

Abstract
The strongly anion-selective porin channel Omp32 from the bacterium Delftia acidovorans differs from other unspecific porins by its pronounced selectivity for anions and its particularly small channel cross-section. Multinanosecond molecular dynamics simulations of chloride ion movement in this pore protein suggest that translocated anions interact intimately with the charges of a “basic ladder”, whose dynamics lead the anions in a stepwise manner through the constriction zone of the channel. The ladder-steps comprise the central clustered arginine groups and flanking basic residues at its exoplasmic and periplasmic sides. The computed free energy profile of ion movement in and around the constriction zone shows a corresponding succession of free energy minima and barriers. A number of polar atoms from other amino acids contribute to the coordination of Cl− at certain sites and to its temporary immobilization in the channel. A special binding site occurs at the transition of the constriction zone to the periplasmic funnel, binding the chloride ion over significant lengths of time. The results from our MD study offer a possible explanation for the nonlinear conductance properties and unusual salt-dependent characteristics of Omp32 observed earlier in experimental measurements.
INTRODUCTION
Porins are channel proteins in the outer membrane of Gram-negative bacteria, mitochondria, and chloroplasts that enable the passage of hydrophilic solutes, water, and ions across the hydrophobic core of the membrane. Bacterial outer membrane porins are classified into channels specific for the uptake of nutrients, such as maltose or sucrose, and unspecific pore proteins (Schirmer, 1998; Koebnik et al., 2000). Unspecific porins have repeatedly been described as passive, water-filled channels facilitating the diffusion of ions and polar molecules up to the size of trimeric oligosaccharides (Jap and Walian, 1996; Schulz, 1996; Robertson and Tieleman, 2002).
Omp32 is the major outer membrane porin of the bacterium Delftia acidovorans (formerly Comamonas acidovorans) that preferentially grows on organic acids (Willems et al., 1992). It occurs as a heterohexamer, consisting of the homotrimeric porin proper and an intimately associated homotrimeric polypeptide whose atomic structure is as yet unknown (Zeth et al., 2000). Similarly to other porins, it forms a 16-stranded β-barrel, while two loops fold into the channel and, together with a protrusion from β-strand 2, shape the interior of the channel. The infolding loops decrease the channel's cross-sectional area to <25 Å2 toward the membrane center in the crystal structure, the smallest constriction found among all structurally known porins (Zeth et al., 2000). Like in other porins, this narrowest region contains a cluster of three closely adjacent arginine groups. The cluster was calculated by continuum electrostatics to carry three positive charges at physiological pH, despite the mutual proximity of the arginine side-chains, due to stabilization by interaction with buried glutamate groups (Zachariae et al., 2002). Other unspecific porins, such as the moderately cation-selective porin OmpF or the weakly anion-selective PhoE from Escherichia coli, possess a cluster of acidic groups at the opposite side of the constriction zone (Cowan et al., 1992), which is not present in Omp32. This gives Omp32 a unique positive potential in the pore and selectivity for anions (Zachariae et al., 2002). Electrophysiological experiments performed on Omp32 embedded in planar lipid bilayers yielded unusually asymmetric current-voltage curves and conductance characteristics that differ considerably from other porins (Mathes and Engelhardt, 1998).
To shed light on the structure-function relationship of protein channels, a wide range of computational techniques have been applied (Tieleman et al., 2001; Karplus and McCammon, 2002). A number of computational studies on ion passage through a rigid protein channel of wild-type and mutant OmpF filled with a continuum water model have been carried out using diffusional Brownian dynamics (BD) simulations (Schirmer and Phale, 1999; Im et al., 2000; Phale et al., 2001). In these simulations, good agreement between measured and calculated selectivity ratios was achieved. Recently, a comparison of results from continuum BD and three-dimensional Poisson-Nernst-Planck simulations and explicit solvent and membrane molecular dynamics (MD) simulations on OmpF was published (Im and Roux, 2002a). Trajectories from MD simulations suggested the formation of ion pairs in the constriction zone of OmpF to be crucial for the transduction of chloride ions in a 1 M KCl solution (Im and Roux, 2002b). In the same study it was observed that K+ and Cl− ions follow two well-separated pathways through the pore on average, which was also found in BD studies (Schirmer and Phale, 1999).
The considerable difference in the structural and electrostatic properties of Omp32 as compared to OmpF gives rise to the expectation that the properties of ion passage through the Omp32 channel also deviate from those found in OmpF. The high selectivity for anions in Omp32 and the fact that at comparably low ionic strength, reflecting physiological conditions predominantly anions enter the pore, led us to perform MD simulations with chloride ions moving in the highly charged pore of Omp32. Our work focuses on the mechanism of anion translocation in the particularly narrow constriction region of Omp32, whose low diameter and high charge density dominates the characteristics of ion-protein interactions. To our best knowledge, this study represents the first MD simulation of an anion selective porin. By choosing an appropriate simulation system of an Omp32 monomer we were able to carry out multiple multinanosecond simulations under varying conditions and potential of mean force (PMF) calculations. The studies consistently revealed that anion translocation follows a multistep mechanism hitherto unknown in porins.
METHODS
The crystal structure of a monomer of Omp32 was used in our simulations (Zeth et al., 2000; entry 1E54 in the Protein Data Bank). The constriction zone, and the periplasmic and exoplasmic channel vestibules were filled with TIP3P water molecules (Jorgensen et al., 1983). The hydrophilic periplasmic and exoplasmic exterior walls were surrounded by two slabs of TIP3P water molecules with a thickness of 35 Å and 25 Å at the exoplasmic and periplasmic side, respectively, representing the thicker polar region of the lipopolysaccharide headgroups of the exoplasmic lipid layer. To mimic the low-permittivity internal region of the membrane, this space was left free from water molecules (20 Å). A cylinder of 15 Å radius around the central axis of the pore with spherically-shaped caps which included the entire internal structure of the channel was simulated to move freely, while the major part of the rigid β-barrel scaffold was fixed. The simulation system comprised a total of 3739–3740 free atoms (2039 protein atoms, 1699 water atoms, and 1–2 chloride ions) and 21,136 fixed atoms. The ionization state of the protein calculated in Zachariae et al. (2002) was used except for Glu 158, which was assumed to be charged due to interaction with a calcium ion in the hexamer.
In explicit-membrane MD simulations of porins, the outer β-barrel architecture was observed to exhibit only low flexibility as compared to the internal loops (Tieleman and Berendsen, 1998; Im and Roux, 2002b). Therefore we chose this simplified model of the channel, focusing on the dynamics of the internal pore matrix, which we considered to constitute the most important degrees of freedom with regards to the movement of ions in the channel. By this restriction of the system we were able to carry out multiple multinanosecond simulations. This was necessary inasmuch as our experiments showed that the translocation of anions through Omp32 occurs on a long timescale of up to 32 ns. Thereby, it was also possible to perform simulations of ionic motion under varying conditions such as the application of different external electric fields, different starting points for the ions or the effects of additional anions in the channel, and the calculation of a free energy profile for anions around the constriction zone of Omp32.
After 30 ps of preequilibration, chloride ions were placed at different positions near the extracellular or periplasmic entrance regions of Omp32, at z-coordinates near 16 and −16 Å, respectively (a z-coordinate of 0 Å was assigned to the point with the most constricted pore cross-section in the crystal structure). A number of simulations, especially those incorporating a second ion, were started from states in which an anion was bound to certain basic residues near the channel constriction zone.
After 100 steps of steepest descent and 200 steps of ABNR minimization, the simulation runs were started with an initial temperature of 250 K and continuously heated up to 310 K (physiological temperature; D. acidovorans is a facultative pathogen). The simulations then continued at a temperature of 310 K, which was kept constant using the temperature coupling algorithm of Berendsen et al. (1984) with a coupling decay time of 15 ps. The initial phase of equilibration therefore covers the first 50 ps of each simulation. Simulated times varied between 2 and 18 ns. The combined length of our simulations amounts to ∼50 ns.
Simulations were performed using CHARMM version 28 (Brooks et al., 1983), together with the PARAM22 all-hydrogen force field (MacKerrell et al., 1998). A cutoff of 14 Å was used for nonbonded interactions. Bond lengths were kept constant by the SHAKE algorithm (Ryckaert et al., 1977), and the integration time step was 2 fs. Calculations were run on a Beowulf Linux cluster using parallel code on eight CPUs.
The umbrella sampling PMF simulations (Torrie and Valleau, 1977) were carried out using CHARMM's umbrella sampling module. A total of 41 independent simulations of 110 ps length each were started from corresponding equilibrated geometries taken from a trajectory of anion translocation through the constriction of Omp32 obtained from an unbiased production run. The equilibration time in the new potential was 10 ps. The umbrella potential constrained the motion of the anion to planes parallel to the system's x- and y-axes with a force constant of 10 kcal mol−1 Å−2. In the z-direction, the center of the biasing potential was shifted by 0.5 Å for each window. Due to large overlap (each window covered ∼1.2 Å of sampling in z-direction), combination of the PMF pieces was straightforward. To ensure adequate sampling, the PMF calculations were restricted to ∼20 Å around the constriction zone, where a sufficiently narrow cross-section area and an unambiguously defined anion translocation pathway is given. The ion distribution was determined using bins 0.05 Å wide.
Scanning force simulations (Grubmüller et al., 1996) were performed using a spring constant of 4.2 N/m for the spring between the tip and the chloride ion. The virtual tip to which the substrate was attached was pulled with a constant velocity of 0.00625 Å/ps along 25 Å of the pore of Omp32.
For the calculation of the average solvent accessible cross-section area of the channel, 110 structural frames from one simulation run (one frame each 20 ps) were used together with the program CROSS (A. Philippsen, Biozentrum Basel) and a solvent probe radius of 1.5 Å.
RESULTS
Fig. 1 illustrates that the arginine cluster in Omp32 is oriented at an angle to the perpendicular cross-section of the pore, unlike the basic cluster in OmpF. This leads to an increased longitudinal character with respect to the channel axis. According to this, as well as the fact that the electrostatic potential is positive throughout the entire pore of Omp32 (Zachariae et al., 2002), the mechanism of ion translocation was expected to differ from that of OmpF. In the following, we looked at the mechanism of anion translocation in Omp32 pores occupied by one or two Cl− ions. The crystal structure of Omp32 exhibits a single anion bound close to the constriction zone (Zeth et al., 2000). Therefore, a computational model with a single anion in the inner channel of Omp32 lent itself as an appropriate starting point for MD studies. From Poisson-Boltzmann calculations, the partition coefficients for chloride and potassium ions into the channel were estimated according to Dieckmann et al. (1999) to be ∼4 and 2 × 10−2, respectively. Thus, a channel system containing one or two chloride ions and no cations approximately corresponds to a bulk ionic concentration of 25–50 mM.
FIGURE 1.

Orientation and location of the arginine cluster in the constriction zones of the strongly anion-selective porin Omp32 (A), and the moderately cation-selective porin OmpF (B). The figure was produced using DINO (A. Philippsen, DINO: visualizing structural biology, http://www.dino3d.org).
Extracellular vestibule
A total of 10 simulations were started from different chloride positions in the extracellular entrance region of Omp32, which showed a distinct pathway leading the Cl− ions toward the constriction zone. In each case, the pathway involved coordination by the amide nitrogen atoms of residues Asn-130 and Gln-306 in the extracellular vestibule. In our simulations, the Cl− ion was not observed to be attracted by the positive charges of Arg-119, Arg-26, Arg-92, or Lys-74, that are also located in the external vestibule. Binding to one of these residues would have prevented fast transfer toward the constriction zone, which was ensured by the weaker temporary interaction with the polar groups. The first binding event usually occurred with the side chain of Lys-308 after only ∼100–500 ps (please note that the Nζ atom of Lys-308 has a z-coordinate of ∼5.2 Å in the crystal structure; see Figs. 2 A and 3 A). The anion remains bound to Lys-308 for ∼0.2–0.7 ns, while the flexibility and thermal motion of the long side chain ensures that the bound anion can move within a region defined by z-coordinates of ∼3–7 Å (Fig. 2, A–B, and E–F). Owing to this mobility, the Cl− ion can temporarily approach Arg-133, the first basic residue of the arginine cluster in the constriction zone. In general, one of these approaches leads to a transition from a bound state at Lys-308 to a bound state at Arg-133. This crossover reaction has a perpendicular component with respect to the channel axis and can be looked at as the initial step of ion translocation.
FIGURE 2.

Chloride trajectories in the channel of Omp32. (A, B) Immobilization of the anion at specific binding sites at basic residues in the channel (“basic ladder”) from two different MD simulations. (C) Longer timescale of the trajectory from B, showing the anion to switch between Arg-38 and Arg-75 in the most preferred binding region of the pore. (D) Translocation of the chloride ion from Arg-38 toward Arg-89. (E) Chloride ion trajectory of a simulation with an applied electric field of 100 mV pulling the ion toward the periplasmic exit. (F) Trajectory of the Cl− ion from a simulation yielding fast transfer from the exoplasmic to the periplasmic side.
FIGURE 3.
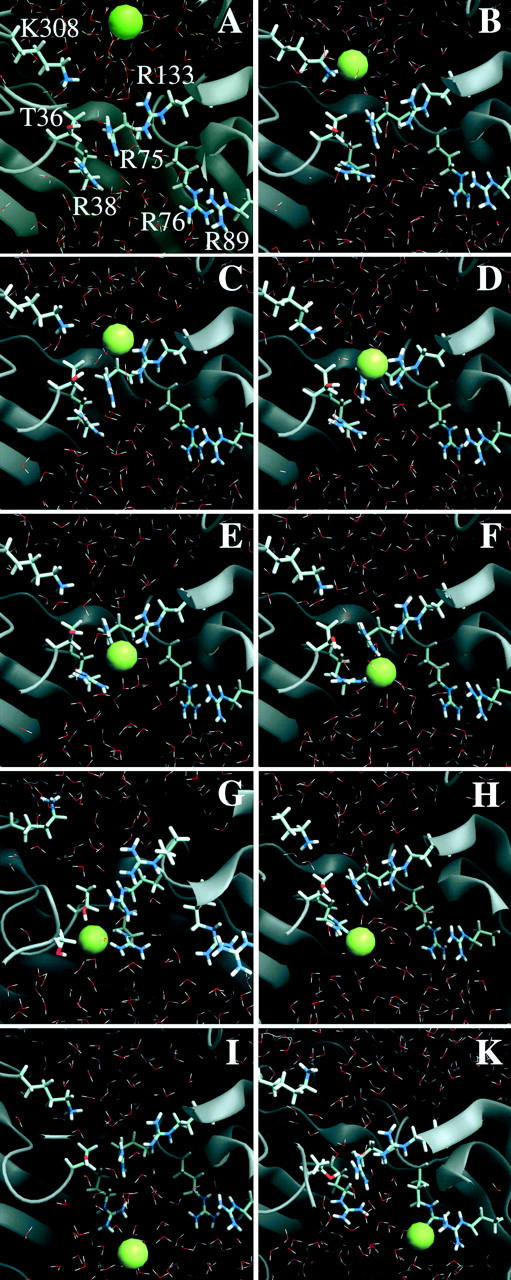
Views of representative snapshots from a trajectory of chloride translocation along the “basic ladder”. (A) Chloride ion approaching Lys-308, (B) chloride ion binding to Lys-308, (C) transfer to Arg-133, (D) joint binding by Arg-75 and Arg-133, (E) binding by Arg-75, and (F) joint binding by Arg-38 and Arg-75. (G) Coordination of the anion at the most preferred binding region involving atoms from Arg-38, Arg-75, Thr-36, and Ser-108. (H, I) Dynamics of the Arg-38 side chain, taking the anion further into the periplasmic funnel; and (K) binding site at Arg-89. The figure was produced using DINO (A. Philippsen, DINO: visualizing structural biology, http://www.dino3d.org).
Constriction zone
Fig. 3 depicts representative snapshots from a characteristic trajectory of ion motion near the Omp32 constriction zone. Starting at the first state bound to the constriction zone residue Arg-133, there were a number of possible distinct binding sites at the arginine cluster. We refer to these multiple binding sites including Lys-308 and two further arginine groups that are not part of the cluster (see below) as basic ladder. The sites at the arginine cluster usually involve anion binding to the terminal guanidinium nitrogen atoms of one or two of the clustered arginine side chains. Preferred binding sites were at Arg-133 (Fig. 3 C), coordinated jointly by Arg-133 and Arg-75 (Fig. 3 D), bound to Arg-75 (Fig. 3 E) including interaction with the neighboring groups, and bound to Arg-38 (Fig. 3, G–H). As can be seen from Fig. 2, A–F, and Fig. 3, these states are clearly distinguishable from each other, and the lifetime of bound states at a specific site easily extends to nanoseconds. Release from one site invariably involves transfer and rebinding to a neighboring site. The specific lifetime of a certain state appears to be governed by random thermal fluctuations, and deviations from the average lifetime are large (from 30 ps to more than 2 ns). A clear tendency toward binding at residues Arg-75 and Arg-38 can be observed; however, transitions from Arg-133 to Lys-308, or from Arg-75/Arg-133 to Arg-133 were also possible. In any case, the sites of immobilization were the same irrespective of the direction of ion translocation.
The succession of closely adjacent basic residues reaching through the narrowest part of the channel apparently functions like the steps of a “basic ladder” for the translocation of the chloride ion through the constriction zone. As Fig. 3, E–F, illustrate, the transitions are facilitated by the dynamics of the groups that bring side chains closer together, especially when the anion is situated between them. The translocation mechanism thus appears to function by an intricate interplay of side-chain dynamics and ionic movement. As described for MD studies of other porins (Tieleman and Berendsen, 1998; Im and Roux, 2002b), the average minimum cross-section area in the pore is reduced in our simulations to 12 ± 3 Å2 (as compared to a value of 25 Å2 in the crystal structure). The narrowest point has an average z-coordinate of −0.7 Å in the simulations. Owing to the pronouncedly high density of positive charges in and near the constriction zone of Omp32, the anion shows a strong tendency to remain in that area for an extensive range of time, as depicted in Fig. 2 C from our longest simulation. The Cl− anion switched between Arg-75 and Arg-38 for nearly 18 ns without ever leaving this area. The immobilization of the chloride anion near Arg-75 and Arg-38—a region which has also been calculated to be the center of the electrostatic potential well in continuum electrostatics calculations (Zachariae et al., 2002) – is further enhanced by coordination with the side-chain oxygen (Fig. 3 G, and Fig. 4) and the backbone nitrogen atom from Thr-36 (Fig. 4), and, less importantly, with the hydroxyl group from Ser-108. The chloride ion has a higher degree of flexibility within the x-y plane at the same z-position here. Concomitantly, the pore widens considerably near Arg-38 (solvent-accessible area of ∼80–150 Å2), such that the number of water molecules solvating the chloride ion increases here from its minimum value found between Arg-133 and Arg-75. Fig. 5 therefore shows a clear peak in the anion coordination number mainly arising from the high number of protein contacts, whereas the total coordination number remains nearly equal throughout all other parts of the inner channel. The first solvation shell of the chloride anion is halved from six water molecules to a minimum of three water molecules in the constriction zone, replacing water coordination with contacts to nitrogen and oxygen atoms from the protein matrix. A coordination number of 6–7 is in accord with experimental and calculated values for Cl− in aqueous solutions (Ohtaki and Radnai, 1993). In the vestibule regions, on average, only one water molecule is replaced by coordination with Asn-130 and Gln-306 (extracellular side) and Ser-108 (periplasmic side).
FIGURE 4.

Most preferred chloride ion binding site of Omp32 near z = −6 Å. The distances from Cl− to its protein contacts are: 3.1 Å (Arg-38 Nη2), 3.1 Å (Thr-36 Oγ), 3.1 Å (Thr-36 N), 4.2 Å (Ser-108 Oγ), and 4.3 Å (Arg-75 Nη1). The figure was produced using DINO (A. Philippsen, DINO: visualizing structural biology, http://www.dino3d.org).
FIGURE 5.

Coordination number of the Cl− ion in the inner channel of Omp32. The summation curve shows a clear peak in the binding region.
An observation which was repeatedly made in our simulations is that a continuous water chain through the pore is not constantly present in the narrow constriction zone of Omp32. Even when a Cl− ion is not bound in that area, the water chain is frequently disrupted for significant lengths of time.
Translocation out of the constriction zone
Leaving the constriction zone is an energetically unfavorable process for anions due to the strong positive potential created by the clustered arginine charges. Nevertheless, our simulations suggest two possible pathways of disconnecting and moving into the periplasmic entrance funnel. First, transitions from Arg-38 to Arg-89, one of a cluster of two arginine residues, displaced from Arg-38 by ∼11 Å in the crystal structure, were observed to take place spontaneously after detaching from the most preferred binding site (Figs. 2 D, and 3 K). This transition is facilitated by the flexibility of the side chain of Arg-38, which is greatly enhanced compared to the other clustered arginine groups since it is located where the constriction zone ends, and the channel cross-section abruptly increases forming the periplasmic vestibule region. Second, the increased dynamics of Arg-38 can also act to carry the anion further toward the periplasmic rim of the porin (Fig. 3 I). Subsequently, it was observed to temporarily diffuse away from Arg-38 into the periplasmic funnel.
In one of our simulations, a relatively fast transfer from the exoplasmic side through the constriction zone into the periplasmic vestibule was observed. Due to lower lifetimes especially of the bound states at Arg-75 and Arg-38, the ion translocation through the constriction zone occurred in little more than 3 ns (Fig. 2 F). Although it is less clearly evident in the earlier stages of the trajectory, the pattern of close binding and detaching from the specific basic and polar groups of the pathway was unchanged.
Throughout all of our simulations, the binding distances between chloride and its bonding partner base were in the range of 3.0–3.7 Å, with an average value of 3.2 Å.
Periplasmic vestibule
Simulations that were started from periplasmic positions of a Cl− ion were also performed. Its motion in the periplasmic funnel was, similar to the exoplasmic vestibule region, dominated by contacts with uncharged groups. In this case, the hydroxyl groups of the side chains of Ser-103 and Ser-108 provided the coordination contacts for the Cl− ion. The Cl− anion was transferred from Ser-103 first to Ser-108, from where it eventually reached Arg-38. The pathway was, however, less definite than along the constriction zone “basic ladder”.
PMF profile of ion translocation
The PMF profile of the ionic pathway along the “basic ladder” of Omp32 is shown in Fig. 6. Immobilization of the chloride ion at specific sites along the z-axis is corroborated by the free energy curve, exhibiting a succession of free-energy minima whose positions are in very good agreement with the binding sites observed in the simulations (Fig. 2). The energy wells are separated by moderate to low direction-dependent free-energy barriers, ranging from ∼5 to 1 kcal/mol. Local energy wells can be associated with ion binding to Lys-308 (z = 6–7 Å), Arg-133 (z = 3.5 Å), and one to two positions jointly coordinated by Arg-133 and Arg-75 (z = 0 Å, z = −2.5 Å). The observation of two preferred binding sites near the periplasmic vestibule region is reflected by the presence of two nearly equivalent global energy wells in the PMF profile near z = −6 Å and z = −3.5 Å. They are separated by an energy barrier of ∼3 kcal/mol. Inward ion translocation through the narrowest part of the pore, from z = 0 Å to z = −3.5 Å, involves an energy barrier of slightly less than 2 kcal/mol, whereas movement in the opposite direction is obstructed by a significantly larger activation free energy.
FIGURE 6.

Potential of mean force profile of chloride ion translocation through the constriction of Omp32.
A large energy barrier corresponds to ion transfer between Arg-133 and the first joint coordination site by Arg-75 and Arg-133. This reaction involves movement of the Arg-75 side chain in the especially narrow environment. The immobilization site at z = −8.5 (the outer face of Arg-38) is only slightly stabilized against ion transition to the neighboring global minimum by a low activation barrier of ∼1 kcal/mol. Depopulation of this state toward the periplasm can also occur via the perpendicular crossover reaction from Arg-38 to Arg-89 which is not covered in this PMF study. Overall, the free energy difference between the first bound state at Lys-308 and the global free energy minimum at Arg-38 amounts to ∼6.5 kcal/mol. The tendency toward binding at Arg-38 at the transition to the periplasmic vestibule can be explained in terms of the PMF shape, since the twofold global minimum of the PMF is significantly shifted toward the periplasmic funnel (cross-section minimum at −0.7 Å in the simulations).
Scanning force simulations, in which a chloride ion was pulled through the constriction zone of Omp32, also support the view that multiple binding sites exist in this porin in showing a succession of positive and negative force regions. The force needed to pull the chloride ion out of the most favored binding region was calculated by this method to be ∼325 pN (data not shown).
Multiple ion simulations and electric fields
To test the influence of a second Cl− ion in the channel of Omp32, calculations were performed in which a further ion was introduced into the channel at the same starting position, whereas the first anion was bound in the preferred binding region of the constriction zone. In accord with the single-ion simulations, the further ion moved quickly toward the central area of the pore. However, instead of pushing the first ion out of the constriction zone (Suenaga et al., 1998), the second anion remained associated with the polar groups forming the ionic pathway of the external vestibule (Asn-130 and Gln-306) for a significantly longer period of time. The time spent by the Cl− ion bound to Lys-308 is also considerably extended. After switching over to Arg-133, the Cl− ion was subsequently observed to become repelled again from that location and to move back up the funnel by ∼10 Å. Therefore, our results suggest that the entry of the second ion into the constriction zone is decelerated, if not inhibited, whereas the first ion is bound in that area. The first anion apparently needs to detach from the constriction zone by one of the presented mechanisms first to allow entry of the second ion. This behavior can be ascribed to the reduced flexibility of the Arg-75 and Arg-133 side chains with respect to motion toward Lys-308. While they are attracted by the first ion, situated in the constriction zone, their dynamics is more restricted.
To investigate the effects of the application of an external electric field of a similar strength that is used in planar membrane experiments, we repeated our calculations with electric fields of 0.6–2 × 107 V/m, corresponding to a transmembrane potential of ∼30–100 mV. We tested both transmembrane polarizations. As depicted in Fig. 2 E, which is taken from a simulation with a field attracting anions to the periplasmic side of the membrane, the pattern of ionic movements in the channel is unchanged. The pathway of ion translocation remains unaltered, and the lifetime of the respective bound states up to the preferred binding site is not systematically reduced in our simulations.
DISCUSSION
The simulations presented in this work are, to our best knowledge, the first molecular dynamics studies of an anion-selective channel protein. As opposed to the only porin examined by means of MD simulations, OmpF (Im and Roux, 2002b; Tieleman and Berendsen, 1998; Suenaga et al., 1998), the constriction zone of Omp32 contains only basic groups and does not exhibit an electrostatic cross-field. This, together with additional basic groups in the funnel regions, creates a very positive potential in the pore of Omp32 (Zachariae et al., 2001). The clustered arginines of Omp32 are aligned in a direction with a more longitudinal character with respect to the z-axis of the pore than those of OmpF, giving rise to a different role in the ion translocation mechanism, and creating the central part of a “basic ladder”. Our calculations, performed with one and two anions in the channel, should reflect a rather characteristic situation in Omp32, corresponding to a low bulk concentration of ∼50–25 mM. This concentration likely represents physiological conditions, because D. acidovorans is a soil bacterium and is known as an opportunistic pathogen (Bofill et al., 1996). Our data do not give information on the translocation of cations yet. However, it is unlikely that cations add significantly to the translocation of anions by, e.g., screening charges, because negatively charged amino-acid residues are not present in the channel. Nevertheless, one could imagine that the principle of ion pairing described for OmpF (Im and Roux, 2002b) may also play a role for cation translocation in the case of Omp32.
It is useful to consider some of the limitations of our study, the most serious being the restricted system used in the simulations and the use of a cutoff for the long-range electrostatic interactions due to this. However, the entire Omp32 system does not only consist of the porin trimer but also of a trimeric polypeptide, whose atomic structure is not yet solved. Both together form a stable heterohexameric complex in the outer membrane (Zeth et al., 2000). When the structure of this functionally unknown polypeptide is solved, further calculations should help to give a more complete look at the properties and energetics of ion conductance. It is quite unlikely to observe structural transitions of the protein caused by using a cutoff radius because the outer region of the system is kept rigid. With such a system setup, neglecting long-range electrostatic interactions should, if at all, only affect the relative energetics of the chloride ion felt inside the pore (see Fig. 6).
Single ion BD simulations, performed on wild-type and mutant porin OmpF, were able to reproduce experimentally obtained selectivities to a very large extent (Schirmer and Phale, 1999). However, the identical computational protocol appeared to fail on the especially narrow, highly and uniformly charged Omp32, yielding only very few productive ion translocations that did not reproduce the experimental values (unpublished results). We ascribe the failure of simple BD calculations on Omp32 to the special features of ion translocation in this channel involving the dynamics of particular amino-acid side chains and the fact that the minimum solvent-accessible area is as small as 12 Å2 on average in the pore.
According to our MD simulations, Omp32 is not a simple diffusion channel, but interacts strongly with anions such that ion conduction is an activated process. As judged from our MD and free energy simulations, there are several binding sites for chloride ions at basic residues in and near the constriction zone, and ion translocation is accomplished by a unique mechanism of “ion handover” between these sites. The transfer steps involve energy barriers of ∼1–5 kcal/mol. Bernèche and Roux (2001) calculated somewhat lower energy barriers in the range of 2–3 kcal/mol for the concerted mechanism of K+ ion conduction in an umbrella sampling study of the KcsA channel, for which multiple cation binding sites were observed (Morais-Cabral et al., 2001).
The PMF profile is markedly asymmetrical with respect to the pore center, and the barrier heights vary significantly with the direction of ion translocation. The PMF drops to a twofold minimum in the inner pore of Omp32, shifted toward the periplasmic vestibule, exactly where the ion was located in the x-ray structure (Zeth et al., 2000). We suggest that this might account for the unusual asymmetry in experimental current-voltage curves of Omp32 (Mathes and Engelhardt, 1998). The asymmetry in conductance (i.e., the ratio of conductance values at −50 mV and 50 mV) is illustrated by a factor of 1.45 for 3 mM KCl and 1.33 for 10 mM (Mathes and Engelhardt, 1998), which indicates a significant effect. The observation that the translocation of anions may take 18 ns or longer according to our simulations is also in agreement with experimental data. The conductance of Omp32 at 100 mM KCl and a voltage of 100 mV is ∼180 ± 30 pS for one trimer (Mathes and Engelhardt, 1998), corresponding to an apparent transition time of 23–32 ns per ion.
At the main binding site in Omp32, the chloride ion not only interacts with two charged arginine groups, but also with 1–2 oxygen atoms from Thr-36 and Ser-108 as well as the backbone nitrogen atom from Thr-36. In that, it shows a feature already described for the chloride binding site of the ClC chloride channel, where the oxygen atoms of Tyr-445 and Ser-107 and two main-chain nitrogen atoms from Ile-356 and Phe-357 are coordinating the chloride ion (Dutzler et al., 2002). In the ClC channel, however, only polar groups interact with the chloride ion.
A “push out” mechanism similar to that described for OmpF by Suenaga et al. (1998) was not observed in our simulations. In fact, entry of a further anion was impeded by an anion present in the constriction zone, which reduced the flexibility of side chains. The strong binding of single anions, which is also apparent from the PMF profile, could provide a way to explain the saturation effects observed in electrophysiological measurements on Omp32 (Fig. 7). The curve shows a clear difference in the conductivity-concentration dependence of Omp32 embedded in planar lipid bilayers and measured in KCl solutions as compared to OmpF (Benz et al., 1978) or the porin from Rhodobacter capsulatus (Przybylski et al., 1996) and other porins not displayed here. The relative conductance of Omp32 first decreases rapidly with an increase in ionic strength, approaching a saturation level, whereas it remains nearly constant for OmpF and the R. capsulatus porin. In highly concentrated KCl solutions, Omp32 exhibits a distinctly lower conductivity than the other two porin channels.
FIGURE 7.

Salt concentration-dependent conductance of Omp32. The pore activity is expressed as a relative value calculated from the porin conductance (nS) at a certain KCl concentration over the respective specific conductance (nS/nm) of the salt solution. The ratio can be taken as a measure for the length of a virtual conductor of a certain resistance and cross-sectional area, indicating that the Omp32 channel is particularly well-conducting at low KCl concentrations. For comparison, the relative conductance values of the porins OmpF and from Rhodobacter capsulatus are displayed. (Data taken from references Benz et al., 1978; Przybylski et al., 1996; Mathes and Engelhardt, 1998; and Schirmer and Phale, 1999.)
The results from the simulations suggest that the travel time of anions through Omp32 is only moderately affected by increasing the concentration of ions, and that the average time of ion passage is mainly governed by the energy barriers of the ion translocation process. This feature would reveal itself by a decrease in relative conductance, as was observed in the experiment. The fact that Omp32 shows a larger conductivity than other porins in low concentrations of KCl may be ascribed to the high number of positive charges in the pore. By the strongly positive potential, ions are more readily attracted toward the channel, especially in lower-concentrated ionic solutions, than toward a more dipolar pore.
Conductivity measurements of the moderately cation selective OmpF in NaCl solutions, however, appear to show a behavior similar to Omp32 (Schirmer and Phale, 1999), which could be an effect of the smaller ion radius and the stronger interactions of Na+ with charged residues (Fig. 7). To this, it should be noted that Suenaga et al. (1998) also described the temporary binding of single Na+ ions to an acidic residue (Asp-113) in the constriction of OmpF. Ion immobilization (if not “ion handover”) may, thus, also be of relevance for the translocation of the preferred ion type in other porins.
We did not find significant effects upon application of external electric fields in the range of 30–100 mV in our simulations. Since the Donnan potential across the outer membrane is not expected to be larger (Sen et al., 1988), this suggests that a field originating from asymmetric charge distribution in lipid headgroups on both membrane sides would not affect the mechanism of ion translocation substantially.
Another interesting aspect of the porin Omp32 is its putative function as an uptake channel for organic acids, the preferred C-source of D. acidovorans (Willems et al., 1992). Although binding of the carboxyl groups to the clustered arginine charges is easily imaginable, the question arises how the hydrocarbon portion of the molecules may affect their transport. Interestingly, the residues Leu-94, Ala-109, and Ile-132 are located in the constriction zone such that they may create a corresponding hydrophobic environment for this part of the substrate. Computational studies involving physiological substrates to test this assumption are currently under way.
Acknowledgments
We thank Tilman Schirmer for his generous help with Brownian dynamics calculations.
This work was supported by a grant of the Deutsche Forschungsgemeinschaft (DFG EN144/3-1).
References
- Benz, R., K. Janko, W. Boos, and P. Läuger. 1978. Formation of large, ion-permeable membrane channels by the matrix protein (porin) of Escherichia coli. Biochim. Biophys. Acta. 511:305–319. [DOI] [PubMed] [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- Bernèche, S., and B. Roux. 2001. Energetics of ion conduction through the K+ channel. Nature. 414:73–77. [DOI] [PubMed] [Google Scholar]
- Bofill, L., M. Wessolossky, E. Vicent, M. Salas, J. Besso, A. Merentes, R. Istúriz, M. Guzmán, and J. Murillo. 1996. Septic shock due to Comamonas acidovorans: a most unusual association. Infect. Dis. Clin. Pract. 5:73–74. [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comp. Chem. 4:187–217. [Google Scholar]
- Cowan, S. W., T. Schirmer, G. Rummel, M. Steiert, R. Ghosh, R. A. Pauptit, J. N. Jansonius, and J. P. Rosenbusch. 1992. Crystal structures explain functional properties of two E. coli porins. Nature. 358:727–733. [DOI] [PubMed] [Google Scholar]
- Dieckmann, G. R., J. D. Lear, Q. Zhong, M. L. Klein, W. F. DeGrado, and K. A. Sharp. 1999. Exploration of the structural features defining the conduction properties of a synthetic ion channel. Biophys. J. 76:618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Grubmüller, H., B. Heymann, and P. Tavan. 1996. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 271:997–999. [DOI] [PubMed] [Google Scholar]
- Im, W., S. Seefeld, and B. Roux. 2000. A grand canonical Monte Carlo-Brownian dynamics algorithm for simulating ion channels. Biophys. J. 79:788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im, W., and B. Roux. 2002a. Ion permeation and selectivity of OmpF porin: a theoretical study based on molecular dynamics, Brownian dynamics, and continuum electrodiffusion theory. J. Mol. Biol. 322:851–869. [DOI] [PubMed] [Google Scholar]
- Im, W., and B. Roux. 2002b. Ions and counterions in a biological channel: a molecular dynamics simulation of OmpF porin from Escherichia coli in an explicit membrane with 1 M KCl aqueous salt solution. J. Mol. Biol. 319:1177–1197. [DOI] [PubMed] [Google Scholar]
- Jap, B. K., and P. J. Walian. 1996. Structure and functional mechanism of porins. Physiol. Rev. 76:1073–1088. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Karplus, M., and J. A. McCammon. 2002. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9:646–652. [DOI] [PubMed] [Google Scholar]
- Koebnik, R., K. P. Locher, and P. van Gelder. 2000. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol. Microbiol. 37:239–253. [DOI] [PubMed] [Google Scholar]
- MacKerell, Jr., A. D., D. Bashford, M. Bellott, R. L. Dunbrack Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Mathes, A., and H. Engelhardt. 1998. Nonlinear and asymmetric open channel characteristics of an ion-selective porin in planar membranes. Biophys. J. 75:1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais-Cabral, J. H., Y. Zhou, and R. MacKinnon. 2001. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 414:37–42. [DOI] [PubMed] [Google Scholar]
- Ohtaki, H., and T. Radnai. 1993. Structure and dynamics of hydrated ions. Chem. Rev. 93:1157–1204. [Google Scholar]
- Phale, P. S., A. Phillipsen, C. Widmer, V. P. Phale, J. P. Rosenbusch, and T. Schirmer. 2001. Role of charged residues at the OmpF porin channel constriction probed by mutagenesis and simulation. Biochemistry. 40:6319–6325. [DOI] [PubMed] [Google Scholar]
- Przybylski, M., M. O. Glocker, U. Nestel, V. Schnaible, M. Bluggel, K. Diederichs, J. Weckesser, M. Schad, A. Schmid, W. Welte, and R. Benz. 1996. X-ray crystallographic and mass spectrometric structure determination and functional characterization of succinylated porin from Rhodobacter capsulatus: implications for ion selectivity and single-channel conductance. Protein Sci. 5:1477–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, K. M., and D. P. Tieleman. 2002. Orientation and interactions of dipolar molecules during transport through OmpF porin. FEBS Lett. 528:53–57. [DOI] [PubMed] [Google Scholar]
- Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of Cartesian equations of motion of a system with constraints—molecular dynamics of n-alkanes. J. Comp. Phys. 23:327–341. [Google Scholar]
- Schirmer, T. 1998. General and specific porins from bacterial outer membranes. J. Struct. Biol. 121:101–109. [DOI] [PubMed] [Google Scholar]
- Schirmer, T., and P. S. Phale. 1999. Brownian dynamics simulation of ion flow through porin channels. J. Mol. Biol. 294:1159–1167. [DOI] [PubMed] [Google Scholar]
- Schulz, G. E. 1996. Porins: general to specific, native to engineered passive pores. Curr. Opin. Struct. Biol. 6:485–490. [DOI] [PubMed] [Google Scholar]
- Sen, K., J. Hellman, and H. Nikaido. 1988. Porin channels in intact cells of Escherichia coli are not affected by Donnan potentials across the outer membrane. J. Biol. Chem. 263:1182–1187. [PubMed] [Google Scholar]
- Suenaga, A., Y. Komeiji, M. Uebayasi, T. Meguro, M. Saito, and L. Yamato. 1998. Computational observation of an ion permeation through a channel protein. Biosci. Rep. 18:39–48. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., and H. J. C. Berendsen. 1998. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoyl-phosphatidylcholine bilayer. Biophys. J. 74:2786–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman, D. P., P. C. Biggin, G. R. Smith, and M. S. P. Sansom. 2001. Simulation approaches to ion channel structure-function relationships. Q. Rev. Biophys. 34:473–561. [DOI] [PubMed] [Google Scholar]
- Torrie, G. M., and J. P. Valleau. 1977. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comp. Phys. 23:187–199. [Google Scholar]
- Willems, A., P. de Vos, and J. de Ley. 1992. The genus Comamonas. In The Prokaryotes, Vol. III. A. Balows, H. G. Trüper, M. Dworkin, W. Harder, and K. H. Schleifer, editors. Springer-Verlag, New York, NY. pp.2583–2590.
- Zachariae, U., A. Koumanov, H. Engelhardt, and A. Karshikoff. 2002. Electrostatic properties of the anion selective porin Omp32 from Delftia acidovorans and of the arginine cluster of bacterial porins. Protein Sci. 11:1309–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeth, K., K. Diederichs, W. Welte, and H. Engelhardt. 2000. Crystal structure of Omp32, the anion-selective porin from Comamonas acidovorans, in complex with a periplasmic peptide at 2.1 Å resolution. Structure. 8:981–992. [DOI] [PubMed] [Google Scholar]