

Abstract
The photovoltaic signal associated with the primary photochemical event in an oriented bacteriorhodopsin film is measured by directly probing the electric field in the bacteriorhodopsin film using an ultrafast electro-optic sampling technique. The inherent response time is limited only by the laser pulse width of 500 fs, and permits a measurement of the photovoltage with a bandwidth of better than 350 GHz. All previous published studies have been carried out with bandwidths of 50 GHz or lower. We observe a charge buildup with an exponential formation time of 1.68 ± 0.05 ps and an initial decay time of 31.7 ps. Deconvolution with a 500-fs Gaussian excitation pulse reduces the exponential formation time to 1.61 ± 0.04 ps. The photovoltaic signal continues to rise for 4.5 ps after excitation, and the voltage profile corresponds well with the population dynamics of the K state. The origin of the fast photovoltage is assigned to the partial isomerization of the chromophore and the coupled motion of the Arg-82 residue during the primary event.
INTRODUCTION
Bacteriorhodopsin (BR) is a trans-membrane protein found in the archae Halobacterium salinarum (Oesterhelt and Stoeckenius, 1971). The protein functions as a light-driven proton pump in the outer membrane of the organism, and provides a photosynthetic source of energy when the oxygen concentration drops below that capable of sustaining respiration (Birge, 1981; Mathies et al., 1991; Ebrey, 1993; Song and El-Sayed, 1998; Lanyi and Pohorille, 2001). The cell membrane of H. salinarum contains purple membrane (PM) fragments that are ∼5-nm thick. The main constituent of these PM fragments is BR, which arranges itself into a hexagonal lattice of trimers with a lattice constant of ∼6 nm. The trimeric arrangement of BR in PM fragments enables the protein to absorb light with equal efficiency in all polarizations. The chromophore of light-adapted BR is an all-trans retinal cofactor, bound to the protein via a protonated Schiff base linkage to lysine-216. The positive charge of the chromophore is localized primarily in the imine region of the chromophore and is stabilized by two nearby aspartic acid residues (Asp-85 and Asp-212). A positively charged residue, Arg-82, interacts with Asp-85 and Asp-212 to form a quadrupole and these three residues are all critical in the wavelength regulation of the chromophore (Ren et al., 2001).
Upon the absorption of light, there is a shift of electron density from the β-ionone ring of the chromophore toward the Schiff base (Fig. 1). This shift is comparable to a 2.6-Å displacement of a single electron down the polyene chain (Birge et al., 1999), and is in the opposite direction of the initial photovoltage spike that is observed in BR (Trissl, 1990). This charge shift is so short in duration that no experimental method has yet been able to observe the voltage associated with the excitation process. But this charge shift is a critical component of the primary event because it sets in motion events that lead to the photoisomerization of the chromophore (Zadok et al., 2002). In particular, the charge shift makes the imine linkage more negative and establishes a repulsive interaction between this region of the chromophore and the two nearby aspartic acid residues (85 and 212; see Fig. 1). This repulsion triggers a torsional photoisomerization about the 13-cis double bond of the chromophore (Mathies et al., 1988; Birge, 1990; Song et al., 1993). It is generally believed that the initial photovoltaic signal (B1) reflects the photoisomerization of the chromophore during the primary event (Rayfield, 1994; Groma et al., 1995; Hsu et al., 1996).
FIGURE 1.
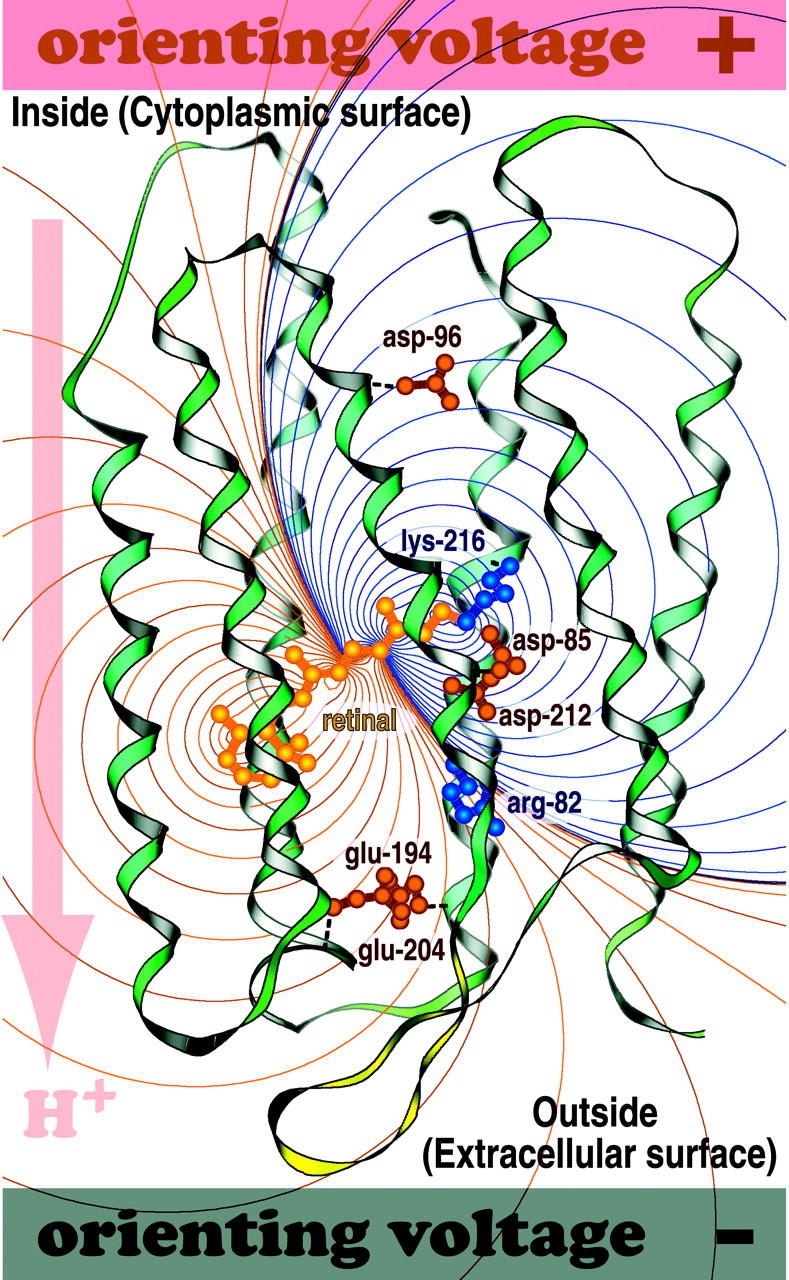
The purple membrane containing bacteriorhodopsin will spontaneously orient in an applied field and attach to the positively charged surface as schematically shown here. By convention, a photovoltage that goes in the same direction as the proton pump is assigned to be positive. The electrostatic field generated by excitation of the chromophore is shown with more positive regions marked with red lines and more negative regions marked with dark blue lines.
The high quantum efficiency, large oscillator strength, broad-band absorptivity, thermal stability, and fast photovoltaic response of the protein make it an attractive material as the photoactive element in high-speed photoreceivers (Birge, 1990). Such applications include high-speed tracking, motion, and edge detection as well as high resolution imaging (Birge, 1990; Miyasaka et al., 1992; Takei et al., 1991; Chen and Birge, 1993; Fukuzawa et al., 1996). The significant potential of BR as a light-transducing material was recently shown in a hybrid protein-semiconductor monolithically integrated transimpedance photoreceiver (Bhattacharya et al. 2002). In this device, the photovoltage generated by the protein biases the gate of an amplifying field effect transistor, creating a photocurrent signal that is further amplified as a current or voltage signal in subsequent transistor-based amplifying stages. The optoelectronic integrated circuit uses the protein as a photodetector, demonstrating performance characteristics comparable to, or better than, all-semiconductor optoelectronic integrated circuit photoreceivers (Xu et al., 2001). But none of the devices studied to date tests the ultimate capability of the protein to provide an ultrafast response. To establish the full potential of such integrated devices and circuits for high speed applications, it is necessary to characterize the dynamics of the initial (B1) photovoltage transient (Blaurock and Stoeckenius, 1971; Khorana et al., 1979; Czege et al., 1982; Kononenko et al., 1987; Henderson et al., 1990). This study determines the initial (fast) photovoltaic response of the protein by using a technique with an effective 3-dB-bandwidth of 350 GHz, a temporal resolution roughly six times greater than previous studies. The results provide a perspective on the ultimate device capability of the protein as well as insights into the molecular origins of the fast photovoltaic signal.
The kinetics of the fast (B1) photovoltaic signal have been studied previously by using a variety of techniques (Groma et al., 1984, 1995; Trissl, 1985; Simmeth and Rayfield, 1990; Rayfield, 1994). Simmeth and Rayfield (1990) have reported a time constant of <5 ps determined from the transient signal generated by pulsed laser excitation and observed on a high-speed digital oscilloscope. However, the pulse width of the exciting laser in these experiments was 3 ps and the K-connector used to connect the BR sample cell to the measurement circuitry of the oscilloscope limited the bandwidth of the latter to 70 GHz, which is proportional to an ∼5-ps rise time (Rayfield, 1994). From a similar measurement, Groma and coworkers have reported a time constant in the range of 2.5–5 ps (Groma et al., 1995). The latter studies benefited from the use of a much shorter laser pulse width of 150 fs, but the temporal response of the measurement was limited by the use of an SMA connector (18 GHz) and a 40-GHz sampling head. The measurements reported here use a 500-fs laser and an ultrafast electro-optic (E-O) sampling technique (Whitaker et al., 1994), wherein the photoinduced electric field generated by BR is directly probed. The technique is limited by the laser pulse width and yields a temporal resolution of better than 1 ps, which converts to an effective 3-dB bandwidth of 350 GHz. Our apparatus is shown in Fig. 2.
FIGURE 2.

A schematic diagram of the electro-optical measurement system used to observe the photovoltage associated with the excitation of a thin film of BR by a 500-fs pulse width laser. The pump beam (532 nm, SHG of 1060 nm light using nonlinear BBO crystal) and probe beam (1060 nm), generated from the same 500-fs pulse train, are directed onto the sandwiched ITO/oriented bacteriorhodopsin film/electro-optic crystal with a controllable optical delay. The induced birefringence in the electro-optic crystal by the electric field of the photo-pumped bacteriorhodopsin film, as shown in the inset, is translated into the intensity variation after the probe beam propagates through the polarizer/analyzer pair. The light signal is detected with an InGaAs photodetector using lock-in techniques and plotted as a function of delay time. The sample has a surface area of 10 × 10 cm2. The intensities of pump and probe beams are 50 mW/cm2 (∼35 μJ/cm2 pulse energy density) and 300 mW/cm2 (∼200 μJ/cm2 pulse energy density), respectively.
In a nonlinear E-O crystal, the induced birefringence, or electro-optic effect, causes unequal refractive indices, nx and ny, along the privileged directions x and y for a wave propagating in the z-direction. The polarized components of a wave traveling along x- and y-directions will travel with different propagation constants and a phase difference, or phase retardation. The electric field-dependent phase retardation is given by
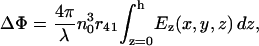 |
(1) |
where n0 is the quiescent refractive index of the E-O crystal, r41 is its linear electro-optic (Pockels) coefficient, Ez is the applied field, and h is the thickness of the crystal. Thus, the linearly polarized incident light will be elliptically polarized at the output of the E-O crystal. By placing the crystal between crossed polarizers, the change in direction of polarization is translated into an intensity variation proportional to the applied field. The light intensity detected by the photodiode is given by:
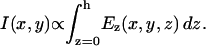 |
(2) |
If the E-O crystal is placed in close proximity of the BR sample, the transient photovoltage developed in it upon photoexcitation will be transmitted to the E-O crystal. Scanning the laser (photoexcitation) beam along the surface of BR will deliver an intensity distribution proportional to the electric field generated inside the BR film.
When the photoexcitation process of the protein is synchronized with the field probing technique, it is possible to make time-resolved measurements on the charge displacement time during the early stages of the photochemical cycle of the light sensitive protein. A short femtosecond laser pulse is divided into excitation and probe beams. By varying the optical delay between the two beams, the sampling probe pulses can probe the amplitude, in a voltage form, created by the interaction between the excitation beam and the BR sample under test (Bhattacharya, 1997).
MATERIALS AND METHODS
Oriented BR films, 10-μm thick and obtained by electrophoretic deposition of PM fragments, were used for the experiments (Varo, 1981). The BR film had an optical density of ∼2 at 570 nm and was placed in direct contact with the E-O crystal, Bi4Ge3O12 (Bismuth-Germanium-Oxide, or BGO). Any air gaps >1 μm between the BGO crystal and the protein film will seriously attenuate the electric field penetrating the BGO crystal. Atomic force microscopy images indicated that the surface roughness of our BR film was less than 0.2 μm. The BGO crystal used in the experiment is 100-μm thick and has a surface area of 10 × 10 mm2 normal to the 〈001〉 direction. It has cubic symmetry (23), a refractive index of 2.55 at λ = 1.06 μm, a dielectric constant of 40, a linear electro-optic coefficient of 3.4 × 1012 m/V, a resistivity of 8 × 1011 Ω × cm, and a loss tangent of 0.0035. A highly reflective coating, at both wavelengths of 1060 nm and 532 nm, was placed on the protein-contacting side of the BGO crystal. The BGO crystal-BR film pair is sandwiched between two glass plates coated with indium tin oxide (ITO), as shown in Fig. 2. The ITO provides an optically transparent conductive coating that allows the two glass plates to be connected via a conducting shunt. This arrangement forces the entire voltage differential generated by the protein photoexcitation to be imposed across the BGO crystal.
The temporal resolution of our apparatus is limited by the pulse-width of the laser (500 fs) and the transit time of the probe pulse through the BGO crystal. The influence of the E-O crystal geometry on the measurement resolution is determined by the convolution of the optical probe pulse and the BR electrical field signal as they propagate though the E-O crystal. When the propagation of these two signals are parallel, the temporal resolution is merely the time for the optical sampling pulse to travel across the active region of the crystal, which is ΔTlimit = ∼1.3 ps. This transit time would mean that the fastest exponential formation time that our system could reliably measure is τlimit = ΔT/e = ∼0.5 ps. Although deconvolution can extend the resolution, and we explore that issue below, it is important to note that the relevant temporal variables measured in this study are longer than the limits imposed by these values (e.g., ΔTmeas = 4.5 ps, τform = 1.68 ps).
Measurements are made with a passively mode-locked 1.06-μm regenerative amplifier delivering 500 fs, 0.025 mJ pulses with a repetition rate of 1.4 KHz. As we discuss below, we are using a much higher repetition rate than is optimal for studying a protein with a 10-ms photocycle. While lower repetition rates could be achieved by using a pulse-picker, lower rates will decrease the signal-to-noise ratio below an acceptable level. Each laser pulse provides a single measurement which must be extracted from the noise by using a lock-in amplifier, and there is a repetition rate below which analog lock-in detection is no longer effective at extracting the signal. For the present study, rates <1 KHz would not have provided an adequate signal-to-noise ratio. We discuss this issue in more detail below and demonstrate that the accuracy of our measured formation time observed will not be diminished by the choice of repetition rate.
The photocycle of the protein is initiated by using a separate pump beam at 532 nm which is generated with a nonlinear BBO crystal. The 1.06-μm probe beam is passed through a variable delay line and the sample is placed between crossed polarizer and analyzer. The accuracy of the delay time is limited by the pulse width of the incident light, which is 500 fs. Detection of the transmitted probe signal is done by using a cooled InGaAs photodiode and lock-in technique for increased sensitivity. For mapping the field distribution in the BR film and BGO crystal, 532 nm light is deflected by a gold-plated mirror servoactuated by a galvanometer. User interfacing with the drive circuitry of the galvanometer is achieved by using a computer-interfaced scan controller. The scan resolution is determined by the incremental motion of the scanning mirror, as well as the integration time of the lock-in amplifier. A typical spatial resolution of 300 μm is achieved in our measurement.
RESULTS AND DISCUSSION
The temporal photoelectric response characteristics of the BR films were first measured with a 10-ns pulse YAG laser and a fast (250 MHz) oscilloscope, to confirm their suitability for electro-optic measurements. For these measurements the BR film is sandwiched between two ITO contact glass plates. A typical photoresponse characteristic, after each laser pulse (with a 10-Hz repetition rate), is shown in Fig. 3 a and is consistent with the earlier observations (Keszthelyi and Ormos, 1980; Gergely et al., 1993). It is evident that the dried BR films generate a large photovoltage, which is necessary for the E-O measurement. The differential responsivity exhibited in the data is characteristic of BR and is believed to be due to rapid light-induced charge displacement in the membrane (Varo, 1981; Bhattacharya, 1997), although other effects for the manifestation of the differential signal have also been proposed (Robertson and Lukashev, 1995; Hong, 1997).
FIGURE 3.

(a) Measured photoelectric response characteristics of dried film of BR oriented between two ITO plates. The fast (B1) and slow (B2 and B3) components are associated with both structural changes in the protein as well as net translocation of the proton. The rise and decay time of B1 could not be accurately measured due to the limited bandwidth of the measurement system. The photoelectric response of the BR film as a function of scanning distance is shown in b. The pump and probe beams were focused during the mapping process to achieve a spatial resolution of 300 μm. The inset indicates that, in the electro-optic sampling measurement, the detected light intensity is directly proportional to the electric field inside the BGO crystal.
It is important to confirm that the probe beam does not saturate the BGO crystal and introduce nonlinearity in the response. To verify linearity, we passed the probe beam through the BGO crystal in the absence of the BR film and an AC voltage signal was applied to it. The detected signal increases linearly with the voltage amplitude, as shown in the inset of Fig. 3 b. Spatial mapping of the photoelectric field was performed by scanning the 532-nm photoexcitation beam across the surface of the BR film, while keeping the 1.06-μm probe beam incident onto the same point on the BGO crystal. The varying separation between the excitation region and the infrared probe beam on the sample provides quantitative information about the field distribution inside the photoexcited BR film. Fig. 3 b shows a one-dimensional photoelectric field distribution inside the BR film, when it is excited with a Gaussian laser beam. As expected, the electric field also exhibits a Gaussian profile.
The time-resolved E-O measurements were carried out with an incident (average) maximum pump beam intensity of 50 mW/cm2 (∼35 μJ/cm2 pulse energy density) and (average) maximum probe beam intensity of 300 mW/cm2 (∼200 μJ/cm2 pulse energy density). The transmitted probe light detected with lock-in amplification is plotted against delay time. Fig. 4 shows the normalized BR photoelectric temporal response, with the raw data shown in the insert (a). The data were fit via least-squares methods to an exponential formation and decay function to yield a formation rate of 1.68 ps and an initial decay rate of 28.1 ps. The photovoltaic signal continues to rise for 4.5 ps after excitation and represents the limiting temporal response of a photodetector based on the native BR protein.
FIGURE 4.

The photovoltaic signal from BR at ambient temperature measured using the system shown in Fig. 2 is plotted after normalization and inversion (original data shown in a, inset). The data are least-squares fit to an exponential formation and decay product function and the time constants and error function are indicated.
We note that the measured transient signal was always biased by a fixed background signal. When the initial pump pulse excites the BR sample, there are a large number of BR molecules available for charge displacement. As noted in Methods, the 1.4 KHz repetition rate was kept at the lowest possible value consistent with a viable signal-to-noise ratio. At ambient temperature, the photocycle of light-adapted BR requires ∼20 ms for >98% of the protein to return to the initial resting state. If we were required to maintain all the molecules in the resting state, we would need to use a repetition rate of 50 Hz or less. By using a 1.4-KHz repetition rate, the vast majority of the excited BR molecules will still be in the photocycle when the next pulse arrives. Thus, the measured transient signal will be biased by a fixed background signal that represents the steady-state photovoltage associated with the collection of protein molecules still going through the photocycle. The background signal has two origins. First, intermediates continuing through the photocycle will create a photovoltage associated with the B2 and B3 processes as shown in Fig. 3 a. Second, these intermediates are photoactive and upon excitation can generate new photovoltage transients. The question we now address is whether this background signal will influence our measurements.
We minimized the number of molecules remaining in the photocycle as well as the magnitude of the background signal by using dried BR films. These films have reduced hydration levels, although the key biological water remains present. Dried BR films have been shown to have a modified and partially truncated photocycle that forms very little L and virtually no N or O intermediates (Ganea et al., 1997). Thus, the majority of protein molecules remaining in the photocycle will be in the M state (λmax = 410 nm). The key advantage for this study is that the M state has a very low absorptivity at the excitation wavelength of 532 nm. Thus, we will not have significant background photovoltaic signals associated with the reverse photoreaction of M to BR. In addition, the pulse energy of our photoexcitation is quite low (∼35 μJ/cm2) and each pulse will activate <0.3% of the molecules in the film.
Simulations indicate that these background signals will contribute to the DC offset and ringing that is observed in the signal shown in Fig. 4, but will have no significant impact on the shape of the B1 formation signal. Thus, we are confident that our measured formation times are reliable. In contrast, we predict a small impact on the B1 decay signal at longer time. We consider the B1 decay values measured here to be approximate.
Although the background signal discussed above will have no impact on the B1 formation time measured in this study, the B1 signal that we measure is limited by the temporal resolution of the apparatus. The finite size of the E-O (BGO) crystal and the pulse width individually impose a 0.5-ps resolution limit on the calculated exponential formation time (see above). We explore the impact of this experimental constraint by using deconvolution and three different kinetic models to assign the formation time. We view over-deconvolution as worse than none at all, so we have taken the conservative approach of deconvolving with reference to a 500-fs Gaussian excitation pulse. The results are presented in Table 1. In general, deconvolution decreases the exponential formation time by 0.07 ± 0.02 ps. (Deconvolution has no significant impact on the decay time.) We note that this shift is comparable to the experimental error, but suggest that the best estimate of the formation time is the deconvolved value of 1.61 ± 0.04 ps.
TABLE 1.
Least-squares temporal analysis of the B1 photovoltaic signal
| τform (ps), without deconvolution | τform (ps), with deconvolution | Equation used for fitting* |
|---|---|---|
| 1.724 | 1.643 | 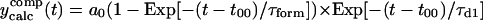 |
| 1.636 | 1.581 | 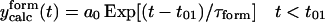 |
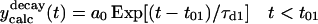 |
||
| 1.68 ± 0.05 | 1.61 ± 0.04 | 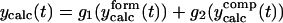 |
The first equation is a composite equation, 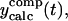 that fits the formation and decay using a product of the two exponential terms shown. In this equation, t00 is the onset time, τform is the exponential formation time (shown in the left column), and τd1 is the decay time (28.5 ps, 28.2 ps with deconvolution). The composite equation is assumed to be zero when t < t00. The second equation set uses a split equation with the formation term used when t < t01 and the decay term used when t ≥ t01. In this equation, t01 is the fitted time at maximum intensity of the signal and τd1 is the decay time (34.8 ps, 34.4 ps with deconvolution). The third equation is a linear combination of the above two fits with g1 and g2 calculated by the formulas
that fits the formation and decay using a product of the two exponential terms shown. In this equation, t00 is the onset time, τform is the exponential formation time (shown in the left column), and τd1 is the decay time (28.5 ps, 28.2 ps with deconvolution). The composite equation is assumed to be zero when t < t00. The second equation set uses a split equation with the formation term used when t < t01 and the decay term used when t ≥ t01. In this equation, t01 is the fitted time at maximum intensity of the signal and τd1 is the decay time (34.8 ps, 34.4 ps with deconvolution). The third equation is a linear combination of the above two fits with g1 and g2 calculated by the formulas 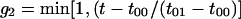 and g1 = 1 − g2.
and g1 = 1 − g2.
A comparison of the kinetics of the photovoltage with previous studies of the primary event kinetics is shown in Fig. 5. Note that the temporal axis is presented using a log scale. Previous studies of the photokinetics of the K state yield a maximum concentration of this species between 3 and 5 ps (Sharkov et al., 1985; Polland et al., 1986; Dobler et al., 1988). The assignment of the formation kinetics is complicated by the presence of the J precursor, which has a shifting absorption spectrum and is generally believed to be a vibrationally hot ground state species (Mathies et al., 1988; Pollard et al., 1989). Assignment of the decay kinetics of the K state is complicated by the presence of the KL intermediate, the population of which is difficult to rigorously assign due to its spectral similarity to K (Milder and Kliger, 1988; Doig et al., 1991). We have plotted the range of experimental results for the K state kinetics in Fig. 5. We note that the formation of the fast photovoltage is in excellent agreement with the observed formation kinetics of the K state. Thus, we can conclude with confidence that the formation of the K state is responsible for the onset of the B1 photovoltage. In contrast, our photovoltaic signal decays more rapidly than aggressive estimates for the formation of the KL state. We conclude that the decay rate of the photovoltaic signal is due in part to relaxation processes of the protein indirectly coupled or precedent to the formation of the KL state.
FIGURE 5.

The B1 photovoltaic signal (purple) is compared to the population kinetics of the I460*, J, K, and KL states. The range of experimental assignments of the K state population dynamics is indicated in yellow. Note that the formation kinetics of the K state and the B1 photovoltage coincide within experimental error.
The recent crystallographic study of the trapped K state carried out by Schobert et al. (2002) provides an opportunity to explore the molecular origins of the B1 photovoltage. Starting with the BR and K structures in the protein data bank file 1MOP, we added and energetically minimized the hydrogen atoms following standard procedures (Ren et al., 2001). Atomic charges were calculated by using the AM1 Hamiltonian and the linearized semiempirical methods available in MOPAC 2000 (Stewart, 1996). The electrostatic field generated by the formation of the K state was calculated by generating a charge difference map as shown in Fig. 6. We note that only residues that change position or charge density in going from the BR to the K state will contribute to the photovoltage. The key residues with respect to a differential charge shift are shown in Fig. 6. The crystal structure of the K state predicts a photovoltage three times larger than that induced via the excitation process (Fig. 1) and in the opposite direction (Fig. 6). The K state depicted in Schobert et al. (2002) predicts only a partial isomerization of the chromophore, and the chromophore geometry differs from the light-adapted resting state of the protein only in the imine region (see Fig. 6). Nevertheless, the partial isomerization is sufficient to induce a calculated negative photovoltage associated with the chromophore. This photovoltage is significantly enhanced by the coordinated motion of Arg-82 in the upwards direction toward the two aspartic acid residues 85 and 212. The charge profile of the K state is thus represented as distortion in the [LYS216(+):ASP85(−):ASP212(−):ARG82(+)] quadrupole with the two negative charges remaining fixed while the two positive charges shift upward. The charge shift swamps that associated with the initial excitation process (Fig. 1) because it is orthogonal to the membrane surface and has a significantly longer lifetime.
FIGURE 6.
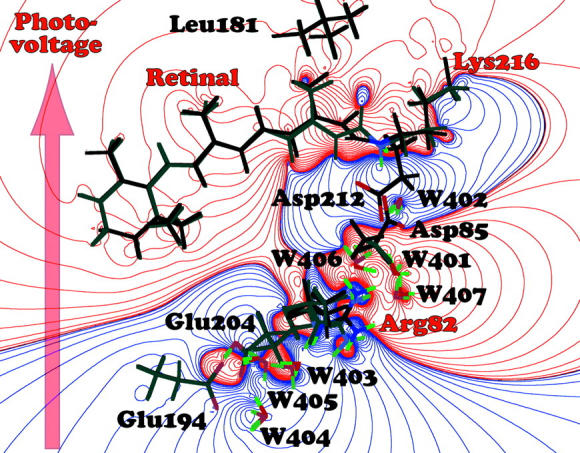
Electrostatic difference map associated with the formation of the K state. The dark bonds indicate K state residues and the lighter gray bonds indicate BR residues, and some residues do not move enough to be visible in this figure. The arrow indicates the net shift in positive charge, and by convention, the resulting photovoltage is assigned to be negative because it is in the opposite direction as the proton pumping direction (Fig. 1). The major contributors to the charge shift are labeled with red letters. The crystallographic data are from protein data bank entry 1M0P (Schobert et al., 2002).
COMMENTS AND CONCLUSIONS
The B1 photovoltaic signal has been resolved with a bandwidth of better than 350 GHz. The exponential formation time of 1.68 ± 0.05 ps [τ(deconvolved) = 1.61 ± 0.04 ps] and the voltage profile as a function of time corresponds within experimental error to the observed population dynamics of the K state. Insights into the molecular origin of the signal is obtained by generating an electrostatic map of the K state relative to the BR resting state based on the crystallographic studies of Schobert et al. (2002). This map indicates that both the partial isomerization of the chromophore and the motion of Arg-82 in response to the chromophore motion are the major contributors to the photovoltage. The importance of Arg-82 was unexpected.
In closing we return to the issue of the ultimate speed of BR-based photoreceivers. The rapid rise time is less important than the signal duration (Δt = 4.5 ps) in determining the response time of the photoreceiver, and the latter represents an important speed limitation of devices based on the native protein. But previous studies have demonstrated that mutations within the binding site can have a profound impact on the kinetics of the primary event (Song et al., 1993). What remains is a systematic study of new mutants with the goal of improving the temporal response.
Acknowledgments
The authors thank Professors Supriyo Datta and Jeffrey Stuart for useful discussions, and Adrienne Stiff for help in the scan measurements.
This work is supported by the Army Research Office (Multidisciplinary University Research Initiative Program), under grant DAAD19-99-1-0198; and the National Science Foundation, under grant EIA-0129731.
References
- Bhattacharya, P. 1997. Semiconductor Optoelectronic Devices. Prentice Hall, New Jersey.
- Bhattacharya, P., J. Xu, G. Varo, D. L. Marcy, and R. R. Birge. 2002. Monolithically integrated bacteriorhodopsin-GaAs field-effect transistor photoreceiver. Optics Lett. 27:839–841. [DOI] [PubMed] [Google Scholar]
- Birge, R. R. 1981. Photophysics of light transduction in rhodopsin and bacteriorhodopsin. Annu. Rev. Biophys. Bioeng. 10:315–354. [DOI] [PubMed] [Google Scholar]
- Birge, R. R. 1990. Photophysics and molecular electronic applications of the rhodopsins. Annu. Rev. Phys. Chem. 41:683–733. [DOI] [PubMed] [Google Scholar]
- Birge, R. R., N. B. Gillespie, E. W. Izaguirre, A. Kusnetzow, A. F. Lawrence, D. Singh, Q. W. Song, E. Schmidt, J. A. Stuart, S. Seetharaman, and K. J. Wise. 1999. Biomolecular electronics: protein-based associative processors and volumetric memories. J. Phys. Chem. B. 103:10746–10766. [Google Scholar]
- Blaurock, A. E., W. Stoeckenius. 1971. Structure of the purple membrane. Nature. (London) New Biol. 233:152–155. [DOI] [PubMed] [Google Scholar]
- Chen, Z., and R. R. Birge. 1993. Protein based artificial retinas. Trends Biotechnol. 11:292–300. [DOI] [PubMed] [Google Scholar]
- Czege, J., A. Der, and L. Zima'nyi. 1982. Restriction of motion of protein side chains during the photocycle of bacteriorhodopsin. Proc. Natl. Acad. Sci. USA. 79:7273–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobler, J., W. Zinth, W. Kaiser, and D. Oesterhelt. 1988. Excited-state reaction dynamics of bacteriorhodopsin studied by femtosecond spectroscopy. Chem. Phys. Lett. 144:215–220. [Google Scholar]
- Doig, S. J., P. J. Reid, and R. A. Mathies. 1991. Picosecond time-resolved resonance Raman spectroscopy of bacteriorhodopsin's J, K, and KL intermediates. J. Phys. Chem. 95:6372–6379. [Google Scholar]
- Ebrey, T. G. 1993. Light energy transduction in bacteriorhodopsin. In Thermodynamics of Membrane Receptors and Channels. M. B. Jackson, editor. CRC Press, Boca Raton, FL. pp.353–387.
- Fukuzawa, K., K. Yanagisawa, and H. Kuwano. 1996. Photoelectrical cell utilizing bacteriorhodopsin on a hole array fabricated by micromachining techniques. Sens. Actuat. B. 30:121–126. [Google Scholar]
- Ganea, C., C. Gergely, K. Ludmann, and G. Váró. 1997. The role of water in the extracellular half channel of bacteriorhodopsin. Biophys. J. 73:2718–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gergely, C., C. Ganea, G. Groma, and G. Varo. 1993. Study of the photocycle and charge motions of the bacteriorhodopsin mutant D96N. Biophys. J. 65:2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groma, G. I., J. Hebling, C. Ludwig, and J. Kuhl. 1995. Charge displacement in bacteriorhodopsin during forward and backward BR-K phototransition. Biophys. J. 69:2060–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groma, G. I., G. Szabo, and G. Varo. 1984. Direct measurement of picosecond charge separation in bacteriorhodopsin. Nature. 305:557–558 (Lett.). [Google Scholar]
- Henderson, R., J. M. Baldwin, T. A. Ceska, F. Zemlin, E. Beckmann, and K. H. Downing. 1990. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 213:899–929. [DOI] [PubMed] [Google Scholar]
- Hong, F. T. 1997. Molecular sensors based on the photoelectric effect of bacteriorhodopsin. FEBS Lett. 109:189–193. [Google Scholar]
- Hsu, K. C., G. W. Rayfield, and R. Needleman. 1996. Reversal of the surface charge asymmetry in purple membrane due to single amino acid substitutions. Biophys. J. 70:2358–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keszthelyi, L., and P. Ormos. 1980. Electric signals associated with the photocycle of bacteriorhodopsin. FEBS Lett. 109:189–193. [Google Scholar]
- Khorana, H. G., G. E. Gerber, W. C. Herlihy, C. P. Gray, R. J. Anderegg, K. Nihei, and K. Biemann. 1979. Amino acid sequence of bacteriorhodopsin. Proc. Natl. Acad. Sci. USA. 76:5046–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononenko, A. A., E. P. Lukashev, S. K. Chamorovksy, A. V. Maximychev, S. F. Timashev, L. N. Chekulaeva, A. B. Rubin, and V. Z. Paschenko. 1987. Oriented purple-membrane films as a probe for studies of the mechanism of bacteriorhodopsin functioning. II. Photoelectric processes. Biochim. Biophys. Acta. 892:56–67. [Google Scholar]
- Lanyi, J., and A. Pohorille. 2001. Proton pumps: mechanism of action and applications. Trends Biotechnol. 19:140–144. [DOI] [PubMed] [Google Scholar]
- Mathies, R. A., C. H. Brito Cruz, W. T. Pollard, and C. V. Shank. 1988. Direct observation of the femtosecond excited-state cis-trans isomerization in bacteriorhodopsin. Science. 240:777–779. [DOI] [PubMed] [Google Scholar]
- Mathies, R. A., S. W. Lin, J. B. Ames, and W. T. Pollard. 1991. From femtoseconds to biology: mechanism of bacteriorhodopsin's light-driven proton pump. Annu. Rev. Biophys. Biophys. Chem. 20:491–518. [DOI] [PubMed] [Google Scholar]
- Milder, S. J., and D. S. Kliger. 1988. A time-resolved spectral study of the K and KL intermediates of bacteriorhodopsin. Biophys. J. 53:465–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka, T., K. Koyama, and I. Itoh. 1992. Quantum conversion and image detection by a bacteriorhodopsin-based artificial photoreceptor. Science. 255:342–344. [DOI] [PubMed] [Google Scholar]
- Oesterhelt, D., and W. Stoeckenius. 1971. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nature (London) New Biol. 233:149–152. [DOI] [PubMed] [Google Scholar]
- Polland, H. J., M. A. Franz, W. Zinth, W. Kaiser, E. Kolling, and D. Oesterhelt. 1986. Early picosecond events in the photocycle of bacteriorhodopsin. Biophys. J. 49:651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard, W. T., C. H. B. Cruz, C. V. Shank, and R. A. Mathies. 1989. Direct observation of the excited-state cis-trans photoisomerization of bacteriorhodopsin: multilevel line shape theory for femtosecond dynamic hole burning and its application. J. Chem. Phys. 90:199–208. [Google Scholar]
- Rayfield, G. 1994. Photodiodes based on bacteriorhodopsin. In Molecular and Biomolecular Electronics. R.R. Birge, editor. American Chemical Society, New York. pp.561–575.
- Ren, L., C. H. Martin, K. J. Wise, N. B. Gillespie, H. Luecke, J. K. Lanyi, J. L. Spudich, and R. R. Birge. 2001. Molecular mechanism of spectral tuning in sensory rhodopsin II. Biochemistry. 40:13906–13914. [DOI] [PubMed] [Google Scholar]
- Robertson, B., and E. P. Lukashev. 1995. Rapid pH change due to bacteriorhodopsin measured with a tin-oxide electrode. Biophys. J. 68:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobert, B., J. Cupp-Vickery, V. Hornak, S. O. Smith, and J. K. Lanyi. 2002. Crystallographic structure of the K intermediate of bacteriorhodopsin: conservation of free energy after photoisomerization of the retinal. J. Mol. Biol. 321:715–726. [DOI] [PubMed] [Google Scholar]
- Sharkov, A. V., A. V. Pakulev, S. V. Chekalin, and Y. A. Matveetz. 1985. Primary events in bacteriorhodopsin probed by subpicosecond spectroscopy. Biochim. Biophys. Acta. 808:94–102. [Google Scholar]
- Simmeth, R., and G. W. Rayfield. 1990. Evidence that the photoelectric response of bacteriorhodopsin occurs in less than 5 picoseconds. Biophys. J. 57:1099–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, L., and M. A. El-Sayed. 1998. Primary step in bacteriorhodopsin photosynthesis: bond stretch rather than angle twist of its retinal excited-state structure. J. Am. Chem. Soc. 120:8889–8890. [Google Scholar]
- Song, L., M. A. El-Sayed, and J. K. Lanyi. 1993. Protein catalysis of the retinal subpicosecond photoisomerization in the primary process of bacteriorhodopsin photosynthesis. Science. 261:891–894. [DOI] [PubMed] [Google Scholar]
- Stewart, J. J. P. 1996. Application of localized molecular orbitals to the solution of semiempirical self-consistent field equations. Int. J. Quant. Chem. 58:133–146. [Google Scholar]
- Takei, H., A. Lewis, Z. Chen, and I. Nebenzahl. 1991. Implementing receptive fields with excitatory and inhibitory optoelectrical responses of bacteriorhodopsin films. Appl. Optics. 30:500–509. [DOI] [PubMed] [Google Scholar]
- Trissl, H.-W. 1990. Photoelectric measurements of purple membranes. Photochem. Photobiol. 51:793–818. [PubMed] [Google Scholar]
- Trissl, H. W. 1985. Primary electrogenic process in bacteriorhodopsin probed by photoelectric measurements with capacitive metal electrodes. Biochim. Biophys. Acta. 806:124–135. [Google Scholar]
- Varo, G. 1981. Dried oriented purple membrane samples. Acta Biol. Acad. Sci. Hung. 32:301–310. [Google Scholar]
- Whitaker, J. F., H.-J. Cheng, D. Craig, and G. A. Mourou. 1994. Optical sampling for high-speed electronics. Inst. Phys. Conf. Ser. 135:393–402. [Google Scholar]
- Xu, J., P. Bhattacharya, and G. Varo. 2001. A photoreceiver based on the monolithic integration of oriented bacteriorhodopsin and GaAs MODFETs. Proc. Lasers Electro-Optics Soc. 2001:833–834. [Google Scholar]
- Zadok, U., A. Khatchatouriants, A. Lewis, M. Ottolenghi, and M. Sheves. 2002. Light-induced charge redistribution in the retinal chromophore is required for initiating the bacteriorhodopsin photocycle. J. Am. Chem. Soc. 124:11844–11845. [DOI] [PubMed] [Google Scholar]