

Abstract
Ion channels can be gated by various extrinsic cues, such as voltage, pH, and second messengers. However, most ion channels display extrinsic cue-independent transitions as well. These events represent spontaneous conformational changes of the channel protein. The molecular basis for spontaneous gating and its relation to the mechanism by which channels undergo activation gating by extrinsic cue stimulation is not well understood. Here we show that the proximal pore helix of inwardly rectifying (Kir) channels is partially responsible for determining spontaneous gating characteristics, affecting the open state of the channel by stabilizing intraburst openings as well as the bursting state itself without affecting K+ ion-channel interactions. The effect of the pore helix on the open state of the channel is qualitatively similar to that of two well-characterized mutations at the second transmembrane domain (TM2), which stabilize the channel in its activated state. However, the effects of the pore helix and the TM2 mutations on gating were additive and independent of each other. Moreover, in sharp contrast to the two TM2 mutations, the pore helix mutation did not affect the functionality of the agonist-responsive gate. Our results suggest that in Kir channels, the bottom of the pore helix and agonist-induced conformational transitions at the TM2 ultimately stabilize via different pathways the open conformation of the same gate.
INTRODUCTION
Ionic fluxes across biological membranes are mediated via ion channels. The process controlling ionic fluxes through ion channels is broadly termed “gating”. A “gate” is a molecular element within the channel protein, which enables or disables ionic flow. A given channel may possess several different gates, and for ions to flow through the channel, all the gates have to be open simultaneously. Some gates open and close in response to biological signals such as voltage, intracellular messengers, pH, or other proteins. However, even when fully activated, ion channels switch stochastically between open and closed states, with a kinetic pattern that is unique for each channel and serves as its signature. This phenomenon will be referred to throughout this work as “spontaneous gating”. In K+ channels, a wealth of information concerning gating phenomena, in combination with the recent crystal structures for several conformations of bacterial channels (Doyle et al., 1998; Zhou et al., 2001; Jiang et al., 2002), provides a solid basis for studying spontaneous gating.
Mutations at the selectivity filter and close to it were shown to affect ion permeation as well as single-channel gating (Lu et al., 2001a,b; Proks et al., 2001; Chapman et al., 1997; Choe et al., 1998, 2001; Zheng and Sigworth, 1998; Espinosa et al., 2001). It was thus suggested that the selectivity filter may in itself be a gate, and short-lived blocking by permeating ions was shown to be the mechanism of this kind of gating, commonly termed “fast gating” (Choe et al., 1998, 2001).
In addition to the selectivity filter region, control of channel gating has been associated with other channel parts. For example, in K+ as well as in cyclic nucleotide gated (CNG) channels, functional studies have shown that a bending and/or rotation of the TM2/S6 transmembrane helix couples the sensing of voltage (for Kv channels) or intracellular ligands (for Kir and CNG channels) to channel opening (Holmgren et al., 1998; Tucker et al., 1998; Perozo et al., 1999; Loussouarn et al., 2000; Flynn and Zagotta, 2001; Johnson and Zagotta, 2001; Sadja et al., 2001; Yi et al., 2001; Jiang et al., 2002; Jin et al., 2002). The pore helix has also been shown to respond to the activated state of CNG channels (Liu and Siegelbaum, 2000). A comparison between the crystal structures of the bacterial channels KcsA (which is presumably in its closed state) and MthK (which is presumably in its open state) (Jiang et al., 2002), supports the notion that activation gating occurs through a bending and rotation of TM2/S6, which opens the intracellular entrance to the permeation pathway. On the single-channel level, activation gating is usually manifested as changes in the duration of interburst closed times, (Choe et al., 1997; Drain et al., 1998; Tucker et al., 1998) a phenomenon that is commonly termed “slow gating”. Despite all this available data, the phenomenon of spontaneous gating, and particularly its connection to external cue-dependent activation gating, has not been well characterized.
Inwardly rectifying K+ channels (Kir) have been used as a tool for studying spontaneous gating. They possess a prototypic K+ channel structure, with two transmembrane domains and a pore-loop, and are believed to be similar in structure to KcsA (Doyle et al., 1998). Some are gated by external cues whereas others are constitutively active (Stanfield et al., 2002). Despite a high degree of similarity in sequence, especially at the pore region, they present a wide array of single-channel kinetics phenotypes (Choe et al., 1999; Yakubovich et al., 2000; Pessia et al., 2001; Proks et al., 2001). Chimeric approaches were used to identify the core region (TM1 through TM2) as the main region responsible for spontaneous gating phenotype (Slesinger et al., 1995). The contribution of large domains within the core region to single-channel kinetics has also been examined (Choe et al., 1999). Additionally, a glutamine to glutamate mutation at position 140 in Kir2.1 was shown to reduce the mean open time of the channel, due to the introduction of a frequent brief closed state (Guo and Kubo, 1998). However, the mechanism by which these domains influence spontaneous gating has not been established.
The G-protein-coupled potassium channel, GIRK1/4 or Kir3.1/3.4, is a classical example of a channel that posses an intimate relationship between gating by external cues and complex spontaneous gating. Kir3.1/3.4 is mainly gated by the Gβγ subunits of the G-protein and exhibits a very low open probability, even when coexpressed with a saturating concentration of Gβγ (Sakmann et al., 1983, Sadja et al., 2001). Single-channel openings are brief, with a mean open duration of a few milliseconds. Channel openings occur in the form of bursts, which are grouped as rapid openings and closings of the channel. These bursts are separated by distinct periods of long channel closing (Sakmann et al., 1983). Binding of Gβγ stabilizes the channel in the bursting state. However, the bursting phenomenon in itself is not a result of Gβγ gating and is rather an intrinsic property of the channel (Ivanova-Nikolova and Breitwieser, 1997; Yakubovich et al., 2000; Sadja et al., 2001). Ligand binding, e.g., Gβγ, does not only affect interbursting kinetics, as the open state within bursts is also stabilized (Ivanova-Nikolova and Breitwieser, 1997; Nemec et al., 1999; Sadja et al., 2001). Thus Kir3.1/3.4 channel presents an excellent model for studying the possible relationship between spontaneous and agonist-induced gating.
In the present work we took an advantage of Kir2.1 (Kubo et al., 1993), a constitutively active channel (with no external cue-dependent activation gating), that displays a radically different kinetic signature than Kir3.1/3.4. Kir2.1/Kir3.1 chimeras were used as an initial step to screen for regions that control spontaneous gating. We found that the bottom of the pore helix is a major determinant responsible for stabilizing both the open state and bursting activity, without affecting the interaction of the channel with permeating K+ ions. Furthermore, we found that this control of channel gating is additive to the control by Gβγ subunits. The molecular mechanism for spontaneous gating and its relation to activation gating in K+ channels will be discussed.
MATERIALS AND METHODS
Mutagenesis
Chimeras and point mutations in Kir2.1 were constructed using a standard two-step PCR. The PCR products were subcloned back into the parental gene, using silent restriction sites that were previously engineered into the mouse Kir2.1 gene (Kubo et al., 1993). For Kir3.1/3.4, mutagenesis was performed using the Pfu PCR method (Promega, Madison, WI; Stratagene, La Jolla, CA). In all cases, the oligonucleotide used for each mutation carried a silent restriction site for screening purposes. Positive clones were verified by sequencing.
RNA preparation
Capped cRNA was transcribed using a home-assembled cRNA transcription kit (Stratagene, and Pharmacia, Piscataway, NJ). The cRNA integrity and concentration were determined by running an aliquot on a formaldehyde gel.
Solutions
ND96 solution contained (concentrations in mM) 96 NaCl, 2 KCl, 1 CaCl2, 1 MgCl2, 5 HEPES pH = 7.4. Nominally calcium-free ND96 solution was prepared as above, but without CaCl2. For two electrode voltage clamp recordings, the bath solution, which will be referred to as 90 K solution, contained: 90 KCl, 10 HEPES, 2 MgCl2, and pH = 7.4 (KOH). For carbachol-gating measurements, 3 μM carbachol (Sigma, St. Louis, MO) was freshly diluted from a frozen 3 mM stock. For cell-attached patch-clamp recordings, both the bath and pipette solution contained 90 K. When necessary, the patch pipette also contained 50 μM GdCl3, aimed at blocking the endogenous mechano-sensitive channels (Yang and Sachs, 1989).
For Kir2.1(PH2), Kir2.1(Q140E) and Kir2.1(T141A), some of the recordings were carried using a pipette solution containing 90 KCl, 10 HEPES, 1 EGTA, 3 MgCl2 (the free [Mg2+] was 2 mM, calculated using the program Bound And Determined 4.30 (S. Brooks)) pH = 7.4. For inside-out patch-clamp recordings, the pipette solution contained 30, 90, 150, 220, or 300 mM KCl, 10 HEPES, 2 MgCl2, pH = 7.4. In the solutions containing 30 and 90 mM KCl, the cationic concentration was brought up to 150 mM using NMDG. The bath solutions contained identical KCl and NMDG concentrations as the pipette solutions and in addition, 10 HEPES, 10 EGTA, 20 Na2H2P2O7, 5 NaF, 0.1 Na3VO4, 5 NaOH, pH = 7.4.
Oocyte preparation
Female Xenopus laevis frogs were anaesthetized by immersion in water containing 0.15% (w/v) tricaine (Sigma) for 10–20 min. The anaesthetized frogs were placed on ice and several ovarian lobes were surgically removed under sterile conditions. After the operation, frogs were placed in shallow fresh water and were monitored until they regained consciousness. Adequate time for healing was allowed between procedures. Oocytes were defolliculated by shaking in Ca2+-free ND96 solution containing 2 mg/ml type 1 collagenase (Worthington, Lakewood, NJ) for ∼1 h at room temperature. The oocytes were then washed in ND96 solution. Healthy looking stages 5–6 oocytes were selected, and then microinjected with ∼50 nl cRNA of the various channel mutants. After injection, oocytes were maintained at 18°C. Currents were recorded 1–7 days post injection.
Electrophysiology
Currents through the expressed channels were recorded by the two-electrode voltage clamp technique using a CA1-B amplifier (Dagan, Minneapolis MN) (Reuveny et al., 1994). Electrodes were filled with 3 M KCl and had a resistance of 0.1–0.6 MΩ. Data acquisition and analysis were done using pCLAMP 6.04, pCLAMP 8.1, and pCLAMP 9.0 (Axon Instruments, Union City, CA). Open probability was calculated using NPo2.13 (J. Sui, freely available on the Axon Instruments, Union City, CA web site).
Single-channel currents were measured by the patch-clamp technique (Hamill et al., 1981) in either the cell-attached or in the inside-out configurations, using an Axopatch 200B amplifier (Axon Instruments). Current traces were low-pass filtered at 5 kHz, digitized at 94.4 kHz (VR-10B analog-to-digital converter by Instrutech), and stored on a VCR tape. For analysis, data segments of interest were transferred to computer disk, digitized at 5 kHz, and, using Digidata 1200B (Axon Instruments), low-pass filtered at 1 kHz using an eight-pole Bessel filter (Frequency Devices, Haverhill, MA).
Dwell-time analysis
Single-channel transitions were detected by the 50% threshold crossing criterion. For dwell-time analysis, current records were interpolated before event detection using the cubic spline function with an interpolation factor of 10, resulting in an effective sampling rate of 50 KHz (using Clampfit 9). Events were log-binned at 12–15 bins per decade. The y ordinates of the histograms were square-root transformed for improved display of the multiple components (Sigworth and Sine, 1987).
Histograms were usually fitted using least-square minimization. The F criterion was used to determine the number of exponential components where visual inspection was insufficient.
Burst analysis
Bursts are defined as groups of openings separated by closures that are longer than a defined critical time, tc. The critical time was calculated by numerically solving the following equation (Colquhoun and Sakmann, 1985):
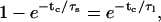 |
where τs is the time constant for the short closings within bursts and τl is the time constant for closings between bursts. Since Kir3.1/3.4 displays multiple closed states (see Tables 2 and 3, Supplementary Material, and also Yakubovich et al., 2000), τs and τl were chosen such that τl was at least 10 times larger than τs (For more information, see Supplementary Material). For burst-duration analysis, current records were analyzed without interpolation using pClamp6. To calculate the number of events per each burst, the first 5000–10,000 events for each patch were analyzed for burst duration and number of openings per burst using Microsoft Excel. The burst durations for bursts containing more than a single opening were subsequently log-binned and fitted using pClamp6, in a similar way to the dwell-time histograms.
Unless otherwise indicated, data presented is the mean ± SE of the mean. Statistical significance was set at p < 0.05 using t-test or one-way analysis of variance.
RESULTS
Two members of the inwardly rectifying K+ channel superfamily, Kir2.1 and Kir3.1/3.4, display radically different single-channel kinetics (Fig. 1 A) and thus were used to study the molecular basis of intrinsic gating. We were first interested in characterizing the kinetic signature of Kir2.1 and Kir3.1/3.4 wild-type channels expressed in Xenopus oocytes. Kir2.1 exhibited long openings, with a distribution that was fitted as a sum of three exponentials, with decay times of τo1 = 0.8 ± 0.06 ms, τo2 = 52.2 ± 0.3 ms, and τo3 = 262 ± 0.02 ms at −100 mV (Fig. 1 B). In contrast, Kir3.1/3.4, coexpressed with a saturating concentration of Gβγ, exhibited short-lived flickery openings that were frequently grouped into bursts. The open-time distribution for Kir3.1/3.4 was fitted as a sum of three exponentials, with decay times of τo1 = 0.4 ± 0.16 ms, τo2 = 1.9 ± 0.4 ms, and τo3 = 6.6 ± 0.3 ms at −100 mV (Fig. 1 C). It thus appears that the main difference between the two channels is in the time constants of the second and third components of the open-time distribution. Furthermore, the tendency of the channel to dwell in the longest open state also differed substantially between the two channels: at −100 mV, the fractional area under the curve for the long open component was 0.83 for Kir2.1 versus 0.28 for Kir3.1/3.4 + Gβγ. The two channels also exhibited radically different open probabilities (Po): at −100 mV, for Kir2.1, Po = 0.85 ± 0.01, whereas for Kir3.1/3.4 coexpressed with Gβγ, Po = 0.007 ± 0.003.
FIGURE 1.

Characterization of single-channel kinetics of Kir3.1/3.4 + Gβγ and Kir2.1 (A) Single-channel traces of Kir2.1 and Kir3.1/3.4 + Gβγ. The dotted line denotes the closed channel current level. The currents were recorded at −100 mV. Pipette solution contained 90 K with 50 μM Gd3+. (B) Open-time distribution for Kir2.1 at −100 mV. (C) Open-time distribution for Kir3.1/3.4 + Gβγ at −100 mV. For B and C, the bars are binned data events and the solid lines are curve fits. The arrows point to the peaks of the exponential fit.
A chimeric approach to single-channel gating in Kir channels
Having established the differences in single-channel kinetics between Kir3.1/3.4 and Kir2.1, a chimeric approach was used to identify the molecular elements responsible for these differences. Since the channel core region (TM1 through TM2) was shown to be the main determinant for single-channel kinetics (Slesinger et al., 1995; Choe et al., 1999), we arbitrarily divided this region into seven parts. Chimeras were constructed by replacing Kir2.1 regions with the corresponding parts of Kir3.1 (Fig. 2 A). Single-channel currents were then recorded for each chimera and the average open time and open probability were measured at −100 mV. All chimeras had open-time distributions that could be described as a sum of three exponentials, as in their Kir2.1 and Kir3.1/3.4 parent channels (data not shown). Both parameters, as well as the single-channel amplitude, varied among the different chimeras, suggesting that single-channel behavior is fine-tuned by many sites within the channel protein. The open probability, but not the single-channel amplitude, correlated with the average open times (Fig. 2, B–D). The average open time and open probability for one particular chimera, PH2, which contained two amino acid substitutions at the bottom of the pore helix (Fig. 2, E), showed the largest degree of resemblance to the Kir3.1/3.4 phenotype. Single Kir2.1(PH2) channels (Fig. 3, A versus B and C) exhibited bursts of short openings, with an average open time of 9.1 ms Po = 0.04 ± 0.01 at −100 mV (Fig. 3 A). Kir2.1(PH2) burst-duration distribution was fitted as a sum of three exponentials with decay times of τb1 = 13.9 ± 0.2 ms, τb2 = 121 ± 0.1 ms, and τb3 = 828 ± 0.2 ms (Fig. 3 D). It is unlikely that the bursts exhibited by Kir2.1(PH2) are the result of an acquisition of a new short-lived closed state that punctuates the Kir2.1 channel openings, as was shown for Kir2.1(Q140E) (Guo and Kubo, 1998). First, in such a case, we should expect to see a similarity between the burst-duration distribution for Kir2.1(PH2) and the open-time distribution for the wild-type Kir2.1. Our results show that they are not similar. Second, the reduction in the open probability (Fig. 3 E) displayed by Kir2.1(PH2) is too large to be explained by a mere acquisition of a short-lived closed state. In fact, the main reason for the observed reduction in the open probability of the Kir2.1(PH2) channel is the prolongation/acquisition of a long closed state (data not shown).
FIGURE 2.

Chimeric approach to study spontaneous gating in Kir channels. (A) Schematic diagram of the Kir2.1/Kir3.1 chimeras amino acid sequence. The location of the first and second transmembrane domains, M1 and M2, and the pore region, H5, are shown above the chimeras. The name of each chimera is indicated at its left, and the number of amino acid substitutions in each is indicated in parenthesis. (B) The average open time for each channel or chimera at −100 mV. (C) Open probability at −100 mV (D) Single-channel current level at −100 mV for each channel or chimera. (E) Left, a 3D model of Kir2.1 backbone based on the published structure of KcsA (Doyle et al., 1998). Q140 and T141 side chains are shown as space fill. Right, comparison between the amino acid sequences of the pore regions of Kir2.1, 3.1, and 3.4. The two amino acids composing the PH2 region are framed.
FIGURE 3.

Kir2.1(PH2) displays Kir3.x-like gating. (A) Single-channel trace of Kir2.1(PH2) at −100 mV. The bottom trace is an expansion of a 500-ms segment (indicated by a horizontal bar) from the upper trace. (B) A 500-ms trace of a single Kir2.1 channel at −100 mV. (C) A 500-ms trace of a single Kir3.1/3.4 channel at −100 mV. The dotted line denotes the closed channel current level. (D) Burst-duration distribution for Kir2.1(PH2). The bars are binned events, the solid line is a curve fit. (E) Po at −100 mV for the parental Kir2.1, Kir3.1/3.4 + Gβγ, Kir2.1(PH2), Kir2.1(Q140E), and Kir2.1(T141A).
The PH2 chimera contains only two amino acid substitutions: Q140E and T141A. T141 in Kir2.1 was shown to affect the affinity for extracellular Ba2+ ion block (Zhou et al., 1996; Alagem et al., 2001). Since single-channel gating of Kir channels was shown to be influenced by even trace amounts of extracellular Ba2+ ions, (Choe et al., 1998), we recorded single-channel currents in the presence of 1 mM EGTA in the pipette solution to chelate traces of contaminating Ba2+ originating from the KCl salt. The open-time distributions were virtually identical under the two conditions. The only difference in kinetic parameters observed in the presence of EGTA was the vanishing of a component of long closings (results not shown) as was shown before (Choe et al., 1998). We can therefore conclude that the differences in single-channel gating between Kir2.1 and Kir2.1(PH2) are not caused by different affinities for Ba2+ ions.
To further explore the relative contribution of the two amino acid substitutions in PH2 to single-channel gating, single-channel currents were recorded for Kir2.1(T141A) and Kir2.1(Q140E). Both mutants exhibited an open-time distribution that was the sum of a few exponential components (data not shown). The average open time for Kir2.1(Q140E) was 45.6 ms and the open probability was Po = 0.32 ± 0.07 at −100 mV. The average open time for Kir2.1(T141A) was 102.2 ms and the open probability was Po = 0.84 ± 0.05 at −100 mV (Fig. 3 E). Taken together, the above results suggest that these two sites act in combination to determine the open-state stability of Kir2.1.
The effect of the reverse mutations on Kir3.1/3.4
We were interested to test whether the PH2 region controls gating in Kir3.1/3.4 as well as in Kir2.1. Single-channel currents were recorded for Kir3.1(E141Q A142T)/3.4(E147Q) coexpressed with Gβγ (referred to as Kir3.1/3.4(PH2) hereby) (Fig. 4 A). As in the wild-type, the open-time distribution of this mutant could be described as a sum of three exponentials (see Table 1, Supplementary Material). Unexpectedly, τo1 and τo2 were similar between the wild-type and the mutant channel, and additionally, only a slight increase in the mean duration of the longest openings was observed for this mutant: τo3 = 9.7 ± 0.1 ms at −100 mV. However, the probability of Kir3.1/3.4(PH2) to be found in the longest-opening state (as measured by the fractional area under the plot) increased considerably, from 0.28 in Kir3.1/3.4 to 0.45 in Kir3.1/3.4(PH2) (Fig. 4 D).
FIGURE 4.

Reverse PH2 chimera shows increase in open times and burst durations. This phenotype can be attributed to E141Q\E147Q. (A–C) Single-channel traces (left), open-time distributions (middle), and burst-duration distributions of bursts containing more than one opening (right) at −100 mV for (A) Kir3.1/3.4(PH2) + Gβγ, (B) Kir3.1/3.4(EQ) + Gβγ, and (C) Kir3.1/3.4 (A/T) + Gβγ. For an easy comparison, the fits of the open-time distributions (D) and burst-duration distributions (E) for the wild-type and the mutants were normalized such that the area under the plot equals 1 and superimposed.
In addition to the moderate stabilization of the open state, the reverse PH2 mutations produced a large effect on the bursting pattern of Kir3.1/3.4. Since Kir3.1/3.4(PH2) has increased the tendency of the channel to be found in the longest open state, we were first interested in determining whether it has a similar effect on the bursting states as well. We therefore examined whether the Kir3.1/3.4(PH2) chimera exhibits a larger tendency toward firing bursts over isolated single openings. The bursting probability Pburst was defined as the probability of the channel to have more than one opening within a burst. Pburst was calculated as the fraction of bursts containing more than one opening for each channel. For the wild-type, Pburst was measured to be 0.34 + 0.03, indicating that most bursts contained only a single opening. For the PH2 chimera, however, the majority of bursts contained multiple openings, with Pburst = 0.59 ± 0.02. Second, we examined the effect of the Kir3.1/3.4(PH2) chimera on the burst-duration distribution of the channel. We therefore constructed burst-duration histograms for bursts containing more than one opening. Kir3.1/3.4(PH2) exhibits new long bursting states along with an increased tendency to be found in longer bursting states as compared to the wild-type (Fig. 4 E, see also Table 5, Supplementary Material). Our results so far suggest that the mutations in Kir3.1/3.4(PH2), located in the pore helix, are mainly involved in the stabilization of the bursting mode.
We have shown that in Kir2.1, the combination of the two mutations, Q140E and T141A, is required to produce a Kir3.x-like phenotype. To test whether a combination of two mutations might also be required for the Kir3.1/3.4(PH2) phenotype, single-channel currents were recorded for the individual point mutations Kir3.1(E141Q)/3.4 (E147Q) (hereby referred to as Kir3.1/3.4(EQ)) and Kir3.1(A142T)/3.4 (hereby referred to as Kir3.1/3.4(AT)) (Fig. 4, B and C). Our results show that in Kir3.1/3.4, the PH2 phenotype could be attributed to the E141Q/E147Q mutation alone. The Kir3.1/3.4 (AT) mutant was similar to the wild-type in its single-channel gating phenotype (Fig. 4, D and E).
The effects of the pore helix mutations on gating are additive to those of TM2 mutations
In a previous work, we described mutations in the second transmembrane domain of Kir3.1/3.4, which eliminate Gβγ gating, thus producing constitutively active channels (Sadja, et al., 2001). On the single-channel level, these TM2 mutants showed an increase both in burst duration and in opening duration similar to the Kir3.1/3.4(PH2) mutant. We were therefore interested in finding whether these effects on single-channel kinetics, originating from two physically different areas, are operating through the same mechanism. Therefore, we compared the single-channel opening pattern of two such TM2 mutations, Kir3.1(S170P)/3.4(S176P) and Kir3.1(C179A)/3.4(C185A) (hereby referred to as Kir3.1/3.4(SP) and Kir3.1/3.4(CA), respectively) to that of the combined mutants Kir3.1(E141Q A142T S170P)/3.4(E147Q S176P) and Kir3.1(E141Q A142T C179A)/3.4(E147Q C185A) (hereby referred to as Kir3.1/3.4(PH2 SP) and Kir3.1/3.4(PH2 CA), respectively). The mutant channels carrying combined pore helix and TM2 mutations displayed single-channel openings and bursts that were considerably longer than either of the single mutations alone, alongside an increase in the tendency to be found in the long burst states. (Fig. 5, A–J). The combined pore helix and TM2 mutants also displayed an increase in Pburst (Fig. 6 A). The stabilization of the open and bursting states was reflected in the channel's open probability: Both combined pore helix and TM2 mutants increased Po above the levels of the wild-type and each of the corresponding PH2 or TM2 mutants (Fig. 6 B). Interestingly, Kir3.1/3.4(PH2 SP) displayed a particularly high open probability, 0.72 ± 0.04, which is comparable to the Po value of Kir2.1. In summary, the effects of the pore helix and the two TM2 mutations on gating are additive, and therefore, the pore helix and two TM2 residues may operate through independent mechanisms to stabilize the open/bursting state of the channel.
FIGURE 5.

Effect of the PH2 region on gating is additive to the effect of the S170P/S176P and C179A/C185A mutations in the TM2. All the recordings were made at −100 mV. (A–B) Single-channel recordings of (A) Kir3.1/3.4(SP) and (B) Kir3.1/3.4(PH2 SP). The dotted lines mark closed channel current levels. (C–D) Burst-duration distributions (of bursts containing more than one opening) for Kir3.1/3.4(SP) and Kir3.1/3.4(PH2/SP), respectively. The gray bars are binned events and the solid line is a curve fit. (E) For an easy comparison, the fits of the burst-duration distributions for the wild-type and the mutants were normalized such that the area under the plot equals 1 and superimposed. (F–G) Single-channel recordings of Kir3.1/3.4(CA) and Kir3.1/3.4(PH2/CA), respectively. The dotted lines mark closed channel current levels. (H–I) Burst-duration distributions (of bursts containing more than one opening) for Kir3.1/3.4(CA) and Kir3.1/3.4(PH2/CA), respectively. The gray bars are binned events and the solid line is a curve fit. (J) For an easy comparison, the fits of the burst-duration distributions for the wild-type and the CA and PH2 CA mutants were normalized such that the area under the plot equals 1 and superimposed.
FIGURE 6.
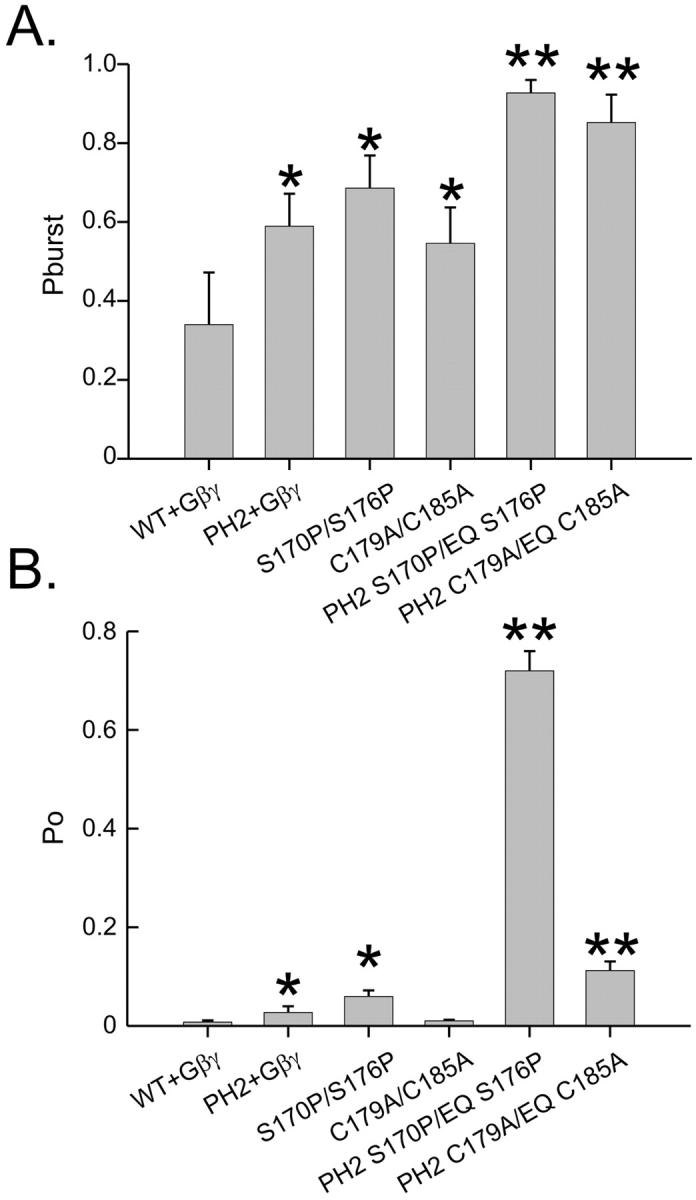
PH2 region and the two TM2 mutations increase the probability of bursting and open probability in an additive manner. (A) Probability of entering a bursting state once the channel opens. (B) Open probability. The single asterisks indicate significant difference from the wild-type. Double asterisks indicate significant difference of combined PH2 and TM2 mutants over the levels of either of the respective PH2 and TM2 mutants.
The PH2 region is not energetically coupled to residues 170/176 and 179/185
Another approach to test whether the pore and TM2 regions affect gating through two independent mechanisms would be to test whether the effects of these two regions on the energies of the open and closed states are coupled. We thus performed a double mutant cycle analysis (Horovitz and Fersht, 1990), where the energy difference between the open and closed states of the channel, ΔG0 (measured in kBT units), was calculated using:
 |
The changes in the ΔG caused by mutations were then calculated as,
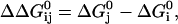 |
where i is the initial form and j is the additional mutation. ΔΔGij0 values were calculated for each step in the cycle, and compared with those obtained for the opposite side of the cycle. Differences in ΔΔGij0 of more than one kBT unit should indicate energy coupling. The resulting cycles are shown below. The ΔΔGij0 values for I = PH2, j = SP, and for I = PH2, j = CA are 1.98 and 0.35, respectively.
These values indicate no energy coupling between the pore helix and Kir3.1(179)/Kir3.4(185) in the TM2 region. In the case of Kir3.1/(170)/Kir3.4(176), the value of 1.98 kT units suggests some nonadditivity. However, this value has a large inherited error due to ∼45% error in the open probabilities for the Kir3.1/Kir3.4 and for the Kir3.1/3.4(PH2) channels. We therefore conclude that this 1.98 kT ΔΔG value (1.17 kCal/mol at 25°C) may not indicate significant coupling. Values in this range were also considered nonsignificant in a recent report describing paired mutations affecting gating in Shaker channel (Yifrach and MacKinnon, 2002).
Stabilization of the open state by E141Q/E147Q does not affect Gβγ-mediated gating
Since there is no significant energy coupling between positions 170/176 and 179/185 and the pore helix in stabilizing the open state of the channel, and since the TM2 mutations stabilize the channel in a Gβγ independent state, we should expect that activation gating by Gβγ should not be affected by the pore helix mutations. To verify this point, Kir3.1/3.4, Kir3.1/3.4(PH2), Kir3.1/3.4(EQ), and Kir3.1/3.4(AT) were coinjected into Xenopus oocytes along with RNA for the muscarinic receptor type 2 (m2R). The functionality of the Gβγ gate was then tested by measuring current induction with 3 μM carbachol, an agonist of the m2R. As expected from the above hypothesis, carbachol-mediated gating of Kir3.1/3.4(PH2), as that of the two point mutations, was not different than that of the wild-type Kir3.1/3.4 (Fig. 7). These results again support a separate role for the pore helix and Gβγ- induced activation gating, in stabilizing the open state of the channel.
FIGURE 7.

Agonist activation of the channel is not affected by the pore helix mutations. Muscarinic type 2 receptor activation by 3 μM carbachol of Kir3.1/3.4 and its mutants at −80 mV. Carbachol induction levels were determined as fold increase in current over the basal current before the application of carbachol.
The 141/147 position does not affect the interaction of the channel with permeating K+ ions
Guo et al. (1998) have hypothesized that in Kir2.1, Q140 (corresponding to E141/E147 in Kir3.1/3.4) controls gating of Kir2.1 by interacting with permeating K+ ions. Others have observed a permeant ion effect on “fast gating” (short-lived closings of the permeation pathway) (Reuveny et al., 1996; Choe et al., 1998). The selectivity filter and residues in its close proximity have been shown to affect “fast gating” (Lu et al., 2001a; Proks et al., 2001), therefore suggesting that permeation and gating may be coupled at the selectivity filter. We did not observe any effect of Kir3.1/3.4(PH2) on the duration of fast intraburst closings (see Tables 2 and 3, Supplementary Material). However, since position 141/147 is close to the selectivity filter, we were interested in selectively determining the effect of the EQ mutation on K+ permeation. To measure the ability of K+ ions to interact with the permeation pathway, we recorded single-channel currents under various symmetrical K+ concentrations (Fig. 8 A), and measured the dependence of the single-channel conductance on K+ concentrations. As seen in Fig. 8, B and C, there is no substantial difference in current amplitude and K+-dependent conductance changes between the wild-type and the E141Q/E147Q mutant channels. We have also tested the selectivity of this mutant (K+ versus Na+) and found no significant difference to the wild-type channel (not shown). These results may suggest that the gating effects seen with the E141Q/E147Q mutant are not related to a change in K+ ion-channel interactions.
FIGURE 8.

Potassium ion-channel interaction is not affected by the pore helix mutation E141Q/E147Q. (A) Current traces recorded at a holding potential of −80 mV from inside-out patches containing Kir3.1/3.4 channels under symmetrical K+ concentrations (in mM) 30 (top), 90 (middle), and 300 (bottom). (B) Current-to-voltage plot of single Kir3.1/3.4 (circles) and Kir3.1/3.4(EQ) (triangles) at 30 (open symbols) and 300 mM K+ (filled symbols). The solid lines are linear regressions to the data. (C) The dependence of single-channel conductance on the symmetrical K+ concentrations. The solid line is a curve fit. Data are presented as mean ± SD.
The open state and the bursting state are coupled
Our results so far show that in Kir3.1/3.4, all our mutants, either at the pore helix or at the TM2 domain, affect single-channel gating by stabilizing both the open and the bursting states of the channel. We were interested in finding the correlation between open-state stabilization and burst-duration stabilization for Kir3.1/3.4. Plotting average burst duration for each of the channel mutants as a function of its open-state components shows that the average burst duration exhibits a strong correlation with the longest open-time component, τo3—the larger the time constant, the longer the mean burst duration (Fig. 9 A). It was important to establish whether the increase in burst duration is due to the stabilization of the bursting state itself or whether it is a mere consequence of the more stable long open state within bursts. Thus, the fractional increase (relative to Kir3.1/4 wild-type) in τo3, and in the average number of openings per burst for each mutant, were calculated. Our results show that the increase in the average number of openings per burst is the main determinant for the increase in burst duration (Fig. 9 B). Since burst durations are correlated with τo3, and since the increased stability of the bursting state is mainly due to an increase in the number of openings per burst, we were interested to find out whether the stability of the open state (τo3) correlates with the number of intraburst openings. Indeed, Fig. 9 C shows a strong correlation between the two, therefore suggesting that in inwardly rectifying K+ channels, the stability of the intraburst open state and the stability of the bursting state are strongly coupled.
FIGURE 9.

Open-time and burst duration are coupled in Kir3.1/3.4. (A) Average burst duration at −100 mM plotted as function of each mean open-time component. The solid line is a linear regression. (B) Fold increase over wild-type in τ03 and number of openings per burst for the various mutants. (C) Number of openings per burst plotted as function of τo3. The solid line is a linear regression.
DISCUSSION
Our work demonstrates the importance of the pore helix region in controlling spontaneous gating of Kir channels. From our results presented above, it is apparent that the amino acid composition at the bottom of the pore helix dictates the stability of the open state, therefore partially underlying the spontaneous gating characteristics of the channel. The control of spontaneous gating by the bottom of the pore helix mainly affects burst durations without affecting K+ ion-channel interactions. In addition, we have been able to demonstrate that activation gating control by Gβγ binding, and spontaneous gating control at the pore helix, are separate and possibly independent processes.
Fast gating, manifested as rapid transitions between open and closed states within bursts, has mainly been associated with regions that interact with permeating K+ ions. Mutations that affect single-channel mean open time usually also affect the interaction of ions with the permeation pathway, either by affecting the ability of the selectivity filter to select for K+ ions (Zheng and Sigworth, 1997), or by changing the rate of ion conduction (Lu et al., 2001a; Proks et al., 2001). From our results it seems that the E141Q/E147Q mutation, which has a major effect on single-channel kinetics, had no effect on K+ permeation of Kir3.1/3.4, as selectivity and the dependence of single-channel conductance on symmetrical K+ concentrations is virtually identical between the wild-type and this mutant channel. These results are supported by previous work demonstrating that the side chain at this position plays no role in permeation: Dart et al. (1998) have found Kir2.1(Q140C) to be unavailable for silver modification. Lancaster et al. (2000) showed that Kir3.1(E141) does not play a role in Ba2+ and polyamine block of Kir3.1/3.4. Moreover, once aligning Kir3.1/3.4 on the recently resolved KcsA structure (Zhou et al., 2001), it appears that the glutamate side at the 141/147 position faces the TM2 helix (probably in its closed state (Perozo et al., 1999)). Additionally, we observed no effect of the 141/147 position on the mean duration of short intraburst closings (see Tables 2 and 3, Supplementary Materials), leading us to suggest that the 141/147 position does not play a role in the classical process of K+ dependent fast gating (Choe et al., 1998, 2001). Other residues, located closer to the selectivity filter, seem to play a role in this process in the closely related KATP channel (Proks et al., 2001). In conclusion, it seems that the bottom of the pore helix is involved in the stabilization of a physical gate with a mechanism that is independent of ion permeation.
What is the interplay between activation gating transitions and open-state stabilization by the pore helix? On the single-channel level, the effect of the pore helix mutation on gating was qualitatively similar to those of two TM2 mutations that rendered the channel constitutively active by stabilizing the open conformation of the activation gate. We interpret this data as stabilization of the open state of the same gate/s by the pore helix mutation E141Q/E147Q and TM2 mutations. However, the bottom of the pore helix and the two TM2 mutations seem to stabilize the open state of the spontaneous gate via two different pathways, as the E141Q/E147Q mutation does not interfere with activation gating. Moreover, the effects of the pore helix and TM2 mutations on gating were additive, and a partial mutant cycle analysis indicates no significant energy coupling between PH2 and the 170/176 or 179/185 residues.
We would like to interpret our data in light of the emerging structural information concerning K+ channel activation gating. It has been established that the bending of the TM2 helix underlies activation gating in G-protein gated Kir channels (Sadja et al., 2001; Jin et al., 2002) as well as in other K+ channels. However, recent data in the field shows that for Kir2.1, Kir6.2, and CNG channels, access to the pore from the intracellular side of the membrane is possible in the closed state. (Craven and Zagotta, 2002; Philips et al., 2002; Xiao and Yang, 2002) (but see Note added in proof). Given the high degree of similarity between Kir channels, it is very likely that the bundle crossing does occlude ion permeation in the closed Kir3.1/3.4 as well. Our study provides clues to suggest that the conformation of the TM2 helix does not solely underlie the open state (and in particular, the bursting state) stability of Kir3.1/3.4: First, the S170P\S176P mutant, in which the TM2 helix is kinked to constitutively mimic the Gβγ-bound state of the channel, in which the bundle crossing at the cytoplasmic end of the TM2 is dilated, still displays bursting activity and, in fact, spends most of its time in a closed state. Second, our results show that the pore helix affects the stability of the spontaneous gate without affecting the conformation of the TM2 helix, as evidenced by the lack of effect on activation gating. Our study thus supports the notion that the spontaneous gate and the TM2 helix bundle crossing are not structurally identical. We propose that the conformational changes at the TM2 induced by agonist binding at the intracellular side of the channel may only serve as a relay of signaling, to stabilize the opening of a physical gate located elsewhere in the channel. Further studies are required to identify the position of the spontaneous gate of Kir channels, and the mechanism by which the TM2 and pore helix influence its open-state stability.
The relationship between spontaneous and activation gating for other K+ channels, such as voltage-gated channels, where the bundle crossing at the TM2/S6 was shown to form a physical barrier for ion permeation (Liu et al., 1997; del Camino and Yellen, 2001; Rothberg et al., 2002) will need to be examined in detail in the future. It is interesting to note that for the KcsA bacterial K+ channel, recent data (Cuello and Perozo, 2002), suggest that a conformational coupling exists between the TM2 and pore helix, where the bottom of the pore helix responds to the activation state of the TM2 helix, but does not affect proton-dependent activation gating. It seems, thus, that for KcsA, some interplay exists between the pore helix and activation gating at the TM2.
We have shown that the open-state stability is strongly coupled to the bursting state stability in Kir3.1/3.4. We cannot yet offer a plausible explanation to this intriguing result. However, it seems that this phenomenon is not unique to G-protein-coupled channels. ATP was shown to affect both open-state and bursting stability in the pancreatic KATP channel (Li et al., 2002). BK channels also exhibit a link between intraburst and interburst gating (Magleby and Pallotta, 1983). These findings suggest that the mechanism controlling spontaneous gating may indeed be conserved among different K+ channels.
How can we explain why spontaneous gating in the constitutively active channel Kir2.1 was affected by a combination of two pore helix mutations, whereas for Kir3.1/3.4, only one mutation is sufficient? We have shown that in Kir2.1 the residues at positions 140 and 141 cooperatively participate in single-channel gating. In Kir3.1/3.4, however, only the position corresponding to Kir2.1 Q140 is sufficient for controlling single-channel gating. This suggests that despite the high extent of sequence homology between the two channels, subtle differences exist in the mechanisms by which the gating process is controlled. The asymmetry in the pore of Kir3.1/3.4 at the 142/148 position may be the reason why this region has different effects in the two channels. Asymmetry in the pore region of Kir3.1/4 was previously shown to affect K+ selectivity (Silverman et al., 1998) and Ba2+ block (Lancaster et al., 2000). This fact also may explain the unique single-channel kinetics of Kir3.4 homotetramers that do not burst (Corey and Clapham, 1998; Guo and Kubo, 1998). Another possibility is the fact that Kir2.1 is a constitutively active channel, and has no apparent control by external cues. This may suggest that the TM2 in Kir2.1, or in other constitutively active K+ channels, adopt a slightly different conformation that affect the stability of the open state differently.
Here we report of a critical domain in K+ channels that controls spontaneous gating in K+ channels, through influencing the stability of the open state, and concomitantly, the stability of the bursting state. This control of channel gating is independent of K+ permeation as well as agonist gating. The mechanism by which this domain influences channel gating may be conserved among K+ channels.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Figure 10.

Figure 11.

Acknowledgments
Note added in proof: Recently, using methanosulfonate accessibility to the water-filled cavity of Kir6.2 channel, it has been shown that TM2 bundle crossing can occlude the accessibility of methanosulfonate reagents, providing that the channel displays very low probability of opening (<0.01) (Phillips et al., 2003). This thus suggests that the bundle crossing may indeed form a physical gate for potassium ions.
We thank Rona Sadja, Inbal Riven, and Shachar Iwanir for reading the manuscript, and Ruth Meller and Elisha Shalgi for technical assistance.
This work was supported in part by grants to E.R. from the Israeli Academy of Sciences (633/01) and the Minerva Stiftung Foundation (Munich).
References
- Alagem, N., M. Dvir, and E. Reuveny. 2001. Mechanism of Ba(2+) block of a mouse inwardly rectifying K+ channel: differential contribution by two discrete residues. J Physiol. 534:381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, M. L., H. M. VanDongen, and A. M. VanDongen. 1997. Activation-dependent subconductance levels in the drk1 K channel suggest a subunit basis for ion permeation and gating. Biophys. J. 72:708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe, H., L. G. Palmer, and H. Sackin. 1999. Structural determinants of gating in inward-rectifier K+ channels. Biophys. J. 76:1988–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe, H., H. Sackin, and L. G. Palmer. 1998. Permeation and gating of an inwardly rectifying potassium channel. Evidence for a variable energy well. J. Gen. Physiol. 112:433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe, H., H. Sackin, and L. G. Palmer. 2001. Gating properties of inward-rectifier potassium channels: effects of permeant ions. J. Membr. Biol. 184:81–89. [DOI] [PubMed] [Google Scholar]
- Choe, H., H. Zhou, L. G. Palmer, and H. Sackin. 1997. A conserved cytoplasmic region of ROMK modulates pH sensitivity, conductance, and gating. Am. J. Physiol. 273:F516–F529. [DOI] [PubMed] [Google Scholar]
- Colquhoun, D., and B. Sakmann. 1985. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol. 369:501–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey, S., and D. E. Clapham. 1998. Identification of native atrial G-protein-regulated inwardly rectifying K+ (GIRK4) channel homomultimers. J. Biol. Chem. 273:27499–27504. [DOI] [PubMed] [Google Scholar]
- Craven, K. B., and W. N. Zagotta. 2002. Discerning the closed size of the rod cyclic nucleotide-gated channel smokehole. Biophys. J. 82:274a.11751315 [Google Scholar]
- Cuello, L. G., and E. Perozo. 2002. Fast gating transitions determined by dynamic rearrangements in the selectivity filter of KcsA. Biophys. J. 82:174a. [Google Scholar]
- Dart, C., M. L. Leyland, P. J. Spencer, P. R. Stanfield, and M. J. Sutcliffe. 1998. The selectivity filter of a potassium channel, murine kir2.1, investigated using scanning cysteine mutagenesis. J Physiol. 511:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Camino, D., and G. Yellen. 2001. Tight steric closure at the intracellular activation gate of a voltage-gated K(+) channel. Neuron. 32:649–656. [DOI] [PubMed] [Google Scholar]
- Doyle, D. A., J. Morais Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Drain, P., L. Li, and J. Wang. 1998. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 95:13953–13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa, F., R. Fleischhauer, A. McMahon, and R. H. Joho. 2001. Dynamic interaction of S5 and S6 during voltage-controlled gating in a potassium channel. J. Gen. Physiol. 118:157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn, G. E., and W. N. Zagotta. 2001. Conformational changes in S6 coupled to the opening of cyclic nucleotide-gated channels. Neuron. 30:689–698. [DOI] [PubMed] [Google Scholar]
- Guo, L., and Y. Kubo. 1998. Comparison of the open-close kinetics of the cloned inward rectifier K+ channel IRK1 and its point mutant (Q140E) in the pore region. Receptors Channels. 5:273–289. [PubMed] [Google Scholar]
- Hamill, O. P., A. Marty, E. Neher, B. Sakmann, and F. J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Holmgren, M., K. S. Shin, and G. Yellen. 1998. The activation gate of a voltage-gated K+ channel can be trapped in the open state by an intersubunit metal bridge. Neuron. 21:617–621. [DOI] [PubMed] [Google Scholar]
- Horovitz, A., and A. R. Fersht. 1990. Strategy for analysing the co-operativity of intramolecular interactions in peptides and proteins. J. Mol. Biol. 214:613–617. [DOI] [PubMed] [Google Scholar]
- Ivanova-Nikolova, T. T., and G. E. Breitwieser. 1997. Effector contributions to G beta gamma-mediated signaling as revealed by muscarinic potassium channel gating. J. Gen. Physiol. 109:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- Jin, T., L. Peng, T. Mirshahi, T. Rohacs, K. W. Chan, R. Sanchez, and D. E. Logothetis. 2002. The subunits of G proteins gate a K+ channel by pivoted bending of a transmembrane segment. Mol. Cell. 10:469–481. [DOI] [PubMed] [Google Scholar]
- Johnson, J. P., Jr., and W. N. Zagotta. 2001. Rotational movement during cyclic nucleotide-gated channel opening. Nature. 412:917–921. [DOI] [PubMed] [Google Scholar]
- Kubo, Y., T. J. Baldwin, Y. N. Jan, and L. Y. Jan. 1993. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 362:127–133. [DOI] [PubMed] [Google Scholar]
- Lancaster, M. K., K. M. Dibb, C. C. Quinn, R. Leach, J. K. Lee, J. B. Findlay, and M. R. Boyett. 2000. Residues and mechanisms for slow activation and Ba2+ block of the cardiac muscarinic K+ channel, Kir3.1/Kir3.4. J. Biol. Chem. 275:35831–35839. [DOI] [PubMed] [Google Scholar]
- Li, L., X. Geng, and P. Drain. 2002. Open state destabilization by ATP occupancy is mechanism speeding burst exit underlying KATP channel inhibition by ATP. J. Gen. Physiol. 119:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J., and S. A. Siegelbaum. 2000. Change of pore helix conformational state upon opening of cyclic nucleotide-gated channels. Neuron. 28:899–909. [DOI] [PubMed] [Google Scholar]
- Liu, Y., M. Holmgren, M. E. Jurman, and G. Yellen. 1997. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 19:175–184. [DOI] [PubMed] [Google Scholar]
- Loussouarn, G., E. N. Makhina, T. Rose, and C. G. Nichols. 2000. Structure and dynamics of the pore of inwardly rectifying K(ATP) channels. J. Biol. Chem. 275:1137–1144. [DOI] [PubMed] [Google Scholar]
- Lu, T., A. Y. Ting, J. Mainland, L. Y. Jan, P. G. Schultz, and J. Yang. 2001a. Probing ion permeation and gating in a K+ channel with backbone mutations in the selectivity filter. Nat. Neurosci. 4:239–246. [DOI] [PubMed] [Google Scholar]
- Lu, T., L. Wu, J. Xiao, and J. Yang. 2001b. Permeant ion-dependent changes in gating of Kir2.1 inward rectifier potassium channels. J. Gen. Physiol. 118:509–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magleby, K. L., and B. S. Pallotta. 1983. Burst kinetics of single calcium-activated potassium channels in cultured rat muscle. J Physiol. 344:605–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemec, J., K. Wickman, and D. E. Clapham. 1999. Gβγ binding increases the open time of IKACh: kinetic evidence for multiple Gβγ binding sites. Biophys. J. 76:246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo, E., D. M. Cortes, and L. G. Cuello. 1999. Structural rearrangements underlying K+-channel activation gating. Science. 285:73–78. [DOI] [PubMed] [Google Scholar]
- Pessia, M., P. Imbrici, M. C. D'Adamo, L. Salvatore, and S. J. Tucker. 2001. Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J Physiol. 532:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, L. R., D. Enkvetchakul, and C. G. Nichols. 2003. Gating dependence of inner pore access in inward rectifier K+ channels. Neuron. 37:953–962. [DOI] [PubMed] [Google Scholar]
- Philips, L. R., G. Loussouarn, and C. G. Nichols. 2002. Cold draft through the teepee: MTSEA access to the inner vestibule of ATP closed Kir6.2 channels implies gating does not occur at the smokehole. Biophys. J. 82:590a. [Google Scholar]
- Proks, P., C. E. Capener, P. Jones, and F. M. Ashcroft. 2001. Mutations within the P-loop of Kir6.2 modulate the intraburst kinetics of the ATP-sensitive potassium channel. J. Gen. Physiol. 118:341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuveny, E., Y. N. Jan, and L. Y. Jan. 1996. Contributions of a negatively charged residue in the hydrophobic domain of the IRK1 inwardly rectifying K+ channel to K(+)-selective permeation. Biophys. J. 70:754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuveny, E., P. A. Slesinger, J. Inglese, J. M. Morales, J. A. Iniguez-Lluhi, R. J. Lefkowitz, H. R. Bourne, Y. N. Jan, and L. Y. Jan. 1994. Activation of the cloned muscarinic potassium channel by G protein beta gamma subunits. Nature. 370:143–146. [DOI] [PubMed] [Google Scholar]
- Rothberg, B. S., K. S. Shin, P. S. Phale, and G. Yellen. 2002. Voltage-controlled gating at the intracellular entrance to a hyperpolarization-activated cation channel. J. Gen. Physiol. 119:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadja, R., K. Smadja, N. Alagem, and E. Reuveny. 2001. Coupling Gβγ-dependent activation to channel opening via pore elements in inwardly rectifying potassium channels. Neuron. 29:669–680. [DOI] [PubMed] [Google Scholar]
- Sakmann, B., A. Noma, and W. Trautwein. 1983. Acetylcholine activation of single muscarinic K+ channels in isolated pacemaker cells of the mammalian heart. Nature. 303:250–253. [DOI] [PubMed] [Google Scholar]
- Sigworth, F. J., and S. M. Sine. 1987. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys. J. 52:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman, S. K., H. A. Lester, and D. A. Dougherty. 1998. Asymmetrical contributions of subunit pore regions to ion selectivity in an inward rectifier K+ channel. Biophys. J. 75:1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesinger, P. A., E. Reuveny, Y. N. Jan, and L. Y. Jan. 1995. Identification of structural elements involved in G protein gating of the GIRK1 potassium channel. Neuron. 15:1145–1156. [DOI] [PubMed] [Google Scholar]
- Stanfield, P. R., S. Nakajima, and Y. Nakajima. 2002. Constitutively active and G-protein coupled inward rectifier K+ channels: Kir2.0 and Kir3.0. Rev. Physiol. Biochem. Pharmacol. 145:47–179. [DOI] [PubMed] [Google Scholar]
- Tucker, S. J., F. M. Gribble, P. Proks, S. Trapp, T. J. Ryder, T. Haug, F. Reimann, and F. M. Ashcroft. 1998. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 17:3290–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, J., and J. Yang. 2002. Where is the activation gate for PIP2 gating of Kir2.1 channels? Biophys. J. 82:180a. [Google Scholar]
- Yakubovich, D., V. Pastushenko, A. Bitler, C. W. Dessauer, and N. Dascal. 2000. Slow modal gating of single G protein-activated K+ channels expressed in Xenopus oocytes. J Physiol. 524:737–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. C., and F. Sachs. 1989. Block of stretch-activated ion channels in Xenopus oocytes by gadolinium and calcium ions. Science. 243:1068–1071. [DOI] [PubMed] [Google Scholar]
- Yi, B. A., Y. F. Lin, Y. N. Jan, and L. Y. Jan. 2001. Yeast screen for constitutively active mutant G protein-activated potassium channels. Neuron. 29:657–667. [DOI] [PubMed] [Google Scholar]
- Yifrach, O., and R. MacKinnon. 2002. Energetics of pore opening in a voltage-gated K(+) channel. Cell. 111:231–239. [DOI] [PubMed] [Google Scholar]
- Zheng, J., and F. J. Sigworth. 1997. Selectivity changes during activation of mutant Shaker potassium channels. J. Gen. Physiol. 110:101–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, J., and F. J. Sigworth. 1998. Intermediate conductances during deactivation of heteromultimeric Shaker potassium channels. J. Gen. Physiol. 112:457–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H., S. Chepilko, W. Schutt, H. Choe, L. G. Palmer, and H. Sackin. 1996. Mutations in the pore region of ROMK enhance Ba2+ block. Am. J. Physiol. 271:C1949–C1956. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.