

Abstract
Fluorescence resonance energy transfer (FRET) between mutant green fluorescent proteins (GFP) provides powerful means to monitor in vivo protein-protein proximity and intracellular messengers. However, the leading FRET pair of this class (CFP/YFP) entails suboptimal donor excitation by Argon lasers, thereby hindering FRET imaging on many confocal microscopes. Further challenges arise from the large spectral overlap of CFP/YFP emission. By contrast, DsRed, along with other members of a growing family of red-shifted sea coral fluorophores, features spectra that could obviate such limitations, using DsRed as FRET acceptor, and GFP or CFP as donor. Nonetheless, DsRed suffers from slow chromophore maturation, which confounds quantitative FRET. Here, we develop strategies minimizing the resulting complexity: 1), Pulsed activation of inducible promoters, driving expression of DsRed-tagged molecules, yields a uniform bolus of mature fluorophore; 2), The 33-FRET detection algorithm, adapted for CFP/DsRed and GFP/DsRed, proves insensitive to distortion by slow maturation. We thus show that DsRed supports strong FRET in CFP-DsRed or GFP-DsRed concatemers. These results reveal the promise of sea coral fluorophores like DsRed as FRET partners with GFP or CFP.
INTRODUCTION
Lubert Stryer transformed the theory of fluorescence resonance energy transfer (FRET) (Förster, 1948) into the realm of biological promise (Stryer and Haugland, 1967), bringing forth the notion that an optical ruler with molecular resolution could be constructed by quantifying FRET between suitably matched fluorophores attached to interacting biomolecules. FRET initially fostered much progress in defining biomolecular dimensions and interactions, primarily in the in vitro setting (Clegg, 1992; Wu and Brand, 1994). Now, FRET between genetically encoded fluorophores is revolutionizing widespread detection of protein-protein interactions in situ, as they occur in single, living cells (Erickson et al., 2001; Janetopoulos et al., 2001; Siegel et al., 2000; Vanderklish et al., 2000). Because fluorescent fusion proteins, comprised of such fluorophores and molecules of interest, can be expressed intracellularly from cDNAs engineered via straightforward molecular biology, FRET experiments have become convenient and feasible across a wide spectrum of systems and molecules. The more traditional approach of using chemistry to specifically label molecules is generally a more invasive and case-specific endeavor, with far more restricted feasibility.
Among the genetically encoded fluorophores, two green fluorescent protein (GFP) color mutants—CFP and YFP—have emerged as the leading donor/acceptor pair for FRET experiments (Miyawaki et al., 1997). These fluorophores afford reasonable spectral separation and brightness, while not requiring potentially harmful ultraviolet excitation. Nonetheless, the spectral properties of this pair are suboptimal for FRET in two regards, thus limiting the full promise of experiments using GFP color mutants. First, the high degree of overlap between emission spectra for cells expressing CFP and YFP (Fig. 1, top row) entails substantial “cross talk” of CFP emission in the YFP detection channel (Gordon et al., 1998), thereby complicating quantification of FRET. Second, FRET experiments with these fluorophores require excitation of CFP, a difficult proposition with the repertoire of lasers available on most confocal microscopes: Argon ion (458, 488, and 514 nm; Fig. 1, dashed lines), dual-gas Krypton/Argon (488, 568, and 657 nm), green HeNe (543 nm), and red HeNe (633 nm). Some groups have successfully employed the 458-nm line of the Argon laser to excite CFP while accepting the tradeoff of substantial cross-excitation of YFP (Rizzo et al., 2002). Nonetheless, the commonly available lasers are generally more suitable for FRET experiments involving archetypical donor/acceptor pairs like fluorescein/rhodamine (Fig. 1, bottom row). Several confocal microscope manufacturers now offer excitation sources capable of efficient CFP excitation, such as Krypton ion lasers (413 nm), violet laser diodes (405 nm), or HeCd lasers (442 nm), but these sources are often lacking on standard instrument configurations, and adding them involves considerable expense.
FIGURE 1.

Spectral properties favoring DsRed as a FRET partner with GFP or CFP. Excitation (thick lines) and emission (thin lines) spectra collected from suspensions of cells expressing the indicated donor or acceptor fluorophores. Dashed lines indicate the 458- and 488-nm lines of an Argon ion laser.
A potential solution to these challenges has come with the discovery of a growing class of sea coral fluorescent proteins that have dramatically shifted emission spectra toward longer wavelengths, some with genuinely red emission (Labas et al., 2002; Matz et al., 1999). The advantages of this red shift for FRET experimentation are illustrated by considering a leading exemplar of this class—DsRed, a GFP analog from Discosoma coral (Matz et al., 1999). FRET pairs comprised of CFP/DsRed or GFP/DsRed manifest superb wavelength separation of donor and acceptor emission spectra (Fig. 1, rows 2–3), implicating minimal donor emission cross talk in the acceptor emission channel. For example, a 600-nm longpass emission filter would sensitively report DsRed emission with ∼0 contribution from either GFP or CFP. Such selective detection of DsRed emission would greatly simplify quantification of FRET. Furthermore, the spectral characteristics of GFP/DsRed (Fig. 1, second row) mimic those of fluorescein/rhodamine, thus permitting efficient excitation of the donor (GFP) by a standard Argon laser (488-nm line).
A major challenge to realizing these benefits is the slow maturation of DsRed (>48 h to reach 90% of maximal fluorescence) (Baird et al., 2000) and other sea coral chromophores (Labas et al., 2002). Although targeted mutagenesis has recently provided DsRed variants with accelerated maturation, it is unclear whether this approach will prove widely successful with the larger family of sea coral fluorophores. Moreover, the engineered DsRed constructs generated to date suffer from unfavorable spectral characteristics for FRET application and/or substantial reductions in brightness (Bevis and Glick, 2002; Campbell et al., 2002). In the one case tested (Campbell et al., 2002), the attenuated brightness was severe enough to preclude practical FRET detection. Hence, developing general means to overcome the complications of slow maturation presents as an enormously important goal. Here, we develop several practical strategies, thus improving substantially the prospects of employing DsRed as a FRET partner with GFP or CFP.
MATERIALS AND METHODS
Molecular biology
Mammalian expression plasmids pEGFP-N3 and pDsRed-N1 were purchased from Clontech (Palo Alto, CA). ECFP and EYFP were PCR amplified from yellow cameleon-2 (Miyawaki et al., 1997) (gift from R.Y. Tsien) and subcloned into the pEGFP-N3 vector. ECFP and EYFP were also subcloned into pZeoSV2 (Invitrogen, Carlsbad, CA), with the weak SV40 promoter and no SV40 replication origin. The enhanced mutants EGFP, ECFP, and EYFP are referred to using the more concise names GFP, CFP, and YFP throughout. Starting material for the CFP–DsRed concatemer was a CFP–YFP concatemer subcloned into pcDNA3 (Invitrogen) by Kpn I and Xba I. CFP–YFP/pcDNA3 incorporates a 25-residue interfluorophore linker (SGSSSGSSSLAGIEGRSSSGSSSGS) containing Nhe I and BamH I sites. CFP–DsRed/pcDNA3 was generated by amplifying DsRed using the forward and reverse oligos: 5′-CGGGATCCGTGCGCTCCTCCAAGAACG-3′ and 5′-GCTCTAGATTACAGGAACAGGTGGTGGC-3′. The amplified fragment was digested and ligated into CFP–YFP/pcDNA3 using BamH I and Xba I, thus replacing YFP with DsRed while preserving the linker. GFP–DsRed/pcDNA3 was constructed in turn by amplifying GFP using forward and reverse oligos: 5′-GGGGTACCGCCACCATGGTGAGC-3′ and 5′-GCTGCTAGCGAGCTAGAGCCGGAGCTAGAGCCAGACTTGTACAGCTCGTCC-3′. The amplified fragment was digested and ligated into CFP–DsRed/pcDNA3 using Kpn I and Nhe I, which replaced CFP with GFP. CFP–DsRed was also subcloned into pIND, for use with the ecdysone-inducible mammalian expression system (Invitrogen). All constructs were verified by sequencing and fluorescence spectroscopy.
Fluorescence spectra
HEK293 cells were transfected by calcium-phosphate precipitation with cDNA encoding fluorescent protein. Three days posttransfection, the cells were washed twice with PBS, then harvested by gentle trituration in PBS with 0 mM Ca2+ and 2 mM EDTA. Cells were pelleted, resuspended in 0 mM Ca2+ Tyrode's (pH 7.4), and loaded into a 1-cm cuvette for analysis. Fluorescence excitation and emission spectra were obtained using an SPF-500C spectrafluorometer (SLM Instruments, Rochester, NY); excitation bandwidth was 2 nm and emission bandwidth was 10 nm. Raw spectra were corrected for background emission by subtracting similar spectra obtained on the same day from untransfected cell suspensions. Optical density at the excitation peak was <0.10.
FRET measurements
33-FRET measurements were performed as described previously (Erickson et al., 2001) on a Nikon Eclipse TE300 microscope (Nikon USA, NY). 33-FRET filter cubes for CFP/YFP (excitation, dichroic, emission, company): CFP (D440/20M, 455DCLP, D480/30M, Chroma, Brattleboro, VT); YFP (500DF25, 525DRLP, 530EFLP, Omega Optical, Brattleboro, VT); FRET (440DF20, 455DRLP, 535DF25, Omega Optical). 33-FRET filter cubes for CFP/DsRed: CFP (D440/20M, 455DCLP, D480/30M, Chroma); DsRed (540AF30, 570DRLP, 575ALP, Omega Optical); FRET (440DF20, 455DRLP, 580DF30, Omega Optical). 33-FRET filter cubes for GFP/DsRed: GFP (475AF20, 500DRLP, 510AF23, Omega Optical); DsRed (540AF30, 570DRLP, 575ALP, Omega Optical); FRET (475AF20, 500DRLP, 580DF30, Omega Optical). Experimentally determined RD1, RD2, RA values for CFP/YFP, CFP/DsRed, and GFP/DsRed FRET pairs are shown in Table 1.
TABLE 1.
33-FRET parameters for various FRET pairs
| Donor/Acceptor | RD1 | RD2 | RA |
|---|---|---|---|
| CFP/YFP | 0.2090 ± 0.0006 (n = 30) | 0.0036 ± 0.0002 (30) | 0.0319 ± 0.0001 (25) |
| CFP/DsRed | 0.0259 ± 0.0003 (25) | −0.0008 ± 0.0003 (25) | 0.0302 ± 0.0002 (30) |
| GFP/DsRed | 0.0289 ± 0.0004 (20) | 0.0000 ± 0.0002 (20) | 0.1584 ± 0.0025 (6) |
For donor dequenching experiments, measurements were performed using the CFP cube before and after 30 min of intense illumination using a custom acceptor photobleaching cube (Chroma), consisting of a D535/50× excitation filter and a 100% mirror (instead of the dichroic); this bleaching cube spared the CFP chromophore in control experiments. The ratiometric FRET method (Fig. 4 B) was applied to data collected previously with the 33-FRET method (Fig. 4 A) by simply dividing the FRET cube measurement by the donor cube measurement. All data are reported as mean ± SEM, except where noted.
FIGURE 4.

33-FRET proves insensitive to heterogeneous DsRed maturation. (A) Relationship between FRET measurements (FR, FRET Ratio) by 33-FRET and relative amount of mature DsRed. Top axis, FA/FDA, ratio of DsRed and CFP cube measurements. Bottom axis, fmature, normalized metric for relative amount of mature DsRed. Open symbols, cells continuously expressing CFP–DsRed concatemer under control of a CMV promoter system. Closed symbols, cells in which pulsed induction of CFP–DsRed expression was employed (Fig. 3) to select for cells with predominantly mature DsRed. Horizontal red line indicates average FR for cells with pulsed CFP–DsRed expression. Insensitivity of 33-FRET to DsRed maturation is illustrated by the stability of FR measurements to the right of the dashed vertical line, drawn at fmature = 0.05. Horizontal dashed line indicates FR = 1, or EEFF = 0. (B) Relationship between FRET measurements (F580/F480) by the ratiometric method and relative amount of mature DsRed. Bottom axis same as for A. Red line indicates best fit to data by linear regression. (C) Relationship between FRET measurements (EEFF, FRET efficiency) by CFP dequenching and relative amount of mature DsRed. Bottom axis same as for A. Red line indicates best fit to data by linear regression. Horizontal dashed line indicates EEFF = 0. Bottom color bar indicates the approximate relationship between fmature and cell color, as visualized by a 515-nm longpass filter cube.
Inducible expression of CFP–DsRed
Six plates of HEK293 cells were cotransfected by calcium-phosphate precipitation with cDNA encoding CFP–DsRed/pIND and pVgRXR (Invitrogen) as described (Brody et al., 1997). One day posttransfection, the cells were induced by adding muristerone to a final concentration of 1 μM. Two days posttransfection, the cells were washed three times with PBS then bathed in fresh media. The plates of cells were then divided into two groups of three plates: muristerone was reapplied to one group (+/+), whereas the other group (+/−) remained untreated. The cells were visualized fours days posttransfection using a standard fluorescein cube (470DF35, 505DCLP, 515EFLP, Chroma) on a Nikon Eclipse TE300 microscope. This cube enabled visual “fingerprinting” of cells according to the color (Fig. 2, A–B): cells expressing only CFP appear green; cells expressing only DsRed appear orange-red; and cells containing both CFP and DsRed vary in color from green to yellow to orange-red depending on the relative amounts of CFP and mature DsRed. To discount week-to-week variance, data in Fig. 2 C was pooled from two experiments done in separate weeks. Pictures acquired by a Nikon Coolpix 995 digital camera.
FIGURE 2.

Pulsed expression enriches for mature DsRed. (A) Continuous expression of CFP–DsRed concatemer results in dramatic cell color heterogeneity, as visualized by a filter cube having a 470 ± 17.5-nm excitation filter and 515-nm longpass emission filter. With this cube, cells expressing CFP–DsRed vary in color from green (left) to yellow (middle) to orange-red (right), depending on the relative amounts of CFP and mature DsRed. (B) Wide-field view using 515-nm longpass filter cube of cells expressing CFP–DsRed under control of an inducible promoter system. Cells were transfected on “day 1.” Panels show cells, as visualized on day 4, after receiving either continuous treatment with inducing agent on days 2 and 3 (denoted as +/+), or a onetime pulse of inducing agent on day 2 only (+/−). Pulsed induction selects for cells with predominantly mature DsRed, based on their orange-red appearance. (C) Average population counts from (+/+) and (+/−) plates assayed in parallel four days posttransfection, confirming selection for orange-red cells with pulsed expression. Differences between (+/+) and (+/−) for both green-yellow counts and orange-red counts were significant, P < 0.01.
RESULTS
Spectral properties favoring DsRed as a FRET partner with GFP or CFP
Visual inspection of spectra previewing the prospects for CFP/DsRed and GFP/DsRed FRET (Fig. 1) already raised the potential advantages of spectral separation and convenient excitation. More important for the feasibility of deploying these fluorophore pairs for FRET is the question of coupling strength, determined in large part by the overlap between donor emission and acceptor excitation spectra. In this regard, the overlap region appears substantial for both CFP/DsRed and GFP/DsRed pairs (Fig. 1). The specific impact of this overlap can be assessed by calculating a quantitative metric proportional to the cube root of coupling strength—the Förster distance, R0, equal to the donor-acceptor separation at which FRET is 50% efficient. Explicit determination of R0 required that we first compute the overlap integral, J, based on the spectra in Fig. 1 (rows 1–3). The value for J conveys the extent of spectral overlap between the donor emission spectrum and acceptor excitation spectrum, as provided by (Lakowicz, 1999)
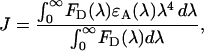 |
(1) |
where FD is the fluorescence emission spectrum of the donor, ɛA is the molar extinction coefficient of the acceptor in M−1 cm−1, and J has dimensions of M−1 cm−1nm4. The equation Förster derived for R0 (in Å) in its simplified form is then (Lakowicz, 1999)
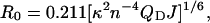 |
(2) |
where κ2 is the orientation factor, QD is the donor quantum yield (QCFP = 0.40; QGFP = 0.60) (Patterson et al., 2001), Na is Avogadro's number, and n is the index of refraction of water at 25°C (n ∼ 1.334). For convenience, we assumed that the relative dipole orientations of the donor and acceptor fluorophores rapidly randomize, thus making κ2 = 2/3 (Lakowicz, 1999). Completing the calculation of J requires knowledge of the maximum extinction coefficient for DsRed, thus permitting appropriate scaling of our DsRed excitation spectra to compute ɛA(λ). Here, there is inexplicable scatter in reported values, with the originally reported maximum extinction coefficient for DsRed being ɛDsRed = 22,500 M−1 cm−1 (Matz et al., 1999), whereas more recently determined coefficients vary among values of 52,000 M−1 cm−1 (Bevis and Glick, 2002), 72,500 M−1 cm−1 (Patterson et al., 2001), and 75,000 M−1 cm−1 (Baird et al., 2000). Using the smallest determination of ɛDsRed = 22,500 M−1 cm−1, we determined R0 values for the pairing of CFP/DsRed and GFP/DsRed to be 41.7 Å and 47.1 Å, respectively. These values closely match those determined previously for spectra obtained from measurements of purified proteins (Patterson et al., 2000). At the other extreme, using the largest ɛDsRed determination (75,000 M−1 cm−1), we determined R0 values for CFP/DsRed and GFP/DsRed to be 50.9 Å and 57.6 Å, respectively. Both minimum and maximum R0 estimates are comparable to or even larger than the R0 reported for CFP/YFP of 49.2 Å (Patterson et al., 2000), indicating that DsRed is capable of supporting robust FRET coupling with either CFP or GFP. The comparatively large R0 = 57.6 Å value would be especially advantageous for “long ranger” FRET-based interaction screens between candidate binding partners tagged with GFP and DsRed, because legitimate binding interactions that happen to place fluorophores beyond FRET range would be minimized.
Pulsed expression of DsRed molecules enriches for a mature population of chromophore
A major challenge for using DsRed in FRET experiments is the reportedly slow maturation of the DsRed chromophore. In the course of maturation, the chromophore proceeds through an intermediate state, which exhibits a faint green fluorescence, before achieving a brilliant red-fluorescent form (Baird et al., 2000; Mizuno et al., 2001; Wiehler et al., 2001). In many practical applications, the immature species can be considered nonfluorescent because the magnitude of its fluorescence emission is <1% of that demonstrated by the mature chromophore (Baird et al., 2000). This slow and heterogeneous maturation process can be demonstrated explicitly by constitutive expression of engineered CFP–DsRed concatemers. The CFP fluorophore, which matures rapidly and uniformly in <10 h (Greenbaum et al., 2002), serves as a reporter for the existence of a concatemer, even when the associated DsRed moiety is in its immature, essentially nonfluorescent form. The CFP emission from such a concatemer would appear green through the 515-nm longpass emission filter used in Fig. 2 A (left). However, when the associated DsRed fluorophore matures, the concatemer would appear red, being dominated by the red fluorescence of DsRed (Fig. 2 A, right). Cells with a mixture of (im)mature DsRed would appear yellow-orange (Fig. 2 A, middle). Inspection of a widefield view (Fig. 2 B, left) confirms ample representation of all the predicted forms. Such heterogeneous DsRed maturation could significantly complicate FRET experiments.
To alleviate such heterogeneity, we tested whether time-gated expression of CFP–DsRed would permit full DsRed maturation of a bolus of expressed concatemers. CFP–DsRed was subcloned into an ecdysone inducible expression plasmid (pIND; Invitrogen), and transfected into HEK293 cells. On the next day, cells were exposed to ecdysone agonist (1 μM muristerone) to induce expression, and then washed after 24 h. Four to five days after transfection, cells were nearly all red colored (Fig. 2 B, right), indicating that already expressed CFP–DsRed had time to fully mature. This scenario contrasts sharply with the marked color heterogeneity present when muristerone was continuously present (Fig. 2 B, left), as described above. Averages from several transfections fully confirmed these trends (Fig. 2 C). Hence, pulsed expression of DsRed-based molecules provides an excellent approach to enrich for mature DsRed, thereby simplifying the task of employing this fluorophore in FRET applications.
33-FRET algorithm proves insensitive to heterogeneous DsRed maturation
In many instances, however, it may be either inconvenient or even unfeasible to employ an inducible promoter, such as with transgenic animals, which often employ constitutive expression of recombinant molecules. In these cases, we suspected that our recently developed three-cube FRET (33-FRET) algorithm (Erickson et al., 2001) would provide accurate quantification of FRET despite heterogeneous DsRed maturation. This capability is expected because 33-FRET relies on sensitized acceptor emission; thus, the functionally nonfluorescent, immature DsRed (Baird et al., 2000) would not impact quantification of FRET by this assay (see Discussion). To explore this conjecture experimentally, we describe the algorithm, and then test how it fares in detecting FRET within an engineered concatemer of CFP and DsRed.
33-FRET is a practical single-cell FRET assay based on sensitized acceptor emission (Clegg, 1992). Three core principles underlie the approach: 1), FRET alters the amplitudes but not the shapes of the individual donor and acceptor emission spectra; 2), measuring the emission spectrum at one wavelength indicates the proper scaling for the entire spectrum at all wavelengths; and 3), careful selection of optical filters permits near spectral selection of donor and acceptor. The goal of this method is to compute the FRET ratio (FR), equal to the fractional increase in acceptor fluorescence emission due to FRET. FR is calculated as the ratio of acceptor emission in the presence of donor (FAD) to acceptor emission in the absence of donor (FA). The procedure for determining FR entails sequential intensity readings from three distinct filter cubes, conveyed as SCUBE(SPECIMEN), where CUBE denotes a particular filter cube and SPECIMEN indicates whether the cell is expressing donor only (D), acceptor only (A), or both (DA). We previously described the 33-FRET method for the CFP/YFP pair (Erickson et al., 2001); here we explicitly extend this procedure to the CFP/DsRed pair. This illustrative case can be easily adapted for the GFP/DsRed pair.
For concreteness, consider an isolated cell expressing the concatemer shown in Fig. 3 A, where CFP and DsRed are connected by a flexible, 25-residue linker. Fig. 3 B shows the resulting emission spectrum with excitation at 440 nm. The characteristic double-humped shape reflects the superposition of underlying CFP and DsRed spectra. In regard to the DsRed component, SFRET(DA) (number 1) is the sum of two parts: CFP emission (number 4); and DsRed emission (number 2), a portion of which is due to direct excitation. Dissecting these components relies on SCFP(DA) and SDsRed(DA), signals from filter cubes that permit optical isolation of the CFP and DsRed signals for a particular cell expressing both fluorophores. SCFP(DA), which excites CFP and DsRed but measures fluorescence where only CFP emits (number 5), is multiplied by a predetermined constant (RD1) to determine the contribution of CFP emission at 580 nm (number 4). Subtracting this from SFRET(DA) leaves FAD, according to
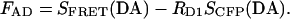 |
(3) |
FIGURE 3.

DsRed FRET detection by 33-FRET. (A) Illustration of key excitation and emission wavelengths for 33-FRET analysis of the CFP–DsRed concatemer. (B) Dissection of 580-nm emission with 440-nm excitation. Graph, overall emission spectrum from a single cell expressing CFP–DsRed (thick black line), reflecting underlying CFP (thick gray line) and DsRed (thin black line) spectra. A portion of DsRed emission is due to direct excitation (black dashed line). Points 1–5 are described in the text.
Similarly, multiplying SDsRed(DA), which provides exclusive excitation of DsRed at 540 nm and therefore precludes FRET, by a constant (RA) yields the component of SFRET(DA) due to direct excitation of DsRed, or FA (number 3). Explicitly, this is given by
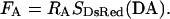 |
(4) |
In cases where the donor and acceptor excitation spectra are not well separated, a small amount of donor excitation can occur with the acceptor cube. This cross talk can be corrected as described before (Erickson et al., 2001) using another constant RD2 and the modified equation
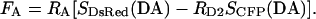 |
(5) |
Finally, FR is computed as the ratio
 |
(6) |
The constants RD1, RD2 and RA are determined in separate cells expressing either donor (CFP) or acceptor (DsRed) alone, and forming the appropriate ratios of measurements as given below.
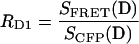 |
(7) |
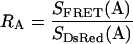 |
(8) |
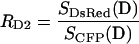 |
(9) |
Recommended filter sets for this method, as well as values for RD1, RA, and RD2, are detailed in the Materials and Methods section. Parameters for CFP/DsRed and GFP/DsRed are included therein. Finally, we can compute the effective FRET efficiency (Erickson et al., 2001), EEFF, by
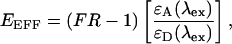 |
(10) |
where EEFF is the effective FRET efficiency and the term in brackets is the ratio of acceptor and donor molar extinction coefficients at a given excitation wavelength, λex.
To test the sensitivity of 33-FRET to heterogeneous DsRed maturation, we applied 33-FRET to CFP–DsRed concatemers, here constitutively expressed under a CMV promoter to produce a heterogeneous population of DsRed. Fig. 2 A (left) illustrates the color heterogeneity encountered in various cells with constitutive expression of concatemer. To gauge approximately the extent of DsRed maturation in a given cell, we used optical measures to approximate fmature, defined as the fraction of concatemers with a mature DsRed moiety. The numerator of such a fraction would be the overall amount of mature DsRed in a cell, which would be proportional to the 580-nm fluorescence emission of DsRed, provided that we could selectively excite DsRed at 540 nm. The desired entity is FA, which can be determined by experimental measures as deduced in Eq. 4 above. The denominator of the sought-after fraction would be the overall amount of concatemer in a cell, which would be proportional to the 580-nm fluorescence emission of CFP due to 440 nm excitation, if we could factor out the partial quenching of CFP fluorescence due to FRET with DsRed. The desired entity would be FD, which is difficult to isolate experimentally. However, we can easily calculate FDA (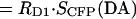 ), which is the 580-nm fluorescence emission of CFP due to 440 nm excitation, including partial quenching of CFP fluorescence due to FRET with DsRed. Given E ∼ 0.3 (shown below in Fig. 4 C), FD will be no more than ∼30% larger than FDA. Hence, the optical index FA/FDA will be nearly proportional to the fraction of concatemers with mature DsRed in a given cell. Determining FR and FA/FDA in multiple cells with variable coloration then permits direct examination of the robustness of the 33-FRET assay in the face of differing degrees of DsRed maturation.
), which is the 580-nm fluorescence emission of CFP due to 440 nm excitation, including partial quenching of CFP fluorescence due to FRET with DsRed. Given E ∼ 0.3 (shown below in Fig. 4 C), FD will be no more than ∼30% larger than FDA. Hence, the optical index FA/FDA will be nearly proportional to the fraction of concatemers with mature DsRed in a given cell. Determining FR and FA/FDA in multiple cells with variable coloration then permits direct examination of the robustness of the 33-FRET assay in the face of differing degrees of DsRed maturation.
Fig. 4 A summarizes the results of such an experiment by plotting FR as a function of FA/FDA. As a reference, measurements were first made on cells enriched for a mature population of CFP–DsRed concatemers, using the pulsed expression strategy (Fig. 2 B, right). Such cells gave rise to FR values clustering around 3.6 and FA/FDA ratios near 2.75 (Fig. 4 A, closed symbols). These determinations established the genuine FRET level for mature concatemers (Fig. 4 A, red line), and helped to normalize the FA/FDA axis to its maximum value of 3.14 (Fig. 4 A, top axis). Normalizing FA/FDA then yields an experimental determination of fmature (Fig. 4 A, bottom axis), as defined above. With these references in mind, it is readily apparent that, except for fmature < 0.05, FR measurements from heterogeneous populations (Fig. 4 A, open symbols) were nearly indistinguishable from the target FRET level for fully mature concatemers. This remarkable convergent behavior directly demonstrates the ability of 33-FRET to extract the legitimate FRET strength despite wide variation in DsRed maturation. Selection of cells with fmature > 0.05 in all subsequent experiments provided a simple means to ensure adequate red-fluorescent signal for robust determination of FR.
By contrast, such immunity to variable DsRed maturation is not shared by FRET-detection methods that rely upon donor (CFP) characteristics. For example, the most common metric for FRET is the simple ratio of fluorescence at acceptor and donor emission wavelengths resulting from ∼440 nm excitation, in this case expressed as F580/F480. Plots of F580/F480 versus fractional maturation (fmature) clearly indicate large apparent changes in FRET coupling with increasing maturation (Fig. 4 B). Donor dequenching is another quantitative FRET detection method (Bastiaens and Jovin, 1996). Here, the enhancement of CFP fluorescence upon total photobleaching of DsRed can be used to specify effective FRET efficiency, EEFF, according to
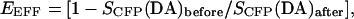 |
(11) |
where and SCFP(DA)before and SCFP(DA)after are CFP emission before and after DsRed photobleaching. Like the simple ratio method, apparent FRET efficiencies determined by donor dequenching varied widely with increasing DsRed maturation (Fig. 4 C), as would be expected from theory (see Discussion).
Overexpression can lead to spurious concentration-dependent FRET
A remaining challenge for engaging DsRed-based FRET experiments, which pertains in general to any FRET experiments where fluorophores are produced from common mammalian expression plasmids, was to exclude the possibility of spurious, concentration-dependent FRET (Lakowicz, 1999), an artifact that is explicitly characterized in Fig. 5. Panel A shows the results for cells expressing both CFP and YFP as separate molecules. For each cell, both FR and FDA were determined. As detailed above, SCFP(DA) (or FDA) is roughly proportional to the concentration of donor (CFP), under the assumption that cells had a roughly uniform volume. The plot shows the cumulative average FR (FRcum) calculated for all cells with SCFP(DA) less than the value on the x axis. For small SCFP(DA), the average FR is essentially unity, as expected for noninteracting CFP and YFP molecules. However, as SCFP(DA) exceeds ∼21,000, FRcum increasingly rises above unity, reaching 1.3 at the highest levels of SCFP(DA). This corresponds to average FR values of ∼1.6 in this high SCFP(DA) regime. This scenario suggests that spurious FRET became appreciable at CFP concentrations corresponding to an SCFP(DA) value of ∼21,000. Thus, for all 33-FRET determinations used for quantification of FRET coupling, we have taken this SCFP(DA) value as a maximum cutoff for inclusion of cells expressing a CFP moiety. Fig. 5 B demonstrates that a similar inclusion criterion can be applied to exclude spurious FRET in the case of two CFP/DsRed configurations, namely CFP/DsRed concatemers (squares) and CFP and DsRed expressed as separate molecules (triangles). In either case, FRcum averaged from cells to the left of the criterion defined a flat plateau region, as expected. By contrast, FRcum progressively climbed above plateau levels as cells to the right of the criterion were included, indicating a contribution of spurious FRET when CFP concentrations became overly elevated. An analogous inclusion criterion proved successful for excluding spurious FRET between GFP and DsRed (not shown).
FIGURE 5.

Overexpression can lead to spurious FRET. (A) Analysis of cumulative average FR (FRcum) versus CFP cube measurements (FDA) for cells coexpressing CFP and YFP with strong CMV promoter (closed circles) and weak SV40 promoter (open circles) (Erickson et al., 2001). Dashed line at FDA = 21,000 indicates cutoff beyond which spurious, concentration-dependent FRET becomes apparent, as indicated by the rise in the FRcum plot. (B) Analysis of FRcum vs. FDA for cells coexpressing CFP and DsRed (triangles) and cells expressing CFP–DsRed (squares).
Another approach for excluding spurious FRET artifacts is to employ expression plasmids with weaker promoters (Erickson et al., 2001), like the SV40 element in pZeoSV2 plasmids. All cells obtained with the pZeoSV2 plasmid fell within the appropriate SCFP(DA) criterion (Fig. 5 A, open circles). By contrast, use of strong CMV-based promoters could easily produce concentrations leading to spurious FRET, as described above (Fig. 5, filled symbols).
Sensitive and selective detection of FRET with GFP/DsRed and CFP/DsRed pairs
With the strategies and criteria developed above, we used 33-FRET to quantify the actual FRET coupling strength within concatemers of CFP–DsRed and GFP–DsRed, both constitutively expressed under the control of a CMV promoter (pcDNA3). In particular, two inclusion criteria were applied: 1), To ensure sufficient DsRed signal, only cells with fmature > 0.05 were included (Fig. 4 A). 2), To exclude contributions from spurious FRET, the SCFP(DA) cutoff (Fig. 5) was applied. The GFP/DsRed concatemer demonstrated unmistakable FRET with an FR ∼ 3 (Fig. 6 A), and the CFP/DsRed concatemer showed an even larger FR ∼ 4 (Fig. 6 B). Expressing the acceptor (DsRed) alone, or in conjunction with unlinked donor (GFP or CFP), gave FR values indistinguishable from unity, showing an appropriate lack of FRET interaction in controls. Clearly, both CFP/DsRed and GFP/DsRed pairs supported robust and well-defined FRET.
FIGURE 6.

Sensitive and selective detection of FRET by 33-FRET. Single cell 33-FRET analysis of FRET between GFP (A) or CFP (B) and DsRed. All cells satisfied two selection criteria: fmature > 0.05 (Fig. 4) and FDA < 21,000 (Fig. 5). *, P < 0.01 versus DsRed alone.
DISCUSSION
FRET using DsRed as acceptor holds important potential advantages for live cell in situ studies. Compared to the current leading FRET pair involving genetically encoded fluorophores (CFP/YFP), there would be far less emission cross talk with both CFP/DsRed and GFP/DsRed pairs (Fig. 1). Moreover, the GFP/DsRed pair would permit efficient excitation on common confocal microscope platforms (Fig. 1). However, slow chromophore maturation and oligomerization have hindered the use of DsRed in FRET applications (Baird et al., 2000; Mizuno et al., 2001). Even though some of these problems may be solved by targeted mutagenesis of DsRed (Bevis and Glick, 2002; Campbell et al., 2002; Knop et al., 2002; Terskikh et al., 2002; Verkhusha et al., 2001; Yanushevich et al., 2002), there is a growing class of red-shifted sea coral fluorophores with potentially useful spectral features, and these molecules also appear to suffer from slow maturation (Labas et al., 2002). It is unclear whether targeted mutagenesis of this class of molecules will prove widely successful in generating variants with accelerated maturation. Therefore, as a complement to targeted mutagenesis efforts, we developed several alternative approaches to overcome the difficulties raised by slow DsRed maturation. In so doing, we have confirmed clear-cut static FRET for CFP/DsRed and GFP/DsRed pairs. These results merit three lines of in-depth discussion: 1), the complementarity of targeted mutagenesis efforts and our own approaches for improving the prospects of DsRed FRET; 2), the theoretical basis for the comparative strengths and weaknesses of the various methods for quantifying DsRed FRET (Fig. 4); and 3), the possibility of concentration-dependent FRET (Fig. 5), an issue relevant to FRET with all genetically encoded fluorophores.
Complementary approaches to overcoming the challenges of DsRed FRET
Apart from niche deployment as a fluorescent timer (Terskikh et al., 2002; Verkhusha et al., 2001), slowly maturing DsRed complicates most applications, and wreaks havoc on quantitative assessment of FRET. In addition, obligate tetramerization of DsRed, with the potential for larger-scale aggregation (Jakobs et al., 2000; Lauf et al., 2001; Mizuno et al., 2001), adds further potential complexity to the assessment of FRET. Though there are anecdotal reports that fusing DsRed to various biomolecules can alleviate aggregation (Lauf et al., 2001), we have observed that all DsRed fusions, as well as DsRed alone, show widely variable tendencies in different cells, ranging from no detectable aggregation to widespread punctate concentrations (not shown). Overall, these challenges may be general for a growing family of red-shifted, sea coral fluorophores (Labas et al., 2002), thus restricting the enormous promise of this entire class of fluorophores for biological application.
One important approach to overcoming these problems is targeted mutagenesis to produce variant fluorophores with accelerated maturation and attenuated oligomerization. Regarding slow maturation, DsRed variants have been engineered with half maturation times less than 1–2 h, such as the T1, T3, and T4 constructs developed by Bevis and Glick (2002). However, compared to DsRed, T1 and T4 are far dimmer, and T3 manifests enhanced green emission that could increase spectral cross talk with potential FRET donors like CFP and GFP. All of these constructs still suffer from obligate tetramerization (Baird et al., 2000; Mizuno et al., 2001). Concerning oligomerization, targeted mutagenesis based on the DsRed crystal structure (Yarbrough et al., 2001) has also provided a monomeric DsRed variant, mRFP1 (Campbell et al., 2002), which exhibits fast maturation akin to T1 and T4. Unfortunately, mRFP1 also showed attenuated brightness that was severe enough to preclude practical detection of FRET (Campbell et al., 2002). In short, though targeted mutagenesis may ultimately produce a bright, monomeric, and rapidly maturing DsRed variant, currently engineered constructs appear too dim or have unfavorable spectral characteristics for practical use in FRET assays. More generally, it is unclear whether this approach will be widely successful with the larger family of sea coral fluorophores, for which crystal structures have yet to be obtained.
This state of affairs motivates the alternative approaches developed in this study to engage the challenges of DsRed. Here, we have demonstrated that quantitative FRET can still be determined despite slowly maturing DsRed, using pulsed DsRed expression and/or a sensitized acceptor emission method (e.g., 33-FRET) to quantify FRET. Although these strategies represent significant advances, there still remains the serious issue of DsRed oligomerization, which could complicate quantification of FRET via intricate second-order mechanisms such as homotransfer among adjacent DsRed molecules (Baird et al., 2000). Moreover, oligomerization could disrupt native targeting or association of molecules tagged with DsRed. In the future, our strategies for engaging slowly maturing DsRed may allow targeted mutagenesis efforts to focus exclusively on producing bright and monomeric DsRed variants, without simultaneously satisfying the call for fast maturation. This relaxation of constraints may hasten the successful engineering of DsRed and other sea-coral fluorophores that permit practical FRET studies with CFP and/or GFP.
In the meantime, our strategies do facilitate certain important applications of CFP/DsRed or GFP/DsRed FRET in live cells. For example, consider an experiment where we pit DsRed-tagged molecules against CFP-tagged molecules, in the context of an in situ screen for binding (Erickson and Yue, 2002). Although DsRed oligomerization could artifactually inhibit interaction, a positive interaction as quantified by an elevated FR is still very meaningful. Though the precise FRET efficiency may be subject to debate, the presence or absence of bona fide FRET should be robustly specified by the 33-FRET approach.
In fact, FR may prove more than simply a reliable “yes/no” indicator of FRET. Our results in Fig. 4 A point to a remarkable possibility—that FRET between nearest CFP and DsRed molecules, such as between fluorophores in a fused CFP–DsRed concatemer, represents the predominant resonance energy transfer process within the larger CFP–DsRed complex, as organized by obligate DsRed tetramerization. This possibility arises upon consideration of the wide range of fmature values seen in Fig. 4 A, suggesting vastly different fractions of (im)mature DsRed moieties in various obligate tetramers (Cotlet et al., 2001; Garcia-Parajo et al., 2001). If appreciable hetero- and homotransfer were to occur among multiple types of fluorophore pairs within a complex, we would expect that FR, a metric of aggregate FRET coupling in the complex, would vary considerably as the mixture of (im)mature DsRed within tetramers changes. Instead, we observe a robust convergence of FR to a single value (red line, Fig. 4 A) that holds for essentially all fmature determinations. This convergence thus strongly implicates the predominance of FRET between immediately adjacent CFP and mature DsRed molecules; this would be the only form of coupling that is invariant with differing fmature. If this simple outcome were to hold for a variety of CFP/DsRed fusion constructs, then FR determinations would quantitatively reflect FRET coupling within individual constructs, rather than among separate constructs comprising a tetramer. Predominance of FRET coupling within concatemer constructs may also explain the convergence of FR despite variation in the degree of larger-scale aggregation.
Theory underlying optimal methods for measuring DsRed FRET
An extensive collection of different FRET detection methods is described in the literature (Selvin, 1995). Selecting the optimal method for a given experiment depends on the specific experimental setup, including which donor/acceptor pair is used. Here, we examine three FRET methods—ratiometric, donor dequenching, and 33-FRET—representing a general class of methods that entail discrete measurements of emission intensities at specific wavelengths. Methods in this class benefit from being easily adapted for the fluorescence microscope, with minimal need for additional equipment. We experimentally determined that 33-FRET uniquely provides stable CFP/DsRed or GFP/DsRed FRET measurements that are largely independent of the relative amount of fully mature DsRed (fmature) (Fig. 4 A). By contrast, ratiometric and donor dequenching FRET measurements vary with DsRed maturation (Fig. 4, B and C). Rather than simply trying all three methods, it would be most convenient to understand in advance which method would be optimal for the donor/acceptor pair being used. In the Results section, we hinted at the basis for such an understanding. Below, we explicitly develop this theory for selecting among these three FRET detection methods, based on an analysis of how each metric is impacted by variations in the relative amount of mature DsRed.
The ratiometric method incorporates a commonly employed FRET metric, R, which is equal to the simple ratio of acceptor to donor emission intensity, recorded near the respective emission peaks. For CFP/DsRed FRET, this translates to the ratio, R = F580/F480, with excitation near 440 nm. An increase in FRET coupling will enhance the acceptor emission peak at the expense of quenching of the donor emission peak, yielding an increase in R. In general, the ratiometric method assumes a fixed (generally 1:1) acceptor to donor expression ratio. Otherwise, changes in relative expression will lead to changes in R that could be mistaken for FRET. In the case of cells expressing the CFP–DsRed concatemer, R will be directly proportional to the ratio of fully mature DsRed to CFP. Thus, any measurement of R will vary in proportion to the relative amount of fully mature DsRed, as indicated by the linear relationship between R and fmature depicted by Fig. 4 B. In sum, the strict dependence of R on the relative amount of mature DsRed renders the ratiometric metric impractical for static FRET measurements on DsRed.
Another method for quantitating FRET is donor dequenching (Bastiaens and Jovin, 1996), where the donor emission peak is recorded before and after acceptor photobleaching. If FRET is present initially, there will be an enhancement, or dequenching, of the donor emission peak after the near-complete elimination of the acceptor. A number of studies have successfully deployed donor dequenching for the CFP/YFP FRET pair (Miyawaki and Tsien, 2000; Pentcheva and Edidin, 2001). However, donor dequenching does not provide a stable measurement of FRET for the CFP/DsRed (Fig. 4 C) or GFP/DsRed pairs (not shown), as predicted by the following reasoning. For steady-state FRET, we are most interested in measuring the true FRET efficiency, E, which conveys the amount of FRET coupling between closely associated donor and acceptor molecules. Donor dequenching instead provides EEFF, which can be related to the true efficiency by (Epe et al., 1983; Erickson et al., 2001)
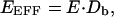 |
(12) |
where Db is the fraction of donor molecules associated with a mature DsRed molecule. In the case of the CFP–DsRed concatemer, a cell with predominantly immature DsRed (fmature ∼ 0) will have a Db value near zero. By contrast, a cell with the majority of its DsRed molecules in the fully mature state (fmature ∼ 1) will have Db near one. In fact, Eq. 12 can be restated as
 |
(13) |
which explicitly depicts the relationship between donor dequenching FRET measurements and the relative amount of mature DsRed. This relationship is borne out by Fig. 4 C, which shows that FRET measurements by donor dequenching rise in direct proportion to increasing fmature. An additional methodological challenge for donor dequenching is the relative resistance of DsRed to photobleaching (Baird et al., 2000). Using continuous illumination with a 150 W Xenon arc lamp and a 535 ± 25 nm filter, we found that ∼30 min of exposure was required to bleach DsRed by 90%; however, pulsed, high-intensity laser illumination may accelerate this process (Mizuno et al., 2001). By comparison, YFP is bleached by >90% in ∼3 min of continuous illumination. Despite the dual challenges of sensitivity to DsRed maturation and resistance to photobleaching, some have made use of donor dequenching for measuring DsRed FRET (Cornea et al., 2001). In sum, FRET measurements based on donor emission will depend on Db, and will thus be challenged by variable DsRed maturation, as shown by the CFP–DsRed experiments.
In contrast to the previous two cases, FRET assays based on 33-FRET are well suited for measuring CFP/DsRed or GFP/DsRed FRET, as indicated by the stability of FR readings across 95% of the range of fmature values (Fig. 4 A). Like donor dequenching, 33-FRET can be used to calculate an EEFF (Eq. 10). However, the EEFF determined by 33-FRET has a different relationship with E, according to (Epe et al., 1983; Erickson et al., 2001)
 |
(14) |
where Ab is the fraction of fully mature DsRed molecules associated with a donor molecule. In the case of the CFP–DsRed concatemer, all fully mature DsRed molecules are directly linked with CFP, thus making Ab = 1. Eq. 14 can thereby be restated simply as EEFF = E, revealing that 33-FRET measurements are not a function of fmature. Important to the theoretical deductions associated with Eq. 14 is the property that CFP molecules linked to immature DsRed molecules will not contribute appreciably to the FRET measurement, because immature DsRed can be considered approximately nonfluorescent from a practical standpoint. Specifically, experimental evidence for the effectively nonfluorescent nature of immature DsRed comes from the shape invariance of normalized excitation spectra (583 nm emission) determined from preparations of DsRed with very different proportions of immature fluorophore (Fig. 1, D and E, in Mizuno et al., 2001). Moreover, the tight clustering of our RA determinations for the CFP/DsRed configuration (0.0325 ± 0.0010, mean ± SD, n = 30 cells, Table 1), obtained from cells with varying proportions of immature DsRed, explicitly confirms the previously reported shape invariance of excitation spectra (Mizuno et al., 2001).
Fig. 4 A provides resounding experimental confirmation of the expectation that EEFF = E, as determined by the 33-FRET algorithm. Indeed, it is only for cells with very little mature DsRed (small fmature) that 33-FRET fails to provide a stable measure of FRET. The divergent estimates of FR at very low fmature values probably result from signal-to-noise issues arising from a lack of sufficient red-fluorescent DsRed signal in those cells, rather than an inherent inability of 33-FRET to extract the actual FR over the small fmature range. In sum, of the three methods examined here, 33-FRET is optimal for CFP/DsRed or GFP/DsRed FRET. Moreover, 33-FRET would be the optimal method in any situation where the acceptor fluorophore matures more slowly than the donor.
A simple yet effective way to overcome the signal-to-noise limitation of 33-FRET for low fmature values is to establish a minimum fmature cutoff point, beyond which the signal is sufficiently strong to provide a stable reading of E. In the present study, taking fmature > 0.05 effectively excluded cells outside of the usable range for 33-FRET (Fig. 4 A). It must be noted that use of the fmature cutoff is only appropriate when the relative expression of donor and acceptor molecules is fixed, a condition that is assured when employing both fluorophores in a unimolecular construct like the CFP–DsRed concatemer. Many of the genetically encoded FRET-based sensors are unimolecular (Miyawaki et al., 1997; Mochizuki et al., 2001), thus making them ideal candidates for application of an fmature cutoff strategy. Ensuring a fixed ratio of donor and acceptor molecules may be more difficult for bimolecular FRET experiments, in which CFP and DsRed are fused to different proteins. In practice, it may be possible to achieve fixed relative expression of separate CFP- and DsRed-tagged molecules using a bicistronic vector that incorporates cDNA encoding both fusion products on a single plasmid (Martinez-Salas, 1999). However, in this case the relative expression of CFP to DsRed may not be 1:1, so an fmature cutoff other than 0.05 would have to be determined experimentally.
There are some notable exceptions when 33-FRET may not be optimal for monitoring FRET with DsRed. For example, FRET detection by the ratiometric method is generally more effective when rapid assessments of dynamic FRET are required. In these cases, the accuracy of FRET determination can be enhanced by preselection of cells with a predominance of fully mature DsRed, using our approximate index for DsRed maturity (fmature). Furthermore, the preference for 33-FRET over donor dequenching is reversed for an experimental system in which the donor, rather than the acceptor, is slow to mature. Here, Ab would vary with fmature, and Db would be independent of fmature, meaning that donor dequenching would provide the most reliable measure of FRET efficiency. For example, if one were to use DsRed as a FRET donor, possibly paired with the HcRed fluorescent protein (Clontech) as acceptor, donor dequenching would be preferred over 33-FRET for establishing a stable measurement of FRET efficiency that does not vary with the relative amount of mature DsRed.
One final point to be gleaned from theoretical comparison of FRET detection methods concerns the determination of the maximal DsRed molar extinction coefficient, ɛDsRed. As mentioned in the Results, there is substantial variation among literature values for ɛDsRed, ranging from a minimum of 22,500 M−1 cm−1 (Matz et al., 1999) to a maximum of 75,000 M−1 cm−1 (Baird et al., 2000). It could well be that the precise ɛDsRed value is rather sensitive to experimental conditions; hence, ɛDsRed should ideally be determined in situ, within the same live cells where actual FRET experiments are being performed. All previous determinations of ɛDsRed have been undertaken in vitro (Baird et al., 2000; Bevis and Glick, 2002; Matz et al., 1999; Patterson et al., 2001), but the theoretical comparison of donor dequenching and 33-FRET methods developed here reveals a simple approach to specify ɛDsRed in situ, as follows. Consider the live-cell, 33-FRET analysis of the CFP–DsRed concatemer, summarized in Fig. 4 A. According to Eq. 10, the convergent FR value of 3.6 (Fig. 4 A, red line) should be related to the effective FRET efficiency EEFF by the ratio of acceptor (DsRed) and donor (CFP) molar extinction coefficients at the excitation wavelength of 440 nm (ɛCFP(440 nm) and ɛDsRed(440 nm), respectively). In discussing Eq. 14, we deduced that Ab = 1, so that EEFF in Eq. 10 could be set equal to E, the actual FRET efficiency between a mature DsRed and CFP, as fused together in a CFP–DsRed concatemer. Because ɛCFP(440 nm) is well established to be 25,100 M−1 cm−1 in situ (Erickson et al., 2001), we could then directly solve for the in situ value for ɛDsRed(440 nm) if we could experimentally determine E. At first glance, this requirement might appear difficult, because donor dequenching data specify EEFF = E · fmature (Eq. 13), rather than E. However, straightforward linear extrapolation of donor dequenching data to fmature = 1 yields an E value of 0.41 (red line, Fig. 4 C). This allows us to solve for an in situ ɛDsRed(440 nm) value of 3,960 M−1 cm−1. Multiplying this by a factor of 10.9, corresponding to ɛDsRed(558 nm)/ɛDsRed(440 nm) as specified by the DsRed excitation spectra (Fig. 1), yields a maximum ɛDsRed value of 43,200 M−1 cm−1, in reasonable agreement with the 52,000 M−1 cm−1 value reported by Bevis and Glick (2002). This approach can be undertaken case-by-case to determine an appropriate maximal ɛDsRed value for each experimental cell system.
Sources of concentration-dependent FRET
A final challenge is that of concentration-dependent, or spurious FRET (Fig. 5). This challenge is not specific to experiments involving DsRed; rather it is a factor that must be considered whenever strong, constitutive promoters are used to drive expression of fluorescent proteins (Miyawaki and Tsien, 2000).
What is spurious FRET, as detected in Fig. 5? One possibility is that spurious FRET readings result from high bulk concentration of expressed fluorophores, which would tend to decrease the average separation between donor and acceptor molecules moving freely in the cytosol. The first-order assumption of FRET experiments is to imagine that FRET will only occur when donor and acceptor fluorophores are brought “close together” by a specific interaction, perhaps by direct binding between proteins to which the fluorophores are fused, or by an engineered linker that explicitly tethers donor and acceptor moieties together. However, donor molecules at high enough concentrations could be on average close to an acceptor molecule, even in the absence of a specific interaction or engineered linker. That such a possibility could be realized in practice is made clear by determining just how high the donor concentration would have to be to precipitate such a scenario.
Consider an acceptor molecule at position r = 0. The probability that a donor molecule resides within distance r and r + dr of the acceptor molecule is expressed as
 |
(15) |
where D is the donor concentration (mol/L), Na is Avogadro's number, and r is in units of Å. The expected FRET efficiency for the coupling between the acceptor and randomly dispersed donors at all distances would then be
 |
(16) |
where the R0 is the Förster distance. Substituting Eq. 15 provides
 |
(17) |
noting that  yields
yields
 |
(18) |
Setting 〈E〉 = 0.02 and solving for D provides D0.02, the critical donor concentration that would support a concentration-dependent FRET efficiency of 0.02 (or 2%), according to
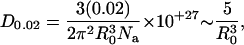 |
(19) |
where R0 is in Å and D0.02 is in mol/L. For the R0 values typical of GFP color mutants and DsRed (∼50 Å) (Patterson et al., 2000), D0.02 ∼ 40 μM. This means that donor concentrations as low as 40 μM would be sufficient to bring donor molecules in close enough proximity to an acceptor molecule to support FRET efficiencies of 0.02. Concentrations in the general range of 40 μM should be achievable for protein expression driven by strong CMV-based expression plasmids (Miyawaki and Tsien, 2000).
Spurious FRET could also arise from the known tendency of highly concentrated GFP-based fluorophores to form concatemers, with Kd ∼ 100 μM (Phillips, 1998; Zacharias et al., 2002). Concatemerization of GFP-based fluorophores, as well as tetramerization of DsRed fluorophores, could act to bring donors and acceptors close together, thus effectively decreasing further the value of D0.02. Recently described efforts to generate monomeric GFP-based fluorophores (Zacharias et al., 2002) and monomeric DsRed (Campbell et al., 2002) could help alleviate the problems posed by fluorophore aggregation, but do not solve the overall challenge of concentration-dependent FRET.
Fig. 5 not only confirms the presence of spurious, concentration-dependent FRET, as predicted by Eq. 19, but also provides important clues about how to control for this confounding feature. Steady-state FRET measurements are often depicted by bar charts, which compare measurements of FRET efficiencies among controls and various experimental conditions. However, it would be difficult to interpret differences among FRET measurements obtained with dramatically different fluorophore concentrations. Equation 19 highlights one convenient method of controlling for concentration-dependent FRET. When calculating FRET based on sensitized acceptor emission, such as with 33-FRET, spurious FRET is a function of donor concentration only. Thus, the most appropriate graphical representation for distinguishing genuine from spurious FRET is to plot E (or FR) versus donor emission SCFP(DA), as was done in Fig. 5. If test data cluster in a locus above that for free fluorophores (compare CFP–DsRed to CFP/DsRed), then the FRET interaction exceeds that expected for spurious FRET. By contrast, spurious FRET as measured by methods based on donor emission, such as donor dequenching, is a function solely of acceptor concentration. The analogous graphical analysis for this case would be E plotted versus acceptor emission (FA). In either case, it is crucial to make comparisons of control and test data for similar fluorophore concentrations, as illustrated in Fig. 5.
Acknowledgments
We thank Rebecca Alvania and Devi Rathod for technical assistance. We also thank Omega Optical, Inc. for allowing us to test multiple filter cubes to optimize 33-FRET for DsRed.
This work was supported by a Whitaker Graduate Studentship (to MGE.) and research grants from the American Heart Association and National Institutes of Mental Health (to DTY).
Michael G. Erickson and Daniel L. Moon contributed equally to this work.
References
- Baird, G. S., D. A. Zacharias, and R. Y. Tsien. 2000. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA. 97:11984–11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiaens, P. I., and T. M. Jovin. 1996. Microspectroscopic imaging tracks the intracellular processing of a signal transduction protein: fluorescent-labeled protein kinase C beta I. Proc. Natl. Acad. Sci. USA. 93:8407–8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevis, B. J., and B. S. Glick. 2002. Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed). Nat. Biotechnol. 20:83–87. [DOI] [PubMed] [Google Scholar]
- Brody, D. L., P. G. Patil, J. G. Mulle, T. P. Snutch, and D. T. Yue. 1997. Bursts of action potential waveforms relieve G-protein inhibition of recombinant P/Q-type Ca2+ channels in HEK 293 cells. J. Physiol. (Lond.). 499:637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 99:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg, R. M. 1992. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 211:353–388. [DOI] [PubMed] [Google Scholar]
- Cornea, A., J. A. Janovick, G. Maya-Nunez, and P. M. Conn. 2001. Gonadotropin-releasing Hormone Receptor Microaggregation. J. Biol. Chem. 276:2153–2158. [DOI] [PubMed] [Google Scholar]
- Cotlet, M., J. Hofkens, S. Habuchi, G. Dirix, M. Van Guyse, J. Michiels, J. Vanderleyden, and F. C. De Schryver. 2001. Identification of different emitting species in the red fluorescent protein DsRed by means of ensemble and single-molecule spectroscopy. Proc. Natl. Acad. Sci. USA. 98:14398–14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epe, B., K. G. Steinhäuser, and P. Woolley. 1983. Theory of measurement of Förster-type energy transfer in macromolucules. Proc. Natl. Acad. Sci. USA. 80:2579–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson, M. G., B. A. Alseikhan, B. Z. Peterson, and D. T. Yue. 2001. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 31:973–985. [DOI] [PubMed] [Google Scholar]
- Erickson, M. G., and D. T. Yue. 2002. FRET-based two-hybrid mapping of the molecular contacts underlying Ca2+ inactivation of L-type Ca2+ channels (abstr.). Biophys. J. 82:107a. [Google Scholar]
- Förster, T. 1948. Intermolecular energy migration and fluorescence. Annalen der Physik. 2:55–75. [Google Scholar]
- Garcia-Parajo, M. F., M. Koopman, E. M. van Dijk, V. Subramaniam, and N. F. van Hulst. 2001. The nature of fluorescence emission in the red fluorescent protein DsRed, revealed by single-molecule detection. Proc. Natl. Acad. Sci. USA. 98:14392–14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, G. W., G. Berry, X. H. Liang, B. Levine, and B. Herman. 1998. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 74:2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum, L., D. Schwartz, and Z. Malik. 2002. Spectrally resolved microscopy of GFP trafficking. J. Histochem. Cytochem. 50:1205–1212. [DOI] [PubMed] [Google Scholar]
- Jakobs, S., V. Subramaniam, A. Schonle, T. M. Jovin, and S. W. Hell. 2000. EFGP and DsRed expressing cultures of Escherichia coli imaged by confocal, two-photon and fluorescence lifetime microscopy. FEBS Lett. 479:131–135. [DOI] [PubMed] [Google Scholar]
- Janetopoulos, C., T. Jin, and P. Devreotes. 2001. Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science. 291:2408–2411. [DOI] [PubMed] [Google Scholar]
- Knop, M., F. Barr, C. G. Riedel, T. Heckel, and C. Reichel. 2002. Improved version of the red fluorescent protein (drFP583/DsRed/RFP). Biotechniques. 33:592–602. [DOI] [PubMed] [Google Scholar]
- Labas, Y. A., N. G. Gurskaya, Y. G. Yanushevich, A. F. Fradkov, K. A. Lukyanov, S. A. Lukyanov, and M. V. Matz. 2002. Diversity and evolution of the green fluorescent protein family. Proc. Natl. Acad. Sci. USA. 99:4256–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy, 2nd. ed. Kluwer Academic/Plenum Publishers, New York. 367–394.
- Lauf, U., P. Lopez, and M. M. Falk. 2001. Expression of fluorescently tagged connexins: a novel approach to rescue function of oligomeric DsRed-tagged proteins. FEBS Lett. 498:11–15. [DOI] [PubMed] [Google Scholar]
- Martinez-Salas, E. 1999. Internal ribosome entry site biology and its use in expression vectors. Curr. Opin. Biotechnol. 10:458–464. [DOI] [PubMed] [Google Scholar]
- Matz, M. V., A. F. Fradkov, Y. A. Labas, A. P. Savitsky, A. G. Zaraisky, M. L. Markelov, and S. A. Lukyanov. 1999. Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotechnol. 17:969–973. [DOI] [PubMed] [Google Scholar]
- Miyawaki, A., J. Llopis, R. Heim, J. M. McCaffery, J. A. Adams, M. Ikura, and R. Y. Tsien. 1997. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 388:882–887. [DOI] [PubMed] [Google Scholar]
- Miyawaki, A., and R. Y. Tsien. 2000. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 327:472–500. [DOI] [PubMed] [Google Scholar]
- Mizuno, H., A. Sawano, P. Eli, H. Hama, and A. Miyawaki. 2001. Red fluorescent protein from Discosoma as a fusion tag and a partner for fluorescence resonance energy transfer. Biochemistry. 40:2502–2510. [DOI] [PubMed] [Google Scholar]
- Mochizuki, N., S. Yamashita, K. Kurokawa, Y. Ohba, T. Nagai, A. Miyawaki, and M. Matsuda. 2001. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 411:1065–1068. [DOI] [PubMed] [Google Scholar]
- Patterson, G., R. N. Day, and D. Piston. 2001. Fluorescent protein spectra. J. Cell Sci. 114:837–838. [DOI] [PubMed] [Google Scholar]
- Patterson, G. H., D. W. Piston, and B. G. Barisas. 2000. Förster distances between green fluorescent protein pairs. Anal. Biochem. 284:438–440. [DOI] [PubMed] [Google Scholar]
- Pentcheva, T., and M. Edidin. 2001. Clustering of peptide-loaded MHC class I molecules for endoplasmic reticulum export imaged by fluorescence resonance energy transfer. J. Immunol. 166:6625–6632. [DOI] [PubMed] [Google Scholar]
- Phillips, G. N. 1998. The three-dimensional structure of green fluorescent protein and its implications for function and design. In: Green Fluorescent Protein: Properties, Applications, and Protocols. M. Chalfie and S. Kain, editors. New York: Wiley-Liss. p 77–96 .
- Rizzo, M. A., M. A. Magnuson, P. F. Drain, and D. W. Piston. 2002. A functional link between glucokinase binding to insulin granules and conformational alterations in response to glucose and insulin. J. Biol. Chem. 277:34168–34175. [DOI] [PubMed] [Google Scholar]
- Selvin, P. R. 1995. Fluorescence resonance energy transfer. Methods Enzymol. 246:300–334. [DOI] [PubMed] [Google Scholar]
- Siegel, R. M., J. K. Frederiksen, D. A. Zacharias, F. K. Chan, M. Johnson, D. Lynch, R. Y. Tsien, and M. J. Lenardo. 2000. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 288:2354–2357. [DOI] [PubMed] [Google Scholar]
- Stryer, L., and R. P. Haugland. 1967. Energy transfer: a spectroscopic ruler. Proc. Natl. Acad. Sci. USA. 58:719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terskikh, A. V., A. F. Fradkov, A. G. Zaraisky, A. V. Kajava, and B. Angres. 2002. Analysis of DsRed mutants. Space around the fluorophore accelerates fluorescence development. J. Biol. Chem. 277:7633–7636. [DOI] [PubMed] [Google Scholar]
- Vanderklish, P. W., L. A. Krushel, B. H. Holst, J. A. Gally, K. L. Crossin, and G. M. Edelman. 2000. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 97:2253–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhusha, V. V., H. Otsuna, T. Awasaki, H. Oda, S. Tsukita, and K. Ito. 2001. An enhanced mutant of red fluorescent protein DsRed for double labeling and developmental timer of neural fiber bundle formation. J. Biol. Chem. 276:29621–29624. [DOI] [PubMed] [Google Scholar]
- Wiehler, J., J. von Hummel, and B. Steipe. 2001. Mutants of Discosoma red fluorescent protein with a GFP-like chromophore. FEBS Lett. 487:384–389. [DOI] [PubMed] [Google Scholar]
- Wu, P., and L. Brand. 1994. Resonance energy transfer: methods and applications. Anal. Biochem. 218:1–13. [DOI] [PubMed] [Google Scholar]
- Yanushevich, Y. G., D. B. Staroverov, A. P. Savitsky, A. F. Fradkov, N. G. Gurskaya, M. E. Bulina, K. A. Lukyanov, and S. A. Lukyanov. 2002. A strategy for the generation of non-aggregating mutants of Anthozoa fluorescent proteins. FEBS Lett. 511:11–14. [DOI] [PubMed] [Google Scholar]
- Yarbrough, D., R. M. Wachter, K. Kallio, M. V. Matz, and S. J. Remington. 2001. Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0-Å resolution. Proc. Natl. Acad. Sci. USA. 98:462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias, D. A., J. D. Violin, A. C. Newton, and R. Y. Tsien. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 296:913–916. [DOI] [PubMed] [Google Scholar]