

Abstract
Conformational transitions are thought to be the prime mechanism of amyloid formation in prion diseases. The prion proteins are known to exhibit polymorphic behavior that explains their ability of “conformation switching” facilitated by structured “seeds” consisting of transformed proteins. Oligopeptides containing prion sequences showing the polymorphism are not known even though amyloid formation is observed in these fragments. In this work, we have observed polymorphism in a 15-residue peptide PrP (113–127) that is known to form amyloid fibrils on aging. To see the polymorphic behavior of this peptide in different solvent environments, circular dichroism (CD) spectroscopic studies on an aqueous solution of PrP (113–127) in different trifluoroethanol (TFE) concentrations were carried out. The results show that PrP (113–127) have sheet preference in lower TFE concentration whereas it has more helical conformation in higher TFE content (>40%). The structural transitions involved in TFE solvent were studied using interval-scan CD and FT-IR studies. It is interesting to note that the α-helical structure persists throughout the structural transition process involved in amyloid fibril formation implicating the involvement of both N- and C-terminal sequences. To unravel the role of the N-terminal region in the polymorphism of the PrP (113–127), CD studies on another synthetic peptide, PrP (113–120) were carried out. PrP(113–120) exhibits random coil conformation in 100% water and helical conformation in 100% TFE, indicating the importance of full-length sequence for β-sheet formation. Besides, the influence of different chemico-physical conditions such as concentration, pH, ionic strength, and membrane like environment on the secondary structure of the peptide PrP (113–127) has been investigated. At higher concentration, PrP (113–127) shows features of sheet conformation even in 100% TFE suggesting aggregation. In the presence of 5% solution of sodium dodecyl sulfate, PrP (113–127) takes high α-helical propensity. The environment-dependent conformational polymorphism of PrP (113–127) and its marked tendency to form stable β-sheet structure at acidic pH could account for its conformation switching behavior from α-helix to β-sheet. This work emphasizes the coordinative involvement of N-terminal and C-terminal sequences in the self-assembly of PrP (113–127).
INTRODUCTION
Misfolded isoforms of the naturally occurring prion protein (PrP) have been shown to be the causative agents in many mammalian neurodegenerative disorders, including Creutzfeldt-Jakob disease in humans, scrapie in sheep, and bovine spongiform encephalopathy in cows (Prusiner, 1997). Prion infectivity is unique in that the pathogenic prion form (PrPSc) is involved in the conversion of the endogenous conformation (PrPc) into the transformed PrPSc. This leads to infusion of transformed prion particles to “seed” the host for production of large amounts of the pathogenic conformation without the influence of foreign nucleic acids (Prusiner, 1982). The “protein-only” hypothesis (Harrison et al., 1997; Prusiner, 1998) asserts further that no extraneous agents are necessary to explain prion's unusual behavior. This hypothesis has been advanced by a number of in vitro experiments involving recombinant and synthetic prion fragments (Forloni et al., 1993).
The mechanism by which PrPc converts to PrPSc is still unknown, although two major models have been proposed (Kelly, 2000). The standard catalytic model (Huang et al., 1996a) proposes that PrPc and PrPSc are distinct in monomeric stable states, but the presence of PrPSc catalyzes the conversion of PrPc to PrPSc. Alternatively, the nucleated polymerization model (Lansbury and Caughey, 1995; Caughey et al., 1995) suggests that PrPSc is intrinsically multimeric, differing from PrPc primarily in its quaternary structure. Changes in secondary and tertiary structure may accompany this change in the polymerization state, as seems to occur with amyloid formation (Kelly, 1998).
The uncertainty about the mechanism of the PrP structural transition is due to the fact that the structure of PrPSc is yet to be understood in atomic detail. The primary structure of PrPc is composed of two structurally different parts: an extended N-terminal segment (residues 23–125) with features of a flexible and disordered peptide chain and a well-defined globular domain (residues 126–231) with three α-helices and two antiparallel β-sheets (James et al., 1997). The gross structure of PrPSc is known to have more β-sheet content (Forloni et al., 1996) than PrPc. A model for the tertiary structure of PrPSc was also suggested (Huang et al., 1996b). Recently, Wille et al. (2002) have proposed a parallel β-helical structure for PrPSc based on their electron microscopic work. This opens up a new experimental possibility of prion's assembly structure in the assembled form. However, the dynamic details of structural transition need novel methods to probe the solution state structure of prion peptide fragments using spectroscopic techniques. Early conformational studies have indicated that a transition from α-helical to β-sheet structure (PrPc-PrPSc) is likely to be the crucial event in prion propagation (Pan et al., 1993; Prusiner, 1998; Prusiner et al., 1998; Petchanikow et al., 2001; Levy and Becker, 2002). To investigate which amino acid sequences feature in the conformational transition from PrPc to PrPSc and PrP amyloid, several groups have analyzed the secondary structure and fibrillogenic properties of synthetic peptides of PrP (Baldwin et al., 1995). Using these peptides, it has been extensively studied and established that consecutive segment of prion protein spanning the residues 106–147 is important for the fibrillogenic properties of the protein (Tagliavini et al., 1991, 1993, 1994; Gioia et al., 1994). In particular, the sequence comprising residues 106–126 of human prion protein, which corresponds to a highly conserved region of PrP, located in the N-terminal segment adjacent to the structurally organized globular domain (Donne et al., 1997), is found to be neurotoxic (Forloni et al., 1994). Moreover, it is also reported that in brain tissues of patients of an Indiana family with Gerstmann-Sträussler-Scheinker disease, most of the fibrillogenic sequence corresponds to residues 106–126 of human PrP (Tagliavini et al., 1991, 1993).
PrP (106–126) consists of an N-terminal polar head (KTNMKHM) followed by a long hydrophobic tail (AGAAAAGAVVGGLG), and its structural characterization is markedly influenced by several physiological factors such as ionic-strength, pH, solvent composition, and other factors (Gioia et al., 1994). This peptide readily forms amyloid fibrils, which are resistant to proteinase K and pronase digestion (Selvaggini et al., 1993); it is neurotoxic and induces activation of astrocytes and microglial cells in vitro (Brown, 1999). The conformational changes of PrP (106–126) as a function of pH are confined to both an alanine-containing peptide sequence, AGAAAAGA (Ragg et al., 1999) and a valine-rich tail part, VVGGLGG. Molecular modeling studies (Huang et al., 1996b) have already indicated that the pathogenical conversion of PrPc into PrPSc might involve the AGAAAAGA region. Further, AGAAAAGA peptide blocked the toxicity of PrP (106–126), suggesting that this sequence is necessary for the interaction of PrP (106–126) with neurons (Brown, 2000). This indicates the possibility that the AGAAAAGA peptide may show a therapeutic opportunity for controlling prion disease. Gasset et al. (1992) have suggested that the hydrophobic part (VVGGLGG) may have no role in determining the secondary structure of PrP (113–127).
We felt it is worthwhile to study the conformational transition of AGAAAAGAVVGGLGG, an amyloid forming fragment, and AGAAAAGA, a fragment known to form nontoxic fibrils and prevent amyloid propagation. To the best of our knowledge, this is the first time that this sequence is subjected to detailed conformational analysis. The main problem faced by the earlier workers was the solubility of this sequence. Both the above peptides are virtually insoluble in aqueous solution. We have preprocessed with an acetonitrile water mixture (1:1) with little formic acid to make a solvated gel of the peptide. Interestingly this gel got dissolved dramatically in aqueous solutions and made this study possible.
Conformational analysis of the above two peptides in structure forming solvents such as 2,2,2-trifluoroethanaol (TFE) was carried out. TFE has been used extensively to enhance the helical propensity by various workers (Nelson and Kallenbach, 1986; Jansanoff and Fersht, 1994; Bodkin and Goodfellow, 1996; Luidens et al., 1996). In this work we have investigated whether and how different physicochemical conditions such as pH and ionic strength or membrane-like environment and concentration influence the conformation and self-assembly of PrP (113–127).
MATERIALS AND METHODS
Peptide synthesis and purification
The peptides PrP (113–127) and PrP (113–120) were synthesized by manual solid phase chemistry, using t-butyloxy carbonyl group (Boc) as the protective group for N-terminal ends, and 1-hydroxy benzotriazole (HOBt) and N, N-dicyclohexyl-carbodiimide as activators of carboxylic ends. The peptides were cleaved from (4-methyl benzhydrylamine) MBHA resin with trifluoromethane sulfonic acid/thioanisole/ethanedithiol/trifluoroacetic acid (1:1:1:7) (Stewart and Young, 1984) and precipitated with cold ether. The composition of peptides was determined by amino acid analysis using the phenyl isothiocyanate (PITC) method. The purified peptide was identified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis. All reagents used in peptide synthesis were of the purest analytical grade; t-butyl carbazide, HOBt, N, N-dicyclohexyl-carbazide, and trifluoroacetic acid were purchased from Aldrich (Seelze, Germany). Boc-amino acids were prepared using standard procedures (Katsoyannis and Schwartz, 1977; Bodanszky and Bodanszky, 1984) and were characterized by thin-layer chromatography and FTIR spectroscopic studies.
Predicted secondary structure
The α-helix and β-sheet propensities were calculated by the Chou and Fasman secondary structure prediction algorithm (Chou and Fasman, 1978) using the program Peptide Companion version 1.24.
Circular dichroism spectroscopy
To investigate the influence of the α-helix stabilizing solvent 1,1,1-trifluoroethanol (TFE) on the secondary structure of the peptides, TFE titrations were carried out. Similar stock solutions of the peptides in a phosphate buffer (50 mM, pH 7) and in neat TFE were prepared. The two stock solutions were mixed in such a way as to give different concentration of TFE. CD spectra have been recorded at a temperature of 300 K and at 0%, 5%, 10%, 15%, 20%, 25%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, and 100% TFE.
To analyze the effects of sodium dodecyl sulfate (SDS) micelles on the PrP (113–127) and PrP (113–120) conformation, CD spectra were collected from a solution of 5% SDS in deionised water. SDS was obtained from SRL (Mumbai, India); TFE was purchased from Aldrich.
CD spectra were recorded in a quartz cell with an optical path of 0.1 cm using a Jasco J-715 (Tokyo, Japan) spectropolarimeter at a scan speed of 50 nm/min. The percentages of the secondary structures of PrP (113–127) and PrP (113–120) were calculated using the SELCON3 program (Sreerama et al., 2000).
FT-IR studies
The FT-IR spectra have been recorded in the Avatar 310 thermonicolet spectrometer (32 scans, resolution: 1 cm−1). The Fourier self-deconvoluted IR spectra (FSD) have been obtained by using the Nicolet software with a reduction factor of 2.5 and a bandwidth of 15 cm−1. All the IR spectra were recorded at 300 K.
TEM studies
Electron microscopy was carried out by using specimen grids that were coated with 0.25% formvar and the carbon and subjected to glow discharge (Prusiner et al., 1983). Five microliters of gel stock of PrP (113–127) was applied to a grid for 1 min before any excess was washed away. Negative staining was performed in uranyl acetate for 10 s (Williams, 1977; McKinley et al., 1986). Specimen grids were examined by using PHILIPS electron microscopy at 80 keV.
RESULTS AND DISCUSSION
The aim of this work is to analyze the intermediate conformational preferences in the polymorphic behavior of AGAAAAGAVVGGLGG. The individual domain conformational preferences, the helix (A) or sheet propensity (B) of each residue for the full sequence has been carried out by the Chou and Fasman method and is depicted in Fig. 1. The results indicate that the 113–127 sequence has two major domains, a helical conformation favoring the 113–118 sequence and a β-sheet forming domain consisting of residues of 118–122, thereby indicating that this peptide has the tendency to form both helical and sheet conformation.
FIGURE 1.

The amino acid sequence, α-helical and β-sheet probability for PrP (113–127) and PrP (113–120). The single and double bar represent the region of PrP peptides having a high probability of β-sheet and α-helical structures, respectively.
Our results indicate that PrP (113–127) in 100% water exhibits β-sheet conformation, whereas it takes α-helical structure in 100% TFE. The smaller N-terminal segment PrP (113–120), an alanine-rich sequence, exhibits random coiled structure in 0% TFE and helical structure in 100% TFE. CD spectra in the region of 260 to 185 nm for PrP (113–127) and PrP (113–120) in different TFE concentrations in aqueous solutions are shown in Fig. 2 and Fig. 3, respectively. The increase in the TFE concentration from 0% to 80% of the PrP (113–127) solution results in a shift of positive maximum from 199.3 to 190 nm, and the value of [θ]222 shifts from −7621 to −10,660 deg cm2 dmol−1 in the CD spectra. This indicates the structural transition from sheet to helical conformation. Around 15% TFE, the CD curves are characteristic of β-sheet plus undefined structure, as characterized by the presence of an isodichroic point at 210 nm (Fig. 2 a). Between 20% and 60% TFE, the spectrum undergoes a major change and exhibits features suggestive of α-helical structure (Fig. 2 b). The maximal helix formation is found to occur at 80% TFE (Table 1) and on further increase in concentration of TFE, the CD spectrum exhibited a significant decrease in the signal intensity (Fig. 2 c; Table 1). The positive maximum is located at 190 nm, with the negative maximum at 205.7 nm and the value of [θ]222 = −10,177 deg cm2 dmol−1 (in 100% TFE). At low TFE concentrations (<15% TFE), the intrinsic preference of the peptide to form β-structure is manifested due to the stabilization of intermolecular hydrogen bonding at this microenvironment. However, at higher TFE concentrations, the hydrophobic interior of the β-sheet structure would be disrupted and the PrP (113–127) then refolds as an α-helix.
FIGURE 2.



CD spectrum of PrP (113–127) in phosphate buffer (0.05 M, pH 7) titrated with neat TFE at 293 K. (a) a, 0; b, 5; c, 10; and d, 15% TFE (v/v). (b) e, 20; f, 25; g, 30; h, 40; i, 50; and j, 60% TFE (v/v). (c) k, 70; l, 80; m, 90; n, 95; and o, 100% TFE (v/v). Concentration of PrP (113–127) is 0.7 mM.
FIGURE 3.

CD spectrum of PrP (113–120) in phosphate buffer (0.05 M, pH 7) titrated with neat TFE at 293 K (0 → 100% TFE (v/v)). Concentration of PrP (113–120) is 0.26 mM.
TABLE 1.
Estimates of secondary structure in PrP (113–127) from analysis of CD spectra
| % of secondary structure for PrP (113–127)
|
||||
|---|---|---|---|---|
| α-helix | β-sheet | β-turn | Random coil | |
| a. %TFE | ||||
| 0 | 5.2 | 44.9 | 16.5 | 31.4 |
| 5 | 5.3 | 44.9 | 18.6 | 29.1 |
| 10 | 3.6 | 45.4 | 21.6 | 32.9 |
| 15 | 3.3 | 40.6 | 25.5 | 32.0 |
| 20 | 5.9 | 36.7 | 24.9 | 32.2 |
| 25 | 6.7 | 34.5 | 26.8 | 38.5 |
| 30 | 14.0 | 26.2 | 23.1 | 29.6 |
| 40 | 24.8 | 23.4 | 21.6 | 31.5 |
| 50 | 31.5 | 18.9 | 19.5 | 29.2 |
| 60 | 34.4 | 16.9 | 19.3 | 28.5 |
| 70 | 36.3 | 11.1 | 20.0 | 31.0 |
| 80 | 38.4 | 9.6 | 18.3 | 31.4 |
| 90 | 35.1 | 11.5 | 20.4 | 30.5 |
| 95 | 36.4 | 12.4 | 19.7 | 30.0 |
| 100 | 36.0 | 15.4 | 19.1 | 29.1 |
| b. Concentration, mM | ||||
| 0.17 | 41.7 | 14.8 | 18.7 | 26.0 |
| 0.86 | 38.0 | 15.8 | 19.6 | 27.0 |
| 1.21 | 33.9 | 18.7 | 18.8 | 28.0 |
| 1.73 | 30.2 | 21.5 | 20.1 | 27.9 |
| 2.25 | 25.0 | 27.0 | 21.4 | 27.4 |
| 2.95 | 24.3 | 29.9 | 20.2 | 26.0 |
| c. 5% SDS solution | 44.7 | 13.6 | 16.4 | 26.6 |
| d. pH | ||||
| 2 | 1.7 | 43.4 | 21.2 | 32.4 |
| 5 | 6.2 | 43.0 | 18.7 | 29.9 |
| 7 | 5.2 | 44.9 | 16.5 | 31.4 |
| 10.6 | 22.5 | 41.6 | 14.8 | 22.0 |
Molecular interactions that stabilize helix formation in peptides are dependent not only upon sequences but also on the molecular environment (Merukta et al., 1990). It has been shown for many peptides that the increase in TFE concentration results in further increase in the degree of helicity (Nelson and Kallenbach, 1986; Lehrman et al., 1990; Bruch et al., 1991; Storrs et al., 1992) or helix formation reaches a plateau (McLeish et al., 1994; Pintar et al., 1994; Wilson et al., 1994; Zhang and Vogel, 1994; Xu et al., 1995). In this study, for the PrP (113–127), helix formation increases with increase in TFE concentration, and it reaches a maximum at 80% TFE. However, further increase in TFE concentration leads to a decrease in helicity of the PrP (113–127), indicating destabilizing interactions that are reinforced in a hydrophobic environment (Fig. 4 a; Table 1).
FIGURE 4.


(a) Plot of degree of helicity of PrP (113–127) (•) and degree of sheet structure (▴) with TFE concentration. A decrease in helicity at high TFE concentration indicating a destabilizing effect in a hydrophobic environment. (b) Plot of degree of helicity of PrP (113–120) (•) and degree of random coiled structure (▴) with TFE concentration. An increase in helicity with increase with TFE concentration, indicating stabilizing interactions that are reinforced in a hydrophobic environment.
These results suggest that TFE stabilizes the helical structure of PrP (113–127), which has the preference for β-sheet conformation in water. TFE is known to be a denaturant and destabilizes hydrophobic regions of the protein. Since the β-sheet structure involves the alignment of noncontiguous regions of the polypeptide chain and is thought to be stabilized by packing of nonpolar residues in the interior of the sheet, it is proposed that TFE can denature such structure. Our titration data, together with the data reported from other groups (Nelson and Kallenbach, 1986; Lehrman et al., 1990; Bruch et al., 1991; Storrs et al., 1992; Chakrabartty et al., 1993; Zhou et al., 1993; McLeish et al., 1994; Pintar et al., 1994; Wilson et al., 1994; Zhang and Vogel, 1994; Albert and Hamilton, 1995; Xu et al., 1995), emphasizes the crucial role of hydrophobic interaction in helix to sheet transition. Further, the titration data for PrP (113–127) suggests a loss of helicity above 80% TFE (in enhanced hydrophobic environment), which is similar to that observed in polyalanine-based peptides by Chakrabartty et al. (1993).
The plot of % β-sheet conformation versus TFE concentration for PrP (113–127) is also shown in Fig. 4 a. It is tacit that between 0% and 10% TFE, there is no noticeable change in the sheet content. However, after TFE attains 15%, there is a rapid decrease in sheet preference and attainment of the minimal value of sheet conformation at 80% TFE. These observations suggest that the conformational transition determined by the hydrophobic or hydrophilic environment is a two-stage process. The loss of helicity is compensated by gain of the sheet structure.
To probe the role of AGAAAAGA in determining the conformation of PrP (113–127), a TFE titration study was carried out on PrP (113–120) (Fig. 3). Unlike the 15-residue sequence, the PrP (113–120) takes random coil conformation in 100% water and helical conformation in 100% TFE (Table 2). In proteins or peptides, random coil conformation is characterized by a negative absorption between 195 and 200 nm (Greenfield and Fasman, 1969). Random coil conformation of PrP (113–120) in 0% TFE is deduced by the strong negative absorption at 195 nm (Fig. 3). On increasing the concentration of TFE, the proportion of random coiled structure of PrP (113–120) decreases gradually with increase in helical conformation. The structural transition of PrP (113–120) from coil to helical structure on increasing the TFE concentration is accompanied by shift of the negative maximum (195 nm) of random coiled structure to π-π* exciton component (204 nm) of helical structure, and the value of [θ]222 increases from −409 to −9248 deg cm−1 dmol−1. The increase of α-helicity is attributable to the increase in hydrophobic stabilizing interaction (Zhou et al., 1993).
TABLE 2.
Estimates of secondary structure in PrP (113–120) from analysis of CD spectra
| % of secondary structure for PrP (113–120)
|
||||
|---|---|---|---|---|
| α-helix | β-sheet | β-turn | Random coil | |
| a. % TFE | ||||
| 0 | 3.2 | 6.2 | 23.7 | 59.6 |
| 5 | 3.5 | 12.6 | 24.0 | 58.1 |
| 10 | 9.3 | 13.1 | 26.3 | 51.7 |
| 15 | 7.7 | 16.4 | 24.5 | 52.4 |
| 20 | 9.6 | 18.7 | 23.1 | 47.9 |
| 25 | 11.9 | 18.6 | 24.8 | 47.9 |
| 30 | 12.4 | 17.6 | 25.4 | 46.3 |
| 40 | 17.5 | 17.4 | 24.5 | 43.2 |
| 50 | 20.7 | 16.3 | 21.8 | 40.9 |
| 60 | 22.0 | 21.4 | 19.0 | 36.9 |
| 70 | 25.9 | 21.7 | 19.8 | 34.9 |
| 80 | 24.7 | 24.3 | 19.2 | 30.7 |
| 90 | 28.4 | 17.7 | 20.4 | 34.0 |
| 95 | 26.8 | 20.7 | 21.1 | 32.0 |
| 100 | 28.5 | 20.9 | 21.0 | 29.6 |
| b. 5% SDS solution | 24.3 | 13.0 | 16.1 | 42.9 |
The CD spectroscopic data for PrP (113–120) in different concentrations of TFE indicate the existence of two conformations, random coil and α-helix. This inference is further supported by the presence of an isodichroic point at 200.5 nm (Fig. 3), where the [θ] intensities are equal. Also, it could be seen from Fig. 4 b that the slope (m = 0.27, R2 = 0.949) of decrease in % random coil structure with increasing the TFE concentration is almost equal to the slope (m = 0.26, R2 = 0.954) of increase in % helicity with TFE concentration. The results could be explained by the TFE property, which is known to stabilize the “nascent helices” (Jansanoff and Fersht, 1994) even in small fragments. Further the SELCON3 analysis reveals that there is an increase in % helicity from 3.2 (0% TFE) to 28.5 (100% TFE) and % unordered form decreases from 59.6 (0% TFE) to 29.6 (100% TFE); the same is given in Table 2. In a fully aqueous environment, the dominant interactions are intermolecular interactions, and in high TFE, the intramolecular interaction predominates (Myers et al., 1998). However, for the peptide PrP (113–120), the lack of β-sheet structure indicates that in aqueous solution such intermolecular association is possible only with more hydrophobic sequences. This is because the hydrophobic sequence will lead to aggregation that will lead to intermolecular interaction.
The importance of aggregation-promoted sheet formation is explored by studying the conformation of PrP (113–127) at different concentrations. The representative CD spectra of PrP (113–127) at 0.17, 0.86, 1.21, and 2.95 mM, are shown in Fig. 5. The amount of secondary structure calculated for the different concentrations of PrP (113–127) in TFE are tabulated in Table 1. It is found that at higher concentration, sheet structure predominates even in TFE, indicating that at these concentrations the molecular crowding effect operates, leading to β-sheet formation.
FIGURE 5.

CD spectrum of PrP (113–127) in a (——) 0.17 mM, b (— —) 0.86 mM, c (···–···–) 1.21 mM, and d (·······) 2.95 mM in TFE.
To further assert the effect of molecular crowding on the structure of PrP (113–127), an interval scan CD study with TFE was carried out. A dilute solution of peptide (0.17 mM) in TFE was sandwiched between two quartz plates, and the solvent TFE was allowed to evaporate slowly. CD spectra were recorded during the removal of TFE from the peptide environment. The interval scan CD spectrum is shown in Fig. 6 a. From the figure it could be noted that removal of TFE molecules around the peptide backbone results in more sheet conformation at the expense of helical and unordered structures. This makes one infer that in absence of a solvent (TFE) environment, PrP (113–127) takes predominantly sheet conformation (Fig. 6 a). A similar study was also performed using an interval scan FT-IR spectrometer, using ZnSe diskettes. The FT-IR spectra taken at different time intervals of time on removal of TFE from the peptide backbone are shown in the inset of Fig. 6 a. In the presence of TFE, PrP (113–127) shows a mixture of helical (1656 cm−1), sheet (1627 cm−1), and unordered (1647 cm−1) conformations, which is consistent with the CD studies, whereas removal of solvent TFE from the peptide backbone results in β-sheet (antiparallel) structure. A notable feature observed was the persistence of α-helical content in the structural transition, indicating the importance of helical turn conformation in the aggregation process. There are reports in favor of the view that most of the proteins may start their life in an α-helical conformation, even if their later state is to be β-sheet, indicating that the helix nucleation is more kinetically stabilized when compared to β-sheet structure. Moreover, the existing sheet structure of PrP (113–127) in the xerogel state (inset of Fig. 6 a) exhibits two types of carbonyls (1637 and 1627 cm−1) in addition to helix (1656 cm−1) and coil (1647cm−1) structures. The conformational transition is also accompanied by increase in absorbance of peak at 1689 cm−1, suggestive of antiparallel sheet conformation.
FIGURE 6.


(a) Interval scan CD and FT-IR spectra (inset) of PrP (113–127), on evolution of TFE. (b) Interval scan CD and FT-IR spectra (inset) of PrP (113–120), on evolution of TFE.
The interval scan CD spectrum of PrP (113–120) on gradual removal of TFE is shown in Fig. 6 b. It is inferred from the figure that removal of TFE molecules around the peptide backbone results in more sheet structure at the expense of unordered structure. The interval scan FT-IR spectra of PrP (113–120) on TFE evolution are shown in the inset of Fig. 6 b, which is in agreement with CD studies. This makes one conclude that in the absence of a solvent (TFE) environment, PrP (113–120) takes dominant sheet conformation, which is indicated by the peak at 1627 cm−1, depicted in Fig. 6 b. In the CD spectrum, the negative band near 219 nm is assigned to the n-π* transition, whereas the positive band near 200 nm is assigned to the π-π* exciton component (Madison and Schellman, 1972). Weakly twisted sheets have n-π* and π-π* maxima of approximately equal magnitude; and for strongly twisted sheets, the π-π* band at 200 nm is much stronger than the n-π* band (Fasman, 1996). From the xerogel CD spectra of both the peptides, it is inferred that the π-π* exciton component of PrP (113–120) (θπ-π*/θn-π* = 4.7) is stronger than that of PrP (113–127) (θπ-π*/θn-π* = 2.7), suggesting PrP (113–120) sheet conformation is more twisted than PrP (113–127). The results indicate that both the peptides take antiparallel β-sheet conformations in the solid state, which are found to be in agreement with ATR-FTIR studies of Gasset et al. (1992).
SDS is an amphipathic molecule extensively used to mimic a membrane-like environment (Rapoport, 1985; Sitaram and Nagaraj, 1993). When dissolved in 5% SDS, PrP (113–127) presented a high content of α-helical structure, as shown by two negative absorptions of nearly the same intensity at 207 and 221 nm and the positive band at 192 nm (Fig. 7). The presence of the β-sheet structure could be deduced from the broadening of absorption peaks at 207 and 221 nm. PrP (113–120) also takes more of a helical structure in 5% SDS (Fig. 7). To extend the observation of the stability of the β-sheet structure of PrP (113–127), up to 5% SDS was added to the solution of PrP (113–127) in 200 mM phosphate buffer, pH 5 or 7. In such a high ionic condition, the existing β-sheet gets converted into an α-helical structure. It should be mentioned that the longer PrP (106–126) was reported to show no changes in the β-sheet content when treated with 5% SDS (Gioia et al., 1994). This is because the β-sheet in the amyloid assemblage is quite stable to be disassembled by SDS micelles. The dissolution of PrP (113–127) assemblage in SDS may be due to lack of KTNMKHM sequence. The hydrophobic part AGAAAAGAVVGGLGG offers a suitable module for interacting with membranes. This pattern has been very frequently observed in peptide fragments involved in membrane translocation of proteins (Rapoport, 1985; Wickner and Lodish, 1985), and a transition toward the β-sheet conformation was observed in signal peptides in a highly hydrophobic environment (Laxma Reddy and Nagaraj, 1989). This environment-dependent structural transition leads us to infer that the PrPc to PrPsc transition occurring in the post-translational process may be driven by the nature of the environment around the PrPc and the formation of intermolecular hydrogen bonding.
FIGURE 7.

CD spectrum of PrP (113–127) (——) and PrP (113–120) (·······), in 5% sodium dodecyl sulfate.
To study the effect of pH on the secondary structure of PrP (113–127), CD spectra were taken at pH 2, 5, 7, and 10.6. The CD spectra are shown in Fig. 8. It is observed that PrP (113–127) takes sheet conformation in all the conditions (pH 2, 5, 7, and 10.6), suggesting the mode of aggregation in aqueous conditions. It should be noted that there is slight variation in the characteristic of sheet conformation in different pHs. The PrP (106–126) was reported to show sheet conformation at pH 5 and less ordered conformation at pH 7 (Gioia et al., 1994). These results indicate the importance of the hydrophobic segment 113–127 in deciding the PrP (106–126) conformation at pH 5. Also, the strength of sheet conformation in different pHs does not change on increasing the strength of the buffer solution, implying that the secondary structure of PrP (113–127) is independent of the ionic strength of the medium, which indicates the negligible role of electrostatic interaction in aggregation.
FIGURE 8.

CD spectrum of PrP (113–127) in pH 2 (·······), pH 5 (— —), pH 7 (···–···–), and pH 10.6 (——).
In aqueous solution at physiological pH, PrP (113–127) readily aggregates, a process that occurs during amyloid formation in prion diseases. PrP (113–127) on dissolution in 50% acetonitrile at pH 4 (adjusted with formic acid unbuffered) forms gel, whereas PrP (113–120) does not. The absence of gel formation in PrP (113–120) in this condition indicates the importance of the tail portion in driving the molecule to form the gel. Gasset et al. (1992) have suggested the tail region VVGGLGG has no role in forming the sheet structure of PrP (113–127). However, the gel formation exhibited by the PrP (113–127) leads us to infer that critical length may be required to form amyloid fibrils, in addition to its hydrophobic character. The morphological property of PrP (113–127) in the gel form was analyzed using electron micrographs, shown in Fig. 9 A. In general, formation of a gel requires the gelator to separate the finely dispersed aggregate particles to make them join together and form a continuous coherent framework throughout the fluid volume (Hermans, 1949). Hence, the development of a three-dimensional network by the gelator to capture small domains of isotropic fluid is critical to gel formation (Kishore et al., 1987; Jayakumar et al., 2000). The size and shape of the building blocks for the network may vary greatly between gels, but all must have an immobile framework in which the fluid is trapped. From Fig. 9 A it is seen that the fibrous texture exists, which is attributed to the network supporting the fluid component in the gel. The schematic representation of the structure of the gel state is shown in Fig. 9 B. The approximate diameter of the fibrillar bundle is around 10 nm. The fibrils appear to be three-dimensional and interlocking and to consist of fibrillar bundles. However, on diluting the gel with water, the fibers of the bundles interwine to immobilize the isotropic fluid, probably gained by surface tension (Fig. 9 C). The schematic representation of the structure developing fibrils is shown in Fig. 9 D. Perhaps as a consequence of the weakness of the attractive forces responsible for their stability, the fibers are seemingly flexible. Also, the driving force for the gel to form fibers may be due to its overall hydrophobicity. The developing fibrils of PrP (113–127) on water are shown in Fig. 9 C. The well-developed fibrils formed from PrP (113–127) were reported by Gasset et al. (1992). It is found that the diameter of the developing fibrils of PrP (113–127) (≈9 nm) is similar to the previous report (Gasset et al., 1992).
FIGURE 9.

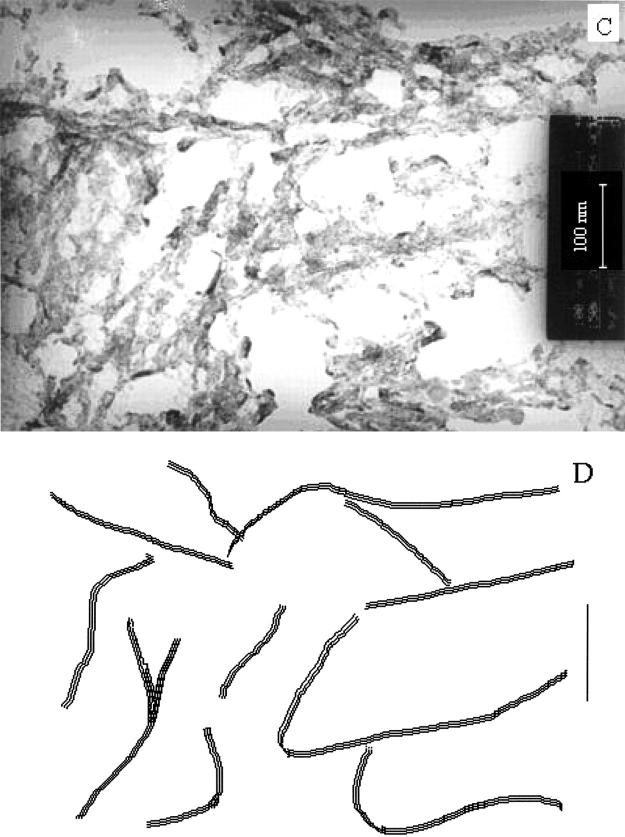
Electron micrographs of PrP (113–127). (A) In the gel form; (B) the schematic representation in the gel form; (C) after diluting with water; and (D) the schematic representation after diluting with water.
CONCLUSION
This work describes the conformational preferences of the prion fragments PrP (113–127) and PrP (113–120) in different microenvironments attained by TFE/water mixtures, 5% SDS and different pH conditions. The TFE titration study of PrP (113–127) shows that hydrophobic environment around the peptide backbone drives the peptide to take the helical conformation. This is further evidenced by the existence of helical conformation in 5% SDS, where the environment around the peptide backbone is hydrophobic. On the other hand, hydrophilic environment around the peptide backbone drives the secondary structure to take the sheet conformation as observed by the existence of sheet conformation on the glass surface. Further, the conformation of PrP (113–127) transitions seems to be a two-stage process involving the helical and sheet structure, where the molecular crowding effect and hydrophobic interaction play a crucial role. By considering both the above conclusion and from the enumeration from the TFE titration, the mechanism of aggregation as shown in Fig. 10 is proposed.
FIGURE 10.

Proposed model for the mechanism of fibril formation of PrP (113–127) (scheme 1) and for PrP (113–120) (scheme 2).
Thus, the major driving forces for the PrP (113–127) to self-assemble are the greater hydrophobicity and intermolecular hydrogen bonding. These support the contention that the ability to form amyloid fibrils is a generic property of polypeptide chains and that absence of fibril in normally functioning organisms is a consequence of the control and regulation mechanisms that have evolved to ensure that proteins find and maintain their correctly folded functional structures (Guijarro et al., 1998; Chiti et al., 1999; Grob et al., 1999). The emerging universality in these properties of amyloid fibrils may be of help in the quest for therapeutic substances that block this misfolding event in the context of human diseases.
Acknowledgments
We thank Dr. T. Ramasamy, Director, CLRI, for his kind permission to publish this work. We also thank Prof. Andrews Chandramohan for taking the TEM photos and Prof. P. Balaram for taking the MALDI-TOF MS spectra.
References
- Albert, J. S., and A. D. Hamilton. 1995. Stabilization of helical domains in short peptides using hydrophobic interactions. Biochemistry. 34:984–990. [DOI] [PubMed] [Google Scholar]
- Baldwin, M. A., F. E. Cohen, and S. B. Prusiner. 1995. Prion protein isoforms, a convergence of biological and structural investigations. J. Biol. Chem. 270:19197–19200. [DOI] [PubMed] [Google Scholar]
- Bodanszky, M., and A. Bodanszky. 1984. The Practice of Peptide Synthesis. Springer-Verlag, New York.
- Bodkin, M. J., and J. M. Goodfellow. 1996. Hydrophobic solvation in aqueous trifluoroethanol solution. Biopolymers. 39:43–50. [DOI] [PubMed] [Google Scholar]
- Brown, D. R. 1999. Prion protein peptide neurotoxicity can be mediated by astrocytes. J. Neurochem. 73:1105–1113. [DOI] [PubMed] [Google Scholar]
- Brown, D. R. 2000. Prion protein peptides: optimal toxicity and peptide blockade of toxicity. Mol. Cell. Neurosci. 15:66–78. [DOI] [PubMed] [Google Scholar]
- Bruch, M. D., M. M. Dhingra, and L. M. Gierasch. 1991. Side chain-backbone hydrogen bonding contributes to helix stability in peptides derived from an alpha-helical region of carboxypeptidase A. Proteins. Struct. Funct. Genet. 10:130–139. [DOI] [PubMed] [Google Scholar]
- Caughey, B., D. A. Kocisko, G. J. Raymond, and P. T. Lansbury. 1995. Aggregates of scrapie-associated prion protein induce the cell-free conversion of protease-sensitive prion protein to the protease-resistant state. Chem. Biol. 2:807–816. [DOI] [PubMed] [Google Scholar]
- Chakrabartty, A., T. Kortemme, S. Padmanabhan, and R. L. Baldwin. 1993. Aromatic side-chain contribution to far-ultraviolet circular dichroism of helical peptides and its effect on measurement of helix propensities. Biochemistry. 32:5560–5565. [DOI] [PubMed] [Google Scholar]
- Chiti, F., P. Webster, N. Taddei, M. Stefani, G. Ramponi, and C. M. Dobson. 1999. Designing conditions for in vitro formation of amyloid protofilaments and fibrils. Proc. Natl. Acad. Sci. USA. 96:3590–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, P. Y., and G. D. Fasman. 1978. Empirical predictions of protein conformation. Annu. Rev. Biochem. 47:251–276. [DOI] [PubMed] [Google Scholar]
- Donne, D. G., J. H. Viles, D. Groth, I. Mehlhorn, T. L. James, F. E. Cohen, S. B. Prusiner, P. E. Wright, and H. J. Dyson. 1997. Structure of the recombinant full-length hamster prion protein PrP(29–231): the N terminus is highly flexible. Proc. Natl. Acad. Sci. USA. 94:13452–13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasman, G. D. 1996. Theory of circular dichroism of proteins. In Circular Dichroism and the Conformational Analysis of Biomolecules. R. W. Woody, editor. Plenum Press, New York. 55.
- Forloni, G., N. Angeretti, R. Chiesa, E. Monzani, M. Salmona, O. Bugiani, and F. Tagliavini. 1993. Neurotoxicity of a prion protein fragment. Nature. 362:543–546. [DOI] [PubMed] [Google Scholar]
- Forloni, G., R. Del Bo, N. Angeretti, R. Chiesa, S. Smiroldo, R. Doni, E. Ghibandi, M. Salmona, M. Porro, L. Verga, G. Giaccone, O. Bugiani, and F. Tagliavini. 1994. A neurotoxic prion protein fragment induces rat astroglial proliferation and hypertrophy. Eur. J. Neurosci. 6:1415–1422. [DOI] [PubMed] [Google Scholar]
- Forloni, G., F. Tagliavini, O. Bugiani, and M. Salmona. 1996. Amyloid in Alzheimer's disease and prion-related encephalopathies: studies with synthetic peptides. Prog. Neurobiol. 49:287–315. [DOI] [PubMed] [Google Scholar]
- Gasset, M., M. A. Baldwin, D. H. Lloyd, J. M. Gabriel, D. M. Holtzman, F. Cohen, R. Fletterick, and S. B. Prusiner. 1992. Predicted alpha-helical regions of the prion protein when synthesized as peptides form amyloid. Proc. Natl. Acad. Sci. USA. 89:10940–10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioia, L. D., C. Selvaggini, E. Ghibaudi, L. Diomede, O. Bugiani, G. Forloni, F. Tagliavini, and M. Salomona. 1994. Conformational polymorphism of the amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. J. Biol. Chem. 269:7859–7862. [PubMed] [Google Scholar]
- Greenfield, N. J., and G. D. Fasman. 1969. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry. 8:4108–4116. [DOI] [PubMed] [Google Scholar]
- Grob, M., D. K. Wilkins, M. C. Pitkeathly, E. W. Chung, C. Higham, A. Clark, and C. M. Dobson. 1999. Formation of amyloid fibrils by peptides derived from the bacterial cold-shock protein cspB. Protein Sci. 8:1350–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guijarro, J. I., M. Sunde, J. A. Jones, I. D. Campbell, and C. M. Dobson. 1998. Amyloid fibril formation by SH3 domain. Proc. Natl. Acad. Sci. USA. 95:4224–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, P. M., P. Bamborough, V. Daggett, S. B. Prusiner, and F. E. Cohen. 1997. The prion protein problem. Curr. Opin. Struct. Biol. 7:53–59. [DOI] [PubMed] [Google Scholar]
- Hermans, P. H. 1949. Chapter XII. Gels. In Colloid Science, Vol. 2. H. R. Kruyt, editor. Elsevier, Amsterdam. 483–651.
- Huang, Z., S. B. Prusiner, and F. E. Cohen. 1996a. Scrapie prions: a three-dimensional model of an infectious fragment. Fold. Des. 1:13–19. [DOI] [PubMed] [Google Scholar]
- Huang, Z., S. B. Prusiner, and F. E. Cohen. 1996b. Structures of prion proteins and conformational models for prion diseases. Curr. Top. Microbiol. Immunol. 207:49–67. [DOI] [PubMed] [Google Scholar]
- James, T. L., H. Liu, N. B. Ulyanov, S. Farr-Jones, H. Zhang, D. G. Donne, K. Kaneko, D. Groth, I. Mehlhorn, S. B. Prusiner, and F. E. Cohen. 1997. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc. Natl. Acad. Sci. USA. 94:10086–10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansanoff, A., and A. R. Fersht. 1994. Quantitative determination of helical propensities from trifluoroethanol titration curves. Biochemistry. 33:2129–2135. [DOI] [PubMed] [Google Scholar]
- Jayakumar, R., M. Murugesan, C. Asokan, and M. Aulice Scibioh. 2000. Self-assembly of a peptide Boc- (Ile)3 -OMe in chloroform and N, N-dimethyl formamide. Langmuir. 16:1489–1496. [Google Scholar]
- Katsoyannis, P. G., and G. P. Schwartz. 1977. The synthesis of peptides by homogeneous solution procedures. Methods Enzymol. XLVII:501–578. [DOI] [PubMed] [Google Scholar]
- Kelly, J. W. 1998. The alternative conformation of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 8:101–106. [DOI] [PubMed] [Google Scholar]
- Kelly, J. W. 2000. Mechanism of amyloidogenesis. Nat. Struct. Biol. 7:824–826. [DOI] [PubMed] [Google Scholar]
- Kishore, R., S. Raghothama, and P. Balaram. 1987. Cystine peptides: the intramolecular antiparallel beta-sheet conformation of a 20-membered cyclic peptide disulfide. Biopolymers. 26:873–891. [DOI] [PubMed] [Google Scholar]
- Lansbury, P. T., and B. Caughey. 1995. The chemistry of scrapie infection: implications of the “ice 9” metaphor. Chem. Biol. 2:1–5. [DOI] [PubMed] [Google Scholar]
- Laxma Reddy, G., and R. Nagaraj. 1989. Circular dichroism studies on synthetic signal peptides indicate beta-conformation as a common structural feature in highly hydrophobic environment. J. Biol. Chem. 264:16591–16597. [PubMed] [Google Scholar]
- Lehrman, S. R., J. L. Tuls, and M. Lund. 1990. Peptide alpha-helicity in aqueous trifluoroethanol: correlations with predicted alpha-helicity and the secondary structure of the corresponding regions of bovine growth hormone. Biochemistry. 29:5590–5596. [DOI] [PubMed] [Google Scholar]
- Levy, Y., and O. M. Becker. 2002. Conformational polymorphism of wild-type and mutant prion proteins: energy landscape analysis. Proteins. 47:458–468. [DOI] [PubMed] [Google Scholar]
- Luidens, M. K., J. Figge, K. Breese, and S. Vajda. 1996. Predicted and trifluoroethanol-induced alpha-helicity of polypeptides. Biopolymers. 39:367–376. [DOI] [PubMed] [Google Scholar]
- Madison, V., and J. Schellman. 1972. Optical activity of polypeptides and proteins. Biopolymers. 11:1041–1076. [DOI] [PubMed] [Google Scholar]
- McKinley, M. P., M. B. Braunfeld, C. G. Bellinger, and S. B. Prusiner. 1986. Molecular characteristics of prion rods purified from scrapie-infected hamster brains. J. Infect. Dis. 154:110–120. [DOI] [PubMed] [Google Scholar]
- McLeish, M. J., K. J. Nielson, L. V. Najbar, J. D. Wade, F. Lin, M. B. Doughty, and D. J. Craik. 1994. Conformation of a peptide corresponding to T4 lysozyme residues 59–81 by NMR and CD spectroscopy. Biochemistry. 33:11174–11183. [DOI] [PubMed] [Google Scholar]
- Merukta, G., W. Lipton, W. Shalongo, S. Park, and E. Stellwagen. 1990. Effect of central-residue replacements on the helical stability of a monomeric peptide. Biochemistry. 29:7511–7515. [DOI] [PubMed] [Google Scholar]
- Myers, J. K., C. Nickpace, and J. M. Scholtz. 1998. Trifluoroethanol effects on helix propensity and electrostatic interactions in the helical peptide from ribonuclease T1. Protein Sci. 7:383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, J. W., and N. R. Kallenbach. 1986. Stabilization of the ribonuclease S-peptide alpha-helix by trifluoroethanol solutions. Proteins. Struct. Funct. Genet. 1:211–217. [DOI] [PubMed] [Google Scholar]
- Pan, K.-M., M. Baldwin, J. Nguyen, M. Gasset, A. Serban, D. Groth, I. Mehlhorn, Z. Huang, R. J. Fletterick, F. E. Cohen, and S. B. Prusiner. 1993. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA. 90:10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petchanikow, C., G. P. Saborio, L. Anderes, M. J. Frossard, M. I. Olmedo, and C. Soto. 2001. Biochemical and structural studies of the prion protein polymorphism. FEBS Lett. 509:451–456. [DOI] [PubMed] [Google Scholar]
- Pintar, A., A. Chollet, C. Bradshaw, A. Chaffotte, C. Cadieux, M. J. Rooman, K. Hallenga, J. Knowles, M. Goldberg, and S. J. Wodak. 1994. Conformational properties of four peptides corresponding to alpha-helical regions of Rhodospirillum cytochrome c2 and bovine calcium binding protein. Biochemistry. 33:11158–11173. [DOI] [PubMed] [Google Scholar]
- Prusiner, S. B. 1982. Novel proteinaceous infectious particles cause scrapie. Science. 216:136–144. [DOI] [PubMed] [Google Scholar]
- Prusiner, S. B. 1997. Prion diseases and the BSE crisis. Science. 278:245–251. [DOI] [PubMed] [Google Scholar]
- Prusiner, S. B. 1998. Prions. Proc. Natl. Acad. Sci. USA. 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner, S. B., M. P. McKinley, K. A. Bowman, D. C. Bolton, P. E. Bendheim, D. F. Groth, and G. G. Glenner. 1983. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell. 35:349–358. [DOI] [PubMed] [Google Scholar]
- Prusiner, S. B., M. R. Scott, S. J. DeArmond, and F. E. Cohen. 1998. Prion protein biology. Cell. 93:337–348. [DOI] [PubMed] [Google Scholar]
- Ragg, E., F. Tagliavini, P. Malesani, L. Monticelli, O. Bugiani, G. Forloni, and M. Salmona. 1999. Determination of solution conformations of PrP106–126, a neurotoxic fragment of prion protein, by 1H NMR and restrained molecular dynamics. Eur. J. Biochem. 266:1192–1201. [DOI] [PubMed] [Google Scholar]
- Rapoport, T. A. 1985. Protein translocation across and integration into membranes. Crit. Rev. Biochem. 20:73–137. [DOI] [PubMed] [Google Scholar]
- Selvaggini, C., L. De Gioia, L. Cantu, E. Ghibaudi, L. Diomede, F. Passerini, G. Forloni, O. Bugiani, F. Tagliavini, and M. Salmona. 1993. Molecular characteristics of a protease-resistant, amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. Biochem. Biophys. Res. Commun. 194:1380–1386. [DOI] [PubMed] [Google Scholar]
- Sitaram, N., and R. Nagaraj. 1993. Interaction of the 47-residue antibacterial peptide seminalplasmin and its 13-residue fragment which has antibacterial and hemolytic activities with model membranes. Biochemistry. 32:3124–3130. [DOI] [PubMed] [Google Scholar]
- Sreerama, N., S. Y. Venyaminov, and R. W. Woody. 2000. Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal. Biol. 287:243–251. [DOI] [PubMed] [Google Scholar]
- Stewart, J. M., and J. D. Young. 1984. Solid Phase Peptide Synthesis. Pierce Chemical Company, Rockford, Il.
- Storrs, R. W., D. Truckses, and D. E. Wemmer. 1992. Helix propagation in trifluoroethanol solutions. Biopolymers. 32:1695–1702. [DOI] [PubMed] [Google Scholar]
- Tagliavini, F., F. Prelli, J. Ghiso, O. Bugiani, D. Serban, S. B. Prusiner, M. R. Farlow, B. Ghetti, and B. Frangiono. 1991. Amyloid protein of Gerstmann-Sträussler-Scheinker disease (Indiana kindred) is an 11 kd fragment of prion protein with an N-terminal glycine at codon 58. EMBO J. 10:513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliavini, F., F. Prelli, L. Verga, G. Giaccone, R. Sarma, P. Gorevic, B. Ghetti, F. Passerini, E. Ghibaudi, G. Forloni, M. Salmona, O. Bugiani, and B. Fragioni. 1993. Synthetic peptides homologous to prion protein residues 106–147 form amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. USA. 90:9678–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliavini, F., F. Prelli, M. Porro, G. Rossi, G. Giaccone, M. R. Farlow, S. R. Dlouhy, B. Ghetti, O. Bugiani, and B. Frangione. 1994. Amyloid fibrils in Gerstmann-Straussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell. 79:695–703. [DOI] [PubMed] [Google Scholar]
- Wickner, W. T., and H. F. Lodish. 1985. Multiple mechanisms of protein insertion into and across membranes. Science. 230:400–407. [DOI] [PubMed] [Google Scholar]
- Wille, H., M. D. Michelitsch, V. Guenebaut, S. Supattapone, A. Serban, F. E. Cohen, D. A. Agard, and S. B. Prusiner. 2002. Structural studies of the scrapie prion protein by electron crystallography. Proc. Natl. Acad. Sci. USA. 99:3563–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, R. C. 1977. Use of polylysine for adsorption of nuclei acids and enzymes to electron microscope specimen films. Proc. Natl. Acad. Sci. USA. 74:2311–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, J. C., K. J. Nielson, M. J. McLeish, and D. J. Craik. 1994. A determination of the solution conformation of the nonmammalian Tachykinin eledoisin by NMR and CD spectroscopy. Biochemistry. 33:6802–6811. [DOI] [PubMed] [Google Scholar]
- Xu, X., L. G. Cooper, P. J. DiMario, and J. W. Nelson. 1995. Helix formation in model peptides based on nucleolin TPAKK motifs. Biopolymers. 35:93–102. [DOI] [PubMed] [Google Scholar]
- Zhang, M., and H. J. Vogel. 1994. The calmodulin-binding domain of caldesmon binds to calmodulin in an alpha-helical conformation. Biochemistry. 33:1163–1171. [DOI] [PubMed] [Google Scholar]
- Zhou, N. E., C. M. Kay, B. D. Sykes, and R. S. Hodges. 1993. A single-stranded amphipathic alpha-helix in aqueous solution: design, structural characterization, and its application for determining alpha-helical propensities of amino acids. Biochemistry. 32:6190–6197. [DOI] [PubMed] [Google Scholar]