

Abstract
Pulmonary surfactant, a lipid/protein complex that lines the air/water interface in the mammalian lung, functions to reduce the work of breathing. Surfactant protein B (SP-B) is a small, hydrophobic protein that is an essential component of this mixture. Structure-function relationships of SP-B are currently under investigation as the protein and its peptide analogs are being incorporated into surfactant replacement therapies. Knowledge of the structure of SP-B and its related peptides in bulk and monolayer phases will facilitate the design of later generation therapeutic agents. Prior infrared reflection-absorption spectroscopic studies reported notable, reversible surface pressure-induced antiparallel β-sheet formation in a synthetic peptide derived from human SP-B, residues 9–36 (SP-B9–36). In the current work, infrared reflection-absorption spectroscopy is applied in conjunction with isotopic labeling to detect the site and pressure dependence of the conformational change. SP-B9–36, synthesized with 13C=O-labeled Ala residues in positions 26, 28, 30, and 32, shifted the β-sheet marker band to ∼1600 cm−1 and thus immediately identified this structural element within the labeled region. Surface pressure-induced alterations in the relative intensities of Amide I band constituents are interpreted using a semiempirical transition dipole coupling model. In addition, electron micrographs reveal the formation of tubular myelin structures from in vitro preparations using SP-B9–36 in place of porcine SP-B indicating that the peptide has the potential to mimic this property of the native protein.
INTRODUCTION
Pulmonary surfactant is a lipid/protein mixture which is suggested to reduce the work of breathing in mammalian lungs by forming a film that modifies the surface properties of the air/alveolar interface. The chemical composition of surfactant is well established. Approximately 90% of the mass is lipid, of which the majority (80–90%) is phospholipid. The most abundant single component is 1,2-dipalmitoylphosphatidylcholine (DPPC) which constitutes ∼40% of the mass. Anionic lipids such as phosphatidylglycerol (PG) constitute ∼10–15% of the lipid mass (Johansson and Curstedt, 1997).
Four surfactant specific proteins (SP-A, SP-B, SP-C, and SP-D) have been identified. Of these, SP-B and SP-C are relatively small, hydrophobic molecules which modify the properties of the surface films. Both proteins enhance the rate of phospholipid adsorption and spreading from the subphase to the air/water interface (Pérez-Gil and Keough, 1998). These observations have led to the design of therapeutic agents for respiratory distress syndrome, a pathological condition resulting from surfactant deficiency. In premature infants, treatments for this disease include natural bovine or porcine extracts, and synthetic preparations of lipid mixtures alone or in combination with isolated SP-B and/or SP-C (Robertson and Halliday, 1998; Johansson et al., 2001). These therapeutic interventions have had mixed success clinically.
The critical nature of SP-B is evident from observations that inherited deficiency of SP-B and SP-B−/−mice develop lethal respiratory disease (Nogee et al., 1994; Clark et al., 1995; Tokieda et al., 1997). Alveolar type II cells in SP-B knockout mice lack well-formed lamellar bodies, which suggests that SP-B plays a role in the generation of this surfactant storage structure (Clark et al., 1995). Functional consequences of this also arise as lamellar bodies are released into the alveolar hypophase, where these densely packed structures are transformed into tubular myelin (TM). TM, the direct precursor of the surface active film, requires both SP-B and hydrophilic, oligomeric SP-A for its in vivo and in vitro formation (Hawgood et al., 1998).
The potential for using SP-B and its peptide analogs for therapeutic intervention has resulted in a plethora of studies geared toward elucidating structure-function relationships for this protein. The mature lipid-associated peptide is 79 amino acids in length, containing ∼50% hydrophobic residues with a net charge of +7 (Weaver and Conkright, 2001; Hawgood et al., 1998). The human sequence is shown in Fig. 1 A. Although the three dimensional structure for the protein has not been reported, sequence homology places SP-B in the saposin-like family for which the NMR structure for NK-lysin has been reported (Liepinsh et al., 1997). Six of the seven cysteine residues in SP-B are highly conserved, forming intramolecular sulfhydryl bridges. A generally accepted structure for the SP-B monomer consists of four to five amphipathic helical segments with a total helical content of ∼50% (Patthy, 1991; Andersson et al., 1995; Weaver and Conkright, 2001).The remaining nonhomologous cysteine residue, Cys48, forms an intermolecular disulfide bond generating a homodimer which is the predominant form of the isolated protein.
FIGURE 1.
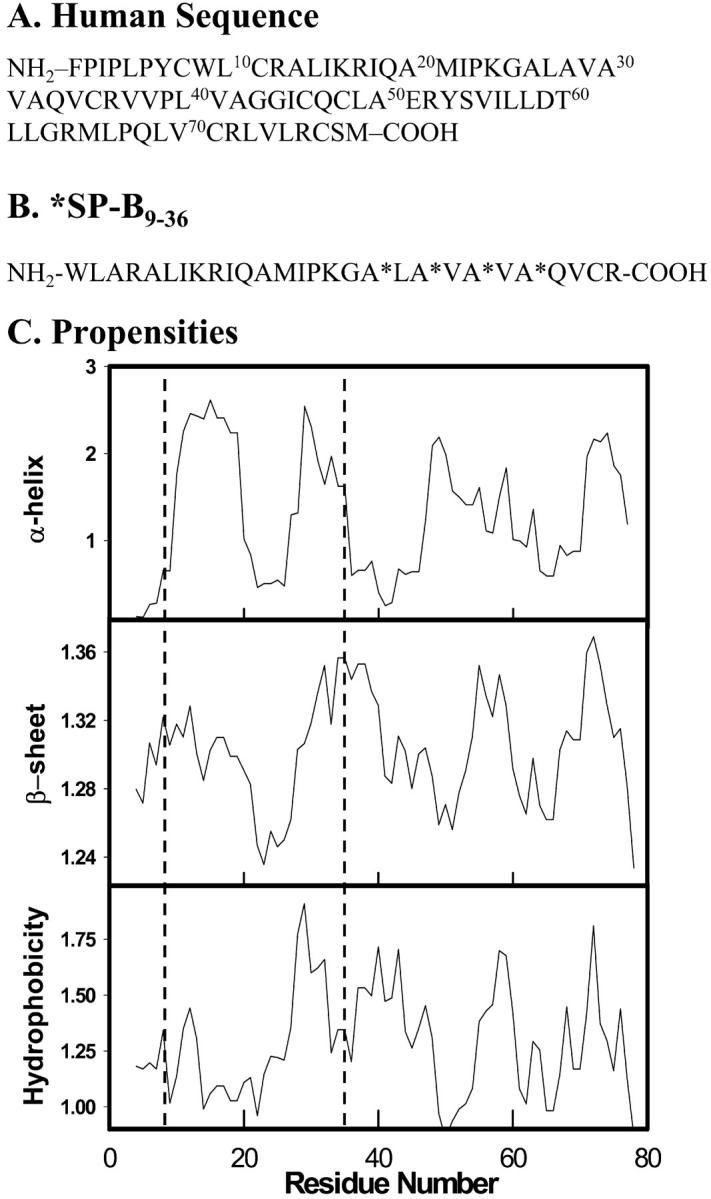
(A) Sequence of human SP-B and (B) the SP-B9–36 peptide with 13C=O labeled Ala residues denoted with (*). (C) Secondary structure predictions and hydrophobicity index for human SP-B calculated using PredictProgram (see text for references). The propensities for the 9–36 peptide are highlighted within the dashed line region.
An inherent property of SP-B is its strong association with lipids, setting this protein apart from other saposin-like members which are only transiently lipid-associated. In the bulk phase, SP-B binds to liposomes, causing destabilization and lipid mixing (Hawgood et al., 1998). These attributes are consistent with the suggested role of SP-B in the packaging of surfactant in lamellar bodies and in the delivery to and dynamics of surfactant at the air/water interface. The protein is thought to interact with lipid headgroups, particularly the anionic phosphatidylglycerol species with the dimer possibly bridging two bilayers or from bilayer to monolayer. While studies have shown that SP-B enhances adsorption and spreading of surfactant phospholipids at air/liquid interfaces, tends to promote respreading of a compressed film, and modulates film composition during dynamic compression and expansion, the molecular mechanisms underlying these actions are poorly understood.
Peptide analogs of SP-B have been employed to better understand the molecular mechanisms of action and to facilitate the design of formulations for therapeutic intervention. This synthetic mimetic approach has been undertaken using two strategies. First, SP-B domains have been modeled with synthetic peptides possessing regions of the native primary sequence producing some useful structural insights. For example, a peptide containing the first 25 residues presumed to consist of the first amphipathic helical segment, SP-B1–25, has been shown to increase the collapse pressure of palmitic acid monolayers (Longo et al., 1993) and induce reversible folding upon collapse and subsequent expansion of surfactant monolayers (Ding et al., 2001). This behavior paralleled that of the full length, native SP-B sequence, and is most likely attributed to an ionic interaction between the net positive charge carried by both the protein and peptide and the anionic nature of the fatty acid monolayer. In addition, SP-B1–25 and its Cys8 S-S-linked homodimer enhanced surface film formation and dynamics with the dimer improving lipid respreadability (Veldhuizen et al., 2000). A second design strategy initiated by Cochrane and colleagues attempts to examine the effects of SP-B's hydrophobic-hydrophilic pattern and charge distribution on surface activity. Analysis of the C-terminal region of SP-B led to the synthesis of peptides such as (KL4)4K (Cochrane and Revak, 1991; Johansson et al., 2001).
The evaluation of structure-function relationships in Langmuir films that model surfactant is limited by the paucity of techniques available for the direct in situ determination of protein structure in aqueous monolayers. Thus, current routine evaluation of synthetic peptide conformation involves structural (for example, nuclear magnetic resonance, circular dichroic, and infrared) measurements in various bulk phase environments coupled with surface activity measurements in monolayers. However, secondary structure determined in bulk phase environments may differ from that found in monolayers, invalidating the correlations drawn between bulk phase structure and surface activity (Dieudonné et al., 1998). It is evident that secondary structure determination of surfactant proteins and related peptides in monolayers is essential for understanding their in vivo function.
In the mid-1980s, Dluhy and co-workers demonstrated the feasibility of acquiring IR reflection-absorption (IRRAS) spectra from aqueous Langmuir films of fatty acids and phospholipids in situ at the air/water interface (Dluhy and Cornell, 1985; Dluhy, 1986). With the success of these measurements, IRRAS provided the means to determine aspects of molecular structure and interactions between monolayer film constituents. A variety of biologically-motivated extensions of this technology ensued. Of those related to pulmonary surfactant, this laboratory used IRRAS to evaluate the conformation of porcine SP-B in the presence and absence of lipids in Langmuir films where the protein was observed to maintain its predominantly α-helical structure (Pastrana-Rios et al., 1995).
IRRAS has also been used to evaluate lung surfactant-related peptides (Bi et al., 2002; Dieudonné et al., 2001). In one prior study, upon which the current investigation is based, this laboratory examined the secondary structure of the synthetic SP-B fragment consisting of native residues 9–36, SP-B9–36 (Dieudonné et al., 2001). A reversible surface pressure-induced conformational change from α-helix ↔ antiparallel β-sheet structure was observed. The current study advances investigation of this peptide in two directions. First, prior IRRAS measurements are extended through the use of 13C isotope labeling to pinpoint the location within the sequence of the surface pressure-induced conformational change in the peptide, SP-B9–36. To our knowledge, this represents the first reported use of peptide isotopic labeling in conjunction with IRRAS measurements. Confidence in the interpretation of spectral changes is strengthened through use of a theoretical model developed to explain anomalous intensities in the solution IR spectra of 13C-labeled peptides in the β-sheet conformation (Brauner et al., 2000). Finally, a possible functional role for this fragment is suggested through the electron microscopy-based observation of tubular myelin formation in bulk phases where native SP-B has been replaced with SP-B9–36.
MATERIALS AND METHODS
Materials
The reagents and solvents used for peptide synthesis were obtained from PerSeptive Biosystems (Framingham, MA). HPLC-grade solvents and salts (Aldrich, Milwaukee, WI and Fisher Scientific, Pittsburgh, PA) were used for the purification of peptides, isolation of surfactant proteins, and preparation of samples. Trizma hydrochloride, HEPES, EDTA, DPPC, egg PG, and sodium chloride were obtained from Sigma (St. Louis, MO). Deuterium oxide (D2O, 99.9%) was purchased from Cambridge Isotope Laboratory (Andover, MA). TAAB 812 resin and sodium cacodylate were purchased from Marivac (Halifax, NS).
Peptide design and synthesis
The primary sequence of the SP-B peptide used in the current experiments (*SP-B9–36) including the position of the isotopically labeled Ala residues (A*) is shown in Fig. 1 B. This region of the native sequence, i.e., residues 9–36, was chosen because it encompasses two predicted helical segments (see Fig. 1 C). Secondary structure predictions in the human sequence were conducted using PredictProgram (Rost and Sander, 1993a,b, 1994). Positioning of the 13C=O labeled residues was based on both the predicted increase in β-sheet propensity in the C-terminus of the peptide (Fig. 1 C) and the enhanced Amide I band intensity distribution observed for alternating 13C=O/12C=O residues in an antiparallel β-sheet structure (Brauner et al., 2000). Replacing one Cys residue (Cys11) in the human sequence with Ala was found to reduce disulfide bond formation to an undetectable level as indicated by mass spectrometry (see following).
Peptide synthesis and purification followed the same protocol as described in Dieudonné et al. (2001). Briefly, the peptide was synthesized using standard procedures for solid phase Fmoc chemistry on a Millipore 9050 Plus synthesizer and was cleaved from the PAC-PEG-PS solid support to obtain a C-terminal carboxylic acid. *SP-B9–36 was purified by reverse-phase HPLC using a YMC ODS-AL semipreparative column (Waters, Milford, MA) with an eluent gradient of water/acetonitrile, both containing 0.1% TFA. The identity and purity of the fraction collected was confirmed by electrospray ionization mass spectrometry. Mass spectrometry analysis was performed at various ionization energies to evaluate dimerization due to disulfide bond formation. Mass spectra at all ionization energies showed only one major component having a molecular weight consistent with monomer *SP-B9–36.
Bulk phase Fourier transform infrared spectroscopy
Fourier transform infrared transmission spectra were collected for the *SP-B9–36 in methanol (∼2 mg/mL) at ambient temperature. A fixed pathlength (100 μm) CaF2 sample cell was used. Spectra were obtained using a Mattson RS-1 (Madison, WI) spectrometer with a mercury-cadmium-telluride detector. Interferograms were collected with a total of 1024 scans acquired at ∼4 cm−1 spectral resolution in eight blocks of 128 scans each, co-added, apodized with a triangular function, and Fourier-transformed with one level of zero filling to yield spectra encoded at 2 cm−1 intervals. Analysis of the Amide I region was made after subtraction of a pure methanol spectrum from the sample.
Monolayer sample preparation, IRRAS measurements, and spectral analysis
Pure *SP-B9–36 peptide was dissolved in chloroform/methanol (4:1, v/v) at ∼0.5 mg/mL and native SP-B was prepared in chloroform/methanol (1:1, v/v) at ∼1 mg/mL.
A D2O-based subphase consisting of 100 mM NaCl and 0.1 mM EDTA in 10 mM Tris buffer at pD 7 was used for all IRRAS experiments. A D2O buffer is used to eliminate the reflectance-absorbance from the H2O bending vibration and to lessen the absorbance from the rotation-vibration bands of water vapor, both of which occur in the conformation-sensitive Amide I spectral region. A custom-designed Langmuir trough (maximum surface area of 86 cm2) constructed by Nima Technology (Coventry, England), equipped with a Model PS4 surface pressure sensor, was used for all experiments. The subphase temperature was held at 21.0 ± 0.5°C. Aliquots of 20–25 μL and 10 μL were spread dropwise on a clean surface for *SP-B9–36 and native SP-B, respectively. A minimum 45-min period was allowed for solvent evaporation and film relaxation/equilibration before compression. All samples were spread with initial pressure <4 mN/m and π-A isotherms were acquired during intermittent compression and expansion. A relaxation period of ∼20 min was allowed between stopping the barrier at desired surface pressure values and IRRAS spectral acquisition. The surface pressure drop during the relaxation period was small, <3 mN/m in the high pressure region and <1 mN/m in the low pressure region.
IRRAS spectra were acquired with a Equinox 55 Spectrometer (Bruker Optics, Billerica, MA) equipped with an external variable angle reflectance accessory, the XA-511. The accessory has been coupled to the Langmuir trough. A detailed description of the instrument has been published (Flach et al., 2001). Briefly, the infrared (IR) beam is directed through the external port in the spectrometer and is reflected by three mirrors in a rigid mount before being focused on the water surface. Computer-driven stepper motors rotate the mirrors to obtain the desired angle of incidence. A wire grid polarizer is placed into the optical path directly before the beam impinges on the water surface. The reflected light is collected at the same angle as the angle of incidence, follows an equivalent mirror path, and is directed onto a narrow band mercury-cadmium-telluride detector. The entire experimental setup is enclosed and purged to keep the relative humidity levels as low and as constant as feasible.
Interferograms were collected with the use of a sample shuttle program to compensate for the residual water vapor rotation-vibration bands in the Amide I region. A total of 1024 scans were acquired at 8 cm−1 resolution, in two blocks of 512 scans each, co-added, apodized with a Blackman-Harris-3-term function, and fast Fourier-transformed with two levels of zero-filling to produce spectral data encoded at 2 cm−1 intervals. Spectra were acquired using s-polarized radiation at a 35° angle of incidence and p-polarized radiation at 40°. Polarized radiation can be used in IRRAS measurements to obtain the orientation of transition dipoles and therefore, tilt angles of particular molecular segments of interest. This topic has been recently elaborated upon in a review by Mendelsohn and Flach (2002), and an article of Bi et al. (2002). IRRAS band intensities measured for spectra obtained using p-polarized radiation are much more sensitive to changes in transition dipole orientation than their s-polarized counterparts. Therefore, spectra acquired using s-polarized radiation generally provide information about molecular structure or conformation as obtained from the band positions (frequencies), whereas the band intensities generated using p-polarization in conjunction with s-polarization provide orientation information.
Data analysis was performed using Grams/32 (Galactic Industries, Salem, NH). Spectra are presented after linear baseline correction. Water vapor subtraction was not necessary. IRRAS spectra of the Amide I region were curve-fit, using two Gaussian bands and limiting the width at half-height for the band found at ∼1597 cm−1 to 25 ± 5 cm−1.
Transition dipole coupling model and spectral simulations
A semiempirical method has been developed to quantitatively examine the complex Amide I contour in the IR spectra of proteins and peptides (Brauner et al., 2000). The method, which calculates Amide I band components for α-helical and antiparallel β-sheet geometries, is based on transition dipole coupling and interactions through valance bonds between adjacent oscillators and through hydrogen bonds. Simulations reproduced the downward frequency shift observed for a single 13C=O substituent in an α-helical peptide. This frequency shift can also be predicted using elementary methods based on the increased mass in an harmonic oscillator approximation. The utility of the semiempirical method was demonstrated by simulations of several 13C=O-labeled β-sheet peptides where an anomalous intensity distribution in Amide I band components (not predicted by elementary methods) was reproduced. In the current study, the majority of the parameters required as input were taken directly from the work of Brauner et al. (2000). A few were modified to obtain a better fit to the experimental IRRAS spectra and are listed here. An initial, unperturbed frequency of 1665 cm−1 was used for both β-sheet and α-helical geometries. Amide I band constituents for both conformations were generated by representing each component as a 30/70 Gaussian/Lorentzian function with a half-width of 12 cm−1 for the β-sheet component band and 18 cm−1 for the α-helical modes. The effect of hydrogen-deuterium exchange on Amide I band positions can be accommodated for in the simulations by adjusting the H-bonding interaction constant and/or the initial, unperturbed frequency, although the calculation does not explicitly require the mass of hydrogen or deuterium.
Surfactant protein isolation and tubular myelin preparation
SP-B was isolated from porcine lavage by the method of Curstedt et al. (1988) and Taneva and Keough (1994). SP-A from porcine lavage was prepared by the method of Haagsman et al. (1987) and Taneva et al. (1995). The concentration of protein was determined by fluorescamine assay (Udenfriend et al., 1972), and amino acid analysis (Sarin et al., 1990) was performed. Lipid concentration was determined by phosphorus assay (Bartlett, 1959; Keough and Kariel, 1987).
DPPC/egg PG (7:3 molar ratio) and 15% (by weight) SP-B or SP-B9–36 were mixed in 1:1 chloroform/methanol, dried under N2, and evacuated overnight in a vacuum desiccator. Two mL of buffer (10 mM Tris, 145 mM NaCl, and 1 mM EDTA, pH 7.4) was added to the dried sample, which was then hydrated at 46°C for 1 h, with frequent vortexing. The sample was centrifuged at 50,000 g for 30 min, the pellet resuspended in 150 μl buffer, and the lipid content determined. SP-A in buffer (5 mM HEPES, pH 7.4) was added, to give a final SP-A concentration of 22% of the phospholipid weight. After a 10–15 min incubation at 37°C, calcium was added to a final concentration of 5 mM. The sample was incubated overnight at 37°C with continuous mixing.
For the isolation of native tubular myelin, rat lungs were lavaged twice with 0.9% NaCl and 5 mM calcium. The lavage was centrifuged at 4°C for 10 min at 150 g to remove cell debris and the supernatant centrifuged at 40,000 g for 15 min. The pellets were resuspended in 500 μl of the remaining supernatant.
Electron microscopy
Gluteraldehyde and osmium tetroxide (OsO4) in a sodium cacodylate buffer were added to the lipid/protein samples to give a final concentration of 2% glutaraldehyde, 1% OsO4, and 0.1 M sodium cacodylate (pH 7.4). After 1 h at room temperature, the sample was pelleted in a microfuge and left overnight. The sample was again centrifuged, supernatant was removed and replaced with 2% uranyl acetate. The sample was held at 4°C overnight. The pellet was then dehydrated using acetone, from 70–100% in 10% steps, before being embedded in TAAB 812 resin. After cutting, the sections were stained with uranyl acetate and lead citrate and viewed using a Zeiss EM 109 electron microscope (Carl Zeiss Canada, Toronto, Canada).
RESULTS
Bulk phase IR spectra of the Amide I region for the unlabeled SP-B9–36 peptide and the 13C-carbonyl group-labeled derivative (*SP-B9–36) acquired in methanol are shown in Fig. 2. Two major Amide I band components are observed for the unlabeled peptide at ∼1657 and 1628 cm−1 which indicate the presence of α-helical and antiparallel β-sheet structures, respectively. The weak high frequency component of the antiparallel β-sheet structure usually observed at ∼1690 cm−1 is obscured by the dominant α-helical band at 1657 cm−1. Isotopic labeling of the peptide changes the Amide I band profile markedly as a new band is observed at 1600 cm−1. Assignment of the low frequency band to antiparallel β-sheet structure in the 13C-labeled region of the peptide is consistent with an anticipated downward isotopic shift. The observed frequency decrease of ∼28 cm−1 is less than that expected for 13C substitution in an isolated C=O stretching frequency (∼37 cm−1) due to the presence of alternating labeled and unlabeled oscillators. This observation will be elaborated on in the Discussion. Some intensity is also observed in the 1620–1630 cm−1 region in the spectrum of the *SP-B9–36 peptide suggesting that a small amount of β-sheet structure may be present in the unlabeled region of this peptide or that additional secondary structure elements are present in the labeled region of the peptide.
FIGURE 2.

Bulk phase IR spectra of the Amide I and II region for the labeled and unlabeled SP-B9–36 peptides in methanol at ∼2 mg/mL.
IRRAS experiments were conducted on monolayers of *SP-B9–36 and compared to native porcine SP-B and the unlabeled SP-B9–36 under similar conditions. Fig. 3 displays IRRAS spectra of all three acquired at a surface pressure of ∼27 mN/m. A large Amide I band constituent assigned to antiparallel β-sheet structure is observed for both the labeled (1597 cm−1) and the unlabeled (1626 cm−1) peptides, whereas the native protein shows much less intensity in the 1626 cm−1 region at the air/water interface. The predominant Amide I component for the three monolayers is found at ∼1645 cm−1, most likely indicative of α-helical or a mix of helix/random coil structures after H→D exchange. The extent of exchange is essentially complete as revealed by the absence of an Amide II band at ∼1550 cm−1.
FIGURE 3.

IRRAS spectra (1750–1540 cm−1 region) of native, porcine SP-B, and the labeled and unlabeled SP-B9–36 peptide monolayers acquired using s-polarization at surface pressure of ∼27 mN/m on a D2O-buffered subphase.
In a previous publication, unlabeled SP-B9–36 was shown to exhibit reversible surface pressure-induced antiparallel β-sheet formation in both the presence and absence of a lipid monolayer (Dieudonné et al., 2001). With the potential to extract the additional structural information provided by specific isotopic labeling, similar IRRAS experiments were conducted using *SP-B9–36. IRRAS spectra of the Amide I region for a *SP-B9–36 peptide monolayer are displayed in Fig. 4 A. The spectra were acquired using s-polarized radiation during the compression and subsequent expansion of the monolayer. The relative intensity of the low frequency component (1597 cm−1) is observed to increase upon compression and decrease as the surface area is expanded. This observation is more clearly displayed in the inset to the figure, where the ratio of the curve fit band areas (1597/1645 cm−1) are plotted as a function of surface pressure. Although there is some scatter in the data, the trend and its reversibility are apparent. In addition, a substantial difference in the widths of the two major Amide I band components is evident in Fig. 4 A. This most likely reflects heterogeneity in the mixture of helix/random coil structures for the 1645 cm−1 constituent and a more homogeneous β-sheet structure for the 1597 cm−1 component.
FIGURE 4.
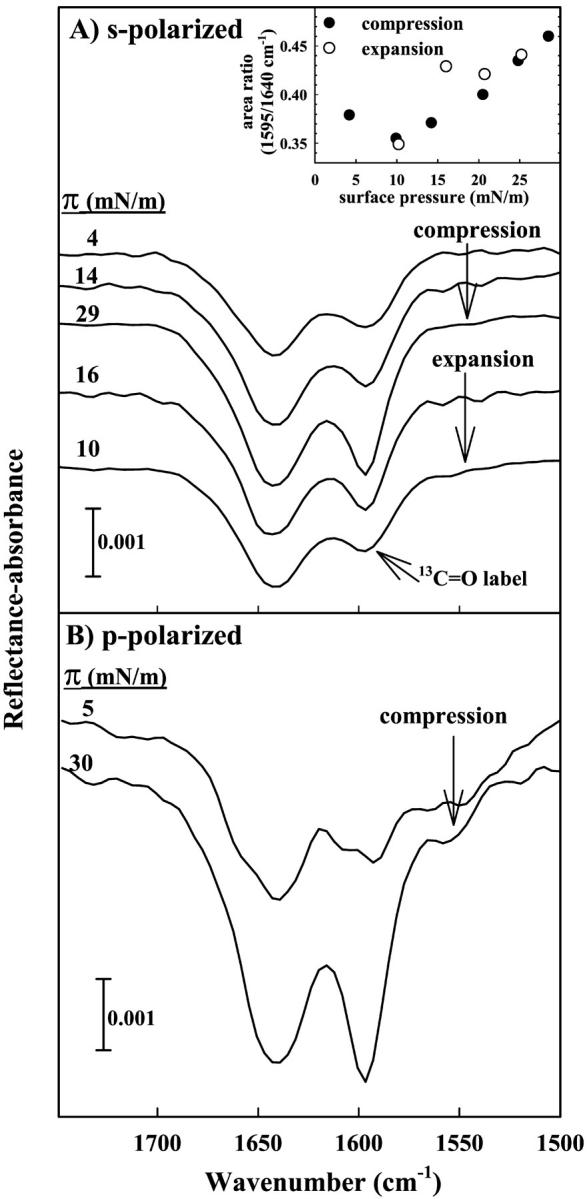
IRRAS spectra of the Amide I region acquired for *SP-B9–36 on a D2O subphase. (A) Spectra are acquired using s-polarization and surface pressure values are noted from top to bottom during compression and subsequent expansion of the monolayer. The inset displays the pressure dependence of the relative area of the curve fit 1597 and 1640 cm−1 Amide I band components during intermittent compression (•) and expansion (○) of the film. (B) Spectra acquired using p-polarized radiation during the compression of an *SP-B9–36 peptide monolayer.
To evaluate whether the observed Amide I intensity variations arise from changes in the orientation of secondary structure elements within the peptide, IRRAS spectra were also acquired using p-polarized radiation (shown in Fig. 4 B). Although the noise level in the spectra acquired using p-polarization is slightly greater than in the s-polarized spectra, the relative intensity changes observed upon compression are essentially the same for both polarizations when comparing spectra at similar surface pressure values. This observation demonstrates that the majority of the intensity variation in the Amide I contour is not due to pressure-induced alterations in orientation as has been previously detailed in Bi et al. (2002). Possible structural factors responsible for this phenomenon are referred to the Discussion.
Electron micrographs of tubular myelin (TM) structures isolated from rat lavage and prepared from native, porcine SP-B, and SP-B9–36 are shown in Fig. 5 A–C, respectively. Surface balance experiments have shown that the presence of meshlike TM structures isolated from surfactant fractions appear to enhance adsorption and spreading when compared to surfactant fractions rich in small vesicles (Goerke, 1998). Although less TM is observed in the micrographs prepared using the 9–36 peptide compared to native SP-B (∼1% vs. 10% of micrograph area counted, respectively), the generation of even small amounts of TM with the peptide suggests that certain properties of the peptide are sufficient to mimic the native protein's role in TM formation.
FIGURE 5.
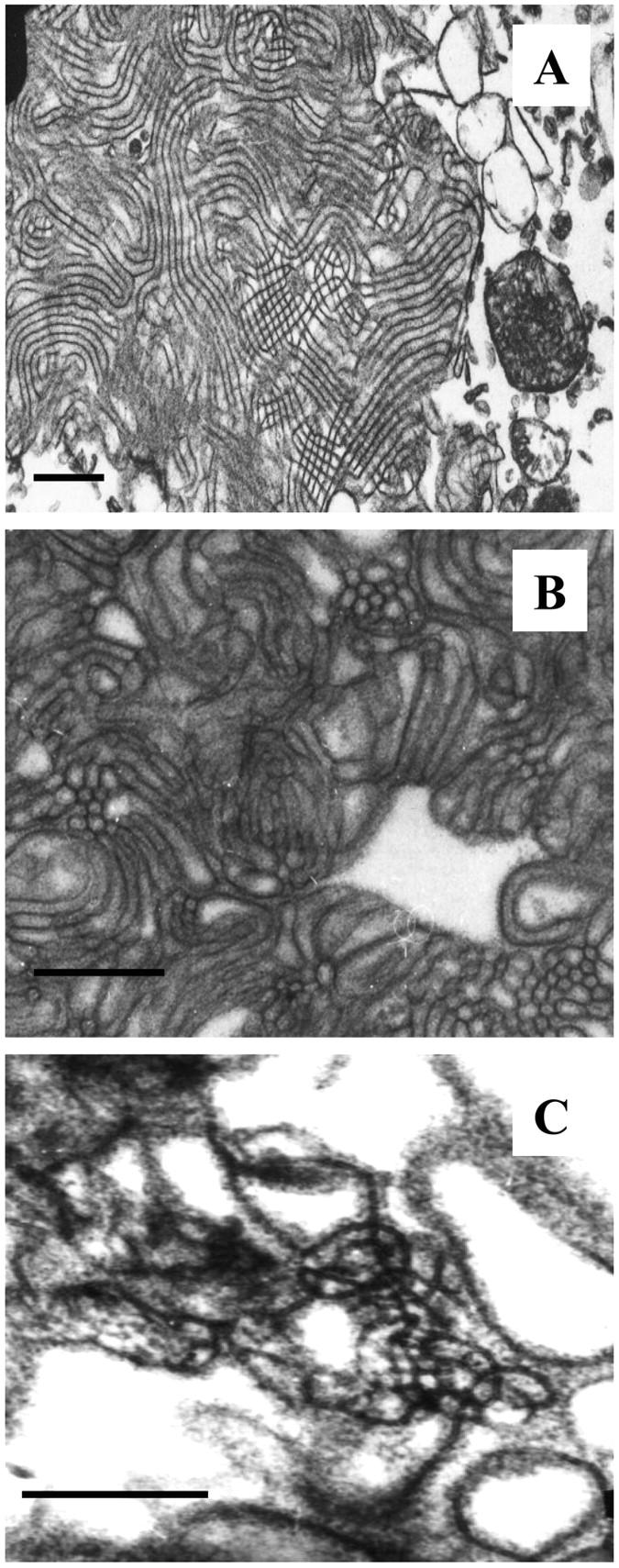
Electron micrographs of (A) tubular myelin isolated from rat lungs, bar = 500 nm; (B) an in vitro preparation of TM using native, porcine SP-B, bar = 500 nm; and (C) TM prepared in vitro using SP-B9–36 in place of native SP-B, bar = 250 nm.
DISCUSSION
With the thought of potential therapeutic agents in mind, many surfactant-based peptides are currently under evaluation. It is important to evaluate these peptides in both bilayer and monolayer environments. Understanding the mechanics of peptide function in lung-related environments will likely aid in the elucidation of intact pulmonary surfactant protein function as well. In the current investigation, IRRAS, in conjunction with isotopic labeling, has been applied to determine the location of particular secondary structure elements within an SP-B-based peptide that is also shown to form the physiologically relevant structure of tubular myelin.
The bulk phase spectra (Fig. 2) immediately identify the position of β-sheet secondary structure within the peptide to the general region of the 13C=O label. The reversible formation of an antiparallel β-sheet structure is evident for SP-B9–36, both from the current spectra and from our earlier investigations.
Several structural issues arising from the current experiments bear comment. The formation of an antiparallel (in contrast to parallel) β-sheet is not surprising, considering that residues 9–20 (the N-terminal of SP-B9–36) exhibit a substantial propensity toward helix formation (see Fig. 1). Indeed, the most intense IRRAS spectral feature (Fig. 3) is assigned to an overlapped contour arising from helix/random secondary structure that is not much altered by isotopic labeling. Thus, the formation of the intermolecular antiparallel β-sheet structure, which is localized to residues beyond residue ∼20 or so, can be rationalized on the basis that steric hindrance induced by the presence of helical/random structures at the N-termini of two strands in close proximity will limit the ability of intermolecular parallel sheets to form.
To understand the spectra in more detail, we have developed a model for simulating Amide I contours using a semiempirical approach which incorporates transition dipole coupling and through-bond interactions within the context of the Wilson FG matrix method (Wilson et al., 1955). This approach produced excellent agreement between calculated and observed Amide I spectra for four isotopic variants (13C in selected residues) of the K2(LA)6 peptide (Brauner et al., 2000).
Fig. 6 A displays simulated spectra of the Amide I region for an antiparallel β-sheet peptide consisting of two strands with 12 amino acid residues each. The dependence of peak position on the placement of 13C=O labels is evident. The frequencies of the simulated major Amide I components are observed at 1626, 1599, and 1585 cm−1 for the unlabeled, alternating labeled/unlabeled, and fully labeled peptides, respectively. The position of the low frequency Amide I component for the unlabeled peptide and peptide with alternating labels are essentially the same as those observed experimentally for the corresponding unlabeled and labeled SP-B9–36 peptides, indicating that the assignment of β-sheet structure to the labeled region of the peptide is indeed correct. It is interesting to note that the full magnitude of the predicted 37 cm−1 frequency shift (based on the harmonic oscillator model of a simple diatomic molecule) is reproduced in the simulated spectrum of the fully labeled antiparallel β-sheet peptide. Simulations also reveal that the attenuation in the magnitude of the frequency shift for the alternating labels is due to coupling between 13C=O and 12C=O oscillators.
FIGURE 6.
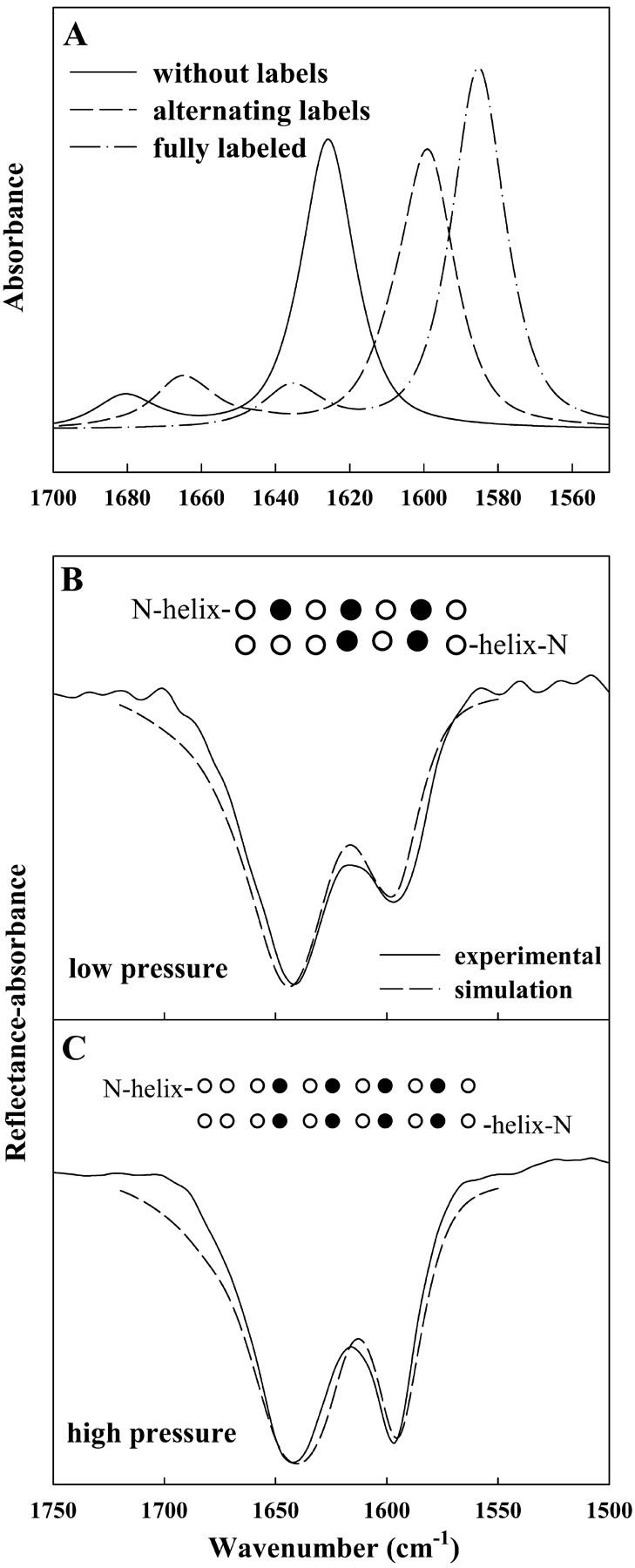
Calculated IR spectra of the Amide I region using a transition dipole coupling method as described by Brauner et al. (2000). (A) Simulations of three peptides, 12 residues in each strand, arranged in an antiparallel β-sheet geometry: (—) all unlabeled residues, (– –) alternating 13C=O/12C=O, and (– · –) all 13C=O labeled residues. (B) Experimental spectrum of *SP-B9–36 acquired at a pressure of 4 mN/m overlaid with an inverted simulated spectrum of two identical peptides with their C-termini in an antiparallel β-sheet geometry. Each peptide consists of 14 residues in an α-helical geometry coupled to an antiparallel β-sheet strand containing seven residues with 13C=O labels as noted (•) in inset. (C) Experimental spectrum acquired at a pressure of 29 mN/m and simulated spectrum similar to B, except that the β-strand portion of each peptide consists of 11 residues, with the labeling differing as noted in the inset. The β-sheet components in the simulated spectra in B and C were multiplied by 1.5 before being added to the helical component.
An interesting consideration in the current work is the nature of the aggregation state (dimers or more extended structures) of these peptides on the aqueous surface. Casual inspection of the Amide I contour cannot reveal the aggregation state as the IR spectra-structure correlations commonly used were established for secondary structure. Determination of higher order structural elements along with elucidation of the effects of isotopic substitution on the Amide I contour require a theoretical approach for spectral simulation. The implicit assumption made in the semiempirical simulations in Fig. 6 is that dimers were the aggregation state, although the nature of the model is such that simulations of higher order aggregates would probably also lead to agreement between the observed and calculated Amide I contour. More recently, Keiderling and his colleagues (Kubelka and Keiderling, 2001) have carried out elaborate (ab initio) calculations to simulate the Amide I contour in isotopically labeled antiparallel β-sheets of various aggregation states of the K2(LA)6 peptides. They conclude that the characteristic intensity enhancement of some components of the contour (observed experimentally in this laboratory) requires sheets comprised of clusters of least five strands to obtain agreement between experimental and simulated results.
To utilize either the ab initio or semiempirical models to explain the IRRAS observations of surface pressure-induced intensity enhancement of the 1600 cm−1 band requires consideration of the following experimental facts, as well as limitations of the theoretical approaches:
The surface pressure-dependent helix/random ↔ β-structure interconversion is reversible. It seems reasonable to suggest that this experimental observation is incompatible with extensive strand formation, which (in bulk phases) would probably lead to gel formation and concomitant irreversibility.
The forces that stabilize secondary structures at the air/water interface are most likely quite different than in bulk phases. Kubelka and Keiderling (2001) note that, for K2(LA)6, “if the strands were to align in an exact antiparallel fashion…the leucine side chains on neighboring strands would point alternatively above and below the β-sheet plane, thus causing no packing hindrance.” They indicate that multistrand formation could easily be accommodated within this model. While these statements are certainly reasonable, they may not be applicable to the SP-B9–36 peptide. In the current instance, steric effects imposed by the presence of helix/random conformations at the N-terminal may hinder the interaction between the individual strands and limit the formation of multistrands.
Both the ab initio calculations and the semiempirical approach assume an idealized geometry for each β-strand. This may be a severe approximation for simulations of the current IRRAS data, as it is unlikely that the small peptide strands interacting here, at the asymmetric environment of the air/water interface, would assume an ideal structure. The effects of structural variations from idealized β-geometries on the results of either the ab initio or semiempirical spectral simulations cannot be easily predicted without extensive computations.
Thus, the nature of the experimental conditions (IRRAS, presence of more than one secondary structure) and limitations on the theoretical calculations (short segments, unknown geometry) would seem to suggest that both theoretical approaches require severe approximations. The spectral simulations discussed below are presented with the above limitations in mind.
To provide an initial explanation for the observed surface pressure-induced changes in the Amide I contour, we utilized the semiempirical model to analyze the relative increase in the 1597 cm−1 component in the Amide I region as a function of surface pressure (see Fig. 4). It is important to apply the model in a way that is consistent with the IRRAS observations, e.g., reversible surface pressure-induced results for both the labeled and unlabeled peptides. The previous report for the unlabeled peptide describes an increase in the relative intensity of the β-sheet Amide I band component versus the helical/unordered component with surface pressure (Dieudonné et al., 2001) similar to that observed in the current investigation. One possible interpretation is represented in Fig. 6, B and C, where simulated spectra that have been inverted are overlaid with experimental IRRAS spectra at low (4 mN/m) and high (29 mN/m) surface pressure. The simplest model to account for the observed spectral changes is an increase in the number of peptide bonds in the β-sheet region with increasing pressure. Simulations carried out with the number of peptide bonds in the β-sheet region of both strands increasing from 14 with five being isotopically labeled (at 4 mN/m) to 22 with eight labels (29 mN/m) reproduced the changes monitored in the experimental spectra. Schematic depictions of the β-sheet region in each case are shown as insets in Fig. 6, B and C. This simple model accounts for the pressure-dependent changes observed in the unlabeled SP-B9–36 peptide as well. We recognize that as our knowledge of the detailed three-dimensional structure of the peptide at the air/water interface increases, more sophisticated models will be applicable for modeling the surface pressure-induced alterations.
A detailed mechanism of the above-described event requires the formation and cleavage of hydrogen bonds as surface area is reduced and subsequently expanded. Similar reversible phenomena involving a lung surfactant SP-C-based peptide were recently reported, although the composition of secondary structure differed in that peptide (Bi et al., 2002). In the SP-C peptide system, the extent of helix hydration varied with surface pressure. Although conditions in both the SP-C and SP-B9–36 peptide monolayer experiments differ substantially from the natural system, surface pressure-induced conformational flexibility in surfactant-based peptides may be relevant to the functional properties of pulmonary surfactant proteins at the air/alveolar interface.
Structural transitions in the peptide may also be important in promoting the formation of tubular myelin. Furthermore, electrostatic interactions between anionic lipids and native SP-B are presumed to play a role. *SP-B9–36 carries the majority of the positive charge (+5) for the native protein (net charge +7). It is possible that the concentration of positive charge is an important factor in the initiation of TM formation. A few synthetic peptides based on N-terminal regions of SP-B have been successful in substituting for the native protein in TM preparations. These include the SP-B1–25 monomer and dimer peptides, along with SP-B11–25 (Keough et al., 2002), all of which carry a +5 charge. However, attempts to generate tubular myelin with a derivative of the 11–25 peptide where all the positively charged residues were replaced with Ser, have not been successful (Keough et al., 2002). An earlier publication on synthetic SP-B1–25 peptides containing amino acid substitutions that altered charge and hydrophobicity reported reduced surface activity with reversal of charge in the peptide and a correlation between helical structure in bulk phase and the degree of interaction with lipids (Bruni et al., 1991). The pressure-dependent secondary structure of SP-B1–25 and its related peptides has not yet been determined at the air/water interface. In the current work, the importance of electrostatic interaction appears clear, whereas establishing a relationship between structure and lipid interaction in both monolayers and TM needs further investigation.
The low abundance of TM in reconstituted systems in conjunction with the required presence of SP-B and SP-A make the investigation of protein structure in TM extremely difficult. The observation of TM in the in vitro preparations with SP-B9–36 in place of the native protein along with isotopic labeling to enhance the resolution of Amide I constituents provides the first step in overcoming these difficulties.
Acknowledgments
This work was supported by a Public Health Service grant (National Institutes of Health, GM29864) to R.M., and an operating grant from the Canadian Institutes of Health Research to K.M.W.K.
References
- Andersson, M., T. Curstedt, H. Jornvall, and J. Johansson. 1995. An amphipathic helical motif common to tumourolytic polypeptide NK-lysin and pulmonary surfactant polypeptide SP-B. FEBS Lett. 362:328–332. [DOI] [PubMed] [Google Scholar]
- Bartlett, G. R. 1959. Phosphorous assay in column chromatography. J. Biol. Chem. 234:466–468. [PubMed] [Google Scholar]
- Bi, X., C. R. Flach, J. Perez-Gil, I. Plasencia, D. Andreu, E. Oliveira, and R. Mendelsohn. 2002. Secondary structure and lipid interactions of the N-terminal segment of pulmonary surfactant SP-C in Langmuir films: IR reflection-absorption spectroscopy and surface pressure studies. Biochemistry. 41:8385–8395. [DOI] [PubMed] [Google Scholar]
- Brauner, J. W., C. Dugan, and R. Mendelsohn. 2000. 13C isotope labeling of hydrophobic peptides. Origin of the anomalous intensity distribution in the infrared Amide I spectral region of β-sheet structures. J. Am. Chem. Soc. 122:677–683. [Google Scholar]
- Bruni, R., H. W. Taeusch, and A. J. Waring. 1991. Surfactant protein B: lipid interactions of synthetic peptides representing the amino-terminal amphipathic domain. Proc. Natl. Acad. Sci. USA. 88:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, J. C., S. E. Wert, C. J. Bachurski, M. T. Stahlman, B. R. Stripp, T. E. Weaver, and J. A. Whitsett. 1995. Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc. Natl. Acad. Sci. USA. 92:7794–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane, C. G., and S. D. Revak. 1991. Pulmonary surfactant protein B (SP-B): structure-function relationships. Science. 254:566–568. [DOI] [PubMed] [Google Scholar]
- Curstedt, T., J. Johansson, J. Barros-Soderling, B. Robertson, G. Nilsson, M. Westberg, and H. Jornvall. 1988. Low-molecular-mass surfactant protein type 1. The primary structure of a hydrophobic 8-kDa polypeptide with eight half-cysteine residues. Eur. J. Biochem. 172:521–525. [DOI] [PubMed] [Google Scholar]
- Dieudonné, D., A. Gericke, C. R. Flach, X. Jiang, R. S. Farid, and R. Mendelsohn. 1998. Propensity for helix formation in the hydrophobic peptides K2(LA)x (x = 6, 8, 10, 12) in monolayer, bulk, and lipid-containing phases. Infrared and circular dichroism studies. J. Am. Chem. Soc. 120:792–799. [Google Scholar]
- Dieudonné, D., R. Mendelsohn, R. S. Farid, and C. R. Flach. 2001. Secondary structure in lung surfactant SP-B peptides: IR and CD studies of bulk and monolayer phases. Biochim. Biophys. Acta. 1511:99–112. [DOI] [PubMed] [Google Scholar]
- Ding, J., D. Y. Takamoto, A. von Nahmen, M. M. Lipp, K. Y. C. Lee, A. J. Waring, and J. A. Zasadzinski. 2001. Effects of lung surfactant proteins, SP-B and SP-C, and palmitic acid on monolayer stability. Biophys. J. 80:2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dluhy, R. A. 1986. Quantitative external reflection infrared spectroscopic analysis of insoluble monolayers spread at the air-water interface. J. Phys. Chem. 90:1373–1379. [Google Scholar]
- Dluhy, R. A., and D. G. Cornell. 1985. In situ measurement of the infrared spectra of insoluble monolayers at the air-water interface. J. Phys. Chem. 89:3195–3197. [Google Scholar]
- Flach, C. R., Z. Xu, X. Bi, J. W. Brauner, and R. Mendelsohn. 2001. Improved IRRAS apparatus for studies of aqueous monolayer films: determination of the orientation of each chain in a fatty-acid homologous ceramide-2. Appl. Spectrosc. 55:1060–1066. [Google Scholar]
- Goerke, J. 1998. Pulmonary surfactant: functions and molecular composition. Biochim. Biophys. Acta. 1408:79–89. [DOI] [PubMed] [Google Scholar]
- Haagsman, H. P., S. Hawgood, T. Sargeant, D. Buckley, W. R. Tyler, K. Drickamer, and B. J. Benson. 1987. The major lung surfactant protein, SP 28–36, is a calcium-dependent, carbohydrate-binding protein. J. Biol. Chem. 262:13877–13880. [PubMed] [Google Scholar]
- Hawgood, S., M. Derrick, and F. Poulain. 1998. Structure and properties of surfactant protein B. Biochim. Biophys. Acta. 1408:150–160. [DOI] [PubMed] [Google Scholar]
- Johansson, J., and T. Curstedt. 1997. Molecular structures and interactions of pulmonary surfactant components. Eur. J. Biochem. 244:675–693. [DOI] [PubMed] [Google Scholar]
- Johansson, J., T. Curstedt, and B. Robertson. 2001. Artificial surfactants based on analogues of SP-B and SP-C. Pediatr. Pathol. Mol. Med. 20:501–518. [DOI] [PubMed] [Google Scholar]
- Keough, K. M., and N. Kariel. 1987. Differential scanning calorimetric studies of aqueous dispersions of phophatidylcholines containing two polyenoic chains. Biochim. Biophys. Acta. 902:11–18. [DOI] [PubMed] [Google Scholar]
- Keough, K., S. Taneva, F. J. Walther, and A. Waring. 2002. Adsorption of lung surfactant protein SP-A to lipid monolayers containing N-terminal segments of surfactant protein SP-B causes segregation of a protein-rich surface phase. Biophys. J. 82:8a. [Google Scholar]
- Kubelka, J., and T. A. Keiderling. 2001. The anomalous infrared Amide I intensity distribution in 13C isotopically labeled peptide β-sheets comes from extended, multiple-stranded structures. An ab initio study. J. Am. Chem. Soc. 123:6142–6150. [DOI] [PubMed] [Google Scholar]
- Liepinsh, E., M. Andersson, J. M. Ruysschaert, and G. Otting. 1997. Saponsin fold revealed by the NMR structure of NK-lysin. Nat. Struct. Biol. 4:793–795. [DOI] [PubMed] [Google Scholar]
- Longo, M. L., A. M. Bisagno, J. A. N. Zasadzinski, R. Bruni, and A. J. Waring. 1993. A function of lung surfactant protein SP-B. Science. 261:453–456. [DOI] [PubMed] [Google Scholar]
- Mendelsohn, R., and C. R. Flach. Infrared reflection-absorption spectroscopy of lipid, peptides, and proteins in aqueous monolayers. In Current Topics in Membranes, Vol. 52. S. A. Simon and T. J. McIntosh, editors. Academic Press, San Diego. pp. 57–88.
- Nogee, L. M., G. Garnier, H. C. Dietz, L. Singer, A. M. Murphy, D. E. deMello, and H. R. Colten. 1994. A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. J. Clin. Invest. 93:1860–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana-Rios, B., S. Taneva, K. M. W. Keough, A. J. Mautone, and R. Mendelsohn. 1995. External reflection absorption infrared spectroscopy study of lung surfactant proteins SP-B and SP-C in phospholipid monolayers at the air/water interface. Biophys. J. 69:2531–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patthy, L. 1991. Homology of the precursor of pulmonary surfactant-associated protein SP-B with prosaposin and sulfated glycoprotein-1. J. Biol. Chem. 266:6035–6037. [PubMed] [Google Scholar]
- Pérez-Gil, J., and K. M. W. Keough. 1998. Interfacial properties of surfactant proteins. Biochim. Biophys. Acta. 1408:203–217. [DOI] [PubMed] [Google Scholar]
- Robertson, B., and H. L. Halliday. 1998. Principles of surfactant therapy. Biochim. Biophys. Acta. 1408:346–361. [DOI] [PubMed] [Google Scholar]
- Rost, B., and C. Sander. 1993a. Improved prediction of protein secondary structure by use of sequence profiles and neural networks. Proc. Natl. Acad. Sci. USA. 90:7558–7562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost, B., and C. Sander. 1993b. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232:584–599. [DOI] [PubMed] [Google Scholar]
- Rost, B., and C. Sander. 1994. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins. 19:55–72. [DOI] [PubMed] [Google Scholar]
- Sarin, V. K., S. Gupta, T. K. Leung, V. E. Taylor, B. L. Ohning, J. A. Whitsett, and J. L. Fox. 1990. Biophysical and biological activity of a synthetic 8.7-kDa hydrophobic pulmonary surfactant protein SP-B. Proc. Natl. Acad. Sci. USA. 87:2633–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneva, S., and K. M. W. Keough. 1994. Pulmonary surfactant proteins SP-B and SP-C in spread monolayers at the air-water interface. 1. Monolayers of pulmonary surfactant protein SP-B and phospholipids. Biophys. J. 66:1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneva, S., T. McEachren, J. Stewart, and K. M. W. Keough. 1995. Pulmonary surfactant protein SP-A with phospholipids in spread monolayers at the air-water interface. Biochemistry. 34:10279–10289. [DOI] [PubMed] [Google Scholar]
- Tokieda, K., J. A. Whitsett, J. C. Clark, T. E. Weaver, K. Ikeda, K. B. McConnell, A. H. Jobe, M. Ikegami, and H. S. Iwamoto. 1997. Pulmonary dysfunction in neonatal SP-B-deficient mice. Am. J. Physiol. 273:L875–L882. [DOI] [PubMed] [Google Scholar]
- Udenfriend, S., S. Stein, P. Bohlen, W. Dairman, W. Leimgruber, and M. Weigele. 1972. Fluorescamine: a reagent for assay of amino acids, peptides, proteins, and primary amines in the picomole range. Science. 178:871–872. [DOI] [PubMed] [Google Scholar]
- Veldhuizen, E. J. A., A. J. Waring, F. J. Walther, J. J. Batenburg, L. M. G. van Golde, and H. P. Haagsman. 2000. Dimeric N-terminal segment of human surfactant protein B (dSP-B 1–25) has enhanced surface properties compared to monomeric SP-B 1-25. Biophys. J. 79:377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, T. E., and J. J. Conkright. 2001. Functions of surfactant proteins B and C. Annu. Rev. Physiol. 63:555–578. [DOI] [PubMed] [Google Scholar]
- Wilson, E. B., Jr., J. C. Decius, and P. C. Cross. 1955. Molecular Vibrations. McGraw-Hill, New York.