

Abstract
The oligomeric states of bovine visual arrestin in solution were studied by small-angle x-ray scattering. The Guinier plot of arrestin at the concentration ranging from 0.4 mg/ml to 11.1 mg/ml was approximated with a straight line, and the apparent molecular weight was evaluated by the concentration-normalized intensity at zero angle (I(0)/conc). Using ovalbumin as a molecular weight standard, it was found that arrestin varied from monomer to tetramer depending on the concentration. The I(0)/conc decreased at high-salt concentration, but was independent of temperature. The simulation analysis of the concentration-dependent increase of I(0)/conc demonstrated that the tetramerization is highly cooperative, and arrestin at the physiological concentration is virtually in the equilibrium between monomer and tetramer. The concentration of arrestin monomer, which is considered to be an active form, remains at an almost constant level even if the total concentration of arrestin fluctuates within the physiological range. The scattering profile of arrestin tetramer in solution was in good agreement with that in the crystal, indicating that the quaternary structure in solution is essentially identical to that in crystal. Small-angle x-ray scattering was applied to a binding assay of phosphorylated rhodopsin and arrestin in the detergent system, and we directly observed their association as the increase of I(0)/conc.
INTRODUCTION
In the animal retina, photons are absorbed by rhodopsin located in the rod outer segments (ROS). Rhodopsin is a typical G-protein coupled receptor having a seven-transmembrane helical segment. Rhodopsin has an 11-cis-retinal chromophore binding to lysine residue at helix VII by Schiff base linkage. The chromophore is isomerized to all-trans form upon photon absorption (Yoshizawa and Wald, 1963; Wald, 1968), and rhodopsin is successively converted to the bleaching intermediates called photorhodopsin, bathorhodopsin, lumirhodopsin, metarhodopsin I, metarhodopsin II, and metarhodopsin III (Shichida and Imai, 1998). Finally, rhodopsin is hydrolyzed into all-trans-retinal and scotopsin, the protein moiety of rhodopsin. The physiologically important intermediate is metarhodopsin II, whose absorption spectrum is located in the near-ultraviolet region. Upon formation of metarhodopsin II, a structural change of the cytoplasmic surface takes place (Imamoto et al., 2000; Resek et al., 1993). As a result, metarhodopsin II interacts with a G-protein, transducin, and activates transducin (Kühn, 1980). After activating ∼100 transducin molecules, serine residues at the C-terminal region of metarhodopsin II are phosphorylated by rhodopsin kinase (Kühn, 1978; Wilden and Kühn, 1982), and arrestin binds to this region (Kühn et al., 1984; Schleicher et al., 1989). Biochemical experiments in vitro have demonstrated that phosphodiesterase activity of ROS membrane is significantly lowered by phosphorylation, and further suppressed by adding arrestin (Wilden et al., 1986). Arrestin can bind to metarhodopsin II even in low phosphorylation stoichiometry, whereas a high phosphorylation level is required for the inactivation of metarhodopsin II by phosphorylation alone (Bennett and Sitaramayya, 1988). Thus arrestin is involved in the regulation of the transducin activation by metarhodopsin II, and it is considered to be one of the key proteins for the shutoff mechanism of the visual transduction. To reveal the mechanism of high time resolution and the wide dynamic range of visual cells, the behavior of arrestin requires investigation.
Bovine rod arrestin is a water-soluble protein of 45 kDa composed of 404 amino acids. The tertiary and quaternary structures of arrestin have been studied by x-ray crystallography at high resolution (Granzin et al., 1998; Hirsch et al., 1999). It has been demonstrated that arrestin crystallizes to form a unique tetramer (Granzin et al., 1998; Hirsch et al., 1999). The arrestin monomer comprises two domains, each of which has a seven-stranded β-sandwich. Arrestin has a long, highly flexible C-terminal chain that interacts with a hinge region and another domain at the N-terminal side. Arrestin tetramer is composed of two asymmetric dimers, each of which is composed of arrestin monomers assuming different conformations: the motif is conserved, but the surface structures that come into contact with the adjacent arrestin molecules are different. Each subunit interacts with the other through tight bonds to form intermolecular β-barrels. Based on these observations, it is proposed that arrestin in the dimer is maintained in an inactive form and thus the oligomerization of arrestin is important for its regulation (Hirsch et al., 1999).
More than 20 years earlier than crystallization, the oligomerization of arrestin in solution had been known (Wacker et al., 1977). It was recently reinvestigated and confirmed by sedimentation analysis (Schubert et al., 1999) and small-angle x-ray scattering (SAXS) (Shilton et al., 2002). It was demonstrated that arrestin is self associated in a concentration-dependent manner. The former showed that arrestin is mainly in the monomer/dimer equilibrium at ∼3 mg/ml, and in nonequilibrating tetramer form at high concentration of ∼10 mg/ml (Schubert et al., 1999) whereas the latter showed scattering profiles are consistent with monomer/dimer equilibrium at 1.3–60 mg/ml. Because the arrestin concentration in the visual cell is estimated to be 2–8 mg/ml (Hamm and Bownds, 1986; Kawamura, 1995), a significant portion of arrestin is considered to be in a dimer form in vivo, suggesting that the dimerization of arrestin has physiological importance. However, the physiological role of arrestin tetramer has been less investigated than dimer, probably because it has been considered to be a nonequilibrating (irreversible) form. Although the tetramer was not detected in the previous SAXS study (Shilton et al., 2002), it was carried out at significantly high-salt concentration (400 mM NaCl), which may affect the oligomeric state of arrestin. This encouraged us to measure the tetramerization of arrestin directly and quantitatively in the physiological condition.
SAXS is a powerful tool to evaluate the shape and size of a scattering particle (Kato et al., 2000). Although the resolution is much lower than crystallography, the protein in solution is subjected to measurement. Radius of gyration (Rg) and scattering intensity at angle of zero [I(0)] are estimated by this method. The former shows the extent of electrons, which correlates with the dimension of the particle, whereas the latter is proportional to the product of the weight concentration and the number of electrons in one particle (i.e., the molecular weight of the particle). Therefore, the association of protein can be detected by the increase of Rg and I(0) divided by weight concentration [I(0)/conc] (Kato et al., 2000). In the present study, the relationship between the apparent molecular weight and the concentration of arrestin was studied in detail to characterize the tetramerization process of arrestin. In addition, binding of arrestin to phosphorylated rhodopsin was directly observed by SAXS in a detergent system.
MATERIALS AND METHODS
Sample preparation
Bovine retinas freshly dissected in the dark were suspended in 40% sucrose in ROS buffer (10 mM MOPS, 30 mM NaCl, 60 mM KCl, 2 mM MgCl2, pH 7.5), shaken vigorously for 1 min, and centrifuged. The supernatant and floating materials were collected, diluted by the equivalent volume of ROS buffer, and centrifuged. The supernatant, which contained the soluble proteins of retina, was used for isolation of arrestin, and the pellet was used for isolation of rhodopsin.
The supernatant was dialyzed against buffer A (10 mM HEPES, pH 7.5) to remove NaCl. Arrestin was purified using DEAE-cellulose (DE-52; Whatman, Kent, UK) and heparin-Sepharose (Amersham Biosciences, Piscataway, NJ) column chromatography (Buczylko and Palczewski, 1993). Purified arrestin was dialyzed against buffer B (10 mM HEPES, 100 mM NaCl, pH 7.5) and concentrated to ∼15 mg/ml by a centrifugal ultrafiltration unit (Centriprep 10; Millipore, Bedford, MA). It was diluted to 0.4–11.1 mg/ml by buffer B and subjected to the experiments. Ovalbumin from chicken egg (43 kDa, A2512; Sigma-Aldrich, St. Louis, MO) was dissolved in buffer B for the control experiment. The concentration of ovalbumin was estimated by the molar extinction coefficient at 280 nm (30,590), which was calculated by using the molar extinction coefficients of Trp (5690) and Tyr (1280) (Gill and von Hippel, 1989).
Crude preparation of ROS obtained by sucrose flotation method was further purified by sucrose stepwise density gradient method (29 and 35% w/v). Rhodopsin in ROS was phosphorylated by endogenous rhodopsin kinase (Wilden and Kühn, 1982) by illuminating in the presence of 5 mM ATP according to the method reported by Ohguro et al. (1993). In this condition, phosphorylation stoichiometry is 3–4 phosphates/rhodopsin (Ohguro et al., 1993). Rhodopsin was then regenerated by adding twofold molar excess of 11-cis-retinal. For the control experiment, ROS was illuminated in the absence of ATP and regenerated similarly (nonphosphorylated rhodopsin). The binding ability of phosphorylated rhodopsin to arrestin upon illumination was confirmed by using a fluorescent probe (Imamoto et al., 2000).
Phosphorylated and nonphosphorylated rhodopsins were selectively solubilized using n-nonyl-β-d-glucopyranoside (NG) according to the method by Okada et al. (1998). Namely, extraction mixture (8 mg/ml rhodopsin, 2% NG, 5 mM Tris-HCl, 0.5 mM MgCl2, 70 mM ZnCl2, pH 7.2) was incubated at room temperature overnight. It was then centrifuged and the supernatant was collected. ZnCl2 was removed by gel filtration column (PD-10, Amersham Biosciences), which had been equilibrated with NG buffer (10 mM HEPES, 100 mM NaCl, 1% NG, pH 7.5). To eliminate x-ray scattering from NG micelle, 20% (w/v) sucrose was added to the sample before the SAXS measurements (see below).
Small angle x-ray scattering
The SAXS measurements were performed at the Beam Line BL10C at the Photon Factory of the High Energy Accelerator Research Organization (Tsukuba, Japan) (Ueki et al., 1985). Sample solution (100 μl) was put in a sample cell (1 mm light path length). The temperature of the sample was maintained at 15°C (detergent system) or 20°C (no detergent system) by circulating temperature-controlled water. The sample was irradiated with the combination of a 1-kw slide projector and a glass cutoff filter (>480 nm, Asahi Techno Glass, Chiba, Japan). The intensity of x-ray scattering was measured by a position-sensitive-proportional counter (PSPC) (Rigaku, Tokyo, Japan). The exposure time was 600 s. The position of each data point was converted to Q using scattering profile of collagen, where Q = 4π sin θ/λ is the amplitude of scattering vector, 2θ is the scattering angle, and λ is the wavelength of x ray (1.488 Å). In the small-angle region, I(Q) is approximated as follows (Guinier and Fournet, 1955; Glatter and Kratky, 1982):
 |
(1) |
where Rg is a radius of gyration. Therefore, the slope and Y-intersection of ln I(Q) versus Q2 (the Guinier plot) give 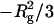 and ln I(0), respectively. In this study, the Guinier plot was fitted by a straight line in the small-angle region where the curvature is negligible. On the other hand, I(0) is expressed as follows:
and ln I(0), respectively. In this study, the Guinier plot was fitted by a straight line in the small-angle region where the curvature is negligible. On the other hand, I(0) is expressed as follows:
 |
(2) |
where N is the number of particles in the unit volume, ρ(r) is electron density at r, r is the distance from the center of gravity of electron, V is the volume of the particle, ρ(solvent) is electron density of solvent, ne is the number of electrons involved in one particle, MW is the molecular weight of the particle, and conc is the weight concentration (mg/ml) of the particle. Therefore, I(0)/conc is proportional to the molecular weight of the particle.
X-ray scattering from NG micelle in the rhodopsin sample was eliminated by adding sucrose at the final concentration of 20% (w/v), by which the electron density of the solvent (ρ(solvent)) was increased to a level comparable to that of NG micelle (Eq. 2).
Calculation of scattering profile from crystal structure
SAXS profile of arrestin in crystal (PDB ID 1CF1) was calculated using CRYSOL software (Svergun et al., 1995), by setting the hydration shell width at 3 Å.
Equilibration models
Tetramerization of arrestin is expressed as follows:
 |
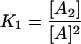 |
(3) |
 |
(4) |
where A is arrestin, K1 and K2 are the association constants for respective equilibria. The monomer/dimer model is equivalent to this model in which K2 = 0. In contrast, K2 considerably greater than K1 gives the cooperative tetramerization model. The total concentration of arrestin (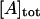 ) is expressed using the concentration of arrestin monomer as follows:
) is expressed using the concentration of arrestin monomer as follows:
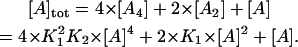 |
(5) |
When K1, K2, and 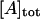 are given, the concentrations of monomer, dimer, and tetramer for each model can be calculated. The observed value of I(0)/conc of the mixture is expressed as follows:
are given, the concentrations of monomer, dimer, and tetramer for each model can be calculated. The observed value of I(0)/conc of the mixture is expressed as follows:
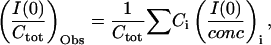 |
(6) |
where i denotes the species of scatterer, Ctot is the total protein concentration, (I(0)/Ctot)Obs is experimentally observed I(0)/conc, and Ci and (I(0)/conc)i are the weight concentration and I(0)/conc for each species, respectively. Because I(0)/conc is proportional to the molecular weight of the particle, (I(0)/conc)di and (I(0)/conc)tetra are two- and fourfold of (I(0)/conc)mono, respectively. (I(0)/Ctot)Obs is described as
 |
(7) |
The best values for (I(0)/conc)mono, K1, and K2 for each model were obtained by a laboratory-designed fitting program. In this study, we examined the noncooperative tetramerization model (K2 = K1), cooperative models (K2 = 10 × K1, K2 = 100 × K1, and K2 = 1000 × K1), and the monomer/dimer model (K2 = 0).
Interaction between arrestin and phosphorylated metarhodopsin II
The SAXS experiment gives the average of I(0) of the particles present in the sample. Therefore, if no interaction between arrestin and rhodopsin is present, I(0)/conc of the mixture of arrestin and rhodopsin should agree with the value calculated from I(0)/conc of arrestin and that of rhodopsin separately recorded as follows (Eq. 6):
 |
(8) |
where (I(0)/conc)arrestin and (I(0)/conc)rhodopsin are I(0)/conc for arrestin and rhodopsin, and concarrestin and concrhodopsin are the weight concentrations of arrestin and rhodopsin in the mixture. The interaction between arrestin and phosphorylated metarhodopsin II was examined by comparing observed and calculated values of I(0)/conc.
RESULTS
Concentration-dependent self association of arrestin
Before the SAXS experiments of arrestin, ovalbumin was used as a standard protein. Fig. 1 a shows the typical SAXS experiment on ovalbumin in the form of the Guinier plot. The traces were linear in Q2 ranging from 0.0005 to 0.005 Å−2. Each trace was fitted by a linear line in the linear region, and ln I(0) and  were given by Y-intersection and slope, respectively. I(0) divided by weight concentration [I(0)/conc] and
were given by Y-intersection and slope, respectively. I(0) divided by weight concentration [I(0)/conc] and  were plotted against weight concentration of ovalbumin (Fig. 2, a and b, respectively). The dependence of I(0)/conc and
were plotted against weight concentration of ovalbumin (Fig. 2, a and b, respectively). The dependence of I(0)/conc and  for ovalbumin on its concentration was negligible, indicating that ovalbumin in solution is monodispersed. The intrinsic I(0)/conc and Rg of ovalbumin were estimated to be 12,000 and 25.5 Å, respectively, from the Y-intersections of Fig. 2.
for ovalbumin on its concentration was negligible, indicating that ovalbumin in solution is monodispersed. The intrinsic I(0)/conc and Rg of ovalbumin were estimated to be 12,000 and 25.5 Å, respectively, from the Y-intersections of Fig. 2.
FIGURE 1.

The Guinier plots of ovalbumin (a) and arrestin (b). The scattering intensity at Q (=4π sin θ/λ) was measured by PSPC, and the logarithm of I(Q) was plotted against Q2. The traces were fitted by linear lines in the linear region, and ln I(0) and  were obtained from the Y-intersection and the slope, respectively. The concentrations of ovalbumin and arrestin are shown in the figure.
were obtained from the Y-intersection and the slope, respectively. The concentrations of ovalbumin and arrestin are shown in the figure.
FIGURE 2.

The concentration-dependent oligomerization of arrestin. I(0)/conc (a) and  (b) were plotted against total concentration. Those for ovalbumin are shown for comparison.
(b) were plotted against total concentration. Those for ovalbumin are shown for comparison.
Similarly SAXS patterns for arrestin were measured at various concentrations. Although the Guinier plots of arrestin at the lower concentrations (0.39 and 0.54 mg/ml) were almost linear up to Q2 = 0.005 Å−2, those at the higher concentrations had an inflection at Q2 = 0.002–0.003 Å−2, and the slope became steeper (Fig. 1 b). It indicates that arrestin in solution shows concentration-dependent association. In fact, it has been reported that arrestin possibly forms dimer, tetramer, and higher-order species (dodecamer) (Granzin et al., 1998; Hirsch et al., 1999; Schubert et al., 1999; Shilton et al., 2002). Because the preparations at high concentrations were the mixture of monomer and oligomer, Guinier plots were curved. Therefore, they were fitted in small Q2 region where the curvature was negligible. I(0) values were estimated by extrapolating them to zero concentration using straight lines, and the apparent Rg values were estimated by their slopes.
The I(0)/conc and apparent 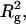 estimated by fitting with straight lines, were plotted against the arrestin concentration (Fig. 2, a and b, respectively). Unlike ovalbumin, I(0)/conc and Rg2 for arrestin dramatically increased as the concentration increased. Rg and I(0)/conc values at 0.39 and 11.1 mg/ml were 30 Å and 14,000, and 48 Å and 40,000, respectively. It is known that I(0)/conc is proportional to the molecular weight. Because the molecular weight of arrestin (45,000) is comparable to that of ovalbumin (43,000), arrestin at the low concentration is mainly in monomer form. Because the increase in I(0)/conc have not reached a plateau at 11.1 mg/ml, the final value of I(0)/conc would be somewhat larger than 40,000, suggesting that arrestin becomes tetramerized at high concentration.
estimated by fitting with straight lines, were plotted against the arrestin concentration (Fig. 2, a and b, respectively). Unlike ovalbumin, I(0)/conc and Rg2 for arrestin dramatically increased as the concentration increased. Rg and I(0)/conc values at 0.39 and 11.1 mg/ml were 30 Å and 14,000, and 48 Å and 40,000, respectively. It is known that I(0)/conc is proportional to the molecular weight. Because the molecular weight of arrestin (45,000) is comparable to that of ovalbumin (43,000), arrestin at the low concentration is mainly in monomer form. Because the increase in I(0)/conc have not reached a plateau at 11.1 mg/ml, the final value of I(0)/conc would be somewhat larger than 40,000, suggesting that arrestin becomes tetramerized at high concentration.
To reveal the process of oligomerization of arrestin, observed increase of I(0)/conc was simulated according to the models of equilibria (see Materials and Methods). Because the sample at 0.39 mg/ml may not be free from oligomer(s), (I(0)/conc)mono value was simultaneously obtained by fitting. The best value for K1 in Eq. 5 for each model was obtained by a laboratory-designed fitting program (Fig. 3 a).
FIGURE 3.

The simulation analysis for the oligomerization of arrestin. (a) The concentration-dependent increase of I(0)/conc of arrestin was simulated assuming K2 = 0 (×0), K2 = K1 (×1), K2 = 10 × K1 (×10), K2 = 100 × K1 (×100), or K2 = 1000 × K1 (×1000). For detail see main text. (b) Concentrations of monomer, dimer and tetramer were calculated according to ×1000 model, and plotted against total concentration of arrestin. The dashed line represents the total concentration (y = x).
First, observed increase of I(0)/conc was simulated by the monomer/dimer model (×0 model). It is obvious that the monomer/dimer model does not reproduce the experimental data even at low concentration because concentration-dependent increase of I(0)/conc in this model is very gentle. Then increase of I(0)/conc was simulated by tetramerization models. The noncooperative tetramerization model, in which the affinity of two dimers is equal to that of two monomers (K2 = K1 = 1.7 × 104 M−1, ×1 model) gave the better result, but showed a significant deviation. Therefore, I(0)/conc was fitted by the cooperative model in which K2 = 10 × K1, K2 = 100 × K1, or K2 = 1000 × K1 (×10, ×100, or ×1000 model, respectively). The best values for K1 were 6.6 × 103 M−1 for ×10 model, 2.6 × 103 M−1 for ×100 model, and 1.1 × 103 M−1 for ×1000 model. The excellent results were obtained in ×100 and ×1000 models. In these models, the best value for (I(0)/conc)mono was 13,000, which agrees with that expected from I(0)/conc and molecular weight of ovalbumin (12,000 and 43,000, respectively). The deviations of experimental values from the simulation curve were very small in whole concentration range, indicating that the oligomerization of arrestin in the physiological concentration is explained by an equilibrium among monomer, dimer, and tetramer. Hereafter the data were analyzed according to ×1000 model, in which the deviation was the smallest.
The concentrations of monomer, dimer, and tetramer were calculated according to ×1000 model and plotted against the total arrestin concentration (Fig. 3 b). Monomer increased proportional to the total concentration at low concentration (<1 mg/ml), whereas the rise became smaller in physiological concentration (2–8 mg/ml). The concentration of dimer is suppressed to a low level in whole concentration range. In contrast to monomer, the increase of tetramer was suppressed at low concentration. However, the increase of arrestin concentration induced further tetramerization at high concentration, resulting in a rise parallel to the total concentration. The arrestin concentration at which half of the total arrestin molecule is in the monomer form was calculated to be 4.9 mg/ml (110 μM).
Higher-order species
Because it is well known that arrestin has a significant tendency to aggregate, the contribution of higher-order species (aggregates), that strongly scatter at small angle, should be taken into consideration for the quantitative analysis of I(0)/conc. To examine the presence of aggregates, the scattering pattern of arrestin tetramer involved in each preparation was calculated and Guinier plotted.
The percentages of monomer, dimer, and tetramer of arrestin at 4.0, 7.9, and 11.1 mg/ml were calculated according to ×1000 model. The scattering patterns of tetramer at these concentrations were calculated by subtracting the scattering patterns at lower concentrations so that the contributions of monomer and dimer were canceled. The contribution of dimer could not be excluded completely, but it was calculated to be less than 5%. These calculated scattering patterns of the tetramer were Guinier plotted (Fig. 4 a). If higher-order species are present in the preparation, it should contribute to the calculated scattering pattern of the tetramer, resulting in the curved Guinier plot. However, the linear regions were clearly observed for these plots in Q2 < 0.0015 Å2 (Fig. 4 a). Using the Rg values of 48 Å (4.0 mg/ml) to 52 Å (11.1 mg/ml) estimated from the slopes, QRg for linear regions were calculated to be 1.9–2.0, which are consistent with the empirical rule that the Guinier plot is linear in QRg < 1.3. These results indicate that the negligible amount of aggregates is present in our preparation.
FIGURE 4.

Scattering profiles of arrestin tetramer calculated by subtracting the contribution of monomer and dimer from the experimental data. (a) Guinier plots of arrestin tetramer calculated using the scattering curves of 11.1, 7.9, and 4.0 mg/ml preparations. (b) 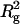 estimated from panel a were plotted against concentration. (c) Entire scattering profile of arrestin tetramer in 11.1 mg/ml preparation (circles) and in crystal (solid line). Note that Rg for solution was 45.6 Å whereas Rg for crystal was 43.6 Å.
estimated from panel a were plotted against concentration. (c) Entire scattering profile of arrestin tetramer in 11.1 mg/ml preparation (circles) and in crystal (solid line). Note that Rg for solution was 45.6 Å whereas Rg for crystal was 43.6 Å.
Quaternary structure of arrestin tetramer in solution
The simulation analysis described above provides only the information about the molecular weight of the oligomer(s) because it is based on I(0)/conc. Then we examined whether or not the quaternary structure of arrestin tetramer in solution is identical to that in crystal.
Rg values of arrestin tetramer estimated from the Guinier plot of calculated scattering patterns (Fig. 4 a) were 48–52 Å, which were larger than that estimated from the crystal structure using a CRYSOL software (Svergun et al., 1995) (44 Å). Rg is possibly dependent on protein concentration because of interparticle interaction. Thus 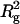 of tetramer estimated at various concentrations of arrestin were plotted against concentration and fitted with a straight line (Fig. 4 b). Rg at concentration of zero was estimated to be 45.6 ± 0.6 Å, which is consistent with that in the crystal. The larger Rg values in solution are therefore explained by the interparticle interaction. In this study, the scattering profiles of the tetramer were calculated by subtracting profiles at low concentration from those at high concentration. Because Rg in the latter is larger than that in the former, Rg values estimated here might be somewhat overestimated.
of tetramer estimated at various concentrations of arrestin were plotted against concentration and fitted with a straight line (Fig. 4 b). Rg at concentration of zero was estimated to be 45.6 ± 0.6 Å, which is consistent with that in the crystal. The larger Rg values in solution are therefore explained by the interparticle interaction. In this study, the scattering profiles of the tetramer were calculated by subtracting profiles at low concentration from those at high concentration. Because Rg in the latter is larger than that in the former, Rg values estimated here might be somewhat overestimated.
The entire scattering curves in solution and in crystal were compared to examine the quaternary structure of arrestin tetramer in solution (Fig. 4 c). I(Q) of tetramer in 11.1 mg/ml sample and that in crystal calculated by CRYSOL (Svergun et al., 1995) were plotted against QRg. They are in good agreement with each other, indicating that the solution structure of arrestin tetramer is essentially identical to crystal structure.
Effects of salt concentration and temperature on the self association of arrestin
The effects of NaCl concentration and temperature on I(0)/conc of arrestin were studied (Fig. 5). SAXS patterns of 5.3 mg/ml arrestin samples containing 10–500 mM NaCl were measured and I(0)/conc and 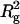 were estimated similarly (Fig. 5 a). Both I(0)/conc and
were estimated similarly (Fig. 5 a). Both I(0)/conc and 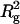 decreased as NaCl concentration increased, indicating that arrestin oligomerization is suppressed by NaCl. The concentration-dependent increase of I(0)/conc was more gentle at high-salt concentration (data not shown), suggesting that salt reduces the cooperativity of tetramerization.
decreased as NaCl concentration increased, indicating that arrestin oligomerization is suppressed by NaCl. The concentration-dependent increase of I(0)/conc was more gentle at high-salt concentration (data not shown), suggesting that salt reduces the cooperativity of tetramerization.
FIGURE 5.

The effect of NaCl concentration and temperature. I(0)/conc and 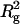 of arrestin were plotted against (a) NaCl concentration (5.3 mg/ml arrestin, 20°C) or (b) temperature (100 mM NaCl, 4.4 mg/ml arrestin).
of arrestin were plotted against (a) NaCl concentration (5.3 mg/ml arrestin, 20°C) or (b) temperature (100 mM NaCl, 4.4 mg/ml arrestin).
SAXS patterns of 4.4 mg/ml arrestin sample containing 100 mM NaCl were measured at 4–30°C. Estimated I(0)/conc and 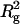 were constant (Fig. 5 b), indicating that the association constant of arrestin is independent of temperature. This finding is consistent with the previous report (Schubert et al., 1999).
were constant (Fig. 5 b), indicating that the association constant of arrestin is independent of temperature. This finding is consistent with the previous report (Schubert et al., 1999).
Interaction of arrestin with phosphorylated rhodopsin
The interaction of arrestin and rhodopsin was analyzed by SAXS in NG system. Our preliminary experiments demonstrated that aggregation of rhodopsin, which hinders the Guinier approximation, is the lowest in the NG system among detergent systems we tested. In addition, one-step purification of rhodopsin is possible in the NG system (Okada et al., 1998). Therefore, NG was used to solubilize rhodopsin.
Rhodopsin was solubilized and suspended in 1% NG buffer. Therefore, SAXS profile of rhodopsin sample is composed of the scattering of NG micelle and that of protein. However, if the electron density of the solvent was increased to the average electron density of NG, scattering from the NG micelle is eliminated (Eq. 2). Our preliminary experiments demonstrated that the sucrose concentration at which scattering of NG micelle is not observed (matching point) was 20% (w/v) (data not shown). As the electron density of protein is higher than that of NG micelle, the scattering from the protein was detectable in this condition, although the intensity was reduced.
Ovalbumin, arrestin, phosphorylated rhodopsin, and nonphosphorylated rhodopsin were suspended in 1% NG buffer containing 20% (w/v) sucrose. First the scattering pattern of each protein in this condition was separately measured (Fig. 6). In the presence of NG, I(0)/conc for ovalbumin was 11,000, whereas that of arrestin was 15,000–20,000 at the concentration ranging from 1 to 4 mg/ml. I(0)/conc for nonphosphorylated rhodopsin in the dark was 10,000, which increased to 20,000 after irradiation (Fig. 6 b). On the other hand, I(0)/conc for phosphorylated rhodopsin in the dark was 15,000, which increased to 21,000 after irradiation (Fig. 6 c). Considering from the molecular weight and I(0)/conc of ovalbumin in NG, I(0)/conc of monodispersed rhodopsin monomer is expected to be ∼10,000. Therefore, some portion of phosphorylated rhodopsin (∼50%) would be in dimer in the dark, and light would dimerize rhodopsin completely irrespective of the phosphorylation state. In fact, rhodopsin crystallized in the NG system is dimerized (Palczewski et al., 2000).
FIGURE 6.

I(0)/conc of arrestin, ovalbumin, and rhodopsin in the presence of 1% NG. (a) Ovalbumin and arrestin. (b) Nonphosphorylated rhodopsin in the dark (•) and after irradiation with yellow light (>480 nm) for 15 s (○). (c) Phosphorylated rhodopsin in the dark (•) and after irradiation with yellow light (>480 nm) for 15 s (○).
The mixtures of arrestin and nonphosphorylated or phosphorylated rhodopsin were then subjected to SAXS measurements (Fig. 7). In the present experiments, the concentration of nonphosphorylated rhodopsin or phosphorylated rhodopsin was fixed at 1 mg/ml, but the concentration of arrestin was changed. The measurements were carried out for four states of rhodopsin (nonphosphorylated/phosphorylated and dark/light) (Fig. 7, a and c). Because of loss of the contrast by sucrose, the Guinier plots for these samples were noisy, but they could be fitted by the straight lines in Q2 < 0.0015 Å−2. I(0) estimated from the Y-intersection of the Guinier plot was divided by total protein concentration [I(0)/Ctotal], and it was plotted against the concentration of arrestin (Fig. 7, b and d).
FIGURE 7.

Interaction between arrestin and rhodopsin. (a) Nonphosphorylated rhodopsin (final concentration, 1 mg/ml) was mixed with arrestin (final concentration, 1, 2, or 4 mg/ml, from bottom to top), and I(Q) was measured in the dark (•) and after irradiation with yellow light (>480 nm) for 15 s (○). They were Guinier plotted and fitted by linear lines. (b) I(0) in the dark (•) and after irradiation (○) were divided by the sum of concentrations of arrestin and rhodopsin as the total concentration (I(0)/conctotal). They were plotted against arrestin concentration. Broken lines indicate I(0)/conc of the mixture of rhodopsin and arrestin calculated from the values of Fig. 6 assuming no interaction between them. (c and d) The same experiments were carried out for phosphorylated rhodopsin.
If no interaction between arrestin and rhodopsin is present, I(0)/conc of the mixture of arrestin and rhodopsin should agree with the value calculated from I(0)/conc of arrestin and that of rhodopsin separately recorded (Fig. 6, a–c) (Eq. 8). The results of calculation are shown in Fig. 7, b and d (dashed lines).
For nonphosphorylated rhodopsin (Fig. 7 b), the observed values of I(0)/conc agreed with the calculated values within the experimental error both in dark and light, indicating that no interactions are present regardless of the activation state of rhodopsin. However, for phosphorylated rhodopsin (Fig. 7 d), significant increase of I(0)/conc was observed in the light condition. These observations indicate that only light-activated phosphorylated rhodopsin interacts with arrestin.
The increase of I(0) was prominent when the molar amount of arrestin was comparable to that of phosphorylated rhodopsin (1 mg/ml). The SAXS data demonstrate the average of the molecular weights of the particles present in the sample. Therefore, if one phosphorylated rhodopsin molecule binds to multiple arrestin molecules, observed I(0)/conc is expected to increase as the ratio of arrestin over phosphorylated rhodopsin increases. However, the trend of the present data was the opposite. Because a limited amount of arrestin could be involved in the complex in our experimental condition, the ratio of free arrestin at the higher concentration would be larger than that at the lower concentration, resulting in the smaller increase of I(0)/conc at the higher concentration. These results are consistent with the idea that arrestin is active in monomeric form.
DISCUSSION
Our SAXS experiments clearly demonstrate that arrestin in the solution is self associated as the concentration increases. Sedimentation analysis using ultracentrifugation has demonstrated the monomer/dimer equilibrium of arrestin at physiological concentration (2–8 mg/ml), and the presence of nonequilibrating tetramer at the high concentration used in crystallography experiments (10 mg/ml) (Schubert et al., 1999). On the other hand, the previous SAXS experiment (Shilton et al., 2002) demonstrated that arrestin is in monomer/dimer equilibrium in wide concentration range (1.3–60 mg/ml). In the present experiment, the relationship between the total concentration of arrestin and its oligomerization states in solution was studied in detail at a physiological salt concentration (100 mM NaCl) and were simulated according to several kinds of equilibration models. In contrast to the previous studies (Schubert et al., 1999; Shilton et al., 2002), the monomer/dimer model did not reproduce the experimental data even at low concentration (Fig. 3 a). However, the model of highly cooperative tetramerization (×100 and ×1000 models) consistently reproduced the experimental data at all concentrations we tested (0.4–11 mg/ml). In this model, arrestin is apparently in equilibrium between monomer and tetramer with only a small amount of dimer formed. Our simulation of the concentration-dependent oligomerization using a monomer-tetramer model gave substantially better result than that using a monomer-dimer model in the previous SAXS measurement (Shilton et al., 2002). As shown in the present study, the oligomeric state of arrestin is sensitive to salt concentration (Fig. 5 a). Because the previous SAXS experiment was carried out at high salt concentration (400 mM NaCl) (Shilton et al., 2002), the difference in salt concentration would account for this discrepancy. However, crystallographic tetramer was formed in the presence of ∼650 mM KCl (Granzin et al., 1998), suggesting that a small amount of tetramer is present even at high-salt concentration.
It is well known that arrestin has a significant tendency to aggregate, which may result in the overestimation of I(0). In fact, higher-order species was detected by sedimentation analysis at 10 mg/ml (Schubert et al., 1999). However, the Guinier plot for tetramer calculated by subtracting the contribution of monomer and dimer showed the linear region even at high concentration (>10 mg/ml) (Fig. 4 a). In addition, ×1000 model consistently reproduced I(0)/conc in whole concentration range (Fig. 3 a). These results indicate that the contribution of the aggregates to the scattering curves was negligible in our experiments.
Rg of arrestin in solution (45.6 Å) was slightly larger than that in crystal (43.6 Å). However, the entire scattering curve of arrestin tetramer in solution was in good agreement with that in the crystal (Fig. 4 c), suggesting that arrestin tetramer in solution assumes essentially identical quaternary structure with the crystallographic tetramer. The monomers would be more tightly arranged in crystal, resulting in slightly smaller dimension. I(0)/conc for arrestin was decreased by increasing the NaCl concentration, whereas it was independent of temperature. Arrestin molecules in crystallographic tetramer interact with neighboring arrestin molecules by electrostatic, hydrophobic, and hydrogen-bonding interactions, with the present results suggesting that electrostatic interaction is mainly responsible for tetramerization.
SAXS was also applied for binding assay of arrestin and phosphorylated metarhodopsin II. Because rhodopsin is a membrane protein, it was solubilized by 1% NG. Because the electron density of NG is higher than water, NG micelle itself scatters x ray. However, the electron density of NG is between that of water and protein, and hence, only the scattering from NG can be eliminated by increasing the electron density of solvent to the same level as NG using sucrose. This method overcame the technical problem, but the properties of arrestin and rhodopsin in the NG system were somewhat different from those in the absence of NG.
In the presence of NG, I(0)/conc of arrestin also increased as its concentration increased (Fig. 6 a). The rise of I(0)/conc relative to total concentration of arrestin in the presence of NG (Fig. 6 a) was steeper than the monomer/dimer model (×0 model in Fig. 3 a), suggesting the formation of tetramer. Because it was comparable to that in the noncooperative model (×1 model in Fig. 3 a), arrestin in the detergent system is in the equilibrium among monomer, dimer, and tetramer, but the percentage of dimer is larger than that in the absence of detergent. NG may inhibit the association by weakening the intermolecular interaction and/or by changing the conformation of arrestin.
I(0)/conc of phosphorylated rhodopsin in the dark and those of phosphorylated and nonphosphorylated rhodopsin after bleaching were 1.5–2 times larger than that of ovalbumin in the NG system (Fig. 6, b and c), suggesting that rhodopsin has a tendency to dimerize. In fact, it has been reported that rhodopsin is present in dimer in the native disc membrane (Fotiadis et al., 2003). However, because two rhodopsin molecules associate upside down in the NG system (Palczewski et al., 2000), crystallographic dimer would be different from native one. Rhodopsin in the crystal is dimerized by contacting the membrane-embedded regions of two rhodopsin molecules (Palczewski et al., 2000). It is thus reasonable to speculate that the rhodopsin in the NG micelle is dimerized in the same manner. Because the cytoplasmic surface of phosphorylated rhodopsin in this dimer is kept open, the dimerization of rhodopsin would not hinder the binding of arrestin. The difference in I(0)/conc between rhodopsin and phosphorylated rhodopsin in the dark is probably due to the difference in the population of dimer. Unlike arrestin, dimerization of rhodopsin was independent of concentration in this range.
NG is the outstanding detergent for the purification and crystallization of rhodopsin (Palczewski et al., 2000; Okada et al., 2000), but it is not necessarily suitable for SAXS. Hence, we tested other detergent systems such as 3-[(3-cholamidopropyl) dimethylammonio] propanesulfonic acid (CHAPS), sucrose monolaurate, n-octyl-β/-D-glucoside, n-dodecyl-β-D-maltoside, and digitonin. In them, no linear region was observed in the Guinier plot: the traces tended to show upward curvature and could not be fitted by a linear line unambiguously, resulting in no quantitative evaluation of I(0) and Rg. This is probably due to nonspecific aggregation. Although the photochemistry of rhodopsin in digitonin is considered to be similar to that in physiological conditions, the electron density of digitonin was as high as protein. A high concentration of sucrose [>50% (w/v)] was thus necessary to eliminate x-ray scattering from the digitonin micelle. In this condition, the scattering intensity of protein was also reduced, and it was not possible to take accurate measurements. In the NG system, while the rhodopsin dimer was formed, I(0) and Rg were estimated quantitatively. This enabled the binding assay in this system. The I(0) of the mixture experimentally observed was compared to that of the calculated one, which demonstrated that the difference in I(0) was prominent when the molar ratio of arrestin over rhodopsin was ∼1, but the difference was reduced as the ratio was increased. Because arrestin is active at very low concentration, arrestin is active in monomeric form. Our observation is consistent with this idea.
Arrestin is located in the rod inner segment in dark adapted retina but moves to the outer segment upon light adaptation (McGinnis et al., 2002). Under dim light condition, splice variant of arrestin, p44, acts as the stop protein (Schröder et al., 2002). Arrestin tetramer would have a physiological role in light condition where metarhodopsin II is successively generated. Due to the equilibrium between monomer and tetramer, change in the concentration of arrestin monomer is small even if the total concentration of arrestin fluctuates within the physiological concentration (Fig. 3 b). If a large amount of rhodopsin in the visual cell is bleached and phosphorylated by an intense light, arrestin monomer is supplied by dissociation of tetramer. In this condition, the concentration of free arrestin monomer, which is considered to be in the active state, is not decreased drastically. The monomer concentration is maintained at the constant level more effectively in the monomer-tetramer model than in the monomer dimer model proposed so far. Hence, the rate of binding of phosphorylated rhodopsin and arrestin is not largely altered. Arrestin is thus tetramerized to quench a large number of phosphorylated rhodopsin and maintain constant efficiency. Arrestin tetramer would thus buffer the concentration of arrestin monomer in light condition.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The experiments at the Photon Factory BL-10C were performed under the approval of the Photon Factory Advisory Committee (Proposal No. 99G336 and 01G363).
References
- Bennett, N., and A. Sitaramayya. 1988. Inactivation of photoexcited rhodopsin in retinal rods: the roles of rhodopsin kinase and 48-kDa protein (arrestin). Biochemistry. 27:1710–1715. [DOI] [PubMed] [Google Scholar]
- Buczylko, J., and K. Palczewski. 1993. Purification of arrestin from bovine retinas. Methods in Neurosci. 15:226–236. [Google Scholar]
- Fotiadis, D., Y. Liang, S. Filipek, D. A. Saperstein, A. Engel, and K. Palczewski. 2003. Rhodopsin dimers in native disc membranes. Nature. 421:127–128. [DOI] [PubMed] [Google Scholar]
- Gill, S. C., and P. H. von Hippel. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182:319–326. [DOI] [PubMed] [Google Scholar]
- Glatter, O., and O. Kratky. 1982. Small Angle X-Ray Scattering. Academic Press, New York.
- Granzin, J., U. Wilden, H. W. Choe, J. Labahn, B. Krafft, and G. Büldt. 1998. X-ray crystal structure of arrestin from bovine rod outer segments. Nature. 391:918–921. [DOI] [PubMed] [Google Scholar]
- Guinier, A., and G. Fournet. 1955. Small Angle X-Ray Scattering. John-Wiley and Sons, New York.
- Hamm, H. E., and M. D. Bownds. 1986. Protein complement of rod outer segments of frog retina. Biochemistry. 25:4512–4523. [DOI] [PubMed] [Google Scholar]
- Hirsch, J. A., C. Schubert, V. V. Gurevich, and P. B. Sigler. 1999. The 2.8 Å crystal structure of visual arrestin: a model for arrestin's regulation. Cell. 97:257–269. [DOI] [PubMed] [Google Scholar]
- Imamoto, Y., M. Kataoka, F. Tokunaga, and K. Palczewski. 2000. Light-induced conformational changes of rhodopsin probed by fluorescent Alexa594 immobilized on the cytoplasmic surface. Biochemistry. 39:15225–15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, R., M. Kataoka, T. Mikawa, R. Masui, N. Nakagawa, H. Kamikubo, and S. Kuramitsu. 2000. Observation of RecA protein monomer by small angle X-ray scattering with synchrotron radiation. FEBS Lett. 482:159–162. [DOI] [PubMed] [Google Scholar]
- Kawamura, S. 1995. Phototransduction, excitation and adaptation. In Neurobiology and Clinical Aspects of the Outer Retina. M. B. A. Djamangoz, S. N. Archer, and S. Vallerga, editors, Chapman & Hall/London. 105–131.
- Kühn, H. 1978. Light-regulated binding of rhodopsin kinase and other proteins to cattle photoreceptor membranes. Biochemistry. 17:4389–4395. [DOI] [PubMed] [Google Scholar]
- Kühn, H. 1980. Light- and GTP-regulated interaction of GTPase and other proteins with bovine photoreceptor membranes. Nature. 283:587–589. [DOI] [PubMed] [Google Scholar]
- Kühn, H., S. W. Hall, and U. Wilden. 1984. Light-induced binding of 48-kDa protein to photoreceptor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Lett. 176:473–478. [DOI] [PubMed] [Google Scholar]
- McGinnis, J. F., B. Matsumoto, J. P. Whelan, and W. Cao. 2002. Cytoskeleton participation in subcellular trafficking of signal transduction proteins in rod photoreceptor cells. J. Neurosci. Res. 67:290–297. [DOI] [PubMed] [Google Scholar]
- Ohguro, H., K. Palczewski, L. H. Ericsson, K. A. Walsh, and R. S. Johnson. 1993. Sequential phosphorylation of rhodopsin at multiple sites. Biochemistry. 32:5718–5724. [DOI] [PubMed] [Google Scholar]
- Okada, T., K. Takeda, and T. Kouyama. 1998. Highly selective separation of rhodopsin from bovine rod outer segment membranes using combination of divalent cation and alkyl(thio)glucoside. Photochem. Photobiol. 67:495–499. [PubMed] [Google Scholar]
- Okada, T., I. Le Trong, B. A. Fox, C. A. Behnke, R. E. Stenkamp, and K. Palczewski. 2000. X-ray diffraction analysis of three-dimensional crystals of bovine rhodopsin obtained from mixed micelles. J. Struct. Biol. 130:73–80. [DOI] [PubMed] [Google Scholar]
- Palczewski, K., T. Kumasaka, T. Hori, C. A. Behnke, H. Motoshima, B. A. Fox, I. Le Trong, D. C. Teller, T. Okada, R. E. Stenkamp, M. Yamamoto, and M. Miyano. 2000. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 289:739–745. [DOI] [PubMed] [Google Scholar]
- Resek, J. F., Z. T. Farahbakhsh, W. L. Hubbell, and H. G. Khorana. 1993. Formation of the meta II photointermediate is accompanied by conformational changes in the cytoplasmic surface of rhodopsin. Biochemistry. 32:12025–12032. [DOI] [PubMed] [Google Scholar]
- Schleicher, A., H. Kühn, and K. P. Hofmann. 1989. Kinetics, binding constant, and activation energy of the 48-kDa protein-rhodopsin complex by extra-metarhodopsin II. Biochemistry. 28:1770–1775. [DOI] [PubMed] [Google Scholar]
- Schröder, K., A. Pulvermuller, and K. P. Hofmann. 2002. Arrestin and its splice variant Arr1–370A (p44). Mechanism and biological role of their interaction with rhodopsin. J. Biol. Chem. 277:43987–43996. [DOI] [PubMed] [Google Scholar]
- Schubert, C., J. A. Hirsch, V. V. Gurevich, D. M. Engelman, P. B. Sigler, and K. G. Fleming. 1999. Visual arrestin activity may be regulated by self-association. J. Biol. Chem. 274:21186–21190. [DOI] [PubMed] [Google Scholar]
- Shichida, Y., and H. Imai. 1998. Visual pigment: G-protein-coupled receptor for light signals. Cell. Mol. Life Sci. 54:1299–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilton, B. H., J. H. McDowell, W. C. Smith, and P. A. Hargrave. 2002. The solution structure and activation of visual arrestin studied by small-angle X-ray scattering. Eur. J. Biochem. 269:3801–3809. [DOI] [PubMed] [Google Scholar]
- Svergun, D. I., C. Baberato, and M. H. J. Koch. 1995. CRYSOL-a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28:768–773. [Google Scholar]
- Ueki, T., Y. Hiragi, M. Kataoka, Y. Inoko, Y. Amemiya, Y. Izumi, H. Tagawa, and Y. Muroga. 1985. Aggregation of bovine serum albumin upon cleavage of its disulfide bonds, studied by the time-resolved small-angle X-ray scattering technique with synchrotron radiation. Biophys. Chem. 23:115–124. [DOI] [PubMed] [Google Scholar]
- Wacker, W. B., L. A. Donoso, C. M. Kalsow, J. A. Yankeelov, Jr., and D. T. Organisciak. 1977. Experimental allergic uveitis. Isolation, characterization, and localization of a soluble uveitopathogenic antigen from bovine retina. J. Immunol. 119:1949–1958. [PubMed] [Google Scholar]
- Wald, G. 1968. Molecular basis of visual excitation. Science. 162:230–239. [DOI] [PubMed] [Google Scholar]
- Wilden, U., and H. Kühn. 1982. Light-dependent phosphorylation of rhodopsin: number of phosphorylation sites. Biochemistry. 21:3014–3022. [DOI] [PubMed] [Google Scholar]
- Wilden, U., S. W. Hall, and H. Kühn. 1986. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc. Natl. Acad. Sci. USA. 83:1174–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa, T., and G. Wald. 1963. Prelumirhodopsin and the bleaching of visual pigment. Nature. 197:1279–1286. [DOI] [PubMed] [Google Scholar]