

Abstract
The Fe(III) → Fe(II) reduction of the heme iron in aquomet-myoglobin, induced by x-rays at cryogenics temperatures, produces a thermally trapped nonequilibrium state in which a water molecule is still bound to the iron. Water dissociates at T > 160 K, when the protein can relax toward its new equilibrium, deoxy form. Synchrotron radiation x-ray absorption spectroscopy provides information on both the redox state and the Fe-heme structure. Owing to the development of a novel method to analyze the low-energy region of x-ray absorption spectroscopy, we obtain structural pictures of this photo-inducible, irreversible process, with 0.02–0.06-Å accuracy, on the protein in solution as well as in crystal. After photo-reduction, the iron-proximal histidine bond is shortened by 0.15 Å, a reinforcement that should destabilize the iron in-plane position favoring water dissociation. Moreover, we are able to get the distance of the water molecule even after dissociation from the iron, with a 0.16-Å statistical error.
INTRODUCTION
Irradiation of heme proteins in solution at cryogenic temperatures is a way to generate intermediate states of the Fe-heme complex (Blumenfeld, 1981; Prusakov et al., 1985; Parak and Prusakov, 1994). Radiolysis of the solvent produces thermalized electrons that can reach the reducible sites of the protein even at low temperature (i.e., the porphyrin group and the histidine residues; see Pin et al., 1989; Le Tilly et al., 1997). Consequently the heme iron Fe(III) is reduced, but at low temperature the protein structure remains frozen in its initial conformation. The relaxation of this intermediate state Mb(II)H2O toward its final, deoxylike state, Mb(II)⋯H2O, has been investigated by Mossbauer spectroscopy as a function of temperature and time (Prusakov et al. 1995), the overall process starting above ∼140 K and being fully completed at ∼200 K. As it was shown first by Mossbauer spectroscopy (Parak and Prusakov, 1994), the Mb(II)H2O state produced after gamma irradiation at 77 K contains Fe(II) low spin; increasing the temperature, molecules relax into the equilibrium state of Mb(II)⋯H2O, with Fe(II) high spin, where the water molecule is dissociated from the iron (Lamb et al., 1998; Engler et al., 2000).
This photo-inducible, irreversible, temperature-dependent dissociation process from Mb(II)H2O to Mb(II)⋯H2O has strong analogies with the widely investigated process of CO dissociation from carbonmonoxy-myoglobin (MbCO) in photolysis experiments (Schlichting et al., 1994; Hartmann et al., 1996; Srajer et al., 1996), with the further advantage of not being limited by rebinding kinetics. Clearly, an accurate description of the detailed mechanisms of the Fe-heme relaxation associated to the ligand dissociation, and their relationships with collective protein motions, would be of outstanding interest for a deeper understanding of such biophysical events that constitute a paradigm for protein dynamics studies.
The photo-reduction process Mb(III)H2O → Mb(II)H2O at low temperature, and the temperature-dependent relaxation Mb(II)H2O → Mb(II)⋯H2O, have been studied by optical spectroscopy and resonance Raman, as well as an x-ray structure at 1.4-Å resolution of Mb(II)H2O that has been reported (Lamb et al., 1998; Engler et al., 2000). The reported Mb(II)H2O structure does not change significantly by that of Mb(III)H2O; the axial distances d(Fe-O) and d(Fe-Nhis) appear shortened by 0.1 and 0.07 Å, a difference comparable with the coordinate error at this resolution. However, whereas the mentioned spectroscopic studies provide poor structural information, during the x-ray diffraction experiment there is no probe of the redox state of the Fe-heme site. The correspondence between the photo-reduced state observed by the different spectroscopies and the extracted x-ray structure is even more uncertain owing to the use of different experimental setup and protocols.
As photo-reduction of metal centers in proteins can be induced after long irradiation under a high flux of x-rays, synchrotron radiation can be used in principle to both induce and study “in situ” redox changes at the metal site by x-ray absorption spectroscopy (XAS). Extended radiolysis of the water solvent under x-ray beam coming from synchrotron radiation is an undesired damage; however, the persistence of diffracting power after x-ray photo-reduction of azurin II crystals (Debenham et al., 1996) has suggested that at cryogenic temperatures, metal photo-reduction (a primary damage) occurs before the loss of crystalline order and protein denaturation (secondary and tertiary damages). This suggestion has been confirmed by more extensive investigations (Teng and Moffat, 2000; Ravelli and McSweeney, 2000; Burmeister, 2000) that have also given an approximate threshold limit for the absorbed x-ray dose below which intense synchrotron radiation x-rays can be used (see Experimental Protocol), at low temperature, for both inducing and probing the photo-reduction effects on aquomet-myoglobin.
Extended x-ray absorption fine structure (EXAFS) spectroscopy and x-ray absorption near edge structure (XANES) spectroscopy are XAS techniques able to investigate the conformations of the metal sites in proteins (Hasnain and Hodgson, 1999) and in particular the Fe site in hemoproteins (Pin et al., 1994) with very high resolution. Very recently a method for performing a full spectral fitting of the XAS low-energy region for structural analysis has been proposed and applied to the K-edge spectra of a number of transition metal compounds (Benfatto and Della Longa, 2001; Benfatto et al., 2002; D'Angelo et al., 2002) and also to interpret the XANES spectra of the low temperature photolysis experiment of MbCO (Della Longa et al., 2001). The possibility of performing quantitative analysis of the XANES spectra to obtain structural and electronic information is very relevant especially for biological systems where the low S/N ratio and the weak scattering power of the light elements C, N, and O surrounding a metal site limit the K-range of the available experimental data. In all these instances, the EXAFS spectroscopy cannot be adequately exploited, since the usable data are very often <200 eV from the absorption edge, in the XANES range.
As a further advantage, XANES is very sensitive to the redox state and the spin state of the metal. Finally, atomic vibrations have negligible effects on XANES, because the Debye-Waller damping terms in the XANES energy region are almost temperature-independent and do not interfere with the structural determinants of the spectrum, so that the interpretation of temperature-dependent XANES experiments is simpler.
The overall process of photo-induced redox change and temperature-dependent photo-dissociation of aquomet-myoglobin can be studied in single XANES experiments, and structurally interpreted. In this article we present a quantitative analysis of both solution and single crystal XANES spectra of the three main states involved: Mb(III)H2O, Mb(II)H2O, and Mb(II)⋯H2O. The relevant, selected structural parameters (essentially the iron-ligand distances) of the Fe-heme are extracted for each main state with an error in the distance determination of 0.02–0.06 Å. This accuracy is comparable to that of the more recently reported x-ray diffraction data.
In this work, the sixth iron ligand is always referred as a water molecule; however, due to the low scattering power of hydrogen, it is not possible to establish by our XAS data whether a water molecule, or an OH− ion, is actually involved in the overall process.
METHODS
Protein and protein crystals
Ferric myoglobin solutions were prepared from lyophilized sperm whale myoglobin purchased before the ban along the method previously described (Della Longa et al., 1998). Monoclinic (P21) single crystals of native sperm whale myoglobin, purchased before the ban, were suspended, immediately before starting the experimental protocol, in a cryoprotectant solution containing mother liquor (3.25 M ammonium sulfate, with 0.05 M phosphate buffer, at pH 6.5) and 20% glycerol, and put in a quartz capillary. The protocol for the crystal orientation and cryogenics has been described in detail in a previous work (Della Longa et al., 1999).
X-ray absorption measurements
Fe K-edge x-ray absorption spectra of photo-reduced Mb in solution, and of the Mb single crystals, were collected in fluorescence mode at the beam line BM32 of the European Synchrotron Radiation Facility (ESRF) running in the two-thirds filling mode at 173 mA, by using an energy-resolving array detector made by 13 Ge elements of very high purity. The energy resolution at the Fe Kα fluorescence (6400 eV) was 170 eV. The x-ray absorption spectra of aquomet-Mb and deoxy-Mb in solution were acquired at the BM8-GILDA beam line of ESRF.
Experimental protocol
Samples of Mb(III)H2O either in solution or in single crystal have been cooled down to 15 K. The spectrum under x-ray irradiation at high intensity (∼1011 photons/s) even at T = 15 K evolves in time; the kinetic curve of the x-ray absorption difference taken at 7120 eV [I(t) − I(0)]/[I(∞) − I(0)] has been used to follow the kinetics of photo-reduction at various temperatures between 15 K and 70 K, the process lasting ∼1 h. In the case of the crystal, the x-ray flux is focused onto the dimension of the crystal (0.2 × 0.2 × 0.2 mm3) and a primary radiation damage (involving photo-reduction of reducible centers and breaking of weak chemical bonds) is more effective. As reported by Teng and Moffat (2000), the irradiation with x-rays at ∼12.4 KeV with a 1012 photons/s flux on a protein crystal profile of 0.1 × 0.1 mm2 at T = 100 K corresponds to an absorbed dose of ∼1.2 106 Gy/h, the primary damage being linearly dependent on the absorbed dose. Above the threshold of ∼107 Gy, excessive, nonlinear damage of the crystal develops, involving loss of crystal order and protein denaturation. The crystal can be considered not affected by secondary and tertiary damage under x-rays irradiation for ∼5–6 h. In our experimental conditions (1011 photons/s at 7 KeV on a 0.2 × 0.2 mm2 crystal profile) the absorbed dose is much less; however, we considered a 6-h time window as an upper limit for our experimental protocol of temperature-dependent XANES. The crystal was cooled at 15 K; orientation at one of the polarization axes took ∼45 min under x-ray irradiation; then, the oriented crystal was irradiated further on, for 1 h, and the last part of the kinetic curve of x-ray absorption difference was followed, as well as for the solution samples. We note here that as we observe saturation well under the upper limit for the primary radiation damage stated by Teng and Moffat, it is possible that some reported x-ray structures could be affected by metal photo-reduction effects. Spectra were acquired for each polarization. Our starting point was the spectrum of Mb(II)H2O at T = 15 K. Then, the x-ray flux was stopped and the temperature was raised maintaining the crystal in the same orientation, waiting for the stabilization at 120 K. Another couple of spectra was acquired for each crystal orientation at the new temperature, and so forth. The spectra between 7100 and 7300 eV, ΔE = 0.5 eV (400 points) have a signal averaging of 3 s/point. The fluorescence count jumped from 200 counts/s/element before the edge (7100 eV), to ∼5000 counts/s/element above the edge (7200 eV), giving a total count jump of ∼195,000 with a noise-to-jump ratio of 6 10−4 before the edge and 2 10−3 after the edge. XANES spectra took only 20 min, so that the entire protocol was carried out in <6 h under x-ray irradiation. In all experimental spectra presented here, the energy is aligned at the absorption threshold of metallic Fe foil.
Polarized XAS spectra of myoglobin
With a structure that has a symmetry axis (symmetry C4, approximately that of the Fe-heme site), it is advantageous during the fit procedure to calculate the XAS components linearly polarized along the symmetry axis (the heme normal), Inormal, and along a direction in the C4 plane (the heme plane), Iheme. Assuming a perfect C4 symmetry, for a certain angle α between the polarization vector ɛ and the heme normal of one myoglobin molecule, the linearly polarized spectrum Iα is given, as a function of Iheme and Inormal , simply by
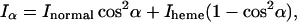 |
and the unpolarized spectrum (i.e., the spectrum in solution) is given by
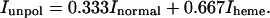 |
As shown in details previously (Della Longa et al., 1999), in the case of the sperm whale P21 single crystal, the following relationships are valid:
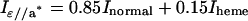 |
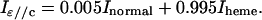 |
In the section dedicated to XANES analysis, theoretical calculations try to fit Inormal and Iheme. According to the formulas given above, the experimental spectrum for Iheme has been assumed identical to Iɛ//c, and the experimental spectrum for Inormal has been obtained as Inormal = (Iɛ//a* − 0.15 Iɛ//c)/0.85. Moreover, when comparing the solution spectra of Mb(II)H2O and Mb(II)⋯H2O with the spectra Iunpol obtained by linear combination of the polarized ones, Iunpol has been obtained as Iunpol = 0.4 Iɛ//a* + 0.6 Iɛ//c.
Computational fitting procedure
We have used the MXAN package (Benfatto and Della Longa, 2001) to extract structural information from the spectra obtained by experiment both in solution and on single crystal. The package works in the framework of the multiple-scattering theory (Natoli et al., 1986; Tyson et al., 1992) within the “muffin-tin” approximation for the shape of the potential. The scattering path operator is calculated exactly without any series expansion to include the XANES region in the calculation and fit. A cluster of 32 atoms, i.e., the porphyrin macrocycle, the histidine imidazole, and the oxygen from the water molecule, has been used. The molecular potential for the nearest atoms, i.e., the FeN5O cluster, has been obtained by spin-unpolarized, self-consistent field method, imposing the formal valence of each atom (Pedio et al., 1994; Diaz-Moreno et al., 2000). Inelastic processes have been taken into account by a convolution with a broadening Lorentzian function, with a width Γ = Γc + Γ(E). The constant part Γc includes the core-hole lifetime and the experimental resolution, while the energy-dependent term represents all the inelastic processes. The functional form of the Γ(E) function follows the universal form of the mean free path in solid (Muller et al., 1982): it is zero below onset energy Es, and it begins to increase from a full width of As. This method introduces three nonstructural parameters. However, to take into account the strong asymmetry of the Fe-heme site, the function Γ(E) used to fit solution samples has a vectorial form with different values of the Es and As parameters for the Inormal and Iheme components of the solution spectrum. As total, five nonstructural parameters: Γc, Esnormal, Esheme, Asnormal, and Asheme are present in the fitting procedure. Theoretically this procedure is justified considering that all the physical quantities involved, in particular the dielectric function associated to the calculation of the self-energy of the system, must have the same symmetry of the geometrical cluster. As a consequence, the mean free path term is largely anisotropic; this is evident comparing the different Es values found for the polarized spectra: ∼14.0 eV for the ɛ//c spectrum and 7.0 eV for the ɛ//a* spectrum.
We have verified that these nonstructural parameters are weakly correlated to the structural determination, their effect resulting just in an increase of a few percentage of the error value reported in the Tables. A constant experimental error corresponding to a noise-to-signal ratio of 0.01 was chosen.
RESULTS
Photo-reduction of met-myoglobin: Mb(III)H2O → Mb(II)H2O
The Fe K-edge XANES spectra of aquomet-myoglobin in solution (Mb(III)H2O) at pH 7, T = 70 K (dotted curve), is shown in Fig. 1. The sample under x-ray irradiation at high intensity evolves in time; we followed the kinetic curve of the x-ray absorption difference spectrum at 7120 eV [I(t) − I(0)]/[I(∞) − I(0)], that reaches saturation in less than 1 h, showing a spectrum with different features in the energy regions P, A, C, D, C2, and E (solid curve of Fig. 1). A red shift is observed of ∼0.6 eV, relative to the first spectrum. This shift is due to reduction of the effective charge of the metal center. The position of the absorption edge depends on the oxidation state of the metal (Yachandra, 1995), more negatively charged metals having inflection points shifted at lower energies. An almost rigid shift of the XANES spectrum without evident changes in shapes are found in redox hemoproteins where the readjustment of the atomic environment that accompanies ligand-metal charge transfer is negligible, as in the case of oxidized and reduced cytochrome-C, for which the spectrum of reduced cytochrome-C appears rigidly red-shifted relative to that of oxidized cytochrome-C (Labhardt and Yven, 1979). Moreover, the red-shift of the XANES edge of ferric N-methylimidazole cytochrome P-450-CAM, relative to that of N-methylimidazole myoglobin (the Fe-heme of the two proteins differing for only one ligand, cystein thiolate in P-450, imidazole nitrogen in myoglobin), has been explained with the electron-releasing character of a cystein thiolate proximal ligand, i.e., by a transfer of electron density toward the Fe (Liu et al., 1995).
FIGURE 1.

Fe K-edge XANES spectra of met-myoglobin at pH 7 at T = 70 K, before (Mb(III)H2O, dashed curve), and after prolonged x-ray irradiation at high intensity (Mb(II)H2O, solid curve), The last spectrum (solid curve) probes the Fe-heme structure in the trapped nonequilibrium state. The inset contains a blowup of the pre-edge features, indicating the formation of a low-spin Fe-heme adduct.
The inset of Fig. 1 contains a blowup of the pre-edge features of the spectra and can be interpreted by a transition from a high spin state (where both t2g and eg iron d-orbitals are partially unoccupied) to a low-spin state (where the eg orbitals are empty and the t2g orbitals are fully occupied) of the iron atom.
These observations fully confirm the formation, after x-ray irradiation, of a photo-reduced, low-spin Fe-heme species—i.e., the same species observed by Mossbauer experiments (Parak and Prusakov, 1994), the last spectrum pertaining to the Fe-heme structure in the ferrous nonequilibrium state Mb(II)H2O.
Thermal relaxation of photo-reduced myoglobin: Mb(II)H2O → Mb(II)⋯H2O
The same experiment has been repeated on an oriented single crystal of aquomet-myoglobin (symmetry P21). Polarized XANES spectra with photon polarization ɛ parallel either to the c-axis, or to the a*-axis of the crystal, were collected. The ɛ//c polarized spectrum probes the structure of the heme plane, and is rather insensitive to the axial environment of the iron. The ɛ//a* polarized spectrum, on the contrary, is strongly sensitive to the axial environment of the iron (fifth and sixth ligand distances and orientations) and rather insensitive to the heme-plane structure. All the details on polarized XANES spectroscopy of myoglobin are reported in previous works (Della Longa et al., 1999, 2001).
In our experimental conditions, it was impossible to acquire the polarized XANES spectra of the starting state Mb(III)H2O, as for the protein in solution. In fact, due to the time needed for the small crystal to be aligned and finely oriented under a focused, intense x-ray beam, the first collected polarized XANES spectrum is already a mixture of Mb(III)H2O and Mb(II)H2O.
The only way to be sure to have completed the photo-reduction process in the crystal was to first reach saturation of the kinetic curve at 7120 eV, and then to compare the polarized spectra with the spectrum of Mb(II)H2O in solution. This comparison is shown in Fig. 2. The polarized spectra of the crystal at the end of the photo-reduction process at T = 70 K are depicted on the top of the figure (dotted curve, ɛ//a* polarized spectrum; solid curve: ɛ//c polarized spectrum). At bottom of the figure, the spectrum of Mb(II)H2O in solution (solid curve) and the 0.4/0.6 linear combination (see Methods) of the ɛ//a* and the ɛ//c polarized spectra, respectively (dotted curve), are compared. The superimposition obtained by these completely independent experiments is almost perfect, showing that the same photo-reduced state is reached in the crystal and in solution. As there is not a crystal crack due to the photo-reduction event, this should be accompanied by only small conformational rearrangements of the protein three-dimensional structure, far from the heme site. Moreover, the good overlapping obtained (Fig. 2, bottom) confirms that both the protein molecules contained in the single unit cell have been reduced homogenously.
FIGURE 2.

Upper curves are ɛ//a* (dotted curve) and ɛ//c (solid curve) polarized spectra of Mb(II)H2O in a single crystal at T = 70 K. Lower curves are the comparison between the spectra of Mb(II)H2O in solution (solid curve) and in the single crystal (dotted curve). This last spectrum is the 0.4/0.6 linear combination of the ɛ//a* and ɛ//c spectra, respectively.
According to the previous investigations, Mb(II)H2O is a nonequilibrium state trapped at low temperature, the water molecule dissociating from the iron when collective motions of the protein become thermally activated, at T > 160 K. In Fig. 3, the temperature-dependent XANES spectra of Mb(II)H2O are shown, the left frame concerning ɛ//a* polarized spectra probing the water dissociation, the right frame concerning ɛ//c polarized spectra probing the heme relaxation. The experimental protocol starts at T = 15 K. From top to bottom, spectra at T = 15 K, 120 K, 160 K, 200 K, and 240 K are presented (solid curves). For each temperature from 120 K to 240 K, the previous spectrum is superimposed (e.g., at 120 K the spectrum taken at 15 K, and so forth) to put in evidence the temperature-dependent spectral changes. From 15 K to 120 K, changes are very small; in particular, there is no evidence of progressive water dissociation by increasing temperature, hence the fraction of Mb(II)⋯H2O should be very small or negligible, as in the experiment by Prusakov et al. (1995). Between 120 K and 160 K, a decrease in amplitude of the ɛ//a* absorption between 7130 and 7150 eV seems a rearrangement that precedes but does not involve the dissociation of the water molecule. It is difficult to interpret these spectral changes because at T = 160 K kinetic and thermal effects probably interfere during the spectral data acquisition. The transition to a deoxylike product (Mb(II)⋯H2O) is identified by large spectral changes in both polarization between 160 K and 200 K, in good agreement with the temperature-dependence of the isomer shifts for the Fe(II) low-spin and Fe(II) high-spin species reported by Mossbauer spectroscopy (Prusakov et al., 1995).
FIGURE 3.

Temperature-dependence of the XANES spectra of photo-reduced myoglobin Mb(II)H2O. (Left frame) ɛ//a* spectra, polarized along the Fe-water bond. The thermally activated water dissociation is probed by the spectral changes of the ɛ//a* spectrum between 200 K and 240 K. (Right frame) ɛ//c spectra, polarized on the heme plane. Here, the relaxation of the Fe-heme system, with the displacement of the iron from the heme plane, is probed between 160 K and 240 K. Between 120 K and 160 K, an evidence for a relaxation of the intermediate Mb(II)H2O is observed in both the polarizations.
Between 200 K and 240 K further changes occur. Different XANES regions evolve at this stage, showing that the transition from the intermediate state Mb(II)H2O to Mb(II)⋯H2O, even at the Fe-heme level, cannot be described by a simple two-state transition, but is a complex dynamical reaction involving a set of structural substates driven by the thermal relaxation of the protein.
The comparison between the starting Mb(II)H2O state at T = 15 K and the final state Mb(II)⋯H2O at T = 240 K is shown in Fig. 4; the ɛ//a* spectra in the upper frame, and the ɛ//c spectra in the middle frame. The changes observed in the ɛ//a* spectra between 15 K and 240 K have strong analogies with those observed in the low temperature photolysis experiment of carbonmonoxy-myoglobin (Della Longa et al., 2001), the spectrum at T = 240 K resembling that of the photoproduct Mb*CO at T = 20 K, the residual differences due to different contributions to the XANES spectra by the CO and the H2O molecules. At the same time, the ɛ//c spectrum at 240 K is shifted by ∼1.5 eV with respect to the spectrum at T = 15 K, a change which is not observed in the photolysis experiment. In the lower frame of Fig. 4, the 0.4/0.6 linear combination of the ɛ//a* and the ɛ//c polarized spectra of Mb(II)⋯H2O, respectively, (dotted curve) is compared with the spectrum in solution of the dithionite reduced deoxy-Mb. There are residual differences in this last comparison that could be due to either still incomplete dissociation or still incomplete thermal relaxation of the Mb(II)⋯H2O state. The changes observed for the ɛ//c polarized spectra at T = 200 and 240 K (last comparison of Fig. 3, right frame) suggest that relaxation of the Mb(II)⋯H2O state could be the process acting at this temperature.
FIGURE 4.
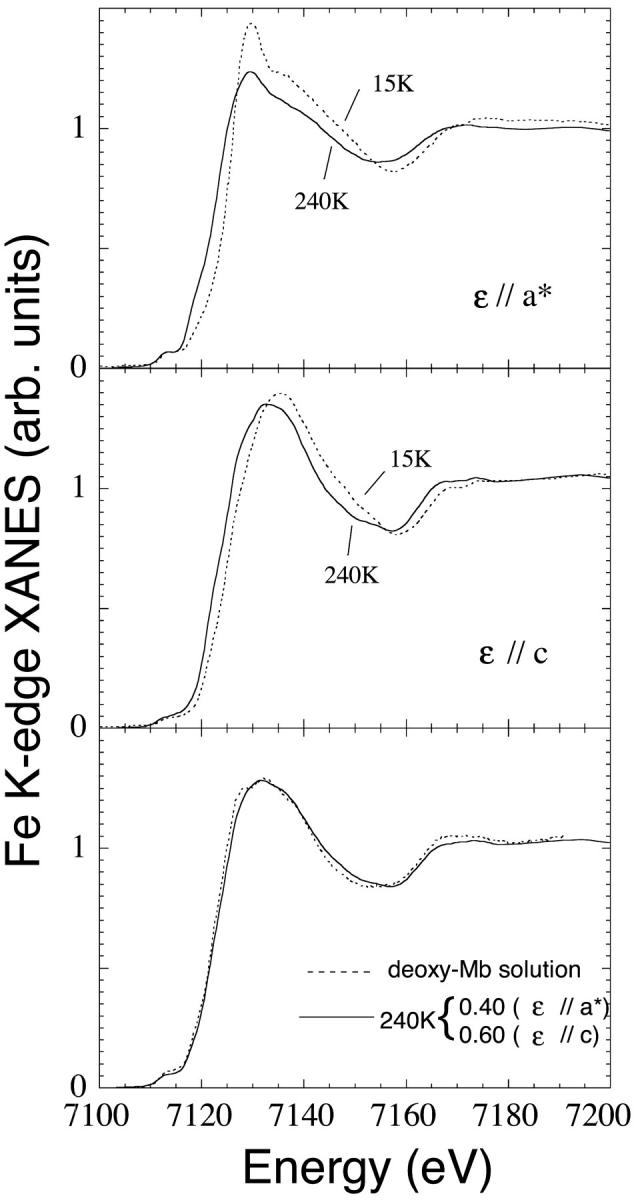
Comparison between the first state Mb(II)H2O at T = 70 K and the final state Mb(II)⋯H2O at T = 240 K of the temperature-dependent dissociation process. (Upper frame) Polarization ɛ//a* along the Fe-water bond. (Middle frame) Polarization ɛ//c on the Fe-heme plane. (Lower frame) Comparison between the spectra of the deoxy-Mb(II) in solution obtained by reduction of met-myoglobin with sodium dithionite (solid curvee) and the Mb(II)—H2O final state of our experiment at T = 240 K in single crystal (dotted curve). This last spectrum is the 0.4/0.6 linear combination of the ɛ//a* and ɛ//c spectra, respectively.
XANES pictures of the myoglobin photo-reduction process: Mb(III)H2O, Mb(II)H2O, and Mb(II)⋯H2O
Starting from a putative atomic configuration of a metal site, we have recently demonstrated that it is possible to use theoretical calculations within the multiple scattering scheme to fit experimental XANES spectra (Benfatto and Della Longa, 2001; Della Longa et al., 2001), by the minimization of χ2 function in the space of n selected structural parameters. The selected parameters for the Fe-heme cluster (32 atoms) are the first shell distances d(Fe-Np), d(Fe-O), d(Fe- Nhis), the tilting angle α between the Fe-O vector and the heme normal, and Fe-heme displacement. During the fit, the imidazole ring of the histidine, and the pyrrolic rings of the porphyrin rigidly follow the motion of the Nhis and Np atoms, respectively.
In Fig. 5, the best fit (solid curves) of the experimental XANES spectra (circles) of Mb(III)H2O (lower curves) and Mb(II)H2O (upper curves) in solution are reported. For an experimental error of 0.012, it was obtained respectively a χ2/n value of 3.36 and 2.46. Here the zero energy corresponds to the theoretical continuum threshold. Alignment of the theoretical and experimental spectra has been obtained with an energy shift of 7116.4 eV and 7115.7 eV, for Mb(III)H2O and Mb(II)H2O, respectively. Interestingly, the 0.7 eV difference corresponds to the observed effect of iron charge on the rising edge position. The energy position of every feature is fitted satisfactorily. The only relevant discrepancy occurs for the intensity of the feature at 60 eV in the case of Mb(III)H2O, due probably to the approximation used for the broadening function fitting the photoelectron energy losses, rather than to some hindered structural parameter. As far as it concerns the nonstructural parameters used in the fitting procedure (the spectral broadening terms Γc, Es, and As; see Methods), in the case of Mb(II)H2O the optimized value of Γc (1.6 eV) is in quite good agreement with the known values of core-hole width (1.2 eV) and experimental resolution (0.5 eV). The optimized values of Esnormal, Esheme, Asnormal, and Asheme (15.2, 6.2, 7.1, and 8.1 eV, respectively) are in reasonably good agreement with the previously reported values of the polarized terms in MbCO (Della Longa et al., 2001). On the contrary, in the case of Mb(III)H2O, the values of Γc (1.25 eV) and Asnormal (3.76 eV) are rather smaller than expected. Hopefully, a more comprehensive expression of the broadening terms is a goal for further theoretical studies.
FIGURE 5.

Best fit of the XANES spectra of Mb(III)H2O and Mb(II)H2O in solution at T = 70 K in terms of the selected structural parameters reported in Tables 1 and 2 (solid lines). The experimental data are plotted in circles.
The structural results relative to Mb(III)H2O are reported in Table 1. The fits are sensitive to the first-neighbor distances Fe-Np, Fe-O, and Fe-Nhis. The Fe displacement from the heme plane, the tilting angle α of the Fe-O vector, and the tilting angle of the histidine residue have been also considered in preliminary runs, but the fit came out poorly sensitive to them.
TABLE 1.
Mb(III)H2O at T = 70 K
| Experiment | Fe–Np | Fe–Nhis | Fe–O |
|---|---|---|---|
| XRD* 1.2 Å | 2.03 | 2.16 | 2.16 |
| XRD† 1.1 Å | 2.03 | 2.13 | 2.13 |
| XRD‡ 1.5 Å | 1.98 | 2.15 | 2.23 |
| EXAFS§ | 2.02 | 2.10 | 2.12 |
| XANES solution | 2.01(2) | 2.14(6) | 2.22(6) |
Comparison between different structural determinations of the Fe-heme site geometry. Distance units, Ångstrom. The resolution of XRD experiments is indicated. The values in parenthesis are the statistical errors as evaluated by the fitting procedure.
PDB code, 1BZ6 (room temperature).
PDB code, 1A6K (T = 90 K).
Engler et al. (2000) (T = 115 K).
Scherk et al. (2001) (T = 40 K).
These data represent the first quantitative determination of the Fe-heme structure in myoglobin in solution by XANES. Table 1 compares our XANES determination with other structural determinations previously reported, including the x-ray structure at 1.2-Å resolution by Popov et al. (1998), the x-ray structure at 1.1-Å resolution by Vojtechovsy et al. (1998), and the 1.5-Å x-ray structure by Engler et al. (2000). An EXAFS determination by Sherck et al. (2000) is also included. The XANES statistical error for the Fe-Np distance is ∼0.02 Å and for the axial distances is ∼0.06 Å, the values being in good agreement with those reported by Popov et al. (1998) and Engler et al. (2000).
The analogous results relative to Mb(II)H2O are reported in Table 2. There is only a previous x-ray structure at 1.4 Å resolution (Engler et al., 2000), reporting a decrease of the axial distances of ∼0.1 Å (Fe-O) and 0.07 Å (Fe-Nhis), going from Mb(III)H2O to Mb(II)H2O, which is within the root-mean-square coordinate error of the data collection. Our XANES determination for Mb(II)H2O reports a shortening of the axial distances by 0.07 Å (Fe-O) and 0.15 Å (Fe-Nhis).
TABLE 2.
Mb(II)H2O at T = 70 K
| Experiment | Fe–Np | Fe–Nhis | Fe–O |
|---|---|---|---|
| XRD* 1.4 Å | not reported | 2.08 | 2.13 |
| XANES solution | 2.03(2) | 1.99(9) | 2.13(3) |
| XANES crystal | 2.03(4)//heme | 1.99(2)//axis | 2.15(4)//axis |
Comparison between different structural determinations of the Fe-heme site geometry. Distance units, Ångstrom. The resolution of XRD experiments is indicated. The values in parenthesis are the statistical errors as evaluated by the fitting procedure.
Then, the fitting procedure has been applied to the polarized XANES experiments on single crystal of Mb(II)H2O at T < 70 K and Mb(II)⋯H2O at T = 240 K. In Fig. 6 the best fits (solid curves) of the polarized XANES spectra with ɛ//-axis and ɛ//-heme (see Methods) are shown, in terms of the structural parameters reported in Table 2 and Table 3. The experimental data are plotted in circles. In the case of Mb(II)H2O, the ɛ//-heme spectra are used to fit the Fe-Np average distance, while the ɛ//-axis spectra are used to fit the other two distances Fe-O and Fe-Nhis. For the species Mb(II)⋯H2O, the fit of the ɛ//-heme spectrum turned out to be sensitive also to the Fe-heme displacement. So the number n of structural parameters fitted was 1,2,2, and 2, and a χ2/n value of 6.65, 2.71, 5.81, and 2.89 was obtained, respectively, for Mb(II)H2O (ɛ//-heme), Mb(II)H2O (ɛ//-axis), Mb(II)⋯H2O (ɛ//-heme), and Mb(II)⋯H2O (ɛ//-axis). The Es and As parameters of the Γ(E) functions used are (15 ± 4) eV and (10 ± 3) eV, respectively. The fit is systematically worse for the ɛ//-heme spectra; here, the approximations of the broadening function seem heavier. Another source of errors is that by using a self-consistent potential, we do not recalculate the potential for any new structure. The effects of systematic errors introduced by a static potential have been reported in our previous work (Benfatto and Della Longa, 2001), and have been verified to determine a worse fit without relevant changes for the numerical results of the fit.
FIGURE 6.
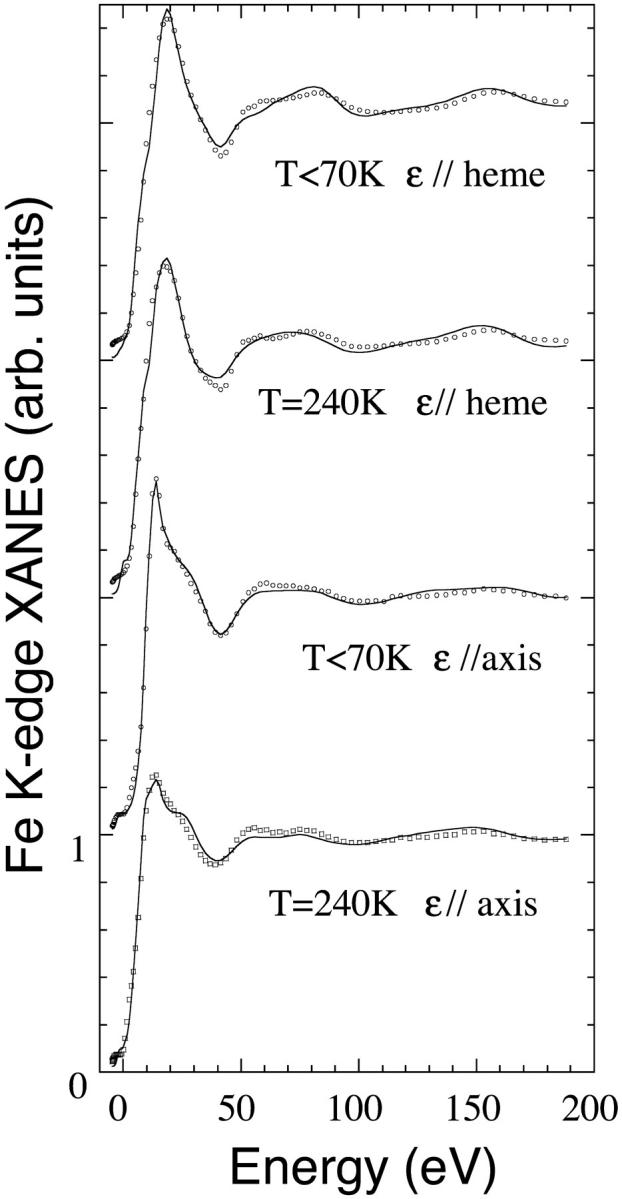
Best fit of the polarized XANES spectra of Mb(II)H2O at T = 70 K and Mb(II)⋯H2O at T = 240 K in terms of the selected structural parameters reported in Tables 2 and 3 (solid lines). The experimental data are plotted in circles.
TABLE 3.
Mb(II)H2O at T > 240 K
| Experiment | Fe–Np | Fe–Nhis | Fe–O | Fe-heme displacement | Tilt |
|---|---|---|---|---|---|
| XRD* 1.1 Å | 2.07 | 2.14 | 3.53 | 0.39 | 30 |
| XRD† 1.1 A | 2.06 | 2.15 | 3.73 | 0.27 | 30 |
| XANES crystal | 2.08(2)//heme | 2.09(3)//axis | 3.67(16)//axis | 0.51(20)//heme | 34(37)//axis |
Comparison between different structural determinations of the Fe-heme site geometry. Distance units, Ångstrom. The resolution of XRD experiments is indicated. The values in parenthesis are the statistical errors as evaluated by the fitting procedure.
Deoxy-Mb. PDB code, 1A6N.
Deoxy-Mb. PDB code, 1BZP.
The results of the XANES fits are summarized in Fig. 7, including a sketch of the three chemical states and the distance and angular determinations. In the case of Mb(II)H2O, the MXAN fit on the protein crystal fully confirm and refine the results extracted from the XANES data on the protein in solution. These data are reported in Table 2. In particular, by comparing the structures of Mb(III)H2O and Mb(II)H2O, a decrease of the axial distances by 0.15 Å (Fe-Nhis), out of our distance error, and by 0.09 Å (Fe-O), within our distance error, is confirmed to accompany the photo-reduction process.
FIGURE 7.

Sketch of the structure of the three main states. The structural data extracted by XANES are reported together with their statistical errors.
In the case of Mb(II)⋯H2O, no x-ray structure has been reported so far from such an experiment, hence we have the MXAN fit results with reported x-ray structures of dithionite-reduced deoxy-Mb (Vojtechovsky et al., 1998; Kachalova et al., 1998). The comparison is shown in Table 3. There is a reasonable agreement as far as it concerns all the parameters, including the long Fe-O distance of (3.67 ± 0.16) Å. This fit is rather sensitive to the Fe-heme displacement, contrary to the hexa-coordinated Mb(III)H2O, due to having lower symmetry and a larger displacement value. The small residual differences between Mb(II)⋯H2O and deoxy-Mb could be due to still incomplete thermal relaxation of the Mb(II)⋯H2O state, as suggested also by the direct comparison between the experimental data of deoxy-Mb and Mb(II)⋯H2O shown in Fig. 4.
DISCUSSION
Due to the inhomogeneity of the protein ensemble, care should be taken in comparing the structures extracted by XANES and x-ray diffraction; the XANES spectrum represents the sum of the signals coming from each protein of the ensemble, whereas XRD probably selects only one or some of the most populated substates. Systematic errors could affect the “single-state” XANES determinations, due to the inhomogeneity of the protein ensemble, rendering the XANES determination out of sense if done on a strongly inhomogeneous ensemble. However, in terms of our experience, the structural sampling made by single-state models seems to adequately fit the XANES experiments only when the structural dispersion of parameters around the main state is small, evaluated approximately by the statistical errors. Out of this range, it is necessary to consider more complex models, including two or more structures, to obtain a satisfactory fit of the spectrum. As a matter of fact, there is no way to fit, within a single-state model, the XANES spectra of Mb(II)H2O at T = 120 K and T = 160 K, containing mixtures of relaxed and unrelaxed structures.
The distance between the iron and the proximal histidine appears shortened in Mb(II)H2O with respect to Mb(III)H2O by ∼0.15 Å. This result is in good agreement with that suggested, but not fully established, by the x-ray diffraction data at 1.4 Å (Engler et al., 2000). This finding supports a dynamic model in which the reinforcement of the Fe-His bond destabilizes the iron in-plane position and favors the water dissociation just when the protein collective motions become thermally activated.
A structural relaxation of the intermediate state >80 K, before water dissociation, has been previously suggested by the observation of a shifting of the Soret band at temperatures >80 K (Lamb et al., 1998). This observation could be connected to our finding that between 120 K and 160 K, a decrease in amplitude of the ɛ//-axis absorption between 7130 and 7150 eV, not related to water dissociation, occurs. As we do not see any evident change in the ɛ//-heme XANES at these temperatures, the relaxation within the intermediate state probed by the shift of the Soret band could consist in electron density transfer from the axial to the heme moiety, associated to a readjustment of the Fe-water, and/or water-distal histidine mutual positions, without any relevant distortion of the heme.
Similarly, we observe XANES changes between 200 K and 240 K, probably due to structural relaxation of the Fe-heme complex in Mb(II)⋯H2O rather than to the final phase of the Mb(II)H2O → Mb(II)⋯H2O temperature-dependent water dissociation. Further work is in progress in our laboratory to try to interpret these structural relaxations, using multistate models in the MXAN fit.
It is well-established, by the previous body of knowledge, that the irreversible transition of photo-reduced aquomet-myoglobin toward its deoxy state is a dynamical process involving a structurally inhomogeneous ensemble of proteins that can relax in time or temperature, either remaining in the same ligated state or changing from the ligated to the unligated state. The present work shows the potentiality of XANES in revealing distinct aspects of the process, so that both dynamical and structural determinations can be provided in the same experiment, for a process that can constitute a true laboratory system for biophysics as well as the photolysis of carbonmonoxy-myoglobin does.
It is important to stress that an EXAFS study, in the same experimental conditions, would require much longer data collection sessions to get a sufficiently high signal-to-noise ratio and a higher absorbed dose; hence, it could not be carried out in the full range of temperatures. Owing to the recently reported ability to extract structural information (Benfatto and Della Longa, 2001; Della Longa et al., 2001), XANES can provide structural models of the Fe-heme, probe the redox state, and give experimental evidences on the thermodynamics of the system, supporting and connecting a large body of experiments from other different unrelated techniques. In particular, owing to the possibility to probe the unbound ligand within a distance of 4–4.5 Å from the iron, XANES could be applied to probe-trapped intermediate states where the ligand resides in docking sites, supporting XRD data in the assessment of pathways for ligand binding (Chu et al., 2001; Brunori and Gibson, 2001).
Finally, we point out the great advantage to work with solution and crystal samples together; in fact it is possible in principle to check in crystals if the conformational rearrangements due to ligand binding or changing of the redox state are affected by the lattice constraints. So if the three-dimensional structure of single intermediate states of a reaction pathway are isolated, we could be able to see if the three-dimensional rearrangement obtained is compatible with solution. This is important especially for those cases (Williams et al., 1997) in which there are great conformational rearrangements involved.
Acknowledgments
Thanks are due to Prof. M. Bolognesi for supplying Mb crystal samples. The authors also thank the staff of the European Molecular Biology Laboratory Grenoble Outstation Laboratory for hospitality and assistance.
A.L.B. was supported by grants from Istituto Nazionale Fisica della Materia, Italy.
References
- Benfatto, M., and S. Della Longa. 2001. Geometrical fitting of experimental XANES spectra by a full multiple scattering procedure. J. Synchrotron Rad. 8:1087–1094. [DOI] [PubMed] [Google Scholar]
- Benfatto, M., P. D'Angelo, S. Della Longa, and N.V. Pavel. 2002. Evidence of distorted fivefold coordination of the Cu2+ aqua ion from an x-ray absorption spectroscopy quantitative analysis. Phys. Rev. B. 65:174205. [Google Scholar]
- Blumenfeld, L. 1981. Problems of Biological Physics. Springer-Verlag, Berlin, Germany.
- Brunori, M., and Q. H. Gibson. 2001. Cavities and packing defects in the structural dynamics of myoglobin. EMBO Rep. 2:674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister, W. P. 2000. Structural changes in a cryo-cooled protein crystal owing to radiation damage. Acta Crystallogr. D. 56:328–341. [DOI] [PubMed] [Google Scholar]
- Chu, K., J. Vojtechovsky, B. H. McMahon, R. M. Sweet, J. Berendzen, and I. Schlichting. 2001. Structure of a ligand-binding intermediate in wild-type carbonmonoxy myoglobin. Nature. 403:921–923. [DOI] [PubMed] [Google Scholar]
- Debenham, M. J., Q. Hao, S. S. Hasnain, F. E. Dodd, Z. H. L. Abraham, and R. R. Eady. 1996. Structure solution of azurin II from Alcaligenes xylosoxidans using the Laue method: possibility to study ‘in situ’ redox changes using x-rays. J. Synchrotron Rad. 3:14–19. [DOI] [PubMed] [Google Scholar]
- D'Angelo, P., M. Benfatto, S. Della Longa, and N. V. Pavel. 2002. Combined XANES and EXAFS analysis of Co2+, Ni2+ and Zn2+ aqueous solutions Phys. Rev. B. 66:064209 /1–7 . [Google Scholar]
- Della Longa, S., A. Arcovito, M. Girasole, J. L. Hazemann, and M. Benfatto. 2001. Quantitative analysis of x-ray absorption near edge structure data by a full multiple scattering procedure: the Fe-CO geometry in photolyzed carbonmonoxy-myoglobin single crystal. Phys. Rev. Lett. 87:155501. [DOI] [PubMed] [Google Scholar]
- Della Longa, S., S. Pin, R. Cortes, A. V. Soldatov, and B. Alpert. 1998. Fe-heme conformations in ferric myoglobin. Biophys. J. 75:3154–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Longa, S., A. Arcovito, B. Vallone, A. Congiu Castellano, R. Kahn, J. Vicat, Y. Soldo, and J. L. Hazemann. 1999. Polarised x-ray absorption spectroscopy of the low temperature photoproduct of carbonmonoxy-myoglobin. J. Synchrotron Rad. 6:1138–1147. [Google Scholar]
- Díaz-Moreno, S., A. Muñoz-Páez, and J. Chaboy. 2000. X-ray absorption spectroscopy (XAS) study of the hydration structure of Yttrium(III) cations in liquid and glassy states: eight- or ninefold coordination? J. Phys. Chem. A. 104:1278–1286. [Google Scholar]
- Engler, N., A. Ostermann, A. Gassmann, D. C. Lamb, V. E. Prusakov, J. Schott, R. Schweitzer-Stenner, and F. Parak. 2000. Protein dynamics in an intermediate state of myoglobin: optical absorption, resonance Raman spectroscopy, and x-ray structure analysis. Biophys. J. 78:2081–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann, H., S. Zinser, P. Komninos, R. T. Schneider, G. U. Nienhaus, and F. Parak. 1996. The x-ray structure determination of a metastable state of carbonmonoxy myoglobin after photodissociation. Proc. Natl. Acad. Sci. USA. 93:7013–7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasnain, S. S., and K. O. Hodgson. 1999. Structure of metal centres in proteins at subatomic resolution. J. Synchrotron Rad. 6:852–864. [Google Scholar]
- Kachalova, G. S., A. N. Popov, and H. D. Bartunik. 1998. PDB code1BZP.
- Labhardt, A., and C. Yven. 1979. X-ray absorption edge fine structure spectroscopy of the active site heme of cytochrome-c. Nature. 277:150–151. [DOI] [PubMed] [Google Scholar]
- Lamb, D. C., A. Ostermann, V. E. Prusakov, and F. Parak. 1998. From metmyoglobin to deoxy myoglobin: relaxations of an intermediate state. Eur. Biophys. J. 27:113–125. [DOI] [PubMed] [Google Scholar]
- Le Tilly, V., S. Pin, B. Hickel, and B. Alpert. 1997. Pulse radiolysis reduction of myoglobin. Hydrated electrons diffusion inside the protein matrix. J. Am. Chem. Soc. 119:10810–10814. [Google Scholar]
- Liu, H. I., M. Sono, S. Kadkhodayan, L. P. Hager, B. Hedman, K. O. Hodgson, and J. H. Dawson. 1995. X-ray absorption near edge studies of cytochrome P-450-CAM, chloroperoxidase, and myoglobin. Direct evidence for the electron releasing character of a cysteine thiolate proximal ligand. J. Biol. Chem. 270:10544–10550. [DOI] [PubMed] [Google Scholar]
- Muller, J. E., O. Jepsen, and J. W. Wilkins. 1982. X-ray absorption spectra: K-edges of 3d transition metals, L-edges of 3d and 4d metals and M-edges of palladium. Sol. State Commun. 42:365–368. [Google Scholar]
- Natoli, C. R., M. Benfatto, and S. Doniach. 1986. Use of general potentials in multiple-scattering theory. Phys. Rev. A. 34:4682–4694. [DOI] [PubMed] [Google Scholar]
- Parak, F., and V. E. Prusakov. 1994. Relaxation of non-equilibrium states of myoglobin studied by Mossbauer spectroscopy. Hyperfine Inter. 91:885–890. [Google Scholar]
- Pedio, M., M. Benfatto, S. Aminpirooz, and J. Haase. 1994. Multiple-scattering analysis of NEXAFS spectra of molecular oxygen anions in differently grown cesium oxides. Phys. Rev. B. 50:6596–6602. [DOI] [PubMed] [Google Scholar]
- Pin, S., B. Hickel, B. Alpert, and C. Ferradini. 1989. Parameters controlling the kinetics of ferric and ferrous heme proteins reduction by hydrated electrons. Biochim. Biophys. Acta. 994:47–51. [DOI] [PubMed] [Google Scholar]
- Pin, S., B. Alpert, A. Congiu-Castellano, S. Della Longa, and A. Bianconi. 1994. X-ray absorption spectroscopy of hemoglobin. Methods Enzymol. 232:266–292. [DOI] [PubMed] [Google Scholar]
- Popov, A. N., G. S. Kachalova, and H. D. Bartunik. 1998. PDB code 1BZ6.
- Prusakov, V. E., R. A. Stukan, R. M. Davidov, and K. Gersonde. 1985. Non-equilibrium state of a monomeric insect hemoglobin induced by gamma-irradiation and detected by Mossbauer spectroscopy. FEBS Lett. 186:158–162. [Google Scholar]
- Prusakov, V. E., J. Steyer, and F. Parak. 1995. Mossbauer spectroscopy on nonequilibrium states of myoglobin: a study of r-t relaxation. Biophys. J. 68:2524–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli, R. B. G., and S. M. McSweeney. 2000. The ‘fingerprint’ that x-rays can leave on structures. Struct. Fold. Des. 8:315–328. [DOI] [PubMed] [Google Scholar]
- Scherk, C. G., A. Ostermann, K. Achterhold, O. Iakovleva, C. Nazikkol, B. Krebs, E. W. Knapp, W. Meyer-Klaucke, and F. Parak. 2001. The x-ray absorption spectroscopy Debye-Waller factors of an iron compound and of met-myoglobin as a function of temperature. Eur. Biophys. J. 30:393–403. [DOI] [PubMed] [Google Scholar]
- Schlichting, I., J. Berendzen, G. N. Phillips, Jr., and R. M. Sweet. 1994. Crystal structure of photolysed carbonmonoxy-myoglobin. Nature. 371:808–812. [DOI] [PubMed] [Google Scholar]
- Srajer, V., T. Y. Teng, T. Ursby, C. Pradervand, Z. Ren, S. I. Adachi, W. Schildkamp, D. Bourgeois, M. Wulff, and K. Moffat. 1996. Photolysis of the carbon monoxide complex of myoglobin: nanosecond time-resolved crystallography. Science. 274:1726–1729. [DOI] [PubMed] [Google Scholar]
- Teng, T. Y., and K. Moffat. 2000. Primary radiation damage of protein crystals by an intense synchrotron x-ray beam. J. Synchotron Rad. 7:313–317. [DOI] [PubMed] [Google Scholar]
- Tyson, T. A., K. O. Hodgson, C. R. Natoli, and M. Benfatto. 1992. General multiple-scattering scheme for the computation and interpretation of x-ray-absorption fine structure in atomic clusters with applications to SF6, GeCl4, and Br2 molecules. Phys. Rev. B. 46:5997–6019. [DOI] [PubMed] [Google Scholar]
- Vojtechovsky, J., J. Berendzen, K. Chu, I. Schlichting, and R. M. Sweet. 1998. PDB codes: 1A6K and 1A6N.
- Williams, P. A., V. Fulop, E. F. Garman, N. F. Saunders, S. J. Ferguson, and J. Hajdu. 1997. Heme-ligand switching during catalysis in crystals of a nitrogen-cycle enzyme. Nature. 389:406–412. [DOI] [PubMed] [Google Scholar]
- Yachandra, V. K. 1995. X-ray absorption spectroscopy and applications in structural biology. Methods Enzymol. 246:638–675. [DOI] [PubMed] [Google Scholar]