

Abstract
Viscotoxins are small proteins that are thought to interact with biomembranes, displaying different toxic activities against a varied number of cell types, being viscotoxin A3 (VtA3) the most cytotoxic whereas viscotoxin B (VtB) is the less potent. By using infrared and fluorescence spectroscopies, we have studied the interaction of VtA3 and VtB, both wild and reduced ones, with model membranes containing negatively charged phospholipids. Both VtA3 and VtB present a high conformational stability, and a similar conformation both in solution and when bound to membranes. In solution, the infrared spectra of the reduced proteins show an increase in bandwidth compared to the nonreduced ones indicating a greater flexibility. VtA3 and VtB bind with high affinity to membranes containing negatively charged phospholipids and are motional restricted, their binding being dependent on phospholipid composition. Whereas nonreduced proteins maintain their structure when bound to membranes, reduced ones aggregate. Furthermore, leakage experiments show that wild proteins were capable of disrupting membranes whereas reduced proteins were not. The effect of VtA3 and VtB on membranes having different phospholipid composition is diverse, affecting the cooperativity and fluidity of the membranes. Viscotoxins interact with membranes in a complex way, most likely organizing themselves at the surface inducing the appearance of defects that lead to the destabilization and disruption of the membrane bilayer.
INTRODUCTION
Thionins are small basic proteins found in seeds, leaves, and stems of a great and varied number of plants. The proteins that belong to the thionin family consist of a polypeptide chain of 45–50 amino acids with 3–4 internal disulfide bonds, show a similar three-dimensional structure, and, except crambin, present a high degree of sequence homology including a similarity of the distribution of the hydrophobic and hydrophilic residues (Florack and Stiekema, 1994). Interestingly, thionins present different toxic activity to fungi, bacteria, and animal and plant cells, which might reflect a role in plant defense against the action of bacteria, fungi, and insects, although their exact biological function is not known (Bohlman and Apel, 1991; Epple et al., 1997; Florack and Stiekema, 1994; García-Olmedo et al., 1998).
Viscotoxins are cationic proteins that belong to thionins type III, comprise 46 amino acids, and are isolated from the leaves and stems of European mistletoe (Viscum album Loranthaceae). They display different toxic activities toward a number of tumor cell lines, suggesting that the different observed cytotoxicity could reflect variations in the three-dimensional structure (Büssing et al., 1999; Jung et al., 1990; Konopa et al., 1980; Orrù et al., 1997; Schaller et al., 1996; Urech et al., 1995). The biological activity of viscotoxins might be related to plant defense since its high expression gives enhanced resistance to pathogens (Holtorf et al., 1998). The amino acid sequence, disulphide bridge arrangement, and distribution in plant tissues of viscotoxins have been recently reviewed and six isoforms, namely A1, A2, A3, B, 1-PS, and U-PS, have been described (Orrù et al., 1997), being viscotoxin A3 (VtA3) the most cytotoxic whereas viscotoxin B (VtB) is the less potent (Büssing et al., 1998, 1999; Schaller et al., 1996; Urech et al., 1995). More recently it has been also found that viscotoxins increase natural-killer cell-mediated killing of tumor cells (Tabiasco et al., 2002), exert a strong immunomodulatory effect on human granulocytes (Stein et al., 1999a,b), and induce apoptosis in human lymphocytes (Büssing et al., 1999). In addition, viscotoxins form complexes with negatively charged DNA (Woynarowski and Konopa, 1980), and recently it has been suggested that the helix-turn-helix motif of viscotoxins might represent a DNA-binding domain (Romagnoli et al., 2000). The NMR structural determination of VtA3 has been reported very recently; the overall shape of VtA3 is very similar to that found for the other members of the thionin family, and is represented by the Greek capital letter gamma (Γ), with two antiparallel pair of α-helices and a short antiparallel β-sheet (Romagnoli et al., 2000). Differences in amino acid sequence between VtA3 and VtB are shown in Fig. 1, along with the three-dimensional structure of VtA3 (Protein Data Bank code 1ED0): all changes in amino acid residues but one are localized in the antiparallel pair of α-helices and in the segment that joins them, and significantly, all changes in charged amino acid residues are localized in one α-helix (Fig. 1).
FIGURE 1.

Comparison of the amino acid sequence of viscotoxins A3 and B, indicating the differences in amino acid positions, as well as their alignment (Clustalw). The three- dimensional structure of viscotoxin A3 (PDB code 1ED0) is also represented, showing the location of the amino acid residues that are different between the two proteins.
Studies of thionin toxicity suggest the requirement for a electrostatic interaction of the positively charged proteins with the negatively charged phospholipids of the membrane leading to major changes in the structure and integrity of the membrane as have been observed in both cell plasma membranes and model membranes (Caaveiro et al., 1998; Huang et al., 1997; Hughes et al., 2000; Wang et al., 1993; Wilson et al., 1997). It is unclear why amphipathic polypeptides such as thionins from mistletoe and other plants sharing a great extent of structural identity show quite different biological behavior. The differences in amino acid positions and charge must be therefore related to their different effects found with plasma and model membranes, but the molecular mechanism behind the toxicity of thionins is far from been elucidated. For example, it has been suggested that the toxicity of β-purothionin could be due to its ability of generating ion channels in cell membranes (Hughes et al., 2000), whereas the toxicity of α-hordothionin and wheat α-thionin would be originated through binding to the membrane surface and disturbance of its organization (Caaveiro et al., 1998; Thevissen et al., 1996). The determination of the specific mechanism of interaction with plasma and model membranes of thionins is therefore crucial to the understanding of their function. Moreover, the study at a molecular level of the biological effect of these membrane-active proteins is important not only from the biological point of view but also from the clinical one, since these proteins could be used as a substitute to overcome the increasing resistance to pathogenic bacteria (Hancock and Scott, 2000). Even more, they could be found of utility if they would show differential toxicity against tumoral cells and therefore be used as potential anticancer drugs. Despite all these studies, little is known about the details of how viscotoxins in particular alter and modify membrane properties. In this work, we have studied the interactions between viscotoxins VtA3 and VtB and model membranes having different phospholipid compositions by using infrared and fluorescence spectroscopies in an attempt to understand their mechanism of action. Since VtA3 is the most potent viscotoxin whereas VtB is the less one, the results provide us with an insight view to the molecular mechanism of action and a hint to the interaction between the protein and the membrane, since they reveal that the interaction of thionins with the membrane might be more complex than a simple electrostatic binding.
MATERIALS AND METHODS
Materials
Viscotoxins A3 and B were prepared and extracted as described previously (Schaller et al., 1996). Briefly, fresh plant material (leaves and stems) from V. album L. was homogenized and extracted in 2% acetic acid. The concentrated extract was subsequently diluted with distilled water and passed through a cation-exchange column. After a washing step, the adsorbed proteins were eluted with 0.1 M HCl. The eluate was neutralized with NaHCO3 and lyophilized. To eliminate impurities, the viscotoxins were taken up in phosphate buffer (0.1 M, pH 7) and fractioned. Elution was carried out with 0.1 M phosphate buffer (pH 7). Viscotoxin-containing fractions detected by high-performance liquid chromatography (HPLC) were pooled, dialyzed against water, and lyophilized. Individual components of the viscotoxin family were finally isolated by HPLC as described (Büssing et al., 1999). To obtain the completely reduced viscotoxins, 2 mg of the proteins were dissolved in 1.5 ml of 0.05 M phosphate buffer (pH 8.5) with 6 M urea by the addition of 60 μl β-mercaptoethanol after introduction of nitrogen. After 6 h at temperature in the dark, the thiol groups were blocked by a 12-fold excess of 2-vinyl pyridine (Sigma, Deisenhofen, Germany) over a period of 2 h in a nitrogen atmosphere. The reduced and blocked viscotoxins, isolated and purified by gel filtration using G25 (Pharmacia, Uppsala, Sweden), contained less than 0.2 mol/mol−1 free thiol groups. Since trifluoroacetate has a strong infrared absorbance at ∼1673 cm−1, which interferes with the characterization of the protein Amide I band (Surewicz et al., 1993), residual trifluoroacetic acid used in the HPLC mobile phase was removed by several lyophilization-solubilization cycles in 10 mM HCl (Zhang et al., 1992). Dimyristoylphosphatidylcholine (DMPC), perdeuterated dimyristoylphosphatidylcholine (DMPCd), dimyristoylphosphatidylglycerol (DMPG), dimyristoylphosphatidylserine (DMPS), dimyristoylphosphatidic acid (DMPA), egg yolk phosphatidylcholine (EPC), egg yolk phosphatidic acid (EPA), and bovine phosphatidylserine (BPS) were obtained from Avanti Polar Lipids (Birmingham, AL). 8-Aminonaphthalene-1,3,6-trisulfonic acid (ANTS), p-xylene-bis-pyridiniumbromide (DPX), 1,6-Diphenyl-1,3,5-hexatriene (DPH), and 1-(4-trimethylammoniumphenyl)-6-phenyl-1,3,5-hexatriene (TMA-DPH) were obtained from Molecular Probes (Eugene, OR). Deuterium oxide (99.9% by atom), Triton X-100, EDTA, and Hepes were purchased from Sigma (St. Louis, MO). All other chemicals were commercial samples of the highest purity available. Water was twice distilled and deionized in a Millipore system (Millipore, Madrid).
Sample preparation
Aliquots containing the appropriate amount of lipid in chloroform/methanol (1:1, v/v) were placed in a test tube, the solvents removed by evaporation under a stream of O2-free nitrogen, and finally traces of solvents were eliminated under vacuum in the dark for more than 3 h. A preweighted amount of freeze-dried protein was suspended by addition of an appropriate volume of 20 mM HEPES, 50 mM NaCl, 0.1 mM EDTA, pH 7.4 buffer (either D2O or H2O, see below). The protein solution was then added to the tube containing the dried lipid to obtain the required specific lipid/protein mole ratio, usually 15:1, unless otherwise stated, and the suspension was vortexed at ∼5°C above the transition temperature of the phospholipid to obtain multilamellar vesicles (MLV). The mixture was freezed/thawed twice to ensure complete homogenization of the sample and maximization of contacts between the protein and the phospholipid and then incubated for 10 min at 55°C with occasional vortexing. The freeze/thaw cycle was repeated again and the suspension was then centrifuged at 15,000 rpm for 15 min at 25°C to remove the protein that was not bound to the phospholipid in the membrane. The freeze/thaw, incubation at 55°C, and centrifugation steps were repeated once more to remove the unbound protein. The pellet was resuspended in either D2O or H2O buffer and used for the measurements. For binding experiments, samples were subjected to the same freeze/thaw cycles as before, centrifuged, and pellets resuspended to a fixed volume with buffer (25 μl) whereas supernatants were collected, lyophilized, and resuspended to the same volume (25 μl). Except for temperatures studies, infrared spectra were obtained at 25°C.
For fluorescence polarization experiments using either DPH or TMA-DPH, MLV containing both protein and phospholipid were used. A few microliters of a stock solution of DPH or TMA-DPH at a concentration of 5 × 10−4 M in N,N′-dimethylformamide were added to the mixture of the MLV suspension and then incubated at 55°C for 60 min for DPH and 20 min for TMA-DPH containing liposomes, respectively. In all cases, the lipid/protein molar ratio was 20:1 and the fluorescence probes/lipid molar ratio was 1:500. Large unilamellar vesicles (LUV) liposomes were used to study vesicle leakage. LUV were prepared by the extrusion method (Hope et al., 1985) using polycarbonate filters with a pore size of 0.1 μm (Nuclepore, Pleasanton, California) using 10 mM HEPES, 20 mM NaCl, 0.1 mM EDTA, pH 7.4 buffer. Buffer used for preparing LUV liposomes for assays of vesicle leakage contained in addition 25 mM 8-Aminonaphthalene-1,3,6-trisulfonic acid and 90 mM p-xylene-bis-pyridiniumbromide. Nonencapsulated fluorescent probes were separated from the vesicle suspension thought a Sephadex G-75 filtration column (Pharmacia) eluted at room temperature with 10 mM HEPES, 130 mM NaCl, 0.1 mM EDTA, pH 7.4 buffer. The phospholipid and protein concentrations were measured by methods described previously (Böttcher et al., 1961; Edelhoch, 1967).
Infrared spectroscopy (IR)
For the infrared measurements, MLVs obtained as described above, were resuspended in ∼50 μl D2O buffer. Pellets were placed in between two CaF2 windows separated by 50-μm Teflon spacers and transferred to a Harrick Ossining demountable cell. Fourier transform infrared spectra were obtained in a Nicolet 520 Fourier transform infrared spectrometer equipped with a deuterated triglycine sulfate detector. Each spectrum was obtained by collecting 250 interferograms with a nominal resolution of 2 cm−1, they were transformed using triangular apodization and, to average background spectra between sample spectra over the same time period, a sample shuttle accessory was used to obtain sample and background spectra. The spectrometer was continuously purged with dry air at a dew point of −40°C to remove atmospheric water vapor from the bands of interest. All samples were equilibrated at the lowest temperature for at least 25 min before acquisition. An external bath circulator, connected to the infrared spectrometer, controlled the sample temperature. Subtraction of buffer spectra taken at the same temperature as the samples was performed interactively using either GRAMS/32 or Spectra-Calc (Galactic Industries, Salem, MA) as described previously (Contreras et al., 2001). Frequencies at the center of gravity, when necessary, were measured by taking the top 10 points of each specific band and fitted to a Gaussian band. Band-narrowing strategies were applied to resolve the component bands in the Amide I′ region. Second-derivative spectra were calculated over a 15-data point range. Fourier self-deconvolution (Kauppinen et al., 1981) of the subtracted spectra was carried out using a Lorentzian shape and a triangular apodization with a resolution enhancement parameter, K, of 2.2, which is lower than log(signal/noise) (Mantsch et al., 1988) and a full width at half-height of 18 cm−1. These parameters assumed that the spectra were not overdeconvolved as was evidenced by the absence of negative side lobes. Protein secondary structure elements were quantified from curve-fitting analysis by band decomposition of the original Amide I′ band after spectral smoothing using the same software stated above (Bañuelos et al., 1995). Briefly, for each component, three parameters were considered: band position, band height, and bandwidth. The number and position of component bands was obtained through deconvolution and in decomposing the Amide I′ band, gaussian components were used. The curve-fitting procedure was accomplished in two steps: in the first one, band position was fixed, allowing width and heights to approach final values, and in the second one, band positions were left to change. When necessary, these two steps were repeated. Band decomposition was performed using SpectraCalc (Galactic Industries, Salem, MA). The fitting result was evaluated visually by overlapping the reconstituted overall curve on the original spectrum and by examining the residual obtained by subtracting the fitting from the original curve. The procedure gave differences of less than 2% in band areas after the artificial spectra were submitted to the curve fitting procedure. The frequency positions of the band centers were independently evaluated by second derivative procedures, being always very close to the positions found by deconvolution. To obtain two-dimensional infrared correlation spectra and detect dynamical spectral variations induced by temperature on the secondary structure of VtA3 and VtB and on the phospholipid, we have obtained two-dimensional synchronous and asynchronous (disrelation) spectra, as described before (Contreras et al., 2001). Correlation calculations have been done over the 1800–1550 cm−1 spectral region. Calculations were done with the use of Mathcad for Windows software.
Fluorescence measurements
Steady-state fluorescence measurements were carried out using a SLM 8000C spectrofluorometer with a 400-W Xe lamp, double emission monochromator, and Glan-Thompson polarizers. Correction of excitation spectra was performed using a Rhodamine B solution and a standard lamp. Typical spectral bandwidths were 4 nm for excitation and 2 nm for emission. All fluorescence studies were carried out using 5 mm × 5 mm quartz cuvettes. The excitation and emission wavelength was 360/362 and 425/450 nm when observing the DPH/TMA-DPH fluorescence, respectively, whereas the excitation and emission wavelength for Tyr was 280 and 310 nm. All the data were corrected for background intensities and progressive dilution. Emission spectra were not corrected for the photomultiplier wavelength dependence. Fluorescence anisotropies were determined according to the equation (Contreras et al., 2001)
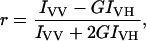 |
where IVV and IVH are the fluorescence intensities and the subscripts indicate the vertical (V) or horizontal (H) orientations of the excitation and emission Glan-Thompson polarizers. The instrumental factor G (G = IHV/IHH) was determined by measuring the polarized components of fluorescence of the protein or probes with horizontally polarized excitation. Leakage was assayed by treating the probe-loaded liposomes (final lipid concentration, 0.1 mM) with the appropriate amounts of protein in a fluorometer cuvette stabilized at 25°C. Changes in fluorescence intensity were recorded with excitation and emission wavelengths set at 350 and 510 nm, respectively. One hundred percent release was achieved by adding to the cuvette Triton X-100 to a final concentration of 0.1% (w/w). Leakage was quantified on a percentage basis according to the equation
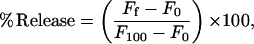 |
Ff being the equilibrium value of fluorescence after protein addition, F0 the initial fluorescence of the vesicle suspension, and F100 the fluorescence value after addition of Triton X-100.
RESULTS
Protein structure and phospholipid binding
The infrared spectra of the Amide I′ region of the fully hydrated wild viscotoxins VtA3 and VtB in D2O buffer at two temperatures, 5 and 60°C, and at pH 7.4 are shown in Fig. 2, A and B, respectively. The spectra are formed by different underlying components that give place to a broad and asymmetric band with a maximum at ∼1646 and 1647 cm−1 for VtA3 and ∼1650 and 1649 cm−1 for VtB at 5 and 60°C, respectively. Protein denaturation is characterized in D2O medium by the appearance of two sharp bands at ∼1685 and 1620 cm−1, due to extended β-strands with strong intermolecular interactions (Arrondo et al., 1993). These bands are not visible in the whole range of temperatures studied so that if aggregation occurred, it was only to a very low extent. The maximum of the band does not change significantly upon increasing the temperature, indicative of the high conformational stability of the protein, as shown previously (Florack and Stiekema, 1994). The infrared spectra of the Amide I′ region of the fully hydrated reduced VtA3 and VtB in D2O buffer at the same temperatures as above are shown in Fig. 2, C and D. The spectra show a maximum at ∼1648 and 1651 cm−1 for reduced VtA3 and ∼1649 and 1652 cm−1 for reduced VtB at 5 and 60°C, respectively. The spectra of the reduced proteins show a small change in the maximum of the bands as the temperature is increased and it is clearly visible an increase of ∼14 cm−1 in the bandwidth at half height compared to the nonreduced proteins, i.e., 50 and 48 cm−1 for the reduced viscotoxins compared to 36 and 34 cm−1 for the nonreduced ones. The increase in bandwidth from the nonreduced to the reduced viscotoxins would indicate either the presence of additional structures contributing to the broader envelope or a greater flexibility of the reduced proteins or both (see below). It can also be observed the appearance of small IR bands at ∼1611–1612 cm−1, which have slightly more intensity than in the nonreduced viscotoxins, which might also indicate the loosening of the three-dimensional structure of the protein due to the breaking the Cys–Cys bonds.
FIGURE 2.

Amide I′ band region of (A) viscotoxin A3, (B) viscotoxin B, (C) reduced viscotoxin A3, and (D) reduced viscotoxin B at 5°C (solid line) and 60°C (dashed line).
To study the proteins when they were effectively bound to the phospholipid model membranes, all samples containing both phospholipid and protein, either wild or reduced, have been prepared by mixing and washing the unbound protein as described in the Materials and Methods section. The infrared spectra in the Amide I′ and C=O region of samples prepared in this way containing different quantities of wild VtA3 and VtB and the same amount of DMPCd/DMPA model membranes are shown in Fig. 3. Upon increasing the quantity of VtA3 from a phospholipid/protein ratio of 100:1, 50:1, and 25:1, the quantity of VtA3 that was bound to the phospholipid membrane increased with a small protein band appearing in the supernatants at higher protein/phospholipid ratios (Fig. 3, A and B). Increasing the quantity of VtB in the same ratios as before, the quantity of protein observed in the supernatants, i.e., unbound, was quite similar to the quantity of the bound one (Fig. 3, C and D). These data would indicate that the binding capacity for both types of proteins, VtA3 and VtB, is different, being the amount of unbound VtB, given the same conditions, always greater than that of VtA3. It can also be observed a small but clear difference at ∼1650 cm−1 for VtA3 that is not observed in the samples containing VtB (Fig. 3, A and C). It should be noted that binding experiments were made at 25°C, so that DMPCd/DMPA membranes could exist in a mixed gel/fluid state at this specific temperature. Therefore binding might be more representative of binding to vesicles in this state than of binding to vesicles in a purely fluid state. It is also interesting to note that, when pure DMPC was used, both VtA3 and VtB were recovered almost completely in the supernatant (not shown), demonstrating therefore that both VtA3 and VtB only bind with high affinity to membranes containing negatively charged phospholipids.
FIGURE 3.

Infrared spectra of the C=O and Amide I′ regions of (A, C, E, and G) pellets and (B, D, F, and H) supernatants, respectively, for samples containing wild VtA3 (A and B), wild VtB (C and D), reduced VtA3 (E and F), and reduced VtB (G and H) in the presence of DMPCd/DMPA at 100:1 (——), 50:1 (·······), and 25:1 (----) phospholipid/protein ratios. All phospholipid mixtures contained a relationship of 1:1.
The infrared spectra in the Amide I′ and C=O region of samples containing different quantities of reduced VtA3 and VtB and the same amount of DMPCd/DMPA model membranes are also shown in Fig. 3. Upon increasing the quantity of reduced VtA3 from a phospholipid/protein ratio of 100:1, 50:1, and 25:1, the quantity of reduced VtA3 that was bound to the phospholipid membrane increased but no protein band appeared in the supernatants at all ratios tested (Fig. 3, E and F). A similar pattern was observed when reduced VtB instead of reduced VtA3 was used (Fig. 3, G and H). Significantly, the Amide I′ band envelope of the reduced proteins in the presence of the phospholipid membranes was completely different to that found in solution. At difference with what was found with the nonreduced proteins, two sharp bands at 1616 and 1685 cm−1 are resolved for both types of proteins, the former band with higher intensity than the latter. The frequency of the bands and their intensity would indicate the presence of extended β-strands with strong intermolecular interactions (Arrondo et al., 1993), in contrast to the nonreduced viscotoxins, where no aggregation was found (Figs. 2 and 3).
Secondary structure of VtA3 and VtB in the presence of phospholipid
To observe the underlying components of the broad Amide I′ band of the proteins, we have applied several enhancements methods, such as self-deconvolution and derivative methods, to the original envelope (Kauppinen et al., 1981) as well as decomposition of the Amide I′ infrared band. The results are observed in Fig. 4. For VtA3 in solution we have identified different component bands having frequencies of ∼1687, 1675, 1663, 1654, 1644, 1632, 1621, and 1611 cm−1, being the 1644 cm−1 band the main one (Fig. 4 A). VtA3, as it has been recently found by NMR, adopts a similar fold to that found in other thionins and consists of two α-helices connected by a turn and a short stretch of an antiparallel β-sheet, having a content of α-helix, β-sheet, and turns of ∼41%, 16%, and 26%, respectively (Romagnoli et al., 2000). If we assume that the bands appearing at 1687 (2%), 1675 (9%), and 1663 cm−1 (15%) are due to turns, the band at 1654 cm−1 (15%) to α-helix, the band at 1644 cm−1 (35%) to either random or α-helix or both, the band at 1632 cm−1 (18%) to β-sheet, and the band at 1621 cm−1 (7%) to aggregated structures (Arrondo et al., 1987, 1993; Ascoli et al., 1997; Vila et al., 2001), then VtA3 in solution, as seen by IR, would have a structure quite similar to that found by NMR. The same number of bands with similar frequencies and intensities have been identified for VtA3 in the presence of negatively charged phospholipid membranes, being the band at ∼1644 cm−1 the main one (Fig. 4 B). Therefore no abrupt change is observed in the structure of VtA3 upon binding to membranes, which should be in accordance with its compact and rigid structure. For VtB in solution we have identified similar component bands as those found for VtA3 (Fig. 4 C). There is no published three-dimensional structure of VtB, either by NMR or x-ray diffraction, but taking into account the differences in primary structure (Fig. 1) and the similar fold found for all thionins, VtB should have a similar three-dimensional structure as that found for VtA3. In the presence of negatively charged phospholipid membranes, the same number of bands with similar frequencies and intensities have been identified for VtB, being the band at ∼1644 cm−1 again the one with greater intensity (Fig. 4 D).
FIGURE 4.

Amide I′ band decomposition of (A and B) VtA3 and (C and D) VtB in solution (A and C) and in the presence of DMPG (B and D), at an original phospholipid/protein molar ratio of 15:1 in D2O buffer at 25°C. The component bands, the envelope, and the difference between the fitted curve and the original spectrum are shown.
As shown above, no dramatic changes in the structure of both proteins on binding to the phospholipid membranes were found. However they show clear differences in binding behavior. To enhance the spectral resolution of the Amide I′ region of VtA3 and VtB in the presence of negatively charged model membranes, we have used two-dimensional infrared correlation spectroscopy to obtain synchronous (synthermal) and asynchronous (asynthermal) spectra, an experimental approach based on the detection of dynamical spectral variations induced by an external perturbation, temperature in our case (Contreras et al., 2001). The synchronous map obtained for VtA3 in the presence of either DMPA or DMPG shows different crosspeaks correlating phospholipid/protein bands, i.e., crosspeaks correlating frequencies 1741/1655 cm−1 (negative) and 1741/1632 cm−1 (positive), whereas for VtB crosspeaks correlating frequencies 1685–1655/1742 cm−1 (negative) and 1742/1630 cm−1 (positive) appeared, but with much less intensity than those for VtA3 (not shown for the sake of briefness). These results would imply that some parts of the proteins with β-structure and α-helix conformation would be involved in binding.
Thermotropic behavior of the lipids from the CH2 stretching vibration
Although it has been shown that the incorporation of transmembrane peptides in the phospholipid palisade of the membrane can affect not only phospholipid chain order but also interchain coupling (Paré et al., 2001), a shift in the frequency of the CH2 symmetric stretching band is a reliable index of the phase behavior of a phospholipid dispersion (Mantsch and McElhaney, 1991). We have studied by infrared spectroscopy the effects of both VtA3 and VtB on the phase transition of different negatively charged phospholipid model membranes (the binding to DMPC membranes was negligible and no effect was observed on this phospholipid).
The temperature dependence of the CH2 symmetric frequency of pure DMPA is shown in Fig. 5, where a highly cooperative change at ∼48°C is observed, corresponding to the gel-to-liquid crystalline phase transition, Tm, of the phospholipid. In the presence of both VtA3 and VtB a small decrease in the Tm values was observed , but no differences were found for both types of proteins (Fig. 5). The temperature dependence of the CH2 symmetric frequency of pure DMPG is also shown in Fig. 5, where a highly cooperative change at ∼23°C is observed, corresponding to the gel-to-liquid crystalline phase transition of DMPG. The most significant result is the reduction in the cooperativity of the gel-to-liquid crystalline phase transition of DMPG induced by both VtA3 and VtB (Fig. 5); however, no shift of the gel-to-liquid phase transition, neither to lower nor to higher temperatures, was observed. It is not possible to study the Amide I′ band of proteins in the presence of phosphatidylserine-containing membranes because of the presence of the strong 1625 cm−1 band of the COO− group of the phospholipid, but it is useful for the study of the acyl chain bands of the phospholipid, such as the CH2 stretching bands. Because it is known that thionins bind with relatively high affinity to phosphatidylserine-containing membranes (Coulon et al., 2002), we have used DMPS in some parts of this work. The temperature dependence of the CH2 symmetric frequency of pure DMPS is observed in Fig. 5, where a highly cooperative change at ∼38°C is observed, corresponding to the gel-to-liquid crystalline phase transition of DMPS. The most significant result is the reduction in the cooperativity of the gel-to-liquid crystalline phase transition of DMPG induced by both VtA3 and VtB, more significant in the last case, as well as the decrease in the frequency values at all temperatures, indicating that the proportion of trans isomers was higher than in pure DMPS. Interestingly, the effect of VtB on DMPS was more evident than that found in the presence of VtA3 (Fig. 5).
FIGURE 5.

Temperature dependence of the CH2 symmetric stretching band frequency of DMPA, DMPG, and DMPS in the pure form (▪) and in the presence of either VtA3 (○) or VtB (▵) at an original phospholipid/protein molar ratio of 15:1.
We have studied other phospholipid bands such as the acyl chain CH2 scissoring and the PO2− double bond stretching bands appearing at 1468 and 1220 cm−1, respectively. The presence of both proteins, VtA3 and VtB, did not induce any significant differences on frequency and width of these bands at different temperatures when compared with the pure phospholipids, indicating that there were no differences in packing and hydration between the pure phospholipid and the mixtures containing the proteins (not shown for briefness).
Steady-state fluorescence anisotropy
The effect of both proteins, VtA3 and VtB, on the structural and thermotropic properties of phospholipid membranes was further investigated by measuring the steady-state fluorescence anisotropy of the fluorescent probes DPH and TMA-DPH incorporated into DMPA, DMPG, and DMPS membranes as a function of temperature (Fig. 6). DPH and TMA-DPH are very useful molecules to study the structural order of the lipid bilayer since the diphenylhexatrienyl moiety of DPH is located at the middle of the bilayer (inner probe) whereas the diphenylhexatrienyl moiety of TMA-DPH extends into the lipid bilayer between the C-5/C-11 carbons of the phospholipid acyl chains (interface probe), reporting essentially structural information on this region of the bilayer (Trotter and Storch, 1989; Mateo et al., 1991). In the case of DMPA membranes, VtA3 and VtB did not affect in a significant way the anisotropy of both DPH and TMA-DPH below and above the phase transition temperature of the phospholipid (Fig. 6, A and B). However, for DMPG membranes, VtA3 and VtB decreased the cooperativity of the anisotropy change for both DPH and TMA-DPH probes, increasing the anisotropy above the gel-to-liquid crystalline phase transition of the phospholipid (Fig. 6, C and D, respectively). It is interesting to note that both probes where similarly affected by both VtA3 and VtB, i.e., the effect at both different depths were similar. The most significant effect was observed for DMPS membranes, since VtA3 and VtB greatly reduced the cooperativity of the transition as detected by both probes, decreasing and increasing the anisotropy of DPH and TMA-DPH below and above the gel-to-liquid crystalline phase transition of the phospholipid and, significantly, VtA3 presented a bigger effect than VtB on the anisotropy change (see Fig. 6, E and F).
FIGURE 6.

Steady-state anisotropy, 〈r〉, of DPH (A, C, and E) and TMA-DPH (B, D, and F) incorporated into (A and B) DMPA, (C and D) DMPG, and (E and F) DMPS model membranes as a function of temperature. Data correspond to vesicles containing pure phospholipid (▪), phospholipid plus VtA3 (○), and phospholipid plus VtB (▵).
Fluorescence anisotropy measurements can provide very useful insights into the dynamics of proteins and peptides when bound to the membrane. The intrinsic fluorescence of the Tyr residues of VtA3 and VtB is rather small, but we have been able of obtaining reliable measurements for both VtA3 and VtB at different lipid/protein ratios in the presence of DMPA. For VtA3 and VtB in solution, we found anisotropy values of ∼0.15–0.17, whereas there was an increase of the anisotropy values for the Tyr residue in the presence of DMPA containing vesicles upon increasing the lipid/protein ratio (not shown for briefness). The limiting value at a high phospholipid/protein ratio seems to be near 0.21–0.23 indicating a significant motional restriction of the Tyr moiety of both proteins in the presence of DMPA membranes (Lakowicz, 1999).
Leakage of vesicle contents
To further explore the possible interaction of VtA3 and VtB with phospholipid model membranes, we studied the effect of both proteins on the release of encapsulated fluorophores using the experimental setup described in Materials and Methods. Fig. 7 shows the results obtained with two different liposome compositions, namely EPC/EPA and EPC/BPS at a molar relationship of 1:1. We had to use unsaturated phospholipids for leakage experiments because of the great variability in the basal fluorescence we found using saturated ones; nevertheless, results were qualitatively similar. It is clearly observed in Fig. 7 that leakage was dependent not only on lipid composition but also on protein, i.e., VtA3 or VtB: leakage was greater for liposomes composed of EPC/EPA than for liposomes composed of EPC/BPS, and the effect observed for VtA3 on leakage was always greater than for VtB (Fig. 7). It is also worth noting that the leakage produced by the reduced forms of both VtA3 and VtB was always lower than that observed for the nonreduced proteins.
FIGURE 7.

Leakage data for LUVs composed of (A) EPC/EPA and (B) EPC/BPS, both phospholipids at a molar ratio of 1:1, in the presence of VtA3 (○), reduced VtA3 (□), VtB (▵), and reduced VtB (▿) at 25°C at different protein/lipid ratios. The insert in B shows representative normalized fluorescence data at 25°C for LUVs containing EPC/BPS and VtA3 (·······), reduced VtA3 (——), VtB (----), and reduced VtB (· · · ·) at a lipid/protein ratio of 25:1. The first change corresponds to the addition of protein whereas the second one corresponds to the addition of detergent (see text for details).
DISCUSSION
There is an increasing interest in the study of the interaction of proteins and peptides with membranes, and specifically for those implicated in defense mechanisms (Hancock and Scott, 2000). Viscotoxins, similarly to other thionins, are basic proteins that have shown different levels of toxicity against diverse types of cells, even tumor cells (Büssing et al., 1999; Schaller et al., 1996; Urech et al., 1995). They have a nearly identical three-dimensional fold to that of other thionins (Romagnoli et al., 2000), and possess a surface with an amphipathic character: a hydrophobic region located on the surface of the helices and a hydrophilic region located in between the helical stem and the β-arm, i.e., defining two separate regions.
A combination of electrostatic interactions with the polar head groups of phospholipids and the presence of hydrophobic interactions with the hydrocarbon chains are a prerequisite for the binding and perturbation of membranes (Zückermann and Heimburg, 2001). As it has been suggested for other thionins, the electrostatic interaction of the positively charged hydrophilic region of viscotoxins play an essential role as their first step upon their interaction with membranes, and subsequently, the insertion of their hydrophobic domain would alter the structure and integrity of the membrane (Caaveiro et al., 1997, 1998; Evans et al., 1989; Huang et al., 1997; Hughes et al., 2000; Thevissen et al., 1996; Wang et al., 1993). It is interesting to note the relationship between the different membrane-active character of the different isoforms of the viscotoxins and their net charge (Orrú et al., 1997). VtA3, the most active, has a net charge of +6 and a pI value of 9.2 whereas VtB, the less potent, has a net charge of +5 and a pI value of 8.8 (Büssing et al., 1998; Schaller et al., 1996; Urech et al., 1995). Even though peptide binding might be reversible and equilibrium might be reached during the centrifugation steps (see above), we have been capable of observing differences in binding capacity for VtA3 and VtB. In the presence of model membranes composed of one pure negatively charged phospholipid, both VtA3 and VtB were bound almost completely. However, in the presence of binary mixtures having one negatively charged phospholipid, either DMPA or DMPG, and one zwitterionic phospholipid, DMPC, the binding of VtA3 was much higher than that of VtB. It should be stressed that in the presence of membranes composed solely of the zwitterionic phospholipid DMPC there was no binding of both VtA3 and VtB, recovering nearly all the protein in the supernatant.
Differences in the primary structure between viscotoxins VtA3 and VtB are localized in the antiparallel pair of α-helices and in the segment that joins them; interestingly, viscotoxin molecules are thought to interact with phospholipid membranes via the two adjacent α-helices (Kelly et al., 1998). In VtA3 positions 23 and 33 are charged residues that are conserved in all thionins as well as position 28 (absent in VtB). Crambin, having a fold identical with that of VtA3 (Romagnoli et al., 2000), has an acidic residue at position 23 (instead of being basic) whereas no more charged residues are found in the rest of the sequence. Since crambin, although having an identical fold, has no effects on membranes (Florack and Stiekema, 1994), differences in biological activity of viscotoxins and hence their membranotropic effects should be related to this region of the proteins. The interaction of peptides and proteins with membrane surfaces involves a number of steps, including the initial binding to the surface, induction or stabilization of a specific secondary structure upon the interaction with the phospholipid, modulation of the phospholipid biophysical properties by binding, and finally insertion of the peptide in the membrane either partially or fully. Differences in toxicity of different viscotoxins could also be related to differences in binding, as we have described here. However, we have observed the binding of nonreduced (natural) and reduced viscotoxins to membranes, but no effects were observed in vivo (Büssing et al., 1999) and much smaller effects where observed for the reduced proteins compared to the nonreduced natural proteins in vitro (this work). Therefore, the binding of viscotoxins to the membranes, although essential to exert their biological activity, is not by itself sufficient to elicit their biological responses. The hydrophobic groups would bind the phospholipids and the polar sides of the helices would interact with the charged groups of the membrane surface positioning the peptide in a specific conformation in the bilayer and therefore causing a perturbation on the membrane structure followed by changes in order and permeability of the phospholipid bilayer and eliciting the biological responses.
After binding, the Tyr residues reside in an environment showing a significant motional restriction, but should remain located at the surface of the membrane, as indicated by the absence of any effect on the fluorescence anisotropy of both inner and interface probes. This motionally restricted environment should be consistent with a location in a region near the membrane interface possibly involved in charge interactions and hydrogen bonding. The binding of the protein to negatively charged membranes but not to zwitterionic ones was confirmed also by infrared since only the Amide I′ band corresponding to the bound protein was present in model membranes composed of negatively charged phospholipid membranes, being absent in membranes formed by DMPC. The difference found on the temperature dependence of the CH2 symmetric frequency of the negatively charged phospholipids in the presence of the proteins might also reflect small differences in their localization.
The infrared spectrum of the Amide I′ region of the fully hydrated proteins in D2O buffer at low and high temperatures are very similar indicating the stability of its conformation in solution, even at the high temperatures we have used. Both proteins, when they are bound to the phospholipid membranes, maintain their conformation without any significant changes, in accordance with their compactness (Lecomte et al., 1987; Park et al., 1999). However, the breaking of the S-S arrangement, which is thought to stabilize a common structure present in small proteins that interact with bilayer membranes (Orrú et al., 1997), changes the scenario: both reduced VtA3 and VtB proteins bind to the membranes with a very high affinity similarly to the nonreduced ones, but reduced ones change significantly their conformation to an aggregated state without any membranotropic activity. The increase in bandwidth of the Amide I′ envelope of the reduced proteins could indicate their greater flexibility, in accordance with the effect of breaking the Cys–Cys bonds that maintain a compact and rigid molecule in the nonreduced native ones (Florack and Stiekema, 1994; Lecomte et al., 1987; Park et al., 1999). Since the reduction of the disulphide bonds of viscotoxins produces a complete loss of viscotoxin activity (Stein et al., 1999a), and the cytotoxicity of viscotoxins against cultured human granulocytes and lymphocytes was only prevented by cleavage of their disulphide bridges (Büssing et al., 1999), the nonreduced unmodified conformation of viscotoxins is essential for its biological activity and binding is necessary, but it is not the essential step to produce the disruption of the membranes. Once the proteins have reached the surface of the vesicle, they destabilize the membrane bilayer and induce the release of its contents. Since the two antiparallel α-helices of viscotoxins are too short to span the bilayer, and the small probability that two α-helices could eventually span the membrane thickness if the S-S bridges were not linking the same positions on the two helixes, the proteins must overall disorganize the membrane bilayer and therefore no more function as a permeability barrier.
Linear dichroism measurements of α-purothionin suggest that the two antiparallel α-helices are highly oriented parallel to the plane of the bilayer (Kelly et al., 1998). Since thionins share the same fold pattern, we could assume that the two antiparallel α-helices of viscotoxins would be oriented parallel to the membrane surface apposing the hydrophobic region located on the surface of the helices to the membrane. As has been commented by Shai (Shai, 1999; Shai and Oren, 2001), amphipathic lytic peptides that might act via the carpet mechanism rather than by the barrel-stave one should be positive, the charges should spread along the peptide chain, and should bind weakly (or not at all) to zwitterionic phospholipids: viscotoxins share these properties. It should be borne in mind that association of nonreduced viscotoxins at the surface of the membrane do not produce aggregation as we have observed by IR; the contrary is true: aggregated reduced viscotoxins do not produce lysis of the membranes. It has been also described for other proteins and peptides that an increase in gel phase disorder and a decrease in the gel to liquid phase transition cooperativity are related to the insertion of the protein into the membrane and spreading the phosphate groups. These results, similarly to what has been described here, might suggest that the perturbation induced by viscotoxins is related to the membrane fluidity and cooperativity changes observed for different phospholipid mixtures as studied here. Viscotoxins would then behave like a detergent (see Caaveiro et al., 1998).
It is commonly accepted that protein-induced leakage requires some type of hydrophobic interaction of the protein with the phospholipid bilayer (Parente et al., 1990). Therefore, it can be inferred that both nonreduced VtA3 and VtB get inserted in the bilayer matrix. Other interactions must occur, apart from the hydrophobic ones, since, as we have demonstrated here, no effect is observed in the presence of zwitterionic phospholipids, reinforcing the idea that, before proper insertion occurs, an electrostatic interaction should occur first. Although no clear mechanism of action might be inferred from these results, it is clear that both VtA3 and VtB alters the membrane structure and therefore the membrane should be their main target. The two-dimensional infrared spectroscopy results show that the main event that takes place upon increasing temperature in mixtures containing both viscotoxins and model membranes corresponds to the main phospholipid phase transition; although there is not a big conformational change in the viscotoxins produced by their interaction with the phospholipid membranes, it demonstrates the existence of a specific interaction of both types of molecules that is localized predominantly in the α-helices of the protein.
The results obtained in this work can be related to the biological reliability of viscotoxins. They are known to interact differently with different types of cells and differences in phospholipid membrane composition might account for the different viscotoxin activity toward different types of cells (Büssing et al., 1999; Konopa et al., 1980; Schaller et al., 1996; Tabiasco et al., 2002; Urech et al., 1995). As commented above, the search for antimicrobial agents is nowadays very intensive since the increasing microbial resistance toward many antimicrobial compounds and the study of antimicrobial peptides and proteins has increased in interest (Hancock and Scott, 2000; Shai and Oren, 2001). However, since many of these antimicrobial peptides perturb and disrupt the cell membranes, they not only are lytic to microbial cells but also to eukaryotic ones. The understanding of their mechanism of action is therefore essential to provide a basis for the design of peptides with a directed action toward specific cell types. The results we have discussed here complement other studies that have been done previously on thionins. Viscotoxins interact in a complex conformationally dependent way with phospholipid membranes, involving a number of processes, being the first one an electrostatic interaction with negatively charged phospholipids and a later (partial) insertion of the protein in the membrane interface leading to the disruption of the membrane. Most likely viscotoxins dispose themselves at the membrane surface as it has been suggested for other proteins that dispose as a carpetlike structure and similarly to other proteins of the thionin family. The exact underlying mechanisms resulting in the membranotropic effects by viscotoxins remain to be characterized, but are obviously dependent on a distinct structural conformation of the different proteins. Further work is being made in our lab to discern that are the specific residues of viscotoxins responsible for the attachment and posterior disruption of the membrane to explore a possible way for their engineering.
Acknowledgments
This work has been supported by grant PM98-0100 from Dirección General de Educación Superior e Investigación Científica (DGESIC), Spain (to J.V.). M.G. and R.P. are recipients of predoctoral fellowships from Consejo Superior de Investigaciones Científicas y Técnicas (CONICET), Argentina-Spain, and Ministerio de Educación y Ciencia, Spain. The financial support of AECI, Programa de Cooperación Científica con Iberoamérica, Spain, is greatly acknowledged.
References
- Arrondo, J. L., H. H. Mantsch, N. Mullner, S. Pikula, and A. Martonosi. 1987. Infrared spectroscopic characterization of the structural changes connected with the E1—E2 transition in the Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 262:9037–9043. [PubMed] [Google Scholar]
- Arrondo, J. L. R., A. Muga, J. Castresana, and F. M. Goñi. 1993. Quantitative studies of the structure of proteins in solutions by Fourier transform infrared spectroscopy. Prog. Biophys. Mol. Biol. 59:23–56. [DOI] [PubMed] [Google Scholar]
- Ascoli, G. A., K. X. Luu, J. L. Olds, T. J. Nelson, P. A. Gusev, C. Bertucci, E. Bramanti, A. Raffaelli, P. Salvadori, and D. L. Alkon. 1997. Secondary structure and Ca2+-induced conformational change of calexcitin, a learning-associated protein. J. Biol. Chem. 272:24771–24779. [DOI] [PubMed] [Google Scholar]
- Bañuelos, S., J. L. R. Arrondo, F. M. Goñi, and G. Pifat. 1995. Surface-core relationships in human low density lipoprotein as studied by infrared spectroscopy. J. Biol. Chem. 270:9192–9196. [DOI] [PubMed] [Google Scholar]
- Bohlman, H., and K. Apel. 1991. Thionins. Annu. Rev. Plant Physiol. Plant Mol. Biol. 42:227–240. [Google Scholar]
- Böttcher, C. S. F., C. M. Van Gent, and C. Fries. 1961. A rapid and sensitive sub-micro phosphorus determination. Anal. Chim. Acta. 203–204.
- Büssing, A., G. Schaller, and U. Pfüller. 1998. Generation of reactive oxygen intermediates (ROI) by the thionins from Viscum album L. Anticancer Res. 18:4291–4296. [PubMed] [Google Scholar]
- Büssing, A., G. M. Stein, M. Wagner, B. Wagner, G. Schaller, U. Pfuller, and M. Schietzel. 1999. Accidental cell death and generation of reactive oxygen intermediates in human lymphocytes induced by thionins from Viscum album L. Eur. J. Biochem. 26:279–287. [DOI] [PubMed] [Google Scholar]
- Caaveiro, J. M., A. Molina, J. M. González-Mañas, P. Rodríguez-Palenzuela, F. García-Olmedo, and F. M. Goñi. 1997. Differential effects of five types of antipathogenic plant peptides on model membranes. FEBS Lett. 410:338–342. [DOI] [PubMed] [Google Scholar]
- Caaveiro, J. M., A. Molina, P. Rodriguez-Palenzuela, F. M. Goñi, and J. M. Gonzalez-Mañas. 1998. Interaction of wheat alpha-thionin with large unilamellar vesicles. Protein Sci. 7:2567–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras, L. M., F. J. Aranda, F. Gavilanes, J. M. González-Ros, and J. Villalaín. 2001. Structure and interaction with membrane model systems of a peptide derived from the major epitope region of HIV protein gp41: implications on viral fusion mechanism. Biochemistry. 40:3196–3207. [DOI] [PubMed] [Google Scholar]
- Coulon, A., E. Berkane, A. Sautereau, K. Urech, P. Rouger, and A. Lopez. 2002. Modes of membrane interaction of a natural cysteine-rich peptide: viscotoxin A3. Biochim. Biophys. Acta. 1559:145–159. [DOI] [PubMed] [Google Scholar]
- Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 6:1948–1954. [DOI] [PubMed] [Google Scholar]
- Epple, P., K. Apel, and H. Bohlmann. 1997. Overexpression of an endogenous thionin enhances resistance of Arabidopsis against Fusarium oxysporum. Plant Cell. 9:509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, J., Y. D. Wang, K. P. Shaw, and L. P. Vernon. 1989. Cellular responses to Pyrularia thionin are mediated by Ca2+ influx and phospholipase A2 activation and are inhibited by thionin tyrosine iodination. Proc. Natl. Acad. Sci. USA. 86:5849–5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florack, D. E., and W. J. Stiekema. 1994. Thionins: properties, possible biological roles and mechanisms of action. Plant Mol. Biol. 26:25–37. [DOI] [PubMed] [Google Scholar]
- García -Olmedo, F., A. Molina, J. M. Alamillo, and P. Rodriguez-Palenzuela. 1998. Plant defense peptides. Biopolymers. 47:479–491. [DOI] [PubMed] [Google Scholar]
- Hancock, R. E., and M. G. Scott. 2000. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acd. Sci. USA. 97:8856–8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtorf, S., J. Ludwig-Müller, K. Apel, and H. Bohlemann. 1998. High-level expression of a viscotoxin in Arabidopsis thaliana gives enhanced resistance against Plasmodiophora brassicae. Plant Mol. Biol. 36:673–680. [DOI] [PubMed] [Google Scholar]
- Hope, M. J., M. B. Bally, G. Webb, and P. R. Cullis. 1985. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta. 812:55–65. [DOI] [PubMed] [Google Scholar]
- Huang, W., L. P. Vernon, L. D. Hansen, and J. D. Bell. 1997. Interactions of thionin from Pyrularia pubera with dipalmitoylphosphatidylglycerol large unilamellar vesicles. Biochemistry. 36:2860–2866. [DOI] [PubMed] [Google Scholar]
- Hughes, P., E. Dennis, M. Whitecross, D. Llewellyn, and P. Gage. 2000. The citotoxic plant protein, α-purothionin, forms ion channels in lipid membranes. J. Biol. Chem. 275:823–827. [DOI] [PubMed] [Google Scholar]
- Jung, M. L., S. Baudino, G. Ribereau-Gayon, and J. P. Beck. 1990. Characterization of cytotoxic proteins from mistletoe ( Viscum album L.). Cancer Lett. 51:103–108. [DOI] [PubMed] [Google Scholar]
- Kauppinen, J. R., D. J. Moffatt, H. H. Mantsch, and D. G. Cameron. 1981. Fourier self-deconvolution: a method for resolving intrinsically overlapped bands. Appl. Spectros. 35:271–276. [Google Scholar]
- Kelly, I., M. Pézolet, and D. Marion. 1998. Quantitative orientation of alpha-helical polypeptides by attenuated total internal reflectance infrared spectroscopy. Biophys. J. 74:A309. [Google Scholar]
- Konopa, J., J. M. Woynarowski, and M. Lewandowska-Gumieniak. 1980. Isolation of viscotoxins. Cytotoxic basic polypeptides from Viscum album L. Hoppe Seylers Z. Physiol. Chem. 361:1525–1533. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy, 2nd ed. Plenum Press, New York.
- Lecomte, J. T., D. Kaplan, M. Llinás, E. Thunberg, and G. Samuelsson. 1987. Proton magnetic resonance characterization of phoratoxins and homologous proteins related to crambin. Biochemistry. 26:1187–1194. [DOI] [PubMed] [Google Scholar]
- Mantsch, H. H., and E. N. McElhaney. 1991. Phospholipid phase transitions in model and biological membranes as studied by infrared spectroscopy. Chem. Phys. Lipids. 57:213–226. [DOI] [PubMed] [Google Scholar]
- Mantsch, H. H., D. J. Moffatt, and H. Casal. 1988. Fourier transform methods for spectral resolution enhancement. J. Molec. Struct. 173:285–298. [Google Scholar]
- Mateo, C. R., M. P. Lillo, J. González-Rodríguez, and A. U. Acuña. 1991. Lateral heterogeneity in human platelet plasma membrane and lipids from the time-resolved fluorescence of trans-parinaric acid. Eur. Biophys. J. 20:41–52. [DOI] [PubMed] [Google Scholar]
- Orrú, S., A. Scaloni, M. Giannattasio, K. Urech, P. Pucci, and G. Schaller. 1997. Amino acid sequence, S-S bridge arrangement and distribution in plant tissues of thionins from Viscum album. Biol. Chem. 378:986–996. [DOI] [PubMed] [Google Scholar]
- Paré, Ch., M. Lafleur, F. Liu, R. N. A. Lewis, and R. N. McElhaney. 2001. Differential scanning calorimetry and 2H nuclear magnetic resonance and Fourier transform infrared spectroscopic studies of the effects of transmembrane α-helical peptides on the organization of phosphatidylcholine bilayers. Biochim. Biophys. Acta. 1511:60–73. [DOI] [PubMed] [Google Scholar]
- Parente, R. A., S. Nir, and F. C. Szoka. 1990. Mechanism of leakage of phospholipid vesicle contents induced by the peptide GALA. Biochemistry. 29:8720–8728. [DOI] [PubMed] [Google Scholar]
- Park, J. H., C. K. Hyun, and H. K. Shin. 1999. Cytotoxic effects of the components in heat-treated mistletoe ( Viscum album). Cancer Lett. 139:207–213. [DOI] [PubMed] [Google Scholar]
- Romagnoli, S., R. Ugolini, F. Fogolari, G. Schaller, K. Urech, M. Giannattasio, L. Ragona, and H. Molinari. 2000. NMR structural determination of viscotoxin A3 from Viscum album L. Biochem. J. 350:569–577. [PMC free article] [PubMed] [Google Scholar]
- Schaller, G., K. Urech, and M. Giannattasio. 1996. Cytotoxicity of different viscotoxins and extracts from the european subspecies of Viscum album L. Phytother. Res. 10:473–477. [Google Scholar]
- Shai, Y. 1999. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta. 1462:55–70. [DOI] [PubMed] [Google Scholar]
- Shai, Y., and Z. Oren. 2001. From “carpet” mechanism to de-novo designed disastereomeric cell-selective antimicrobial peptides. Peptides. 22:1629–1641. [DOI] [PubMed] [Google Scholar]
- Stein, G. M., G. Schaller, U. Pfüller, M. Schiuetzel, and A. Büssing. 1999a. Thionins from Viscum album L: influence of the viscotoxins on the activation of granulocytes. Anticancer Res. 19:1037–1042. [PubMed] [Google Scholar]
- Stein, G. M., G. Schaller, U. Pfüller, M. Wagner, B. Wagner, M. Schietzel, and A. Büssing. 1999b. Characterisation of granulocyte stimulation by thionins from European mistletoe and from wheat. Biochim. Biophys. Acta. 1426:80–90. [DOI] [PubMed] [Google Scholar]
- Surewicz, W. K., H. H. Mantsch, and D. Chapman. 1993. Determination of protein secondary structure by Fourier transform infrared spectroscopy: a critical assessment. Biochemistry. 32:389–394. [DOI] [PubMed] [Google Scholar]
- Tabiasco, J., F. Pont, J. J. Fournié, and A. Vercellone. 2002. Mistletoe viscotoxins increase natural killer cell-mediated cytotoxicity. Eur. J. Biochem. 269:2591–2600. [DOI] [PubMed] [Google Scholar]
- Thevissen, K., A. Ghazi, G. W. De Samblanx, C. Brownlee, R. W. Osborn, and W. F. Broekaert. 1996. Fungal membrane responses induced by plant defensins and thionins. J. Biol. Chem. 271:15018–15025. [DOI] [PubMed] [Google Scholar]
- Trotter, P. J., and J. Storch. 1989. 3-[p-(6-phenyl)-1,3,5-hexatrienyl]phenylpropionic acid (PA-DPH): characterization as a fluorescent membrane probe and binding to fatty acid binding proteins. Biochim. Biophys. Acta. 982:131–139. [DOI] [PubMed] [Google Scholar]
- Urech, K., G. Schaller, P. Ziska, and M. Giannattasio. 1995. Comparative study on the citotoxic effect of viscotoxin and mistletoe lectin on tumour cells in culture. Phytother. Res. 9:49–55. [Google Scholar]
- Vila, R., I. Ponte, M. Collado, J. L. Arrondo, and P. Suau. 2001. Induction of secondary structure in a COOH-terminal peptide of histone H1 by interaction with the DNA. An infrared spectroscopy study. J. Biol. Chem. 276:30898–30903. [DOI] [PubMed] [Google Scholar]
- Wang, F., G. H. Naisbitt, L. P. Vernon, and M. Glaser. 1993. Pyrularia thionin binding to and the role of tryptophan-8 in the enhancement of phosphatidylserine domains in erythrocyte membranes. Biochemistry. 32:12283–12289. [DOI] [PubMed] [Google Scholar]
- Wilson, H. A., W. Huang, J. B. Waldrip, A. M. Judd, L. P. Vernon, and J. D. Bell. 1997. Mechanisms by which thionin induces susceptibility of S49 cell membranes to extracellular phospholipase A2. Biochim. Biophys. Acta. 1349:142–156. [DOI] [PubMed] [Google Scholar]
- Woynarowski, J. M., and J. Konopa. 1980. Interaction between DNA and viscotoxins. Cytotoxic basic polypeptides from Viscum album L. Hoppe-Seyler Z. Physiol. Chem. 361:1535–1545. [DOI] [PubMed] [Google Scholar]
- Zhang, T. P., R. N. A. H. Lewis, R. S. Hodges, and R. N. McElhaney. 1992. Interaction of a peptide model of a hydrophobic transmembrane alpha-helical segment of a membrane protein with phosphatidylcholine bilayers: differential scanning calorimetric and FTIR spectroscopic studies. Biochemistry. 31:11572–11578. [DOI] [PubMed] [Google Scholar]
- Zückermann, M. J., and T. Heimburg. 2001. Insertion and pore formation driven by adsorption of proteins onto lipid bilayer membrane-water interfaces. Biophys. J. 81:2458–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]