

Abstract
We report the crystal structure of a bromide-bound form of the D85S mutant of bacteriorhodopsin, bR(D85S), a protein that uses light energy rather than ATP to pump halide ions across the cell membrane. Comparison of the structure of the halide-bound and halide-free states reveals that both displacements of individual side-chain positions and concerted helical movements occur on the extracellular side of the protein. Analysis of these structural changes reveals how this ion pump first facilitates ion uptake deep within the cell membrane and then prevents the backward escape of ions later in the pumping cycle. Together with the information provided by structures of intermediate states in the bacteriorhodopsin photocycle, this study also suggests the overall design principles that are necessary for ion pumping.
INTRODUCTION
Active transport of ions across the cell membrane is a fundamental energy-driven process that establishes electrochemical gradients for driving a variety of processes, including ATP synthesis and metabolite uptake. Attempts to understand the mechanistic basis of ion pumping from a structural perspective have been hampered by the difficulty of crystallizing integral membrane proteins. As a result, high-resolution structures are available for only a few ion pumps: apart from the proton-pumping electron transport complexes (Iwata et al., 1998, 1995; Soulimane et al., 2000; Tsukihara et al., 1996; Xia et al., 1997; Zhang et al., 1998), these include the sarcoplasmic Ca2+ ATPase (Toyoshima et al., 2000), halorhodopsin (hR) (Kolbe et al., 2000), and bacteriorhodopsin (bR) as well as some of its mutants (Belrhali et al., 1999; Luecke et al., 2000, 1999a,b; Rouhani et al., 2001). Although informative, these studies each lack key information necessary to fully understand the respective mechanisms of these pumps. For example, progress made on the structure of Ca2+ ATPase still shows the protein only in both its resting, substrate-free (Toyoshima and Nomura, 2002) and substrate-bound (Toyoshima et al., 2000) states. Even for bacteriorhodopsin, for which considerable information is available regarding conformational changes that occur during the pumping cycle (Facciotti et al., 2001; Lanyi and Schobert, 2002; Luecke et al., 2000, 1999a; Rouhani et al., 2001; Royant et al., 2000; Sass et al., 2000; Schobert et al., 2002), the question whether this protein is a proton pump or a hydroxyl-ion pump (Oesterhelt and Stoeckenius, 1973; Perkins, 1992) remains under discussion (Betancourt and Glaeser, 2000; Herzfeld and Lansing, 2002; Luecke, 2000).
Like wild-type bR, bR(D85S) contains an all-trans retinal chromophore bound via a protonated Schiff-base linkage to K216 on helix G. In wild-type bR, the ion pumping cycle begins with photoisomerization of the retinal to the 13-cis configuration. The cycle then progresses thermodynamically downhill through a series of spectroscopically identifiable intermediate states that ultimately return the protein to its resting state and the retinal to its initial all-trans configuration. As is wild-type bR, bR(D85S) is also a light-driven ion pump; however, the single amino acid substitution permits the protein to pump anions from the extracellular side of the membrane into the cell and unlike wild-type bR its catalytic cycle does not involve Schiff-base deprotonation (Brown et al., 1996; Sasaki et al., 1995).
As a means to gain more insight about the ion pumping process, we previously crystallized a halide-pumping mutant of bacteriorhodopsin, bR(D85S), in its halide-free state (Rouhani et al., 2001). Here, we describe the crystal structure of bR(D85S) with a bromide ion bound in its ground-state binding site. In conjunction with the previously determined structure of the halide-free state (Rouhani et al., 2001), this study reveals structural changes that occur during the initial substrate-binding step of halide-ion transport, and suggests some design principles that may apply more generally to other ion pumps.
Specifically, these results, in conjunction with the previously solved structures of intermediate states in the wild-type photocycle, suggest that bR(D85S) facilitates active transport via a four-step process. First, the substrate must diffuse into the binding site past a dynamic gate consisting of the side chain of R82. Second, the halide ion makes an ion pair with the protonated Schiff base in the binding site, and in so doing induces a conformational change that latches the dynamic gate in a closed conformation. Third, the energy of the absorbed photon forces a small displacement of the protonated Schiff base, through isomerization of the retinal chromophore. The resulting charge separation raises the potential energy of the substrate to a level higher than it would have on the opposite side of the membrane and thus leads to ion transport. Finally, the protein relaxes back to its ground state.
MATERIALS AND METHODS
Crystallization
Crystals of bR(D85S) were grown in a 1-monooleoyl-rac-glycerol (Sigma, St. Louis) gel. Mutant bacteriorhodopsin was isolated as in Facciotti et al. (2001) and solubilized in 1.2% octylglucoside overnight. The solubilized membrane was then concentrated to 15 mg/ml. A quantity of 10 μl protein was then used to hydrate 10 mg dry monoolein in a 0.2 ml polymerase chain reaction (PCR) tube for each crystallization setup. This mixture was then centrifuged at 10,000 × g at 22°C for 3 h. After centrifugation, 100 μl of the mother liquor consisting of 100 mM sodium acetate, pH 4.6, 200 mM KCl, and 10% PEG 4000 was layered on top of the hydrated gel. Crystals grew to a final size of 5 μm × 30 μm × 120 μm in the period of 3 weeks. Aliquots of the gel were then transferred to a lipase solution (Nollert and Landau, 1998) consisting of 100 mM sodium acetate, pH 4.6, 200 mM KCl, and 50 mg/ml lipase, and allowed to incubate overnight. Individual crystals were then transferred to a solution containing no halide, 100 mM sodium acetate, pH 4.6, and 10% PEG 4000 to remove the bound chloride, after which they were transferred sequentially to cryoprotectant solutions containing 100 mM sodium acetate, pH 4.6, 1.25 M KBr, and 12%, 16%, 20%, and 25% PEG 4000, respectively.
Data collection and model building
Crystals were flash frozen in liquid nitrogen and irradiated on beamline 8.3.1 of the advanced light source (ALS) with a 30-μm collimated x-ray beam. Five crystals were used to collect the halide data set. The halide-bound crystals belong to the C2221 space group with average unit cell dimensions in angstroms (Å) of a = 52.50, b = 121.33, and c = 73.87, whereas the previously described halide-free crystals have a unit cell of a = 51.80, b = 121.30, and c = 85.70. A Quantum 210 CCD detector was used to collect images. Data reduction was performed by the Elves scripts (Holton, 2002). Diffraction data were integrated with MOSFLM (Leslie, 1992) and subsequently scaled with SCALA (Collaborative Computation Project, 1994). Molecular replacement using 1KGB (Facciotti et al., 2001) as the starting search model, without the retinal, water, and lipid molecules, was performed by the program CNS Version 1.1 (Brunger et al., 1998). Refinement with CNS and model building using the program O (Jones et al., 1991) together with annealed simulated omit, |Fo| − |Fc|, and 2|Fo| − |Fc| maps reduced the values of R and Rfree to their final values (Table 1). The final model was analyzed with PROCHECK (Laskowski et al., 1993) and MOLPROBITY (Richardson, 2002) and exhibits good stereochemistry.
TABLE 1.
Data collection and crystallographic refinement
| Data reduction resolution range | 60–2.0 Å |
|---|---|
| Unit cell dimensions (a, b, c) | 52.5, 121.33, 73.87 |
| Total observations | 135,550 |
| Unique structure factors | 16,336 |
| Average I/σ(I)/high res. | 10.75/(3.1) |
| Completeness (%)/high res. | 94.2/(89.8) |
| Rsym(%)/high res.* | 0.127/(0.35) |
| Refinement resolution range | 12–2.0 Å |
|---|---|
| Number of structure factors | 15,316 |
| Number of protein atoms | 1,689 |
| Number of retinal atoms | 20 |
| Number of water molecules | 49 |
| Number of inorganic atoms | 3 |
| Number of lipid atoms | 57 |
| R factor (%)† | 21.51 |
| Rfree (%)‡ | 23.65 |
| Average protein B (Å2) | 21.38 |
| Average retinal B (Å2) | 12.90 |
| Average water B (Å2) | 29.84 |
| Average inorganic B (Å2) | 34.72 |
| Average lipid B (Å2) | 38.15 |
| Deviation from ideal bond lengths (Å) | 0.0070 |
| Deviation from ideal bond angles (°) | 0.953 |
Rsym = Σhkl|Ihkl − 〈Ihkl〉|/Σ(Ihkl) where I is the intensity.
R factor = Σhkl|Fo − Fc|/Σhkl|Fo|, where Fo and Fc are observed and calculated structure factors, respectively.
Rfree = ΣhklT|Fo − Fc|/ΣhklT|Fo|, calculated with the T set (5% of the data) that has been omitted from refinement.
The model has been deposited in the Protein Data Bank with the accession number 1MGY.
RESULTS
Halide binding site
The structure of the halide-bound form of bR(D85S) reveals that bromide ions are bound in two places. In what is clearly the initial substrate-binding site, a bromide ion is located 3.16 Å from the protonated Schiff-base nitrogen of K216, in a position almost identical to that of H2O 402 in wild-type bacteriorhodopsin (Fig. 1). The strong electrostatic interaction with the positively charged Schiff base will contribute a significant fraction to the binding energy. Also located near the bromide are the side-chain groups of residues S85-(OG) and D212-(OD1) as well as H2O 410, all at hydrogen-bonding distances of 2.9 Å, 3.28 Å, and 3.29 Å, respectively. A second bromide ion is located on the cytoplasmic side of the protein, where it forms a complex with the backbone amide nitrogen of D104, OG1 of T107, and water 540 at distances of 3.29 Å, 3.17 Å, and 3.03 Å, respectively.
FIGURE 1.
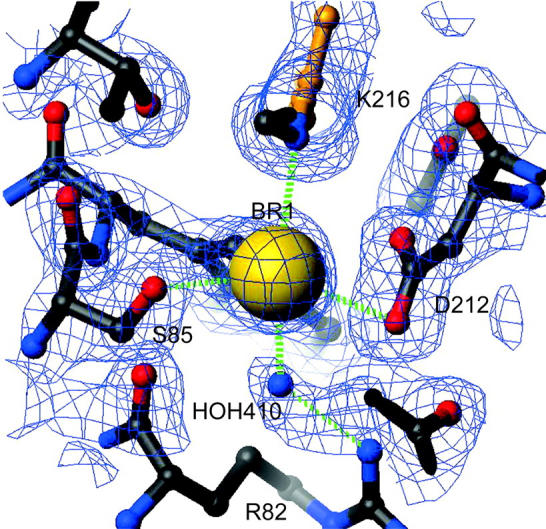
Bromide binding site of bR(D85S). The bromide ion is shown bound near the protonated Schiff base and between residues S85 and D212. The electron density is contoured at 1 σ. Distances to the key residues with which the ion interacts are drawn with green dashed lines. The atoms and bonds shown in gold belong to the retinal. The atom at the binding site was initially modeled by a water molecule. However, after refinement, the B value for the fully occupied molecule was reduced to an unphysical value of 0.25. A bromine atom was subsequently used to model the density feature and refined to a B value of 29.5 and an occupancy of 74%. Another bromide ion (shown in Fig. 2, D) and a potassium ion (not shown) were discovered on the cytoplasmic surface by the same method.
Binding of the substrate anion in close proximity to D212 suggests, as did the observed pH dependence of chloride binding in the bR(D85T) mutant (Brown et al., 1996; Sasaki et al., 1995; Tittor et al., 1997), that residue D212 becomes protonated upon halide binding. This protonation, although necessary for binding of a halide ion, is energetically costly at pH values above the pKa of D212, which is thought to be 6.9 in bR(D85T) when measured in saturating sodium chloride (Tittor et al., 1997).
The halide binding site of bR(D85S) differs from the halorhodopsin-binding site, where D238, the homolog to residue D212 in bacteriorhodopsin, remains negatively charged when a halide ion is in the binding site. This is because the halorhodopsin aspartate does not interact directly with the halide ion, but serves instead as a counterion to the Schiff base and indirectly, through an ordered water molecule, to R108 (Kolbe et al., 2000). Another difference is that in bR(D85S) only one water molecule interacts with the bound bromide ion, whereas three water molecules had been reported in the binding site of halorhodopsin (Kolbe et al., 2000). These differences in polar interactions within the binding site, and the need to protonate D212, are likely to account for the lower halide binding affinity, over a narrower pH range, of the D85S mutant relative to halorhodopsin.
Structural rearrangements caused by halide binding
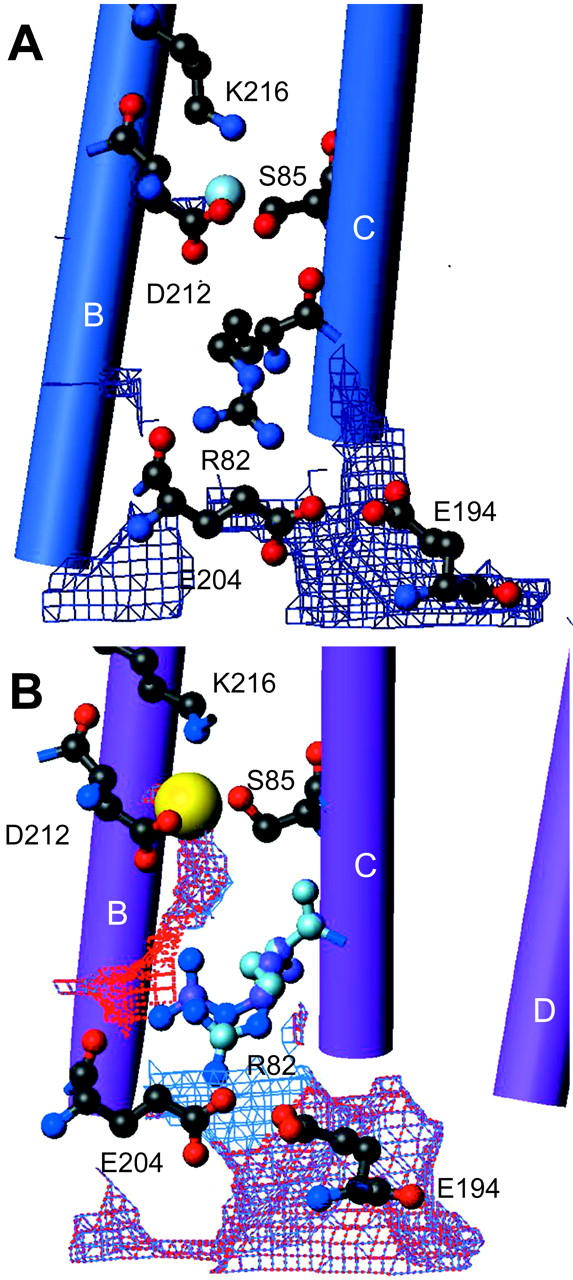
A comparison of the halide-bound and halide-free (Rouhani et al., 2001) forms of bR(D85S) reveals that, after bromide binding, the structure on the cytoplasmic side remains as it was in the halide-free form. On the extracellular side, however, bromide binding induces a variety of side-chain movements (Fig. 2 A). One of the most notable movements involves the side chain of R82, which is best modeled with two distinct and nearly equally occupied conformations (Fig. 3). In what we presume is the functional conformation, CB-CD-CG swing downward in concert with a small shift in backbone position, whereas the guanidinium group itself rotates to a more upward-facing orientation, reminiscent of the conformation adopted in the ground state of wild-type bacteriorhodopsin. In the second rotamer conformation, the side chain of R82 adopts a downward facing structure reminiscent of that adopted in the halide-free form. Residues W138 and W189 also change rotamer conformations between the halide-free and halide-bound states by flipping about χ1 and about both χ1 and χ2, respectively, resulting in a local reorganization of the interhelical hydrogen bonds. In the bromide-bound structure, the OH of Y83 (helix C) accepts a hydrogen bond from the NE1 of W189 (helix F), whereas in the halide-free state Y83 forms a hydrogen bond with the backbone carbonyl of I119 (helix D). The reorganization of Y83 and the formation of its hydrogen bond with W189 is facilitated by rotations about both χ1 and χ2 for W189, during which the residue breaks a π-bond with the indole ring of W138. These changes in hydrogen bonding allow W138 to break a hydrogen bond with the carbonyl oxygen of G122 (helix D) and rotate about χ1 to form a new hydrogen bond with the carbonyl oxygen of P186 (helix F). The net result is that substrate binding eliminates two hydrogen bonds that helix D makes with its neighboring helices, while establishing one new cross-helix-bundle hydrogen bond between helices C and F. These rotamer switches involving R82, W138, and W189 in the bromide-bound form of bR(D85S) regenerate the same bonding arrangement that occurs in the functionally equivalent ground state of wild-type bR.
FIGURE 2.

Conformational changes that occur in bR(D85S) upon binding bromide ions. The figures were generated and models were aligned using the program MOLMOL (Koradi et al., 1996). (A) Changes in rotamer conformation of the R82 side chain. The dark-blue model is the halide-free structure, whereas the pink and light-blue models represent the presumed functional and nonfunctional states of the halide-bound structure, respectively. The little spheres represent water molecules and the bromide-ion substrate. (B) Amino acid side-chain positions in the blue, halide-free state and the helix-helix contacts for selected residues on the extracellular side of the protein. Hydrogen bonds are shown with dashed green lines. (C) The same residues from panel B are shown in the purple, halide-bound state. Note that a wild-type ground-state like connection between Y83 and W189 is established upon halide binding, thus creating a direct interaction between helices C and F. (D) A side view of the protein with cylinder representations of helices. The retinal chromophore is drawn in a gold ball-and-stick representation to help orient the molecule for the reader, and the inorganic ions are drawn as gold spheres. The halide-free model is painted blue whereas the halide-bound representation is painted pink. Note that helix A appears longer, due to the addition of well-ordered residues 4–8. (E) Substrate binding induces a repacking of helices on the extracellular side of the protein. Arrows indicate the direction of helical movements due to anion binding in bR(D85S). (F) The comparison between the positions of helices in bR(D85S), bR(D85S) halide-bound (pink), and ground state of wild-type bR (dark purple).
FIGURE 3.

Stereo view of the 2Fo − Fc electron density around the side chain of R82, contoured at 1 σ. The side chain is shown in the two conformations that best account for difference density features that appeared during model building. Significant density remains in the Fo − Fc map if just the upward facing conformation and its corresponding waters are included in the atomic model. A second conformer was therefore modeled, with its accompanying waters, to account for the residual density features. Two of the most informative density features that are visible in this figure are the extended density accounted for by water molecules and the extra large density feature in which the guanidinium group of the arginine is located. In addition, only weak density is present for the side chain's three-carbon linker, further suggesting that this portion of the residue is highly mobile due to multiple conformations.
An analysis of the positions of the helices on the extracellular side of the protein reveals that larger-scale structural changes also occur upon bromide binding in the bR(D85S) mutant (Fig. 2, D and E). Helices A and B tilt laterally along the elliptical contour of the protein when viewed from the extracellular side. A portion of helix C, between residues 78 and 87 on the extracellular side, also bends in the same manner. In addition, the extracellular sides of helices D and E, from residue 116 to its end and between residues 133 and 144, respectively, tilt in toward the center of the molecule whereas helices F and G change little from their halide-free positions. As a result of the ligand-induced repacking of helices on the extracellular side of the protein, the structure becomes more similar to that found in the ground-state wild-type protein (Fig. 2 F).
DISCUSSION
To further investigate the effects of helix repacking, we calculated the solvent accessible surfaces for both the bromide-free and the bromide-bound forms of bR(D85S). In the classic model for explaining how ion pumps operate, proposed by Jardetzky in the 1960s (Jardetzky, 1966), an energy-driven cycle causes the protein to alternate between inward and outward facing conformations. In its simplest form, one might imagine that these open states provide direct access to the binding site via a static, solvent-accessible tunnel. However, in neither the bromide-free nor the bromide-bound structures is there a true, solvent-accessible tunnel that leads all the way to the binding site (Fig. 4, A and B). Instead, the side chains of R82 and its immediate neighbors cap the short tunnel that does exist. Our analysis suggests, therefore, that refinements need to be made in how the Jardetzky model is used to explain the ability of bR(D85S) to pump anions. We suggest, first of all, that entry of the substrate anion must rely on a fluctuating gate that opens transiently, providing access of substrate to its binding pocket by a mechanism similar to that which permits binding of molecular oxygen to hemoglobin and myoglobin (Karplus and McCammon, 1986).
FIGURE 4.
Solvent accessible surface in bR(D85S). Solvent accessible surfaces were calculated with the program VOIDOO (Kleywegt and Jones, 1994) and displayed in O before being drawn for the figure with MOLMOL. The area shown is confined to the extracellular tunnel of solvent-accessible surface calculated with a probe of radius 1.0 Å. This probe radius is considerably smaller than the typical 1.4-Å probe radius used to represent water, or the 1.95-Å Pauling radius of a bromide ion (Hille, 1984). However, probing with a smaller radius reveals the most likely location of the tunnel that would be opened during the dynamic gating process. (A) The tunnel for bR(D85S) that was crystallized in the blue, anion-free state (Rouhani et al., 2001). (B) The tunnel for the bromide-bound state of bR(D85S); both conformations of the R82 side chain are shown. The blue map is the solvent accessible surface of the presumed functional conformer, whereas the red surface belongs to the alternate conformer. Note that the boundaries for each surface reveal that neither conformer provides a direct substrate-accessible path to the binding site. Since the alternate conformer of R82 resembles the conformer of the halide-free state it is reasonable to suggest that a substrate accessible tunnel, which must open dynamically in the transition between halide-free and halide-bound states, is most simply represented by connecting the two displayed surfaces at their closest points. This approach provides, in our estimation, the most likely path for bromide entry into the binding site.
The dual conformations observed for R82, and its crucial position at the end of the solvent-accessible tunnel on the extracellular surface, suggest that rotamer switching by this residue may play a crucial role in permitting halide ions to reach the binding pocket. Besides contributing steric bulk that blocks the solvent-accessible barrier, R82 may also serve as an anion-pairing carrier arm that escorts the halide ion into the low-dielectric interior of the protein. In this scenario, the guanidium group of R82 forms an ion pair with the anionic substrate and then transports the substrate into the binding site when the side chain switches into an inward facing conformation. A similar ion-pair interaction has been seen between R72 (the homolog to bR's R82) and a partially occupied chloride ion in the structure of sensory rhodopsin II from Natronobacterium pharaonis (Royant et al., 2001). As was shown by Honig and Hubbel (1984), the cost of burying an ion pair in a region of low-dielectric constant is no greater than that of burying a single ion. Thus, the energetic cost associated with transporting an anionic substrate out of the high-dielectric, aqueous medium may have already been prepaid by burying R82 during the protein-folding step.
The use of the buried formal charge of R82 to prepay the Born energy of the anionic substrate during transit to the binding site contrasts with the mechanism used by ion channels to overcome the Born energy barrier for ion transport. The structures of the potassium and chloride channels (Doyle et al., 1998; Dutzler et al., 2002) have revealed that polar peptide-backbone residues of the “pore-loop” sequence are used, rather than buried counterions, to create an environment for a dehydrated ion that is almost equivalent, energetically, to that of its fully hydrated state (Berneche and Roux, 2001).
A key difference between these two mechanisms for lowering the Born energy barrier is that the pore design lacks any way to prevent backflow of the transported ion, and thus it is compatible only with passive (downhill) diffusion of substrate ions. On the other hand, a design that uses rotamer switching of a charged side-chain residue, R82 in the case of bR(D85S), as a flexible gate would allow substrate-induced conformational changes to latch the gate in a single, frozen conformation and thereby prevent backflow during subsequent steps in the pumping cycle. In halorhodopsin, the homologous residue, R108, is also known to block extracellular access to the binding site and would, therefore, also need to move to allow anion access to the binding site (Kolbe et al., 2000). Thus, R108 in halorhodopsin could also serve as a dynamic ion-pairing gate. In addition, the recently published structure of the Ca2+ ATPase in the substrate-free state (Toyoshima and Nomura, 2002) also reveals an access tunnel that is capped by a potential counter ion, in this case the carboxyl group of E309. Upon Ca2+ binding, E309 appears to switch rotamer conformations (Toyoshima et al., 2000) in a way that again suggests this residue plays a role that is functionally equivalent to what we propose for R82 in bR(D85S).
Although we suggest that the bromide binding induced helix repacking could functionally close the extracellular side of the protein, the repacking that is observed does not return all of the helices to the positions occupied in the resting state of wild-type bR. We suspect that the observed repacking thus represents an incomplete closure of the extracellular side in which dual occupancy of rotamer conformations can still exist for the gate, R82. As a result substantial backflow of halide ions could still occur in this mutant. The poor pumping efficiency of bR(D85S), accompanied by a measured component of backflow of chloride ion (Kalaidzidis and Kaulen, 1997), may thus be explained by the differences in helix packing that remain between bR(D85S) and wild-type bR in their respective resting states. The inability of bR(D85S) to completely close the extracellular side is not surprising since a single protein engineering step, (mutating D85 to S) can not be expected to optimize all aspects of halide-ion transport to the extent that evolution has done for halorhodopsin.
The structures of bR(D85S) and hR both reveal that charge-charge interaction is a key component of ion binding in these pumps. The same mechanism of ion binding is also seen in the Ca2+ ATPase, a cation pump, where there is a direct charge-charge interaction between substrate and a cluster of aspartate and glutamate carboxyl groups (Toyoshima et al., 2000). We suggest that direct charge-charge interactions in the binding site could serve an important mechanistic function for ion pumps, beyond merely trapping the substrate in the binding site. The use of externally supplied energy to switch either the position or the orientation of a side-chain counter ion (or in our case the protonated Schiff base of the retinal group), by even a small amount, can raise the potential energy of the substrate ion and even draw it on to the next site.
As a concrete example, we can model what the consequence might be after isomerization of the retinal group in the bR(D85S) mutant by assuming the structural changes to be similar to what occurs in the M intermediate of bacteriorhodopsin. This assumption will only be valid if the reaction coordinate of the halide-pumping mutant, when expressed in terms of a progression of structural changes in the protein, remains similar to that of wild-type bacteriorhodopsin. This is similar to the assumption made when interpreting the structural features of trapped photointermediates of proton-pumping mutants within the context of the wild-type photocycle (Luecke et al., 2000, 1999a).
It is generally accepted that the photon energy is initially stored in the retinal chromophore in the form of torsion of the normally planar polyene chain (Braiman and Mathies, 1982; Lanyi and Schobert, 2002; Schobert et al., 2002; Smith et al., 1985), and that the Schiff-base nitrogen atom is gradually displaced from its original position as this torsion is relaxed (Facciotti et al., 2001; Luecke et al., 1999a). We have therefore used the atomic model of a late M intermediate (Luecke et al., 1999a) to estimate the amount of charge separation that might occur between the protonated Schiff base and the bromide anion in the D85S mutant. Alignment of the late M structure to our atomic model of the bromide-bound resting state of bR(D85S) was carried out in two ways. In the first, the RMSD between the retinal ring of the two models was minimized, whereas in the second, the RMSD between selected CA atoms of the two models was minimized.
According to the first method of alignment, the distance between the halide anion (were it to remain fixed) and the midpoint of the N–H bond of the Schiff-base cation would increase to ∼1.4 times their separation in the ground state, while in the second method of alignment the distance increases by a factor of 1.6 (Fig. 5). If we take even the smaller amount of displacement, the Born energy cost for this small amount of charge separation would actually be ∼40% of the favorable Coulomb interaction that is involved in forming the initial ion pair. The bromide ion could avoid the high cost of this charge separation by moving to a position that is closer to the displaced Schiff-base cation, of course, ultimately moving to the cytoplasmic side of the retinal group. Even then, however, the anion would remain at a higher potential energy than in the resting state, for it would no longer be solvated in this new position by the S85 or D212 polar groups.
FIGURE 5.

Isomerization of the retinal moves the Schiff-base counter ion away from the halide substrate. The bromide ion is shown as a sphere, whereas the model of the all-trans retinal group bound covalently to K216 is shaded dark gray for the bromide-bound form of bR(D85S) and light gray for the model of the D96N late-M intermediate. In addition, the K216-NZ atoms of the D96N late-M intermediate and bromide-bound bR(D85S) models are shown as light- and dark-gray spheres, respectively. The hydrogen atoms and their bonds to K216-NZ were drawn by hand. Distances were measured from the bromide ion to the midpoint of the Schiff-base N–H bond. The models shown here were aligned by minimizing the RMSD of the C1, C2, C3, C4, C5, and C6 atoms of the retinal ring between the two models. This procedure, in contrast to aligning the models by minimizing the RMSD between selected CA atoms, seems to minimize the difference in position between the K216-NZ atoms, thereby minimizing the apparent energetic effect of the charge displacement. The actual charge displacement may thus be larger than that which is reported in the text.
From our observations on bR(D85S), together with information from previously determined structures of pumps and channels, we propose that the active transport of an ion across a cell membrane consists of at least the following four functional transitions, summarized in Fig. 6. i), Three important events occur in the first transition that is shown in Fig. 6. In the first event, substrate entry into the binding site of an ion pump requires the transient conformational opening of a gate that provides access to an internal binding site. We believe that such a gate is formed by R82 in bR(D85S) and its homolog R108 in halorhodopsin. An intriguing possibility is that the guanidinium group of the arginine residue may also act as a Born-energy chaperone for the substrate anion. The internalized substrate ion must next form an ionic interaction with a charged residue in the binding site, a role that is played by the protonated Schiff base in hR and bR(D85S). Finally, substrate binding must induce a conformational change that closes the entrance side of the pump, thereby making it more difficult for transient conformational openings to occur, which would allow backflow of the substrate ion. In bR(D85S), bromide binding induces significant repacking of helices on the extracellular side of the pump that could lock the side chain of R82 in its upward-facing (closed) conformation. ii), Energy consumption must then be coupled to what needs to be no more than a small directional displacement of the charged group with which the substrate ion interacts as a binding partner. In both hR and bR(D85S), this step consists of photon-induced retinal isomerization, which is in effect another instance of rotamer switching. This structural change, although small, can nevertheless have a large effect on the spatial energy landscape for the transported ion, moving it vectorially within the membrane and raising it to a potential energy that is higher than what it will have when it is released on the opposite (in this case cytosolic) side of the cell membrane. The idea that small-scale conformational changes can have quite large energetic consequences with significant functional relevance has previously been suggested as a mechanism for signal transduction across the membrane by the aspartate chemotaxis receptor (Yu and Koshland, 2001). iii), The substrate must then traverse the remaining portion of the protein. In the case of bR(D85S), it is reasonable to suppose that a repacking of helix F and helix G occurs on the cytoplasmic side of the protein, producing an N-like intermediate similar to that observed with the F219L mutant (Vonck, 2000) or the constitutively N-like structure of the D96G/F171C/F219L triple mutant (Subramaniam and Henderson, 2000). The helix packing observed in the N-like structures is believed to increase the access of water molecules to the cytoplasmic side of the Schiff base, and thus may open up a path for the diffusion of the halide ion out to the cytoplasmic side of the membrane. The suggestion that the extracellular side must open to create an exit for the halide ion has also been proposed as a requirement for halorhodopsin's pumping cycle (Kolbe et al., 2000). iv), After the substrate has left, the protein will be free to return to its ground-state conformation.
FIGURE 6.
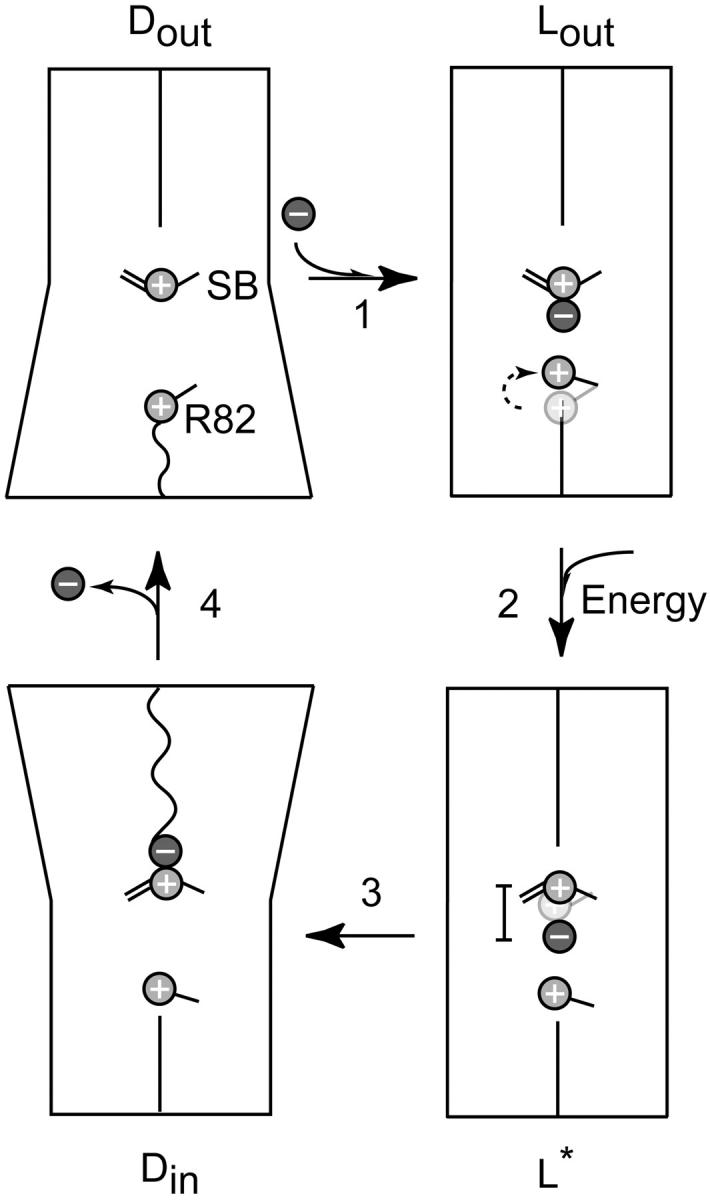
Model of the key functional steps in the ion-transport cycle of bR(D85S). The cartoon schematically shows, via sloping lines, an outward tilt for helices on either the extracellular side or the cytoplasmic side of the protein, producing the two types of dynamic states, (Dout and Din). The presumed increase in the permeability of the corresponding half of the protein is indicated by the transition from a straight to wiggly line crossing that half of the protein. In the first step, the extracellular half of the transport channel starts in a “dynamic” conformation, Dout. The side chain of R82 serves as a dynamic gate that, by fluctuating between inward facing and outward facing conformations, allows entry of a halide ion to the binding site. Substrate binding then induces a conformational transition to the “latched” state, Lout, in which R82 is held tightly in the upward facing position. The movement of R82 is indicated by a change in position of the schematic side chain and a curved dashed arrow. The initial position of the side chain is shown in faint gray. In the second step, photon energy is absorbed by the retinal chromophore and results in a transition to a high-energy state, L*, in which a small displacement of the protonated Schiff base relative to the bound halide raises the potential energy of the halide ion by a large amount. The displacement of the Schiff base is represented in a similar fashion to that of R82, whereas a bar indicates that this displacement increases the distance between the halide ion and the Schiff base. The third step envisions a repacking of helices on the cytoplasmic side of the protein that yields a dynamic state, Din, which allows the anion to exit the protein. Finally, in the fourth step, the protein relaxes back to its ground state, Dout.
Acknowledgments
The authors thank Daniel Facciotti, Elizabeth Facciotti, Daniel Koshland Jr., and Jeremy Thorner for their critical reading of preliminary drafts and for their constructive advice. In addition, we thank Janos Lanyi for providing the cells that express bR(D85S) as well as for his scientific and editorial comments regarding the manuscript. The authors also gratefully acknowledge the generous contributions of James Holton during the course of this work and his support at ALS beamline 8.3.1.
This work was supported in part from a National Institutes of Health grant GM51487.
References
- Belrhali, H., P. Nollert, A. Royant, C. Menzel, J. P. Rosenbusch, E. M. Landau, and E. Pebay-Peyroula. 1999. Protein, lipid and water organization in bacteriorhodopsin crystals: a molecular view of the purple membrane at 1.9 A resolution. Struct. Fold. Des. 7:909–917. [DOI] [PubMed] [Google Scholar]
- Berneche, S., and B. Roux. 2001. Energetics of ion conduction through the K+ channel. Nature. 414:73–77. [DOI] [PubMed] [Google Scholar]
- Betancourt, F. M., and R. M. Glaeser. 2000. Chemical and physical evidence for multiple functional steps comprising the M state of the bacteriorhodopsin photocycle. Biochim. Biophys. Acta. 1460:106–118. [DOI] [PubMed] [Google Scholar]
- Braiman, M., and R. Mathies. 1982. Resonance Raman spectra of bacteriorhodopsin's primary photoproduct: evidence for a distorted 13-cis retinal chromophore. Proc. Natl. Acad. Sci. USA. 79:403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, L. S., R. Needleman, and J. K. Lanyi. 1996. Interaction of proton and chloride transfer pathways in recombinant bacteriorhodopsin with chloride transport activity: implications for the chloride translocation mechanism. Biochemistry. 35:16048–16054. [DOI] [PubMed] [Google Scholar]
- Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography + NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54:905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computation Project. 1994. The Ccp4 suite–programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763. [DOI] [PubMed] [Google Scholar]
- Doyle, D. A., J. Morais-Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Facciotti, M. T., S. Rouhani, F. T. Burkard, F. M. Betancourt, K. H. Downing, R. B. Rose, G. McDermott, and R. M. Glaeser. 2001. Structure of an early intermediate in the M-state phase of the bacteriorhodopsin photocycle. Biophys. J. 81:3442–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzfeld, J., and J. C. Lansing. 2002. Magnetic resonance studies of the bacteriorhodopsin pump cycle. Annu. Rev. Biophys. Biomol. Struct. 31:73–95. [DOI] [PubMed] [Google Scholar]
- Hille, B. 1984. Ionic Channels of Excitable Membranes. Sinauer Associates, Inc., Sunderland, MA.
- Holton, J. M. 2002. http://ucxray.berkeley.edu/∼jamesh/elves/.
- Honig, B. H., and W. L. Hubbell. 1984. Stability of “salt bridges” in membrane proteins. Proc. Natl. Acad. Sci. USA. 81:5412–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata, S., J. W. Lee, K. Okada, J. K. Lee, M. Iwata, B. Rasmussen, T. A. Link, S. Ramaswamy, and B. K. Jap. 1998. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc(1) complex. Science. 281:64–71. [DOI] [PubMed] [Google Scholar]
- Iwata, S., C. Ostermeier, B. Ludwig, and H. Michel. 1995. Structure at 2.8-angstrom resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 376:660–669. [DOI] [PubMed] [Google Scholar]
- Jardetzky, O. 1966. Simple allosteric model for membrane pumps. Nature. 211:969–970. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., J. Y. Zou, S. W. Cowan, and Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 47:110–119. [DOI] [PubMed] [Google Scholar]
- Kalaidzidis, I. V., and A. D. Kaulen. 1997. Cl-dependent photovoltage responses of bacteriorhodopsin: comparison of the D85T and D85S mutants and wild-type acid purple form. FEBS Lett. 418:239–242. [DOI] [PubMed] [Google Scholar]
- Karplus, M., and J. A. McCammon. 1986. The dynamics of proteins. Sci. Am. 254:42–51. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G. J., and T. A. Jones. 1994. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D Biol. Crystallogr. 50:178–185. [DOI] [PubMed] [Google Scholar]
- Kolbe, M., H. Besir, L. O. Essen, and D. Oesterhelt. 2000. Structure of the light-driven chloride pump halorhodopsin at 1.8 angstrom resolution. Science. 288:1390–1396. [DOI] [PubMed] [Google Scholar]
- Koradi, R., M. Billeter, and K. Wuthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51–55. [DOI] [PubMed] [Google Scholar]
- Lanyi, J. K., and B. Schobert. 2002. Crystallographic structure of the retinal and the protein after deprotonation of the Schiff base: the switch in the bacteriorhodopsin photocycle. J. Mol. Biol. 321:727–737. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., M. W. Macarthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK—a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291. [Google Scholar]
- Leslie, A. G. W. 1992. Recent changes to the MOSFLM package for processing film and image plate data. CCP4 and ESF-EACMB Newsletter on Protein Crystallography. 26.
- Luecke, H. 2000. Atomic resolution structures of bacteriorhodopsin photocycle intermediates: the role of discrete water molecules in the function of this light-driven ion pump. Biochimica Et Biophysica Acta-Bioenergetics. 1460:133–156. [DOI] [PubMed] [Google Scholar]
- Luecke, H., B. Schobert, J. P. Cartailler, H. T. Richter, A. Rosengarth, R. Needleman, and J. K. Lanyi. 2000. Coupling photoisomerization of retinal to directional transport in bacteriorhodopsin. J. Mol. Biol. 300:1237–1255. [DOI] [PubMed] [Google Scholar]
- Luecke, H., B. Schobert, H. T. Richter, J. P. Cartailler, and J. K. Lanyi. 1999a. Structural changes in bacteriorhodopsin during ion transport at 2 angstrom resolution. Science. 286:255–260. [DOI] [PubMed] [Google Scholar]
- Luecke, H., B. Schobert, H. T. Richter, J. P. Cartailler, and J. K. Lanyi. 1999b. Structure of bacteriorhodopsin at 1.55 angstrom resolution. J. Mol. Biol. 291:899–911. [DOI] [PubMed] [Google Scholar]
- Nollert, P., and E. M. Landau. 1998. Enzymic release of crystals from lipidic cubic phases. Biochem. Soc. Trans. 26:709–713. [DOI] [PubMed] [Google Scholar]
- Oesterhelt, D., and W. Stoeckenius. 1973. Functions of a new photoreceptor membrane. Proc. Natl. Acad. Sci. USA. 70:2853–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins, G. A. 1992. Trapping the M1 and M2 substates of bacteriorhodopsin for electron diffraction studies. PhD thesis. University of California, Berkeley, Berkley, CA.
- Richardson, J. S. 2002. http://kinemage.biochem.duke.edu/molprobity/.
- Rouhani, S., J. P. Cartailler, M. T. Facciotti, P. Walian, R. Needleman, J. K. Lanyi, R. M. Glaeser, and H. Luecke. 2001. Crystal structure of the D85S mutant of bacteriorhodopsin: model of an O-like photocycle intermediate. J. Mol. Biol. 313:615–628. [DOI] [PubMed] [Google Scholar]
- Royant, A., K. Edman, T. Ursby, E. Pebay-Peyroula, E. M. Landau, and R. Neutze. 2000. Helix deformation is coupled to vectorial proton transport in the photocycle of bacteriorhodopsin. Nature. 406:645–648. [DOI] [PubMed] [Google Scholar]
- Royant, A., P. Nollert, K. Edman, R. Neutze, E. M. Landau, E. Pebay-Peyroula, and J. Navarro. 2001. X-ray structure of sensory rhodopsin II at 2.1-A resolution. Proc. Natl. Acad. Sci. USA. 98:10131–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, J., L. S. Brown, Y. S. Chon, H. Kandori, A. Maeda, R. Needleman, and J. K. Lanyi. 1995. Conversion of bacteriorhodopsin into a chloride ion pump. Science. 269:73–75. [DOI] [PubMed] [Google Scholar]
- Sass, H. J., G. Buldt, R. Gessenich, D. Hehn, D. Neff, R. Schlesinger, J. Berendzen, and P. Ormos. 2000. Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin. Nature. 406:649–653. [DOI] [PubMed] [Google Scholar]
- Schobert, B., J. Cupp-Vickery, V. Hornak, S. Smith, and J. Lanyi. 2002. Crystallographic structure of the K intermediate of bacteriorhodopsin: conservation of free energy after photoisomerization of the retinal. J. Mol. Biol. 321:715–726. [DOI] [PubMed] [Google Scholar]
- Smith, S. O., J. Lugtenburg, and R. Mathies. 1985. Determination of retinal chromophore structure in bacteriorhodopsin with resonance Raman spectroscopy. J. Membr. Biol. 85:95–109. [DOI] [PubMed] [Google Scholar]
- Soulimane, T., G. Buse, G. P. Bourenkov, H. D. Bartunik, R. Huber, and M. E. Than. 2000. Structure and mechanism of the aberrant ba(3)-cytochrome c oxidase from Thermus thermophilus. EMBO J. 19:1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam, S., and R. Henderson. 2000. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 406:653–657. [DOI] [PubMed] [Google Scholar]
- Tittor, J., U. Haupts, C. Haupts, D. Oesterhelt, A. Becker, and E. Bamberg. 1997. Chloride and proton transport in bacteriorhodopsin mutant D85T: different modes of ion translocation in a retinal protein. J. Mol. Biol. 271:405–416. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., M. Nakasako, H. Nomura, and H. Ogawa. 2000. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 angstrom resolution. Nature. 405:647–655. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., and H. Nomura. 2002. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 418:605–611. [DOI] [PubMed] [Google Scholar]
- Tsukihara, T., H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawaitoh, R. Nakashima, R. Yaono, and S. Yoshikawa. 1996. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 angstrom. Science. 272:1136–1144. [DOI] [PubMed] [Google Scholar]
- Vonck, J. 2000. Structure of the bacteriorhodopsin mutant F219L N intermediate revealed by electron crystallography. EMBO J. 19:2152–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, D., C. A. Yu, H. Kim, J. Z. Xian, A. M. Kachurin, L. Zhang, L. Yu, and J. Deisenhofer. 1997. Crystal structure of the cytochrome bc(1) complex from bovine heart mitochondria. Science. 277:60–66. [DOI] [PubMed] [Google Scholar]
- Yu, E. W., and D. E. Koshland, Jr. 2001. Propagating conformational changes over long (and short) distances in proteins. Proc. Natl. Acad. Sci. USA. 98:9517–9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. L., L. S. Huang, V. M. Shulmeister, Y. I. Chi, K. K. Kim, L. W. Hung, A. R. Crofts, E. A. Berry, and S. H. Kim. 1998. Electron transfer by domain movement in stockbroker bc(1). Nature. 392:677–684. [DOI] [PubMed] [Google Scholar]