

Abstract
Interfacial properties of lipid bilayers were studied by 2H nuclear magnetic resonance spectroscopy, with emphasis on a comparison between phosphatidylcholine and sphingomyelin. Spectral resolution and sensitivity was improved by macroscopic membrane alignment. The motionally averaged quadrupolar interaction of interlamellar deuterium oxide was employed to probe the interfacial polarity of the membranes. The D2O quadrupolar splittings indicated that the sphingomyelin lipid-water interface is less polar above the phase transition temperature Tm than below Tm. The opposite behavior was found in phosphatidylcholine bilayers. Macroscopically aligned sphingomyelin bilayers also furnished 2H-signals from the amide residue and from the hydroxyl group of the sphingosine moiety. The rate of water-hydroxyl deuteron exchange could be measured, whereas the exchange of the amide deuteron was too slow for the inversion-transfer technique employed, suggesting that the amide residue is involved in intermolecular hydrogen bonding. Order parameter profiles in mixtures of sphingomyelin and chain-perdeuterated phosphatidylcholine revealed an ordering effect as a result of the highly saturated chains of the sphingolipids. The temperature dependence of the 2H quadrupolar splittings was indicative of lateral phase separation in the mixed systems. The results are discussed with regard to interfacial structure and lateral organization in sphingomyelin-containing biomembranes.
INTRODUCTION
Phosphatidylcholines and sphingomyelins are major constituents of cellular and subcellular bilayer membranes. Phosphatidylcholine is the dominant phospholipid species in most mammalian tissues whereas the sphingomyelin content varies considerably (Yorek, 1993). A large proportion of sphingomyelin is found in the outer leaflet of eukaryotic plasma membranes where it amounts to ∼25% of the total phospholipid in humans and up to 50% in ruminants (Florin-Christensen et al., 2001). The sphingomyelin content is also high in the central nervous system and in the spinal cord. Of particular interest is the observation that the sphingomyelin content increases with age both in the cerebral cortex and in white matter where it reaches 18% of the total phospholipid (Nyberg et al., 1998; Yorek, 1993).
Phosphatidylcholine and sphingomyelin both have phosphocholine headgroups. There are, however, significant differences considering the properties of the interfacial and nonpolar segments of the molecules. Sphingomyelin has only one fatty acid in an amide linkage while the second hydrophobic chain is part of the sphingosine base structure whose interfacial part includes a hydroxyl group at C-3 and a trans double bound between C-4 and C-5 (Scheme 1). The amide plane in sphingolipids was shown to be roughly perpendicular with respect to the average orientation of the hydrophobic chains (Miller et al., 1986; Pascher, 1976; Ruocco et al., 1996).
SCHEME 1.

The interfacial hydroxyl and amide residues of the sphingomyelin molecule are capable of donating and accepting hydrogen bonds while the carbonyl group of the N-acyl chain and the headgroup phosphodiester moiety may act as weak hydrogen bond acceptors. In contrast, there are only hydrogen bond acceptors in the interface of phosphatidylcholines; for example, the fatty acid carbonyls in sn-1 and sn-2 position of the glycerol backbone and the headgroup phosphate. It is tempting to assume that sphingomyelin interacts with itself and with surrounding phosphatidylcholine molecules by forming a network of hydrogen bonds. The amide linkage resides in a region of low dielectric constant, according to available structural data on ceramides (Pascher, 1976) and cerebrosides (Ruocco et al., 1996) which further promotes the formation of strong hydrogen bonds (Barenholz, 1984). This is in line with the low permeability of sphingomyelin bilayers for water and glucose (Hertz and Barenholz, 1975) and with 1H-NMR relaxation time measurements (Schmidt et al., 1977). Similarly, monolayer surface compressional moduli (at a surface pressure of 30mN/m) are indicative of the tighter interfacial packing of sphingomyelins in comparison with chain-matched phosphatidylcholines (Li et al., 2001).
Infrared and Raman spectroscopy was applied to elucidate these structural properties in more detail. However, the results were not conclusive, e.g., it seems unclear whether intra- or intermolecular hydrogen bonds are responsible for the shifts in the amide I and II bands (Hübner and Blume, 1998; Lamba et al., 1991; Villalain et al., 1988). A number of high resolution 1H- and 31P-NMR studies in chloroform solution suggested an intramolecular hydrogen bond between the sphingosine OH group and the phosphodiester moiety of the choline headgroup (Bruzik, 1988; Talbott et al., 2000), in agreement with dynamic differences between the headgroups of phosphatidylcholine and sphingomyelin shown by 14N-NMR spectroscopy (Siminovitch and Jeffrey, 1981).
The transition temperatures Tm of naturally occurring sphingomyelins are close to 37°C (Calhoun and Shipley, 1979) which may result in nonideal mixing and lateral separation of membrane domains under physiological conditions. The involvement of sphingolipids and cholesterol in membrane domains (“rafts”) has been demonstrated (London and Brown, 2000) and the significance for cell signaling and endocytosis is now generally accepted (Anderson and Jacobson, 2002). Sphingolipid binding domains have been recently identified in the amyloid peptide Aβ and in prion and HIV1 proteins (Mahfoud et al., 2002). These findings may be related to a recent report where it was shown that the level of sphingomyelin is increased in the brain of Alzheimer's disease patients (Pettegrew et al., 2001). Further, a relationship was found between the formation of the scrapie prion protein and the sphingomyelin content in neuroblastoma cells (Naslavsky et al., 1999). Thus, an investigation of the properties of the sphingomyelin-water interface is of considerable biomedical interest.
The present study provides a comparison of phosphatidylcholines and sphingomyelins and of mixtures of both lipids with emphasis on interfacial hydration and hydrogen bonding in the vicinity of phase transitions. The role of cholesterol in these systems is beyond the scope of the present work and will be addressed in a later communication. Macroscopically aligned multibilayers were employed (Kurze et al., 2000) and the hydration was kept at or slightly below what is generally considered as “fully hydrated.” This technique avoids problems associated with the heterogeneity of random multibilayer systems (König et al., 1997b; Nagle et al., 1999) and yields maximum sensitivity and resolution. Our results suggest that the fraction of water associated with polar groups in the lipid interface decreases with increasing mole fraction of sphingomyelin in the bilayer.
MATERIALS AND METHODS
Chemicals
Synthetic phospholipids (POPC, POPC-d31, DPPC, DPPC-d62, and α,β-d4-DPPC) were obtained from Avanti Polar Lipids (Alabaster, AL). Sphingolipids (EYSM and BBSM) were also from Avanti or from Sigma-Aldrich (Deisenhofen, Germany). Deuterated solvents were purchased from Cambridge Isotope Laboratories (Promochem GmbH, Wesel, Germany). The lipids were checked for purity by thin layer chromatography before and after NMR measurements.
Sample preparation
Macroscopically aligned phospholipid multibilayers were prepared as described previously (Kurze et al., 2000). Briefly, 30 mg of the lipid or lipid mixture were dissolved in 5 ml of methanol or deuterated methanol (CH3OD). The latter was required when complete exchange of labile protons in sphingomyelins was desirable. The solutions were spread onto 50 ultrathin glass plates (8 × 18 × 0.08 mm; Marienfeld Lab. Glassware, Lauda-Königshofen, Germany) and dried for 20 min under a stream of warm air and then at room temperature for at least 18 h in vacuo (20–30 Pa). The glass plates were stacked on top of each other with gentle pressure and inserted, along with a pair of glass cylinder segments, into an open glass tube (inner diameter 9.8 mm; compare to Fig. 1 in Kurze et al., 2000). Two small paper strips were soaked in D2O and carefully dried to exchange labile hydrogen for deuterium. The strips were attached at the short sides of the glass stacks and a few microliters of D2O were applied onto the paper surface. The tube was rapidly stoppered by two appropriately machined Teflon plugs with silicon O-rings. The membranes were annealed for 8 h at 46°C in the probehead and the annealing process was continuously monitored by 2H-NMR spectroscopy. The hydration step was repeated until the desired hydration was achieved. During long-term acquisitions the tubes were wrapped with parafilm to maintain the hydration level. With this precaution, no changes of the hydration level were observed at 42°C within 5 h. (For further details of the alignment technique, see Kurze et al., 2000.)
FIGURE 1.

2H-NMR spectra of macroscopically aligned multibilayers of DPPC-d62, recorded in the vicinity of the phase transition temperature. (Arrows) Terminal methyl 2H resonances. The bilayers were hydrated with D2O (nw = 23). The corresponding 2H spectra of interlamellar D2O are shown in the inset.
2H-NMR
2H-NMR spectra were recorded using a Varian VXR-400 spectrometer operating at 9.4 T (2H-frequency 61.4 MHz). Spectra of macroscopically aligned samples were acquired using a 10-mm flat wire solenoid. A home-built goniometer, driven by a stepper motor under software control, was used for accurate orientation in the magnetic field at θ = 0° where θ denotes the angle between the normal to the bilayer stack with respect to the field direction (Kurze et al., 2000). Order parameters for individual carbon positions in the sn-1 chain of POPC-d31were obtained according to
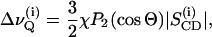 |
(1) |
where χ denotes the quadrupolar coupling constant (170 kHz for a CD bond), P2(cosΘ) the second Legendre polynomial, and 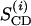 the order parameter of the ith carbon-deuterium bond in the hydrocarbon chain. The quadrupolar echo sequence (Davis et al., 1976) was applied for signal excitation using composite pulses with a 90° pulse width of 7 μs and a pulse spacing of 20 μs. The number of water molecules per lipid headgroup (nw) was obtained by calculating the integral ratio of the respective 2H signals from D2O and from permanently (α,β-d4-DPPC, POPC-d31) or exchange-labeled (EYSM, BBSM) lipids. Integration of the upfield and downfield regions of the 2H NMR spectra, after careful phase adjustment, yielded an estimated error in water content of δnw ≤ 0.5. The recycle delays were chosen so as to avoid saturation.
the order parameter of the ith carbon-deuterium bond in the hydrocarbon chain. The quadrupolar echo sequence (Davis et al., 1976) was applied for signal excitation using composite pulses with a 90° pulse width of 7 μs and a pulse spacing of 20 μs. The number of water molecules per lipid headgroup (nw) was obtained by calculating the integral ratio of the respective 2H signals from D2O and from permanently (α,β-d4-DPPC, POPC-d31) or exchange-labeled (EYSM, BBSM) lipids. Integration of the upfield and downfield regions of the 2H NMR spectra, after careful phase adjustment, yielded an estimated error in water content of δnw ≤ 0.5. The recycle delays were chosen so as to avoid saturation.
The rate constants of labile deuteron exchange were determined using the inversion-transfer technique (Led and Gesmar, 1982). The D2O doublet was selectively inverted using a low-power radiofrequency pulse at the center frequency in the spectrum (90° pulse width, 140 μs). The entire inversion-transfer sequence was πsel-Δ-π/2x-τ-π/2y-τ-acquisition, where the selective 180° pulse, πsel, inverts the D2O signal, and the nonselective 90° pulses, π/2, represent the usual quadrupolar echo sequence. Increasing the delay Δ then results in the expected time variation of those signals that correspond to rapidly exchanging labile deuterons in the lipid interface (compare to Fig. 6). Reference spectra were recorded after each acquisition in the inversion transfer series and signal amplitudes were normalized with respect to the average amplitudes in the reference spectra. The D2O signal was suppressed after signal acquisition by digital filtering (low frequency signal suppression) to minimize baseline distortions and integration artifacts at the OD resonance positions. Thirty five filter coefficients were employed for a sharp filter cutoff (VNMR software, version 5.1). Inversion recovery experiments, i.e., πnonsel-δ-π/2x-τ-π/2y-τ-acquisition, were performed immediately after the inversion transfer series to obtain the spin lattice relaxation time T1z for the interbilayer D2O which was assumed to be independent of the deuteron exchange rate. Nine δ-increments and 64 transients per increment were sufficient for a reliable T1z determination.
FIGURE 6.

Determination of deuteron exchange in the sphingomyelin-water interface (42°C, nw ≈ 29). (A) Typical inversion recovery series of 2H spectra obtained by the pulse sequence πsel-Δ-π/2x-τ-π/2y-τ-acquisition. (B) Variation of the OD signal amplitude with delay time Δ. (C) Arrhenius representation of the temperature dependence of the deuteron exchange rate kOD.
The inversion transfer results were evaluated on the basis of coupled differential equations for a two-side exchange (Led and Gesmar, 1982), e.g., for a deuteron in a hydroxyl group:
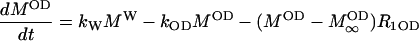 |
(2) |
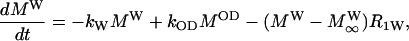 |
(3) |
where MOD, MW, 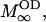 and
and 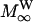 denote the magnetizations and the equilibrium magnetizations, and R1OD, R1W the longitudinal relaxation rates of the OD and water deuterons, respectively. The exchange rate constants kOD and kW refer to the deuteron transfer from D2O to the hydroxyl group in the sphingosine moiety and vice versa. A further condition is provided by the chemical equilibrium, i.e.,
denote the magnetizations and the equilibrium magnetizations, and R1OD, R1W the longitudinal relaxation rates of the OD and water deuterons, respectively. The exchange rate constants kOD and kW refer to the deuteron transfer from D2O to the hydroxyl group in the sphingosine moiety and vice versa. A further condition is provided by the chemical equilibrium, i.e.,
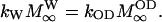 |
(4) |
The results were numerically evaluated as described previously (Kurze et al., 2000).
Differential scanning calorimetry
Measurements were performed using a VP-DSC instrument (Microcal, Northhampton, MA). Scan rates were typically 15°C/h.
RESULTS
Hydration of phosphatidylcholine and sphingomyelin bilayers
Macroscopic alignment of phospholipid membranes has distinct advantages for 2H-NMR spectroscopy, including improved spectral resolution, hydration of the membranes under strict experimental control, and excellent sensitivity for phase transitions. This is shown in Fig. 1 for multibilayers of chain-perdeuterated 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC-d62). The membranes were hydrated with D2O as described in the Methods section and the supporting glass plates were oriented so as to obtain the maximum quadrupolar splittings, indicating that the normal to the membrane surface was parallel to the magnetic field (Θ = 0°, compare to Eq. 1). Before data acquisition the hydration level, typically starting from 30 mol of D2O per mole of phospholipid (nw = 30), was adjusted by slow water evaporation in the magnet to nw = 24 as determined by signal integration. The spectra were recorded at a number of temperatures, including the temperature Tm of the phospholipid main phase transition. Upon cooling from the liquid crystalline state a sudden increase of the 2H line widths and quadrupolar splittings of the deuterated hydrocarbon chains is observed ∼38°C which is close to the transition temperature (37.75°C) reported earlier for DPPC-d62 in excess water (Vist and Davis, 1990), indicating that the multibilayer system can be considered as “fully hydrated.”
The quadrupolar splittings of the interlamellar deuterium oxide (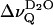 ) vanishes as the temperature reaches Tm, in contrast to the splittings of the phospholipid acyl chains (inset, Fig. 1). This is also shown in Fig. 2, where
) vanishes as the temperature reaches Tm, in contrast to the splittings of the phospholipid acyl chains (inset, Fig. 1). This is also shown in Fig. 2, where 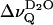 is plotted versus the reduced temperature; for example, (T − Tm)/Tm. Fig. 2 includes data from 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). In the liquid crystalline state
is plotted versus the reduced temperature; for example, (T − Tm)/Tm. Fig. 2 includes data from 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). In the liquid crystalline state 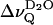 decreases monotonically with decreasing reduced temperature for both phospholipids at full hydration (nw ≈ 24). There is a unique dependence of
decreases monotonically with decreasing reduced temperature for both phospholipids at full hydration (nw ≈ 24). There is a unique dependence of 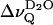 with reference to the reduced temperature although the phase transition temperatures, Tm differ significantly (41°C vs. −5°C for DPPC and POPC, respectively). This homology suggests that the accessibility of polar groups for the interlamellar D2O molecules is very similar in the liquid crystalline state of these lipids.
with reference to the reduced temperature although the phase transition temperatures, Tm differ significantly (41°C vs. −5°C for DPPC and POPC, respectively). This homology suggests that the accessibility of polar groups for the interlamellar D2O molecules is very similar in the liquid crystalline state of these lipids.
FIGURE 2.

2H quadrupolar splittings of interlamellar D2O in multibilayers of DPPC and POPC as a function of the reduced temperature, Tred = (T − Tm)/Tm, where T denotes temperatures in K.
The alignment technique was further employed to explore the interfacial properties of sphingomyelin multibilayers in the vicinity of the phase transition temperature. Natural species from two different sources were analyzed: sphingomyelin from egg yolk (EYSM) and from bovine brain (BBSM). The sphingomyelin samples were first dissolved in the presence of deuterated methanol (CH3OD) before spreading the lipid onto glass plates. This procedure results in complete exchange of the labile hydrogens of the hydroxyl group at C-3 and of the amide residue at C-2 of the sphingosine base by deuterium. 2H-NMR spectra obtained from oriented EYSM multibilayers (nw = 32) are shown in Fig. 3 A where the strong signal from interlamellar D2O has been removed by digital filtering to show more clearly the weak OD and ND resonances. The observation of these signals in the liquid crystalline state of the lipid indicates that chemical exchange among hydroxyl and amide deuterons, as well as exchange with the interlamellar water, is slow on the NMR timescale.
FIGURE 3.

Temperature dependence of 2H-NMR spectra of exchange-labeled EYSM multibilayers. (Left panel) 2H doublets of the hydroxyl and amide residues in the sphingosine backbone. The large D2O doublet signals in the center of the spectra were removed by digital filtering. (Right panel) The corresponding signals from interlamellar D2O (nw ≈ 32).
The assignment of the signals relies on the assumption that rapid rotation of the hydroxyl deuteron about the C=O bond reduces the OD quadrupolar splitting, i.e., the effective splitting corresponds to the projection of the OD vector onto the C-O axis. Further, it has been shown by 13C solid-state NMR spectroscopy that the amide plane is inclined by 35°–52° with respect to the bilayer surface (Ruocco et al., 1996). This would be consistent with an orientation of the N-D bond axis nearly parallel to the membrane surface or, in the present experiment, perpendicular to the magnetic field. Using a typical value for the quadrupolar coupling constant, χ = 240 kHz, for hydrogen bonded amide deuterons in gramicidin A (Prosser et al., 1994), a rigid perpendicular orientation would result in a quadrupolar splitting of 180 kHz. The values obtained experimentally are smaller than this (e.g., 117 kHz at 41°C; see Fig. 3), which may account for motional averaging in the membrane interface as compared to the rigid membrane-bound peptide. Unfortunately, χ strongly depends on the hydrogen bond length, which makes an estimate of quadrupolar splittings uncertain, even if one assumes a rigid perpendicular bond orientation. The quadrupolar splittings of the outer and inner doublet signals decrease between 41°C and 88°C by 8.7% and 16.3%, respectively, which can be attributed to enhanced bilayer fluctuation.
A remarkable feature in Fig. 3 is the temperature dependence of the 2H line widths—i.e., there is a significant broadening of the inner, but not of the outer, doublet components in the high temperature region (T > 70°C). Given the above signal assignment, this differential broadening most probably indicates that chemical exchange of the hydroxyl deuteron with deuterons from interfacial water is more effective than the exchange of the amide deuteron. Below 40°C the amplitudes of both signals decrease simultaneously, which is most likely due to line broadening when the lipid enters the gel state. Eventually, <30°C the signals are no longer detectable.
The spectral features in Fig. 3 are correlated with the phase transition as detected by differential scanning calorimetry (DSC; Fig. 4). The temperature dependence of the signal amplitudes of the ND and OD resonances (normalized with respect to the signal amplitudes obtained above Tm) are shown as solid symbols while the solid lines represent normalized integrals of the heat capacity obtained by DSC, i.e., 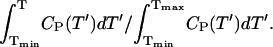 The almost perfect agreement between this data indicates that the 2H signal amplitudes sense the phase transition with great accuracy. The transition is broader for BBSM than for EYSM, which can be attributed to the greater heterogeneity of the fatty acid composition of the former sphingomyelin species.
The almost perfect agreement between this data indicates that the 2H signal amplitudes sense the phase transition with great accuracy. The transition is broader for BBSM than for EYSM, which can be attributed to the greater heterogeneity of the fatty acid composition of the former sphingomyelin species.
FIGURE 4.

The phase transition of (A) fully hydrated EYSM (nw ≈ 32) and (B) BBSM (nw ≈ 41) as detected by 2H NMR in aligned multibilayers and by DSC (multilamellar vesicles). Normalized signal amplitudes (solid symbols), I(t)/Imax, of ND (▴) and OD (▾) resonances, where Imax denotes the amplitudes in the liquid crystalline state. Degree of phase transition obtained by DSC (solid lines). (○) Quadrupolar splittings of interlamellar D2O.
The splittings 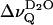 of the D2O signal (that has been removed in Fig. 3, left panel) are also related to the phase transition (open symbols in Fig. 4). Close to the transition temperature,
of the D2O signal (that has been removed in Fig. 3, left panel) are also related to the phase transition (open symbols in Fig. 4). Close to the transition temperature, 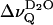 runs through an inflection point for EYSM and through a minimum for BBSM, respectively. In the EYSM sample there is also a minimum ∼51°C, i.e., ≈13°C above the phase transition temperature (≈38°C). Notably,
runs through an inflection point for EYSM and through a minimum for BBSM, respectively. In the EYSM sample there is also a minimum ∼51°C, i.e., ≈13°C above the phase transition temperature (≈38°C). Notably, 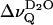 is rather small or zero in the liquid crystalline state of the sphingolipids, in contrast to lecithin bilayers (compare to Fig. 2). Fig. 5 summarizes the temperature dependence of the water splittings obtained at similar hydration for EYSM and DPPC alone and for a EYSM/DPPC mixtures at 2:1 and 1:1 molar ratios. Obviously, sphingomyelin controls the properties of the lipid-water interface in the mixed systems.
is rather small or zero in the liquid crystalline state of the sphingolipids, in contrast to lecithin bilayers (compare to Fig. 2). Fig. 5 summarizes the temperature dependence of the water splittings obtained at similar hydration for EYSM and DPPC alone and for a EYSM/DPPC mixtures at 2:1 and 1:1 molar ratios. Obviously, sphingomyelin controls the properties of the lipid-water interface in the mixed systems.
FIGURE 5.

Comparison of the D2O quadrupolar splittings in the vicinity of the phase transition temperature for DPPC (▴), EYSM (▪), and EYSM/DPPC mixtures at 2:1 (○) and 1:1 (⋄) molar ratio. Hydration nw ≈ 25.
Interfacial deuteron exchange
The chemical exchange of labile deuterons (i.e., the hydroxyl and amid deuterons) was measured as described previously, using an inversion-transfer technique (Kurze et al., 2000). This involves inversion of the D2O signal in the center of the spectrum by a frequency selective low power pulse followed by a variable delay time Δ and a nonselective quadrupolar echo sequence for excitation of the entire spectrum. Magnetization transfer then results in variation of the signal amplitudes of those labile lipid deuterons that exchange with the surrounding water at a rate larger than the spin lattice relaxation rate 1/T1z.
Fig. 6, A and B, show a series of 2H-NMR spectra of aligned EYSM multibilayers (nw = 29) recorded with increasing delay time Δ and the evaluation of the magnetization transfer according to Eqs. 2–4 (see Methods). Again, the central D2O signal was digitally filtered to facilitate observation of the OD and ND signals. The doublet that has been attributed to the sphingosine OD residue varies with increasing Δ whereas the amplitudes of the ND resonances hardly change within the time course of the experiment, indicating that the amide deuteron exchange rate is much smaller than the exchange rate of the hydroxyl deuteron. This result is in qualitative agreement with the temperature dependence of the 2H line broadening shown in Fig. 3.
The rate constant kOD increases with increasing temperature (Fig. 6 C). For EYSM (nw = 26), kOD was 387 ± 65 s−1 at 39°C (close to the phase transition temperature) and 1000 ± 75 s−1 at 55°C. The rather large exchange rate obtained at 55°C is comparable to the rate of proton exchange in pure water (1100 s−1 at 25°C, pH = 7) (Luz and Meiboom, 1964) which argues against strong intermolecular hydrogen bonding of the EYSM hydroxyl group. In contrast, the amide hydrogen seems to be protected from exchange with the interlamellar water. The exchange rate constants at slightly different hydration values follow the same trend (Fig. 6 C) which justifies an evaluation in terms of a single activation energy. Only data for nw ≥ 20 were included in the analysis as membranes at lower hydration values certainly cannot be considered as “fully hydrated.” Linear regression of the entire data set yields an average activation energy of 41 ± 6kJ/mole which is considerably larger than the activation energies reported for proton exchange in pure water (see Discussion).
The exchange rate constants also increased as a function of nw; e.g., for EYSM, at 47°C from 350 ± 20 s−1 to 750 ± 90 s−1 ongoing from nw = 17 to nw = 29 (data not shown). Unfortunately, a determination of kOD at lower hydration values was not feasible as additional D2O signals with unusually large quadrupolar splittings appeared <nw ≈ 15 in pure SM multibilayers, e.g., three signal components could be identified at 47°C at nw = 12. Moreover, the ND and OD signals were no longer detectable, indicating that the phase transition temperature Tm increased >10°C due to dehydration of the membrane interface. A very broad, temperature-dependent component of the water signal persisted above Tm, reaching a quadrupolar splitting Δν > 10 kHz at 57°C (data not shown). Thus, an interpretation of exchange data would be impossible even if inversion of the water magnetization had been achieved.
Binary sphingomyelin/phosphatidylcholine mixtures
Binary mixtures of BBSM and sn-1-d31-palmitoyl-sn-2-oleoyl-glycero-3-phosphocholine (POPC-d31) were studied with regard to hydration, intermolecular packing, and phase transition, again using macroscopically oriented membrane multibilayers. BBSM was chosen here, as a comprehensive investigation of the system BBSM/egg lecithin/water has been published previously (Untracht and Shipley, 1977). Likewise, egg lecithin has a high amount of oleic acid in the sn-2 position and thus closely resembles POPC. Fig. 7 A summarizes the quadrupolar splittings of interlamellar D2O at the same overall hydration (nw ≈ 26) obtained over a temperature range from 20°C to 50°C for pure POPC-d31 and for mixtures of BBSM and POPC-d31 at molar ratios 2:1 and 1:2, respectively. The splittings increase monotonously in this temperature range for POPC-d31 alone and for the mixture containing 33 mol % of BBSM, shown in Fig. 7 A, whereas in the presence of 67 mol % of BBSM, 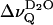 goes through a minimum at 30°C. The minimum can be attributed to a phase transition, in analogy to the results obtained with BBSM alone (Fig. 4). At 46°C, with increasing mole fraction of BBSM, the splittings decrease in a nonlinear fashion, which may reflect nonideal mixing of the system, even in the liquid crystalline state at some distance from the phase transition temperature (Fig. 7 B).
goes through a minimum at 30°C. The minimum can be attributed to a phase transition, in analogy to the results obtained with BBSM alone (Fig. 4). At 46°C, with increasing mole fraction of BBSM, the splittings decrease in a nonlinear fashion, which may reflect nonideal mixing of the system, even in the liquid crystalline state at some distance from the phase transition temperature (Fig. 7 B).
FIGURE 7.

(A) Temperature dependence of the D2O quadrupolar splittings in BBSM/POPC mixtures. (B) D2O splitting as a function of the BBSM mole fraction at 46°C. Hydration, nw ≈ 26.
A characteristic feature of naturally occurring sphingomyelins is the high proportion of saturated long-chain fatty acids (Barenholz and Thompson, 1999). Deuterium order parameter profiles offer a direct proof of the effect of the sphingolipids on the overall packing density in mixed bilayers. Segmental 2H order parameters of the sn-1 chain of POPC–d31 in sphingomyelin/POPC mixtures, normalized with respect to the corresponding order parameter values for POPC-d31 alone, are shown as a function of carbon position in Fig. 8. The signal assignments rely on the usual assumption that the order parameters decrease with increasing distance from the membrane-water interface. Quadrupolar splittings for carbon positions 2–8 were poorly resolved (the order parameter “plateau”) and rather insensitive to the presence of the sphingolipids. Therefore, only resolved resonances are included in Fig. 8.
FIGURE 8.

Order parameters of the POPC-d31 acyl chain in sphingomyelin/POPC mixtures, normalized with respect to the corresponding order parameters of POPC-d31 alone. (×) EYSM/POPC-d31, 2:1 mol/mole, 42°C; (▪) BBSM/POPC-d31, 2:1 mol/mole, 42°C; (•) BBSM/POPC-d31, 1:2 mol/mole, 42°C; and (○) BBSM/POPC-d31, 1:2 mol/mole, 21°C.
Addition of BBSM (BBSM/POPC-d31, 2:1 mol/mole) leads to a remarkable increase of the normalized POPC-d31 order parameters in the middle of the bilayer, with a flat maximum around carbon position 14. Thus, addition of BBSM reduces the probability of chain isomerization which amounts to an increased average projected length of the POPC sn-1 chain as compared to a pure POPC bilayer (Salmon et al., 1987; Schindler and Seelig, 1975). The effect clearly increases with higher molar ratios of BBSM/POPC-d31, i.e., the normalized ordering profile is lower at 1:2 than at 2:1 molar ratio, whereas the temperature dependence seems to be merely small. Interestingly, the flat maximum shifted toward the membrane-water interface when BBSM was replaced with EYSM (Fig. 8, crosses connected by dashed lines), which must be attributed to the shorter acyl chain length in this sphingomyelin species (16:0, 83.9%; 18:0, 6.3%; other, 9.8% in EYSM vs. 18:0, 45.5%; 22:0, 7.2%; 24:0, 23.3%; and other, 14% in BBSM; Jendrasiak and Smith, 2001). An even larger increase of the POPC-d31 order parameters was observed when sphingomyelin was replaced with the synthetic phospholipid DPPC (order parameter ratio ≈ 1.3 at carbon 13 in a 2:1 DPPC/POPC-d31 mixture at 42°C, data not shown). It can be concluded that there is a condensing effect in mixed bilayers containing natural sphingomyelins due to the high abundance of saturated chains. It may also be argued that interfacial hydrogen bonding contributes to the closer lipid packing (Barenholz and Thompson, 1999).
The data in Fig. 8 correspond to virtually fully hydrated membranes (nw ≈ 27) in the liquid crystalline state where the components are completely miscible. Decreasing the interlamellar hydration shifts the phase transition toward higher temperatures and eventually results in lateral phase separation (Untracht and Shipley, 1977). One can expect that in the two-phase region the acyl chains of POPC will return to the more disordered state when sphingomyelin in the gel state separates from the mixture. This is shown in Fig. 9 A where the 2H quadrupolar splittings of the terminal CD3 residue, 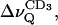 of the POPC-d31 sn-1 chain are plotted as a function of temperature for two different hydration values and three lipid compositions.
of the POPC-d31 sn-1 chain are plotted as a function of temperature for two different hydration values and three lipid compositions.
FIGURE 9.

(A) Quadrupolar splittings of the ω-CD3 group of POPC-d31 in mixed BBSM/POPC-d31 multibilayers. At nw ≈ 26: (▪) POPC-d31 alone; (•) BBSM/POPC-d31, 1:2 mol/mole; (▾) BBSM/POPC-d31, 2:1 mol/mole; and at nw ≈ 11: (□) POPC-d31 alone; (○) BBSM/POPC-d31, 1:2 mol/mole; (Δ) BBSM/POPC-d31, 2:1 mol/mole. (B) 2H spectra of interlamellar D2O obtained in the BBSM/POPC-d31 mixture at nw ≈ 11 (2:1 mol/mole).
For POPC alone, 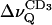 decreases monotonously with increasing temperature, both at full (nw ≈ 26) and reduced (nw ≈ 11) hydration. Addition of BBSM, as expected from Fig. 8, increases the quadrupolar splitting,
decreases monotonously with increasing temperature, both at full (nw ≈ 26) and reduced (nw ≈ 11) hydration. Addition of BBSM, as expected from Fig. 8, increases the quadrupolar splitting, 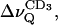 which is more pronounced when nw ≈ 11. Upon lowering the temperature, i.e., when the system approaches the phase transition from the liquid crystalline state,
which is more pronounced when nw ≈ 11. Upon lowering the temperature, i.e., when the system approaches the phase transition from the liquid crystalline state, 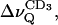 for the BBSM/POPC-d31 2:1 mol/mole mixture, deviates toward the splittings obtained for pure POPC-d31, most probably as a consequence of chain demixing. At low membrane hydration (nw ≈ 11) a slight inflection of the temperature dependence can be also recognized for the 1:2 mixture. A similar behavior was found for the “plateau” values, comprising the methylene deuterons from C-2 to C-7 of the sn-1 chain; i.e., the splittings reach a maximum at 39°C and approach the values observed for POPC-d31 alone ∼20°C (data not shown). Note that for nw ≈ 26 the breakpoint occurs at 31°C (arrow in Fig. 9 A), where
for the BBSM/POPC-d31 2:1 mol/mole mixture, deviates toward the splittings obtained for pure POPC-d31, most probably as a consequence of chain demixing. At low membrane hydration (nw ≈ 11) a slight inflection of the temperature dependence can be also recognized for the 1:2 mixture. A similar behavior was found for the “plateau” values, comprising the methylene deuterons from C-2 to C-7 of the sn-1 chain; i.e., the splittings reach a maximum at 39°C and approach the values observed for POPC-d31 alone ∼20°C (data not shown). Note that for nw ≈ 26 the breakpoint occurs at 31°C (arrow in Fig. 9 A), where 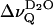 goes through a minimum (compare to Fig. 7). Our results are in excellent agreement with the phase diagram obtained by Untracht and Shipley, which shows a broad two-phase region for a 2:1 mol/mole BBSM/egg lecithin mixture in the same temperature range at a hydration of nw ≈ 11 (Untracht and Shipley, 1977).
goes through a minimum (compare to Fig. 7). Our results are in excellent agreement with the phase diagram obtained by Untracht and Shipley, which shows a broad two-phase region for a 2:1 mol/mole BBSM/egg lecithin mixture in the same temperature range at a hydration of nw ≈ 11 (Untracht and Shipley, 1977).
Lateral phase separation can also be recognized in the D2O spectra at nw ≈ 11 (but not at hydration values nw > 20). A broad second component that increases with decreasing temperature appears <40°C (Fig. 9 B). The narrow component of the composite spectra disappears <30°C. The coexistence of 2H subspectra falls within the temperature range where lipid segregation occurs according to Fig. 9 A. Obviously, the 2H lines are still broadened by diffusional exchange among the domains (compare to the spectrum recorded at 36.5°C). Much narrower lines were observed at lower hydration (nw ≈ 9), indicating that either the domain size increases or the rate of lateral water diffusion decreases (not shown).
DISCUSSION
The hydration of phosphatidylcholines has been thoroughly studied by 2H-NMR spectroscopy, using the motionally averaged quadrupolar interaction of deuterium oxide (Faure et al., 1997; Finer, 1973; Finer and Darke, 1974; Gawrisch et al., 1985; Klose et al., 1992; Salsbury et al., 1972; Volke et al., 1994a,b) or by employing selectively labeled phospholipids (Bechinger and Seelig, 1991; Ulrich and Watts, 1994). However, there are still controversial subjects, e.g., the number of bound water molecules and the related area per lipid, that have only recently been critically reevaluated (Katsaras, 1998; Nagle and Tristram-Nagle, 2000). This uncertainty can be attributed to the inherent inhomogeneity of multilamellar liposomal suspensions that have been frequently used as model systems (König et al., 1997a). As shown in a recent x-ray study, the unit cell spacing increases with temperature in the liquid crystalline state of phosphatidylcholine liposomes (nw ≥ 30), indicating that the interbilayer space imbibes additional water from membrane defects or from the surroundings as a consequence of the onset of bilayer undulations (Costigan et al., 2000). Macroscopic alignment of multibilayers, keeping nw slightly <30, attenuates surface undulations and ensures effective annealing of bilayer defects which justifies the notion that the entire D2O signal reflects water that is in contact with a flat membrane interface. Hence, the average hydration per lipid nw can be conveniently obtained by signal integration.
The small residual quadrupolar splitting, 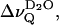 is the result of an effective motional averaging, given the large quadrupolar coupling constant of 220 kHz obtained for deuterium oxide in ice (Soda and Chiba, 1969). Nonetheless,
is the result of an effective motional averaging, given the large quadrupolar coupling constant of 220 kHz obtained for deuterium oxide in ice (Soda and Chiba, 1969). Nonetheless, 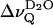 represents an extremely sensitive and potentially useful parameter that has been previously correlated with microscopic properties of the lipid water interface (Bryant et al., 1992a,b; Gawrisch et al., 1992; Volke et al., 1994a,b). It is shown here that the motional anisotropy of the interfacial water (as deduced from
represents an extremely sensitive and potentially useful parameter that has been previously correlated with microscopic properties of the lipid water interface (Bryant et al., 1992a,b; Gawrisch et al., 1992; Volke et al., 1994a,b). It is shown here that the motional anisotropy of the interfacial water (as deduced from 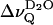 ) reflects a uniform behavior in phosphatidylcholines that differ by as much as 46°C in their respective phase transition temperatures Tm. The temperature dependence of
) reflects a uniform behavior in phosphatidylcholines that differ by as much as 46°C in their respective phase transition temperatures Tm. The temperature dependence of 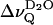 above Tm (Fig. 2) can be attributed to the increasing water-accessible surface area; e.g., for DPPC, the area per lipid changes from 0.48 nm2 at 20°C to 0.64 nm2 at 50°C (Nagle and Tristram-Nagle, 2000). According to x-ray crystallography, the C=O bond of the phosphatidylcholine sn-2 chain is directed with its negative partial charge toward the interlamellar space (Pearson and Pascher, 1979). This results in a dipole potential that, in turn, leads to weak orientation of the water dipoles. The exposure of carbonyls increases with increasing area per lipid which further amplifies the water orientation. This simple electrostatic perspective is in agreement with a recent potential of mean force calculation which yielded a surprisingly narrow orientational distribution of water molecules at a smooth negatively charged surface (Lindahl, 2001). The orientational order parameter of interfacial water was indeed correctly reproduced in a recent molecular dynamics study of a phosphatidylcholine bilayer in the liquid crystalline state (Åman et al., 2003). Two regions along the bilayer normal with negative and positive O-H order parameters were identified, in agreement with an idea introduced in an earlier study on ion and water binding (Lindblom et al., 1976). It remains to be studied whether this sign reversal causes cancellation of the D2O quadrupolar splittings close to the main phase transition.
above Tm (Fig. 2) can be attributed to the increasing water-accessible surface area; e.g., for DPPC, the area per lipid changes from 0.48 nm2 at 20°C to 0.64 nm2 at 50°C (Nagle and Tristram-Nagle, 2000). According to x-ray crystallography, the C=O bond of the phosphatidylcholine sn-2 chain is directed with its negative partial charge toward the interlamellar space (Pearson and Pascher, 1979). This results in a dipole potential that, in turn, leads to weak orientation of the water dipoles. The exposure of carbonyls increases with increasing area per lipid which further amplifies the water orientation. This simple electrostatic perspective is in agreement with a recent potential of mean force calculation which yielded a surprisingly narrow orientational distribution of water molecules at a smooth negatively charged surface (Lindahl, 2001). The orientational order parameter of interfacial water was indeed correctly reproduced in a recent molecular dynamics study of a phosphatidylcholine bilayer in the liquid crystalline state (Åman et al., 2003). Two regions along the bilayer normal with negative and positive O-H order parameters were identified, in agreement with an idea introduced in an earlier study on ion and water binding (Lindblom et al., 1976). It remains to be studied whether this sign reversal causes cancellation of the D2O quadrupolar splittings close to the main phase transition.
An alternative interpretation invoked the Landau theory of phase transitions (Hawton and Doane, 1987; Jähnig, 1981a,b). Within this framework, Hawton and Doane derived a universal relation for the temperature dependence of 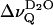 obtained in oriented multilayers of saturated phospholipids, i.e.,
obtained in oriented multilayers of saturated phospholipids, i.e., 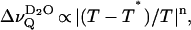 where T* denotes the critical temperature that equals the phase transition temperature Tm for the case of a first-order transition (Hawton and Doane, 1987). Hence, the collapse of the D2O quadrupolar splitting observed upon reaching the phase transition temperature was attributed to area fluctuations which causes fluctuating defects in the membrane-water interface where isotropic motion of the water molecules averages the residual quadrupolar interaction of the water deuterons. It must be noted, however, that this view is still controversial and that it may not be applicable to mixed bilayer systems (vide infra).
where T* denotes the critical temperature that equals the phase transition temperature Tm for the case of a first-order transition (Hawton and Doane, 1987). Hence, the collapse of the D2O quadrupolar splitting observed upon reaching the phase transition temperature was attributed to area fluctuations which causes fluctuating defects in the membrane-water interface where isotropic motion of the water molecules averages the residual quadrupolar interaction of the water deuterons. It must be noted, however, that this view is still controversial and that it may not be applicable to mixed bilayer systems (vide infra).
The hydration of sphingomyelins has not been studied in such detail. Only recently Jendrasiak and co-workers presented a comparative survey of binding isotherms using BET theory for a discrimination of weak and strong water adsorption (Jendrasiak and Smith, 2001). Unfortunately, measurements were performed exclusively at 22°C; i.e., the resulting isotherms corresponded to the gel state for all sphingomyelins studied. Hence, the data obtained by these authors may not be directly comparable with our results. Moreover, the hydration levels attainable by their method at a relative humidity of 100% are low—possibly a consequence of the so-called vapor pressure paradox, a phenomenon which has been recently attributed to temperature gradients within the sample volume (Katsaras, 1998).
The unusually low D2O splitting in the liquid crystalline state of sphingomyelins may be attributed to the average orientation of the amide plane with respect to the membrane surface which involves an in-plane orientation of the C=O bond (Miller et al., 1986; Pascher, 1976; Ruocco et al., 1996). This geometry results in a small effective dipole moment of the carbonyl group (i.e., the dipole moment projected onto the bilayer normal), which excludes strong interaction with a first water layer. The small or negligible contribution of the first water layer to the overall dipole potential of the membrane interface may account for the small 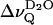 values in the liquid crystalline state of the sphingomyelins (Gawrisch et al., 1992). It may be further assumed that intermolecular hydrogen bonding prevents interlamellar water from entering the interfacial lipid layer. Both conditions, i.e., the average orientation of the carbonyl group and the interfacial hydrogen bond network, seem to make the sphingomyelin lipid-water interface less hydrophilic as compared to the phosphatidylcholine interface. The extent of this interfacial lipid-lipid interaction is likely to depend on the average cross-sectional area of the respective sphingomyelin. This is borne out by the observation that
values in the liquid crystalline state of the sphingomyelins (Gawrisch et al., 1992). It may be further assumed that intermolecular hydrogen bonding prevents interlamellar water from entering the interfacial lipid layer. Both conditions, i.e., the average orientation of the carbonyl group and the interfacial hydrogen bond network, seem to make the sphingomyelin lipid-water interface less hydrophilic as compared to the phosphatidylcholine interface. The extent of this interfacial lipid-lipid interaction is likely to depend on the average cross-sectional area of the respective sphingomyelin. This is borne out by the observation that 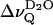 reaches a minimum ∼3°C and 13°C >Tm for BBSM and EYSM, respectively, suggesting that the area per molecule associated with these temperatures is most favorable for the H-bond network. Due to the presence of long acyl chains (45.5%, 18:0; 7.2%, 22:0; and 23.3%, 24:0; Jendrasiak and Smith, 2001), BBSM reaches the critical area close to the phase transition temperature whereas EYSM (83.9%, 16:0) needs a somewhat higher temperature.
reaches a minimum ∼3°C and 13°C >Tm for BBSM and EYSM, respectively, suggesting that the area per molecule associated with these temperatures is most favorable for the H-bond network. Due to the presence of long acyl chains (45.5%, 18:0; 7.2%, 22:0; and 23.3%, 24:0; Jendrasiak and Smith, 2001), BBSM reaches the critical area close to the phase transition temperature whereas EYSM (83.9%, 16:0) needs a somewhat higher temperature.
The packing of the hydrocarbon chains in the gel state may then force the interfacial segments into a conformation with an electrostatic orientation potential that favors the alignment of water dipoles. It must be noted that Gawrisch et al. (1992) observed a significantly smaller water splitting over a large range of hydration values for DPPC than for the ether lipid dihexadecylphosphatidylcholine (DHPC), although the dipole potential for DPPC was larger by 118 mV than that of DHPC (due to the absence of the sn-2 carbonyl group in the ether lipid). Likewise, measurements of the dipole potential in gel state monolayers yielded smaller values for BBSM than for DPPC (328 mV vs. 575 mV; McIntosh et al., 1992) which seems to be inconsistent with the assumption of a simple relation between dipole potential and orientational ordering as measured by the D2O quadrupolar splitting (compare to Fig. 5). A detailed analysis of bond orientations and dihedral angles will be required, e.g., by selective isotope labeling of the interfacial segments, to ascertain the conformational change that sphingomyelin undergoes during the phase transition.
The homogeneous alignment of bilayers between glass plates permits a quantitative assessment of the exchange between interlamellar deuterium oxide and labile amide and hydroxyl deuterons in the lipid-water interface. The deuteron exchange rate kOD of the hydroxyl group of BBSM is ∼600 s−1 at 45°C and nw = 20, which is the same order of magnitude as the value obtained previously for the headgroup hydroxyl deuterons of POPG (kOD ≈ 800 s−1 at 30°C, nw = 20; Kurze et al., 2000). This compares favorably with H3O+/H2O and H2O/OH− hydrogen exchange rates determined for pure water (Luz and Meiboom, 1964; Meiboom, 1961), indicating that the sphingomyelin hydroxyl group has access to the interlamellar hydration layer. It may be noted, however, that the activation energies are 10 kJ/mol and 8.8 kJ/mol, respectively, for the H3O+ and OH− exchange rates versus 41 kJ/mol obtained for the water-sphingomyelin deuteron exchange. A similar activation energy has been reported earlier for the proton exchange in hydrated collagen fibers (Migchelsen and Berendsen, 1973).
In sharp contrast, the amide deuteron exchange is too slow for the inversion transfer technique, suggesting that in the liquid crystalline bilayer the amide deuteron is involved in strong hydrogen bonding. These results are in agreement with the average backbone conformation derived from 13C solid state NMR spectroscopy where it was shown that the sphingosine OH group is directed toward the aqueous phase (Ruocco et al., 1996). On the other hand, strong intermolecular hydrogen bonding of the sphingosine OH group has been deduced from infrared spectroscopy of BBSM bilayers in the gel state (Lamba et al., 1991). The observation that both 2H signals of the labile deuterons broaden upon entering the gel phase supports the assumption of a conformational change resulting in intermolecular hydrogen bonding of the hydroxyl group, in agreement with the earlier results.
A particularly intriguing aspect of the BBSM structure is its chain length asymmetry. Binary mixtures of BBSM and POPC were studied as a paradigm for the interaction among medium chain unsaturated glycerophospholipids and sphingolipids with a long N-acyl chain, a situation prevalent in brain tissue. The phase diagram obtained by Untracht and Shipley (1977) served as a guideline that facilitated the interpretation of the present results. This phase diagram shows complete miscibility of the components above 44°C and the formation of a paratactic molecular compound at 33 mol % lecithin and 66 mol % sphingomyelin <20°C. Lateral phase separation was detected using x-ray diffraction and polarized light microscopy upon cooling below the liquidus line.
In the present study we have focused on the interfacial properties of the BBSM/POPC system in the physiologically relevant temperature range from 20°C to 45°C (neglecting the intermolecular compound). Considering the liquid crystalline state of the mixtures, there are two observations of interest, i.e., the acyl chain order of the selectively deuterated lecithin (POPC-d31) increases and the orientational order of the interfacial water decreases with increasing BBSM concentration in the system. Specifically, the length of the so-called order parameter plateau increases, which amounts to an increasing contribution of the largest quadrupolar splitting in the 2H spectrum (compare to Fig. 1). It is customary to assume that the plateau corresponds to the first methylene segments that flow from the lipid-water interface toward the bilayer center. Here we employed the ratios of segmental order parameters (with reference to the corresponding order parameter values of POPC alone) rather than the usual order parameter profiles (Fig. 8). The ratios are able to demonstrate the ordering effect of sphingomyelin more clearly than a direct comparison of the profiles. Thus, the overall packing density of the lipid mixture increases as a result of the presence of saturated chains, probably in combination with the capability of the sphingolipid to form interfacial hydrogen bonds. The “condensing effect” clearly increases with BBSM concentration while there was no indication of fluid-to-fluid demixing, in agreement with the published phase diagram (Untracht and Shipley, 1977). An analogous behavior has been recently reported for mixtures containing a ceramide (<20 mol %) and POPC where both components were selectively deuterated (Hsueh et al., 2002). It may be noted, however, that in the POPC/ceramide system a metastable solid phase appears when the ceramide content exceeds 20 mol %.
Increasing the mole fraction of sphingomyelin in the mixture also reduces the ordering of interfacial water, probably as a result of the diminished fraction of water molecules in the carbonyl region of the bilayer. Reduced spontaneous transbilayer diffusion of glucose has been observed earlier as a further consequence of the condensing effect of sphingomyelin in mixed phospholipid membranes (Hertz and Barenholz, 1975). It can be also assumed that the presence of sphingomyelin modulates the interaction of amphiphilic peptides and proteins with the membrane interface.
The onset and completion of the phase transition in the 2:1 BBSM/POPC-d31 mixture is reflected by the 2H quadrupolar splittings of the deuterated POPC alkyl chain which can be most easily recognized by the splittings arising from the terminal methyl group at reduced interlamellar hydration (Fig. 9 A). The observation of a single 2H doublet over the temperature range of the transition indicates that almost pure BBSM gel state domains grow out of the mixture when the system crosses the liquidus line. This again suggests that the phase behavior of the BBSM/POPC mixture differs from that of the system ceramide/POPC where a mixed ceramide/POPC gel phase was found below the liquidus line (Hsueh et al., 2002).
Surface domains may also account for the observation of D2O subspectra over the temperature range where phase separation occurs (Fig. 9 B). Such domains have to be sufficiently large so as to preclude signal averaging by random lateral motion of the interlamellar D2O. An order-of-magnitude estimate of the domain size can be made, based on the diffusion equation in two dimensions. Using the diffusion constant for lateral interlamellar water motion of 2.5 × 10−10m2s−1 obtained at the same temperature and hydration in egg phosphatidylcholine (Wassall, 1996), and taking account of the frequency separation of the coexisting D2O signals, this calculation yields a minimum domain area required for the observation of separate water spectra of 6 μm2. This is well within the range of domain sizes determined by epifluorescence microscopy in monolayers of phospholipid/cholesterol or sphingomyelin/cholesterol mixtures (Dietrich et al., 2001; Radhakrishnan et al., 2000).
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, grant BE 828/8.
Abbreviations used: BBSM, bovine brain sphingomyelin; EYSM, egg yolk sphingomyelin; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine.
References
- Åman, K., E. Lindahl, O. Edholm, P. Hakansson, and P. O. Westlund. 2003. Structure and dynamics of interfacial water in an L α phase lipid bilayer from molecular dynamics simulations. Biophys. J. 84:102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, R. G., and K. Jacobson. 2002. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 296:1821–1825. [DOI] [PubMed] [Google Scholar]
- Barenholz, Y. 1984. Sphingomyelin-lecithin balance in membranes: composition, structure, and function relationships. In Physiology of Membrane Fluidity. M. Shinitzky, editor. CRC Press, Boca Raton, FL. pp.131–73.
- Barenholz, Y., and T. E. Thompson. 1999. Sphingomyelin: biophysical aspects. Chem. Phys. Lipids. 102:29–34. [DOI] [PubMed] [Google Scholar]
- Bechinger, B., and J. Seelig. 1991. Conformational changes of the phosphatidylcholine headgroup due to membrane dehydration. A 2H-NMR study. Chem. Phys. Lipids. 58:1–5. [DOI] [PubMed] [Google Scholar]
- Bruzik, K. S. 1988. Conformation of the polar headgroup of sphingomyelin and its analogues. Biochim. Biophys. Acta. 939:315–326. [DOI] [PubMed] [Google Scholar]
- Bryant, G., J. M. Pope, and J. Wolfe. 1992a. Low hydration phase properties of phospholipid mixtures. Evidence for dehydration-induced fluid-fluid separations. Eur. Biophys. J. 21:223–232. [Google Scholar]
- Bryant, G., J. M. Pope, and J. Wolfe. 1992b. Motional narrowing of the 2H NMR spectra near the chain melting transition of phospholipid/D2O mixtures. Eur. Biophys. J. 21:363–367. [DOI] [PubMed] [Google Scholar]
- Calhoun, W. I., and G. G. Shipley. 1979. Fatty acid composition and thermal behavior of natural sphingomyelins. Biochim. Biophys. Acta. 555:436–441. [DOI] [PubMed] [Google Scholar]
- Costigan, S. C., P. J. Booth, and R. H. Templer. 2000. Estimations of lipid bilayer geometry in fluid lamellar phases. Biochim. Biophys. Acta. 1468:41–54. [DOI] [PubMed] [Google Scholar]
- Davis, J. H., K. R. Jeffrey, M. Bloom, and M. I. Valic. 1976. Quadrupolar echo deuteron magnetic resonance spectroscopy in ordered hydrocarbon chains. Chem. Phys. Lett. 42:390–394. [Google Scholar]
- Dietrich, C., L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson, and E. Gratton. 2001. Lipid rafts reconstituted in model membranes. Biophys. J. 80:1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure, C., L. Bonakdar, and E. J. Dufourc. 1997. Determination of DMPC hydration in the La and Lb′ phases by 2H solid state NMR of D2O. FEBS Lett. 405:263–266. [DOI] [PubMed] [Google Scholar]
- Finer, E. G. 1973. Interpretation of deuteron magnetic resonance spectroscopic studies of the hydration of macromolecules. J. Chem. Soc. Faraday Trans. 69:1590–1600. [Google Scholar]
- Finer, E. G., and A. Darke. 1974. Phospholipid hydration studied by deuteron magnetic resonance spectroscopy. Chem. Phys. Lipids. 12:1–16. [DOI] [PubMed] [Google Scholar]
- Florin-Christensen, J., C. E. Suarez, M. Florin-Christensen, M. Wainszelbaum, W. C. Brown, T. F. McElwain, and G. H. Palmer. 2001. A unique phospholipid organization in bovine erythrocyte membranes. Proc. Natl. Acad. Sci. USA. 98:7736–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawrisch, K., W. Richter, A. Möps, P. Balgavy, K. Arnold, and G. Klose. 1985. The influence of water concentration on the structure of egg yolk phospholipid/water dispersions. Stud. Biophys. 108:5–16. [Google Scholar]
- Gawrisch, K., D. Ruston, J. Zimmerberg, V. A. Parsegian, R. P. Rand, and N. Fuller. 1992. Membrane dipole potentials, hydration forces, and the ordering of water at membrane surfaces. Biophys. J. 61:1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawton, M. H., and J. W. Doane. 1987. Pretransitional phenomena in phospholipid/water multilayers. Biophys. J. 52:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz, R., and Y. Barenholz. 1975. Permeability and integrity properties of lecithin-sphingomyelin liposomes. Chem. Phys. Lipids. 15:138–156. [DOI] [PubMed] [Google Scholar]
- Hsueh, Y.-W., R. Giles, N. Kitson, and J. Thewalt. 2002. The effect of ceramide on phosphatidylcholine membranes: a deuterium NMR study. Biophys. J. 82:3089–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner, W., and A. Blume. 1998. Interactions at the lipid-water interface. Chem. Phys. Lipids. 96:99–123. [Google Scholar]
- Jähnig, F. 1981a. Critical effects from lipid-protein interaction in membranes. I. Theoretical description. Biophys. J. 36:329–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jähnig, F. 1981b. Critical effects from lipid-protein interaction in membranes. II Interpretation of experimental results. Biophys. J. 36:347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendrasiak, G. L., and R. L. Smith. 2001. The effect of the choline head group on phospholipid hydration. Chem. Phys. Lipids. 113:55–66. [DOI] [PubMed] [Google Scholar]
- Katsaras, J. 1998. Adsorbed to a rigid substrate, dimyristoylphosphatidylcholine multibilayers attain full hydration in all mesophases. Biophys. J. 75:2157–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose, G., B. König, and F. Paltauf. 1992. Sorption isotherms and swelling of POPC in H2O and 2H2O. Chem. Phys. Lipids. 61:265–270. [Google Scholar]
- König, B., U. Dietrich, and G. Klose. 1997a. Hydration and structural properties of mixed lipid/surfactant model membranes. Langmuir. 13:525–532. [Google Scholar]
- König, B. W., H. H. Strey, and K. Gawrisch. 1997b. Membrane lateral compressibility determined by NMR and x-ray diffraction: effect of acyl chain polyunsaturation. Biophys. J. 73:1954–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurze, V., B. Steinbauer, T. Huber, and K. Beyer. 2000. A 2H NMR study of macroscopically aligned bilayer membranes containing interfacial hydroxyl residues. Biophys. J. 78:2441–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamba, O. P., D. Borchman, S. K. Sinha, S. Lal, M. C. Yappert, and M. F. Lou. 1991. Structure and molecular conformation of anhydrous and of aqueous sphingomyelin bilayers determined by infrared and Raman spectroscopy. J. Mol. Struct. 248:1–24. [Google Scholar]
- Led, J. J., and H. Gesmar. 1982. The applicability of the magnetization-transfer NMR technique to determine chemical exchange rates in extreme cases. The importance of complementary experiments. J. Magn. Reson. 49:444–463. [Google Scholar]
- Li, X. M., M. M. Momsen, J. M. Smaby, H. L. Brockman, and R. E. Brown. 2001. Cholesterol decreases the interfacial elasticity and detergent solubility of sphingomyelins. Biochemistry. 40:5954–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl, E. 2001. Computational Modeling of Biological Membrane and Interface Dynamics. Royal Institute of Technology, Stockholm, Sweden. (PhD thesis.)
- Lindblom, G., N. O. Persson, and G. Arvidson. 1976. Ion binding and water orientation in lipid model membrane systems studied by NMR. Adv. Chem. Ser. 152:121–141. [Google Scholar]
- London, E., and D. A. Brown. 2000. Insolubility of lipids in Triton X-100: physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts). Biochim. Biophys. Acta. 1508:182–195. [DOI] [PubMed] [Google Scholar]
- Luz, Z., and S. Meiboom. 1964. The activation energies of proton transfer reactions in water. J. Am. Chem. Soc. 86:4768–4769. [Google Scholar]
- Mahfoud, R., N. Garmy, M. Maresca, N. Yahi, A. Puigserver, and J. Fantini. 2002. Identification of a common sphingolipid-binding domain in Alzheimer, prion, and HIV-1 proteins. J. Biol. Chem. 277:11292–11296. [DOI] [PubMed] [Google Scholar]
- McIntosh, T. J., S. A. Simon, D. Needham, and C. H. Huang. 1992. Interbilayer interactions between sphingomyelin and sphingomyelin/cholesterol bilayers. Biochemistry. 31:2020–2024. [DOI] [PubMed] [Google Scholar]
- Meiboom, S. 1961. Nuclear magnetic resonance study of the proton transfer in water. J. Chem. Phys. 34:375–388. [Google Scholar]
- Migchelsen, C., and H. J. C. Berendsen. 1973. Proton exchange and molecular orientation of water in hydrated collagen fibers. An NMR study of H2O and D2O. J. Chem. Phys. 59:296–305. [Google Scholar]
- Miller, I. R., D. Chapman, and A. F. Drake. 1986. Circular dichroism spectra of aqueous dispersions of sphingolipids. Biochim. Biophys. Acta. 856:654–660. [DOI] [PubMed] [Google Scholar]
- Nagle, J. F., Y. Liu, S. Tristram-Nagle, R. M. Epand, and R. E. Stark. 1999. Re-analysis of magic angle spinning nuclear magnetic resonance determination of interlamellar waters in lipid bilayer dispersions. Biophys. J. 77:2062–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky, N., H. Shmeeda, G. Friedlander, A. Yanai, A. H. Futerman, Y. Barenholz, and A. Taraboulos. 1999. Sphingolipid depletion increases formation of the scrapie prion protein in neuroblastoma cells infected with prions. J. Biol. Chem. 274:20763–20771. [DOI] [PubMed] [Google Scholar]
- Nyberg, L., R. D. Duan, and A. Nilsson. 1998. Sphingomyelin. A dietary component with structural and biological function. Prog. Coll. Polym. Sci. 108:119–128. [Google Scholar]
- Pascher, I. 1976. Molecular arrangements in sphingolipids. Conformation and hydrogen bonding of ceramide and their implication on membrane stability and permeability. Biochim. Biophys. Acta. 455:433–451. [DOI] [PubMed] [Google Scholar]
- Pearson, R. H., and I. Pascher. 1979. The molecular structure of lecithin dihydrate. Nature. 281:499–501. [DOI] [PubMed] [Google Scholar]
- Pettegrew, J. W., K. Panchalingam, R. L. Hamilton, and R. J. McClure. 2001. Brain membrane phospholipid alterations in Alzheimer's disease. Neurochem. Res. 26:771–782. [DOI] [PubMed] [Google Scholar]
- Prosser, R. S., S. I. Daleman, and J. H. Davis. 1994. The structure of an integral membrane peptide: a deuterium NMR study of gramicidin. Biophys. J. 66:1415–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan, A., T. G. Anderson, and H. M. McConnell. 2000. Condensed complexes, rafts, and the chemical activity of cholesterol in membranes. Proc. Natl. Acad. Sci. USA. 97:12422–12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruocco, M. J., D. J. Siminovitch, J. R. Long, S. K. Das Gupta, and R. G. Griffin. 1996. 2H and 13C nuclear magnetic resonance study of n-palmitoylgalactosylsphingosine (cerebroside)/cholesterol bilayers. Biophys. J. 71:1776–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon, A., S. W. Dodd, G. D. Williams, J. M. Beach, and M. F. Brown. 1987. Configurational statistics of acyl chains in polyunsaturated lipid bilayers from 2H NMR. J. Am. Chem. Soc. 109:2600–2609. [Google Scholar]
- Salsbury, N. J., A. Darke, and D. Chapman. 1972. Deuteron magnetic resonance studies of water associated with phospholipids. Chem. Phys. Lipids. 8:142–151. [DOI] [PubMed] [Google Scholar]
- Schindler, H., and J. Seelig. 1975. Deuterium order parameters in relation to thermodynamic properties of a phospholipid bilayer. Biochemistry. 14:2283–2287. [DOI] [PubMed] [Google Scholar]
- Schmidt, C. F., Y. Barenholz, and T. E. Thompson. 1977. A nuclear magnetic resonance study of sphingomyelin in bilayer systems. Biochemistry. 16:2649–2656. [DOI] [PubMed] [Google Scholar]
- Siminovitch, D. J., and K. R. Jeffrey. 1981. Orientational order in the choline headgroup of sphingomyelin: a 14N-NMR study. Biochim. Biophys. Acta. 645:270–08. [DOI] [PubMed] [Google Scholar]
- Soda, G., and T. Chiba. 1969. Deuterium magnetic resonance study of cupric sulfate pentahydrate. J. Chem. Phys. 50:439–455. [Google Scholar]
- Talbott, C. M., I. Vorobyov, D. Borchman, K. G. Taylor, D. B. DuPre, and M. C. Yappert. 2000. Conformational studies of sphingolipids by NMR spectroscopy. II. Sphingomyelin. Biochim. Biophys. Acta. 1467:326–337. [DOI] [PubMed] [Google Scholar]
- Ulrich, A. S., and A. Watts. 1994. Molecular response of the lipid headgroup to bilayer hydration monitored by 2H-NMR. Biophys. J. 66:1441–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untracht, S. H., and G. G. Shipley. 1977. Molecular interactions between lecithin and sphingomyelin. Temperature- and composition-dependent phase separation. J. Biol. Chem. 252:4449–4457. [PubMed] [Google Scholar]
- Villalain, J., A. Ortiz, and J. C. Gomez-Fernandez. 1988. Molecular interactions between sphingomyelin and phosphatidylcholine in phospholipid vesicles. Biochim. Biophys. Acta. 941:55–62. [DOI] [PubMed] [Google Scholar]
- Vist, M. R., and J. H. Davis. 1990. Phase equilibria of cholesterol/dipalmitoylphosphatidylcholine mixtures: 2H nuclear magnetic resonance and differential scanning calorimetry. Biochemistry. 29:451–464. [DOI] [PubMed] [Google Scholar]
- Volke, F., S. Eisenblätter, J. Galle, and G. Klose. 1994a. Dynamic properties of water at phosphatidylcholine lipid-bilayer surfaces as seen by deuterium and pulsed field gradient proton NMR. Chem. Phys. Lipids. 70:121–131. [DOI] [PubMed] [Google Scholar]
- Volke, F., S. Eisenblätter, and G. Klose. 1994b. Hydration force parameters of phosphatidylcholine lipid bilayer as determined from 2H-NMR studies of deuterated water. Biophys. J. 67:1882–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassall, S. R. 1996. Pulsed field gradient-spin echo NMR studies of water diffusion in a phospholipid model membrane. Biophys. J. 71:2724–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorek, M. A. 1993. Biological distribution. In Phospholipids Handbook. G. Cevc, editor. Marcel Dekker, New York. pp.745–775.