied proteins is titin (Kellermayer et al., 1997; Marszalek et al., 1999; Rief et al., 1997; Tskhovrebova et al., 1997). Carrion-Vazquez et al. (1999) recently reported measurements of a recombinant construct of 8 tethered I27 titin molecules, each with 89 amino acids. We use the titin-unfolding data of Figs. 2 and 3 of (Carrion-Vazquez et al., 1999) to illustrate the utility of the phenomenological approach and the microscopic theory. Because we have access to only limited data, we will average over all eight events assuming that the force-induced unfolding of multiple covalently linked proteins can be treated as the unfolding of a single protein with an average effective pulling spring constant. The fitted apparent rate constant 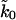 should then be roughly between one and eight times higher than the intrinsic rate constant of a single titin, k0, if the pulling coordinate were an adequate reaction coordinate.
should then be roughly between one and eight times higher than the intrinsic rate constant of a single titin, k0, if the pulling coordinate were an adequate reaction coordinate.
We first need to determine the effective spring constant κs of the linker and AFM system. As pointed out above, and shown in Appendix B, κs is softer than the spring constant of the AFM spring alone. We obtain the effective spring constant κs from the average of the slopes of the pulling force-extension curves before rupture. From Fig. 2 B of Carrion-Vazquez et al. (1999), we estimate an effective spring constant of kBTκs = 10 pN/nm, used here at all pulling velocities. The remaining unknown parameters are the molecular spring constant, κm, the position of the transition state, x‡, and the apparent rate of titin unfolding, 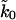 . Fitting of the phenomenological result, Eq. 7, to the force-versus-velocity data extracted from Fig. 3 of Carrion-Vazquez et al. (1999) results in x‡ = 0.18 nm, and
. Fitting of the phenomenological result, Eq. 7, to the force-versus-velocity data extracted from Fig. 3 of Carrion-Vazquez et al. (1999) results in x‡ = 0.18 nm, and 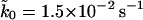 . This rate is larger than the rate of 5 × 10−4 s−1 from chemical-denaturation experiments (Carrion-Vazquez et al., 1999) even after the maximum correction for the number of modules is made.
. This rate is larger than the rate of 5 × 10−4 s−1 from chemical-denaturation experiments (Carrion-Vazquez et al., 1999) even after the maximum correction for the number of modules is made.
Fitting of the analytic approximation to the microscopic theory, Eq. 21, optimized by Eq. 19, gives kBTκm = 900 pN/nm, 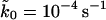 , and x‡ = 0.42 nm, with κm, ln
, and x‡ = 0.42 nm, with κm, ln 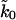 , and the height of the free energy barrier to rupture, ΔG‡ = kBTκm(x‡)2/2, used as fitting parameters. These parameters reproduce not only the experimental data (Carrion-Vazquez et al., 1999) for the average force as a function of pulling speed (Fig. 5), but also the shape of the force distribution not used in the fit (Fig. 6).
, and the height of the free energy barrier to rupture, ΔG‡ = kBTκm(x‡)2/2, used as fitting parameters. These parameters reproduce not only the experimental data (Carrion-Vazquez et al., 1999) for the average force as a function of pulling speed (Fig. 5), but also the shape of the force distribution not used in the fit (Fig. 6).
FIGURE 5.

Average force at unfolding as a function of pulling velocity for an I27 titin construct. Filled circles with error bars show experimental data from Fig. 3 of Carrion-Vazquez et al. (1999). Open squares show results from Brownian dynamics simulations, with statistical errors smaller than the symbol size. The solid line is the result of the microscopic theory obtained by integrating the approximate survival time distribution, Eq. 19. The long-dashed line shows the mean force calculated from the analytic approximation, Eq. 21. The short-dashed line is the phenomenological result for the average force at rupture, Eq. 7. The vertical arrow indicates a pulling speed of k0x‡ where the average force at rupture is approximately zero.
FIGURE 6.

Distribution of forces at rupture (x = x‡) for an I27 titin construct for a pulling velocity of 0.6 μm s−1. Experimental data extracted from Fig. 2 C of Carrion-Vazquez et al. (1999) are shown as blocks. The thick solid line is the analytic expression given in Eq. 17.
Scaling by the number of titin modules gives an intrinsic rate of k0 between 10−5 and 10−4 s−1. This range of corrected rates is below the experimental rate constant of 5 × 10−4 s−1. However, since we are trying to determine three parameters from the limited data in Fig. 5, it is to be expected that there are strong correlations between the fitted parameters. If we fix 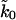 at 8 × 5 × 10−4 s−1, we obtain a reasonable fit to the average forces. Thus, all we can conclude from the data used so far is that the experimental pulling data are roughly consistent with the chemical rate constant.
at 8 × 5 × 10−4 s−1, we obtain a reasonable fit to the average forces. Thus, all we can conclude from the data used so far is that the experimental pulling data are roughly consistent with the chemical rate constant.
To extract a more accurate k0 from pulling measurements one should use not only the mean but the whole distribution of rupture forces (or at least the variance) at each unfolding step as a function of pulling velocity. This entire data set should be fitted globally using our analytic expression Eq. 18 with k0 replaced by (N − i + 1)k0 for the i-th unfolding event. This procedure should yield an accurate estimate of k0 which can then be compared with the result from chemical denaturation. Agreement within experimental error is a necessary but not sufficient condition for the pulling coordinate to be a good reaction coordinate for unfolding.
To show the need to use more information about the probability distribution of rupture forces than just the mean, we analyze the χ2 contour surfaces of the fit. Fig. 7 reveals strong correlations between the fitting parameters, and large uncertainties in the fit. Similar conclusions were reached from fitting of the phenomenological theory to unfolding data for titin I27 mutants (Best et al., 2002; Carrion-Vazquez et al., 1999). In Fig. 7, χF2 is defined as 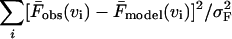 where
where 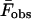 and
and 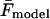 are the measured and calculated average forces. If we assume uniform statistical errors of σF = 10 pN in the measured forces (∼5% relative error), we find that rates as small as
are the measured and calculated average forces. If we assume uniform statistical errors of σF = 10 pN in the measured forces (∼5% relative error), we find that rates as small as 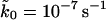 and as large as 10−1 s−1 are consistent with a root-mean-square deviation of 1.2 times the assumed error of 10 pN. The range of parameter values consistent with the pulling data greatly shrinks if we also use measured standard deviations in the forces, extracted from the error bars in Fig. 3 of Carrion-Vazquez et al. (1999). We define
and as large as 10−1 s−1 are consistent with a root-mean-square deviation of 1.2 times the assumed error of 10 pN. The range of parameter values consistent with the pulling data greatly shrinks if we also use measured standard deviations in the forces, extracted from the error bars in Fig. 3 of Carrion-Vazquez et al. (1999). We define 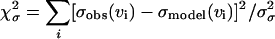 , where σobs and σmodel are the measured and calculated standard deviations. If we assume statistical errors of the standard deviations of σσ = 3 pN (∼10% relative error), we find a small range of parameters consistent with root-mean-square deviations below 1.2 times the assumed errors simultaneously for both means and variances. With that tolerance, we estimate 10−5 s−1≤
, where σobs and σmodel are the measured and calculated standard deviations. If we assume statistical errors of the standard deviations of σσ = 3 pN (∼10% relative error), we find a small range of parameters consistent with root-mean-square deviations below 1.2 times the assumed errors simultaneously for both means and variances. With that tolerance, we estimate 10−5 s−1≤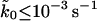 , 12 kcal mol−1 ≤ ΔG‡ ≤ 16 kcal mol−1, and 800 pN nm−1 ≤ kBTκm ≤ 2200 pN nm−1, corresponding to 0.3 nm ≤ x‡ ≤ 0.5 nm. We emphasize that these parameter ranges are based on assumed statistical errors and provide only gross estimates of the uncertainties. We believe that if experimental distributions of rupture forces were to be analyzed separately for each unfolding event, as proposed above, these uncertainties would be substantially reduced.
, 12 kcal mol−1 ≤ ΔG‡ ≤ 16 kcal mol−1, and 800 pN nm−1 ≤ kBTκm ≤ 2200 pN nm−1, corresponding to 0.3 nm ≤ x‡ ≤ 0.5 nm. We emphasize that these parameter ranges are based on assumed statistical errors and provide only gross estimates of the uncertainties. We believe that if experimental distributions of rupture forces were to be analyzed separately for each unfolding event, as proposed above, these uncertainties would be substantially reduced.
FIGURE 7.

Fit of the microscopic theory Eq. 19 to experimental titin pulling data (Carrion-Vazquez et al., 1999). Shown are contour surfaces of χ2 defined as the maximum of independently calculated χF2 and χσ2 for the mean and the standard deviation of the forces, respectively. To determine the maximum range of parameters consistent with a given χ2, the minimum χ2 is shown in projection onto planes of parameters ΔG‡, κm, and apparent rate 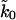 . The vertical shaded area indicates the range of rates
. The vertical shaded area indicates the range of rates 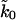 consistent with the chemical-denaturation experiments (Carrion-Vazquez et al., 1999), with the upper and lower limit given by the uncorrected rate, and the rate multiplied by N = 8 to account for the multimodule structure. The thin vertical solid line gives the
consistent with the chemical-denaturation experiments (Carrion-Vazquez et al., 1999), with the upper and lower limit given by the uncorrected rate, and the rate multiplied by N = 8 to account for the multimodule structure. The thin vertical solid line gives the 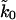 value from a fit of the mean forces using the phenomenological approach. The shaded area in the contour plots outlines the parameter range in which χ2 < 10 simultaneously for average forces and standard deviations, corresponding to a mean-square deviation of less than ∼1.5 times the expected variance. The thick solid line shows the corresponding contour line if only average forces are used.
value from a fit of the mean forces using the phenomenological approach. The shaded area in the contour plots outlines the parameter range in which χ2 < 10 simultaneously for average forces and standard deviations, corresponding to a mean-square deviation of less than ∼1.5 times the expected variance. The thick solid line shows the corresponding contour line if only average forces are used.
With the fitted values of kBTκm = 900 pN nm−1, 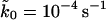 , and x‡ = 0.42 nm, we perform Brownian dynamics simulations of the model in Eqs. 8 and 9. Fig. 5 compares the average force at rupture from experiment (Carrion-Vazquez et al., 1999) and simulation with the phenomenological result, Eq. 7, the full microscopic theory, Eq. 19, and the analytical approximation to the microscopic theory, Eq. 21. The simple harmonic model of force-driven unfolding, Eqs. 8 and 9, is sufficient to reproduce the experimentally measured average forces at unfolding. Both the full microscopic theory, Eq. 19, and its analytic approximation, Eq. 21, are in agreement with the experimental and simulation data for the average forces for pulling velocities between 0.01 and 100 μm s−1. The phenomenological result, Eq. 7, with the parameters of the microscopic model, is seen to be unsatisfactory. Of course, since the available data are almost linear with ln v, one could force fit the data with Eq. 7 but then the resulting parameters would not be meaningful.
, and x‡ = 0.42 nm, we perform Brownian dynamics simulations of the model in Eqs. 8 and 9. Fig. 5 compares the average force at rupture from experiment (Carrion-Vazquez et al., 1999) and simulation with the phenomenological result, Eq. 7, the full microscopic theory, Eq. 19, and the analytical approximation to the microscopic theory, Eq. 21. The simple harmonic model of force-driven unfolding, Eqs. 8 and 9, is sufficient to reproduce the experimentally measured average forces at unfolding. Both the full microscopic theory, Eq. 19, and its analytic approximation, Eq. 21, are in agreement with the experimental and simulation data for the average forces for pulling velocities between 0.01 and 100 μm s−1. The phenomenological result, Eq. 7, with the parameters of the microscopic model, is seen to be unsatisfactory. Of course, since the available data are almost linear with ln v, one could force fit the data with Eq. 7 but then the resulting parameters would not be meaningful.
The estimated free energy barrier to unfolding of about ΔG‡ = 12–16 kcal/mol seems not unreasonable for proteins of ∼100 amino acids. The measured stability of the I27 titin module is ΔG = 7.5 ± 0.3 kcal/mol (Carrion-Vazquez et al., 1999). Within a two-state approximation, we can thus estimate a free energy barrier to folding as ΔGf‡ = ΔG‡ − ΔG of 4–9 kcal/mol. These bounds should tighten considerably when more extensive data are reanalyzed without averaging over all rupture events, as discussed above. It would be of great interest to compare the resulting free energy of activation with that found from the entire free-energy surface, as determined from pulling experiments using our procedure (Hummer and Szabo, 2001) based on a generalization of Jarzynski's theorem (Jarzynski, 1997). Finally, it is interesting that the above range for the height of the free energy barrier to folding is consistent with that recently determined from single-molecule fluorescence measurements on a slightly smaller protein (Schuler et al., 2002).
At the highest pulling speeds (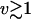 μm s−1), the experimental rupture forces of (Carrion-Vazquez et al., 1999) show an upturn that indicates a transition to the deterministic limit (Fig. 5). However, additional data at even faster speeds would be needed to test the power-law dependence. In another class of experiments, Evans et al. (2001) mechanically probed carbohydrate-selectin bonds. These investigators varied the force-loading rate κsv over four orders of magnitude, and identified two regimes for the dependence of the rupture forces on the loading rate. Interestingly, if the force-versus-loading rate data extracted from Fig. 3 A of Evans et al. (2001) are plotted double-logarithmically, the second regime shows a power-law dependence with exponent 1/2, as predicted here. Although this would explain the transition to larger rupture forces without invoking a second transition state (Evans et al., 2001; Merkel et al., 1999), a more detailed analysis will be needed to address this question conclusively. Such an analysis should include the possibility that at high pulling velocities motions along coordinates other than the pulling direction may dominate rupture.
μm s−1), the experimental rupture forces of (Carrion-Vazquez et al., 1999) show an upturn that indicates a transition to the deterministic limit (Fig. 5). However, additional data at even faster speeds would be needed to test the power-law dependence. In another class of experiments, Evans et al. (2001) mechanically probed carbohydrate-selectin bonds. These investigators varied the force-loading rate κsv over four orders of magnitude, and identified two regimes for the dependence of the rupture forces on the loading rate. Interestingly, if the force-versus-loading rate data extracted from Fig. 3 A of Evans et al. (2001) are plotted double-logarithmically, the second regime shows a power-law dependence with exponent 1/2, as predicted here. Although this would explain the transition to larger rupture forces without invoking a second transition state (Evans et al., 2001; Merkel et al., 1999), a more detailed analysis will be needed to address this question conclusively. Such an analysis should include the possibility that at high pulling velocities motions along coordinates other than the pulling direction may dominate rupture.
CONCLUSIONS
We have developed and tested a new procedure to extract the kinetics of rare molecular events from force-induced mechanical transitions in single molecules, as measured by AFMs, laser tweezers, etc. Our procedure is based on an accurate analytic theory of a simple microscopic model of rupture that includes stochastic fluctuations in the force. It was shown to reproduce the results of simulations without using adjustable parameters. This model predicts that the average force at rupture is a nonlinear function of the logarithm of the pulling velocity. Thus, the experimentally observed upturn in the force-velocity curves at high pulling speeds does not necessarily imply the existence of multiple barriers (Evans et al., 2001; Merkel et al., 1999).
A limiting case of our formalism is the phenomenological description of rupture (Evans and Ritchie, 1997; Evans et al., 1991; Rief et al., 1997, 1998) that is based on an extension of Bell's approximate expression for the rate constant (Bell, 1978) to an explicitly time-dependent force. By comparing its predictions to the results obtained from our microscopic model, we found that if no adjustable parameters are used, the phenomenological formalism works only for pulling velocities well below the experimentally relevant regime.
It is appropriate to summarize here our optimum procedure to analyze pulling experiments using our analytic results. Our formalism involves three adjustable parameters: 1), the intrinsic rate constant (k0) in the absence of external forces; 2), the position of the transition state (x‡); and 3), the height of the free energy barrier (ΔG‡) or, equivalently, the molecular spring constant (κm) that describes the curvature of the free energy surface of the intact state (before rupture) along the pulling coordinate. The remaining parameter is the effective spring constant (κs). It is smaller than the spring constant of the pulling apparatus alone because of softening due to the presence of linker molecules. κs is determined for each pulling velocity by averaging the slope of the force extension curves just before rupture. The three adjustable parameters are then globally determined by fitting the cumulative distributions of rupture forces for all velocities using the analytic expression given in Eq. 18. Estimates for these parameters can be obtained by first fitting the average forces vs. velocity using Eq. 21. If the system consists of N independent units, as for example titin, the above procedure is applied separately to the i-th (i = 1, …, N) rupture event with 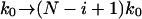 .
.
Because we used only the published data for the mechanical unfolding of an eight-module titin construct (Carrion-Vazquez et al., 1999), we could not illustrate our procedure fully. Specifically, we averaged the data over all rupture events at a given velocity and used our formalism to extract a wide range of apparent intrinsic rate constants 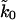 for unfolding that are consistent with AFM pulling experiments. This preliminary analysis showed that the experimental unfolding rate from chemical denaturation (Carrion-Vazquez et al., 1999) falls just above the range of rates we extracted from the pulling experiments, suggesting that for titin the pulling coordinate may indeed be a reasonable reaction coordinate. The question whether the extrapolated and bulk rates are the same, however, is still open and its resolution requires a global fit to the force distribution data. If the pulling coordinate turns out not to be an adequate reaction coordinate, our procedure can still be used to estimate the free energy barrier ΔG‡ along the pulling coordinate, which can be compared with that obtained from molecular dynamics simulations (Balsera et al., 1997; Isralewitz et al., 2001; Lu and Schulten, 1999).
for unfolding that are consistent with AFM pulling experiments. This preliminary analysis showed that the experimental unfolding rate from chemical denaturation (Carrion-Vazquez et al., 1999) falls just above the range of rates we extracted from the pulling experiments, suggesting that for titin the pulling coordinate may indeed be a reasonable reaction coordinate. The question whether the extrapolated and bulk rates are the same, however, is still open and its resolution requires a global fit to the force distribution data. If the pulling coordinate turns out not to be an adequate reaction coordinate, our procedure can still be used to estimate the free energy barrier ΔG‡ along the pulling coordinate, which can be compared with that obtained from molecular dynamics simulations (Balsera et al., 1997; Isralewitz et al., 2001; Lu and Schulten, 1999).
Assessing the adequacy of the simple potential underlying our microscopic model of force-induced rupture is important. This can be accomplished, for instance, by determining the free energy profile along the pulling coordinate (Hummer and Szabo, 2001), or by detailed molecular dynamics simulations (Marszalek et al., 1999). For I27 titin, both molecular dynamics simulations and a small hump in measured force extension curves suggest that force-induced unfolding proceeds in two stages (Marszalek et al., 1999). Recent experiments (Fowler et al., 2002) provide further evidence for an unfolding intermediate in pulling of I27. The intrinsic unfolding rate of a mutant protein mimicking the intermediate was measured as 6.9 × 10−3 s−1 (Fowler et al., 2002), ∼10 times faster than I27 wildtype. Multiple intermediates were suggested to occur in force-induced unfolding of fibronectin III (Gao et al., 2002). If rupturing occurs through a sequence of well-resolved intermediates, our formalism could be applied separately to estimate rates, free energy barriers, and transition-state locations for each of the sequential events.
Acknowledgments
We thank Dr. Jim Hofrichter for insightful discussions.
APPENDIX A: PHENOMENOLOGICAL FORMALISM FOR MULTIPLE COVALENTLY LINKED UNITS
The protein titin consists of multiple independently folded modules covalently connected by short linker peptides. In pulling experiments on natural and synthetic titin constructs, the titin units are sequentially unfolded by mechanical force (Kellermayer et al., 1997; Marszalek et al., 1999; Rief et al., 1997; Tskhovrebova et al., 1997). As the mechanical tension increases, individual titin molecules unfold. With every new unfolding event, the effective linker length grows, transiently releasing tension on the remaining folded proteins. If we assume that unfolding events of sequentially arranged molecules are uncorrelated and irreversible, then we can formulate the phenomenological approach in terms of the first-order kinetics scheme:
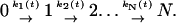 |
(A1) |
State j contains j unfolded modules out of a total of N modules. A similar, reversible kinetic scheme was considered by Evans (2001). Starting from the all-folded state, P0(0) = 1, the relative population of states with a given number of unfolded modules satisfy 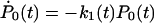 ,
, 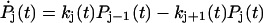 for 1 ≤ j < N, and
for 1 ≤ j < N, and 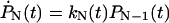 , in analogy to Eq. 2. These kinetic equations can be solved recursively by quadrature,
, in analogy to Eq. 2. These kinetic equations can be solved recursively by quadrature,
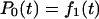 |
(A2a) |
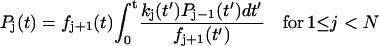 |
(A2b) |
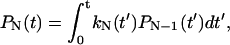 |
(A2c) |
where 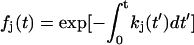 . For N = 1, this reduces to the previous theory, Eq. 3, with S(t) = P0(t). The probability distribution that the j-th unfolding event occurs at time tj* is p(tj*) = kj(tj*)Pj−1(tj*). On average, the j-th unfolding event then occurs at
. For N = 1, this reduces to the previous theory, Eq. 3, with S(t) = P0(t). The probability distribution that the j-th unfolding event occurs at time tj* is p(tj*) = kj(tj*)Pj−1(tj*). On average, the j-th unfolding event then occurs at 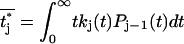 .
.
Within the phenomenological model, the rates kj+1(t) can be determined in analogy to Eq. 1, but accelerated by the number N − j of still folded proteins (Makarov et al., 2001; Zhang et al., 1999):
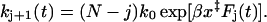 |
(A3) |
The time-dependent force, Fj(t), explicitly depends on the number j of modules already unfolded. This is because after each unfolding event, the linker becomes longer and the effective spring constant softens, as discussed in Appendix B. We can use jL as the contour length of a wormlike-chain model of an anharmonic linker with j unfolded titin modules, Fj(t) = FWLC(vt; jL), where FWLC is given in Eq. C2. Then, Eqs. A2 and A3 provide the analytic solutions to the kinetic simulations of Rief et al. (1998) for the unfolding of titin in the irreversible limit.
APPENDIX B: FLEXIBLE LINKERS
In the simplest model with a linker, the system is described by xm and xl, the molecular and linker coordinates, respectively, with a free-energy surface V(xm,xl,t) = V0(xm) + Vl(xl − xm) + κs(vt − xl)2/2. In general, the higher dimensionality of the potential surface considerably complicates the analysis. However, when the dynamics of the linker coordinate is sufficiently fast, the problem becomes effectively one dimensional. Specifically, the dynamics of the molecular coordinate xm is then governed by the potential of mean force Ve(xm, t) defined by:
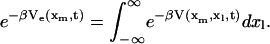 |
(B1) |
The simplest case is when the linker potential is also harmonic, i.e., βVl(xl − xm) = κl(xl − xm)2/2 and the above integral can be evaluated analytically. Within an additive constant, the potential of mean force is then given by
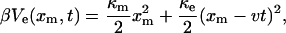 |
(B2) |
which is equivalent to the case without a linker, but with a softened effective pulling spring constant κe = (κs−1 + κl−1)−1 replacing κs. To estimate the effective spring constant κe from a pulling experiment, we calculate the average pulling force as a function of time, 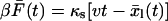 . If we assume equilibrium in the linker coordinate and a “hard” molecular coordinate [κm ≫ κe = (κs−1 + κl−1)−1], then
. If we assume equilibrium in the linker coordinate and a “hard” molecular coordinate [κm ≫ κe = (κs−1 + κl−1)−1], then 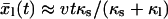 and the force versus extension x = vt curves of individual pulling traces fluctuate about
and the force versus extension x = vt curves of individual pulling traces fluctuate about
 |
(B3) |
Thus, one can estimate κe from the slope of the average force-extension curve before rupture, as illustrated in Fig. 1 C. For slow to intermediate pulling speeds, one can therefore reduce the problem of a harmonic, rapidly relaxing linker to the linker-free case.
APPENDIX C: MULTIMODULE TITIN MODEL WITH WORMLIKE-CHAIN LINKERS
To mimic unfolding of multimodule titin constructs, we simulate a more realistic anharmonic model and compare the results to the microscopic theory, Eq. 21. In this titin model with N independently unfolding modules, each unit i is described by two coordinates: ri(t) is its center of mass and xi(t) its end-to-end extension. For the intramolecular coordinate xi(t), we define two regimes: for xi(t) < x‡, xi(t) moves on a harmonic potential describing the folded well; once xi(t) reaches x‡, the i-th module irreversibly ruptures, and xi(t) moves in the unfolded well described by the approximate wormlike-chain potential (Marko and Siggia, 1995):
 |
(C1) |
with a restoring force:
 |
(C2) |
where x is the extension, lp is the persistence length, and L is the contour length.
Motion occurs only in one dimension, with the N- and C-terminal ends of the i-th titin unit at ri(t) − xi(t)/2 and ri(t) + xi(t)/2, respectively. The C and N termini of adjacent monomers i and i + 1 are held together by harmonic linkers with a spring constant kBTκl = 1000 pN/nm. The molecule is harmonically anchored at the N-terminus of the first module, also with a spring constant kBTκl. Finally, the whole construct is connected to a pulling spring with spring constant kBTκ0 = 50 pN/nm through a wormlike-chain linker. For simplicity, we assume the same contour length L = 28 nm and persistence length p = 0.4 nm for both this linker and the unfolded protein units (Carrion-Vazquez et al., 1999). The tip of the pulling spring is located at rN+1(t), and the force at time t is βF(t) = κ0[vt − rN+1(t)]. The potential surface of the whole system is then given by
 |
(C3) |
σi is one initially; once xi has crossed x‡, σi is irreversibly changed to zero. A schematic representation of the system is shown in Fig. 3. For the dynamics, we assume diffusion on this potential surface with diffusion coefficients Dm for the end-to-end distances xi in the folded well, xi < x‡; after unfolding resulted in a transition to wormlike chain behavior, the diffusion coefficients irreversibly change to Dm∞ = 10 nm2 ms−1. The diffusion coefficients of the center-of-mass coordinates ri and the AFM tip rN+1 are DCM = 100 nm2 ms−1 and Dtip = 50 nm2 ms−1. For the remaining parameters we use kBTκm = 1320 pN/nm, x‡ = 0.335 nm, and k0 = 5.4 × 10−4 s−1, corresponding to Dm = 0.04656 nm2 ms−1.
References
- Balsera, M., S. Stepaniants, S. Izrailev, Y. Oono, and K. Schulten. 1997. Reconstructing potential energy functions from simulated force induced unbinding processes. Biophys. J. 73:1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, G. I. 1978. Models for the specific adhesion of cells to cells. Science. 200:618–627. [DOI] [PubMed] [Google Scholar]
- Best, R. B., S. B. Fowler, J. L. Toca-Herrera, and J. Clarke. 2002. A simple method for probing the mechanical unfolding pathway of proteins in detail. Proc. Natl. Acad. Sci. USA. 99:12143–12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrion-Vazquez, M., A. F. Oberhauser, S. B. Fowler, P. E. Marszalek, S. E. Broedel, J. Clarke, and J. M. Fernandez. 1999. Mechanical and chemical unfolding of a single protein. A comparison. Proc. Natl. Acad. Sci. USA. 96:3694–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, Y., and C. Bustamante. 2000. Pulling a single chromatin fiber reveals the forces that maintain its higher-order structure. Proc. Natl. Acad. Sci. USA. 97:127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembo, M., D. C. Torney, K. Saxman, and D. Hammer. 1988. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc. R. Soc. Lond. B Biol. Sci. 234:55–83. [DOI] [PubMed] [Google Scholar]
- Evans, E. 2001. Probing the relation between force – lifetime – and chemistry in single molecular bonds. Annu. Rev. Biophys. Biomol. Struct. 30:105–128. [DOI] [PubMed] [Google Scholar]
- Evans, E., and K. Ritchie. 1997. Dynamic strength of molecular adhesion bonds. Biophys. J. 72:1541–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, E., and K. Ritchie. 1999. Strength of a weak bond connecting flexible polymer chains. Biophys. J. 76:2439–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, E., D. Berk, and A. Leung. 1991. Detachment of agglutinin-bonded red blood cells. I. Forces to rupture molecular-point attachments. Biophys. J. 59:838–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, E., A. Leung, D. Hammer, and S. Simon. 2001. Chemically distinct transition states govern rapid dissociation of single L-selectin bonds under force. Proc. Natl. Acad. Sci. USA. 98:3784–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin, E. L., V. T. Moy, and H. E. Gaub. 1994. Adhesion forces between individual ligand-receptor pairs. Science. 264:415–417. [DOI] [PubMed] [Google Scholar]
- Fowler, S. B., R. B. Best, J. L. Toca Herrera, T. J. Rutherford, A. Steward, E. Paci, M. Karplus, and J. Clarke. 2002. Mechanical unfolding of a titin Ig domain: Structure of unfolding intermediate revealed by combining AFM, molecular dynamics simulations, NMR and protein engineering. J. Mol. Biol. 322:841–849. [DOI] [PubMed] [Google Scholar]
- Gao, M., D. Craig, V. Vogel, and K. Schulten. 2002. Identifying unfolding intermediates of FN-III10 by steered molecular dynamics. J. Mol. Biol. 323:939–950. [DOI] [PubMed] [Google Scholar]
- Gergely, C., J. C. Voegel, P. Schaaf, B. Senger, M. Maaloum, J. K. H. Horber, and J. Hemmerle. 2000. Unbinding process of adsorbed proteins under external stress studied by atomic force microscopy spectroscopy. Proc. Natl. Acad. Sci. USA. 97:10802–10807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann, B., and H. Grubmüller. 2000. Dynamic force spectroscopy of molecular adhesion bonds. Phys. Rev. Lett. 84:6126–6129. [DOI] [PubMed] [Google Scholar]
- Hummer, G., and A. Szabo. 2001. Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc. Natl. Acad. Sci. USA. 98:3658–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isralewitz, B., M. Gao, and K. Schulten. 2001. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 11:224–230. [DOI] [PubMed] [Google Scholar]
- Izrailev, S., S. Stepaniants, M. Balsera, Y. Oono, and K. Schulten. 1997. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72:1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarzynski, C. 1997. Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 78:2690–2693. [Google Scholar]
- Kellermayer, M. S. Z., S. B. Smith, H. L. Granzier, and C. Bustamante. 1997. Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science. 276:1112–1116. [DOI] [PubMed] [Google Scholar]
- Kramers, H. A. 1940. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica. 7:284–304. [Google Scholar]
- Lu, H., and K. Schulten. 1999. Steered molecular dynamics simulation of conformational changes of immunoglobulin domain I27 interpret atomic force microscopy observations. Chem. Phys. 247:141–153. [Google Scholar]
- Makarov, D. E., P. K. Hansma, and H. Metiu. 2001. Kinetic Monte Carlo simulation of titin unfolding. J. Chem. Phys. 114:9663–9673. [Google Scholar]
- Marko, J. F., and E. D. Siggia. 1995. Stretching DNA. Macromolecules. 28:8759–8770. [Google Scholar]
- Marszalek, P. E., H. Lu, H. B. Li, M. Carrion-Vazquez, A. F. Oberhauser, K. Schulten, and J. M. Fernandez. 1999. Mechanical unfolding intermediates in titin modules. Nature. 402:100–103. [DOI] [PubMed] [Google Scholar]
- Merkel, R., P. Nassoy, A. Leung, K. Ritchie, and E. Evans. 1999. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 397:50–53. [DOI] [PubMed] [Google Scholar]
- Pastor, R. W., R. Zwanzig, and A. Szabo. 1996. Diffusion limited first contact of the ends of a polymer. Comparison of theory with simulation. J. Chem. Phys. 105:3878–3882. [Google Scholar]
- Rief, M., M. Gautel, F. Oesterhelt, J. M. Fernandez, and H. E. Gaub. 1997. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 276:1109–1112. [DOI] [PubMed] [Google Scholar]
- Rief, M., J. M. Fernandez, and H. E. Gaub. 1998. Elastically coupled two-level systems as a model for biopolymer extensibility. Phys. Rev. Lett. 81:4764–4767. [Google Scholar]
- Schuler, B., E. Lipmann, and W. A. Eaton. 2002. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature (Lond.). 419:743–747. [DOI] [PubMed] [Google Scholar]
- Tskhovrebova, L., J. Trinick, J. A. Sleep, and R. M. Simmons. 1997. Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature. 387:308–312. [DOI] [PubMed] [Google Scholar]
- Zhang, B., G. Z. Xu, and J. S. Evans. 1999. A kinetic molecular model of the reversible unfolding and refolding of titin under force extension. Biophys. J. 77:1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwanzig, R. 2001. Nonequilibrium Statistical Mechanics. Oxford University Press, New York.