

Abstract
We investigated how strong cross-bridge number affects sliding speed of regulated Ca2+-activated, thin filaments. First, using in vitro motility assays, sliding speed decreased nonlinearly with reduced density of heavy meromyosin (HMM) for regulated (and unregulated) F-actin at maximal Ca2+. Second, we varied the number of Ca2+-activatable troponin complexes at maximal Ca2+ using mixtures of recombinant rabbit skeletal troponin (WT sTn) and sTn containing sTnC(D27A,D63A), a mutant deficient in Ca2+ binding at both N-terminal, low affinity Ca2+-binding sites (xxsTnC-sTn). Sliding speed decreased nonlinearly as the proportion of WT sTn decreased. Speed of regulated thin filaments varied with pCa when filaments contained WT sTn but filaments containing only xxsTnC-sTn did not move. pCa50 decreased by 0.12–0.18 when either heavy meromyosin density was reduced to ∼60% or the fraction of Ca2+-activatable regulatory units was reduced to ∼33%. Third, we exchanged mixtures of sTnC and xxsTnC into single, permeabilized fibers from rabbit psoas. As the proportion of xxsTnC increased, unloaded shortening velocity decreased nonlinearly at maximal Ca2+. These data are consistent with unloaded filament sliding speed being limited by the number of cycling cross-bridges so that maximal speed is attained with a critical, low level of actomyosin interactions.
INTRODUCTION
Models of Ca2+ regulation of striated muscle contraction differ on whether Ca2+ controls the number of strongly attached cross-bridges or whether there is also regulation of cross-bridge kinetics (Gordon et al., 2000). There is also a question about the role of strongly attached cross-bridges in activating the thin filament. These questions are of particular concern in the control of filament sliding because, during unloaded sliding, the number of strongly attached cross-bridges is substantially reduced and cycling rate is increased in comparison with isometric contraction.
The number of cross-bridges interacting with F-actin in the in vitro motility assay clearly modulates the speed of filament sliding (Uyeda et al., 1990). Speed is thought to decrease because individual cross-bridges act only intermittently. Below a critical heavy meromyosin (HMM) density, there are too few cross-bridges to keep an actin filament moving continuously. Even at high HMM densities, insufficient numbers of cross-bridges may underlie the slowing of regulated F-actin at reduced [Ca2+] observed in motility assays (Gordon et al., 1997; Homsher et al., 1996).
In permeabilized muscle fibers, the number of cross-bridges may similarly modulate velocity of unloaded sarcomere shortening (VU) although the question of whether [Ca2+] modulates VU in the intact sarcomere remains controversial (Gordon et al., 2000). VU varies approximately in proportion to the extent of thin filament activation in permeabilized skeletal muscle fiber preparations where an experimentally controlled fraction of thin filament regulatory units are activated without the dynamic events that normally occur at subsaturating [Ca2+]. These results were observed with partial extraction of TnC and maximal Ca2+ activation of the remaining, unextracted units (Moss, 1986) or activation by substituting a cardiac TnC that is constitutively active in the absence of Ca2+ (Martyn et al., 1994). Partial extraction of troponin complexes activates force without Ca2+ (Moss et al., 1986). Although these various approaches indicated that thin filament activation level modulates VU, a common concern is that thin filament regulatory protein binding sites were not fully occupied.
Complete activation of thin filaments depends on both Ca2+ binding to sTnC and strong cross-bridge binding to the thin filament (Gordon et al., 2000). Although the level of thin filament activation may be reduced when cross-bridge binding is reduced (Vandenboom et al., 1998), the influence on activation may be small (Brenner and Chalovich, 1999). Furthermore, cooperative effects of strong cross-bridges on thin filament activation should be minimal during unloaded filament sliding, when cross-bridge numbers are low and cycling rates are high.
We therefore investigated how Ca2+ controls the unloaded sliding speed of regulated thin filaments in the in vitro motility assay and permeabilized fibers when the number of interacting cross-bridges was varied in complementary ways. First, the density of HMM motors was varied in the motility assay. Second, the proportion of available myosin-binding sites on regulated thin filaments was changed by partial substitution of a sTnC mutant deficient in Ca2+ binding to the N-terminal trigger sites (xxsTnC) (Regnier et al., 2002), reducing the number of Ca2+-activatable regulatory units in both motility assays and in permeabilized rabbit psoas skeletal muscle fibers. The second strategy complements our earlier fiber studies with aTnC (Martyn et al., 1994) and has been used successfully in a broad range of assays by Tobacman and colleagues to investigate Ca2+-regulation of filaments reconstituted with cardiac Tn (Huynh et al., 1996; Morris et al., 2001). Mixtures of WT sTnC and xxsTnC were used such that regulatory units always contained a full complement of troponin. Our results are consistent with the model of Uyeda et al. (1990) in which filament sliding speed is limited by the total number of motors and their duty ratio, but cannot rule out a minor role for cross-bridges in increasing activation of thin filaments. Portions of these data have appeared in abstract form (Liang et al., 2000, 1999).
MATERIALS AND METHODS
Proteins
Troponin and tropomyosin
Regulatory protein isolation was done as described previously (Gordon et al., 1997). Native Tn and the three subunits of Tn (TnC, TnI, and TnT) were purified from rabbit back and leg muscle ether powder as described by Potter (1982). Tropomyosin (Tm) was made from rabbit muscle ether powder as described by Smillie (1982). Purified proteins were analyzed by SDS-PAGE and stored lyophilized at −20°C.
The rabbit skeletal TnC (RsTnC) mutant D27A, D63A, deficient in Ca2+ binding to the two N-terminal trigger sites, was prepared as described in Regnier et al. (2002).
Troponin containing recombinant TnCs was obtained by combining native TnI and native TnT with either WT RsTnC (WT sTn) or xxsTnC (xxsTnC-sTn) according to Potter (1982) as described (Gordon et al., 1997). Native and reconstituted Tn were stored frozen at −80°C.
Actin and regulated thin filaments
F-actin was prepared from rabbit back and leg muscle ether powder as described by Pardee and Spudich (1982). Purified F-actin was stored at 0–4°C for up to 1 month. F-actin was labeled with rhodamine-phalloidin (RhPh) as described by Kron et al. (1991) for visualization by fluorescence microscopy. RhPh F-actin was diluted to 8 nM in actin buffer (AB) (25 mM KCl, 25 mM imidazole, 4 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol (DTT), pH 7.4) (Kron et al., 1991).
Two procedures were used to reconstitute RhPh-labeled, regulated actin. In the first procedure for obtaining well-regulated F-actin (as utilized in Gordon et al., 1997), 400 nM RhPh F-actin was mixed with 150 nM Tm and 100 nM of the desired Tn in AB or, in a limited subset of control experiments, AB plus sufficient CaCl2 to obtain pCa 5. Similar results were obtained with filaments made in the presence or absence of Ca2+. Before infusion into a flow cell, filaments were diluted 1:100 in the same buffer plus 40–150 nM each Tm and Tn. For thin filaments reconstituted in this manner, the relative binding affinity of actin-Tm filaments for xxsTnC-sTn is 3× that for WT sTn at elevated [Ca2+] but their affinities are similar at low [Ca2+], as determined using the method of Huynh et al. (1996) (V. Korman and L. Tobacman, personal communication). We applied the correction factor of 3 when calculating fractional thin filament Ca2+ binding (e.g., Fig. 2) because Ca2+ was included in the relevant assays.
FIGURE 2.

Sliding speed (A) and fraction of filaments moving uniformly (B) in motility assays of RhPh-labeled, regulated F-actin reconstituted with Tm plus mixtures of WT sTn and xxsTnC-sTn. All assays were conducted at pCa 5. Regulated F-actin was reconstituted by adding the desired mixture of regulatory proteins to RhPh F-actin either bound to HMM on the flow cell surface in rigor (solid circles) or in solution, before injection into the flow cell (open triangles). In both instances, tropomyosin and the appropriate mixture of troponins were added to the assay buffer (see Materials and Methods). Fractional thin filament Ca2+ binding was estimated as the fraction of WT sTn bound to F-actin-Tm (see Materials and Methods). In panel A, points are the unweighted mean of means (±SD) for 1–20 flow cells (132 flow cells total, corresponding to 82,897 filament paths) and the line was drawn according to the equation y = x + a(1 − x)x where the nonlinear least squares regression parameter estimate for a, obtained from fitting all of the individual flow cell data, was 0.56 ± 0.03 (SE). In panel B, points are the unweighted mean of means and the line was drawn according to the same equation as in A, except a = 0.20 ± 0.08. Note that there is little change in motility until the fraction of activatable thin filament regulatory units drops below ∼50%.
The second, alternate procedure for reconstitution of regulated, RhPh F-actin was conducted in the motility assay flow cell (see below). RhPh F-actin (no Tm or Tn) was applied to the flow cell in the absence of ATP resulting in rigor binding of RhPh F-actin to HMM on the flow cell surface. Regulation was reconstituted by adding Tm + Tn (1:1; 40–150 nM each) in AB for 3 min before infusion of the motility buffers (MB) that also contained Tm + Tn at the same ratio and concentrations (see below). No correction was made for relative binding affinities of actin-Tm for WT sTn versus xxsTnC-sTn under this second protocol because reconstitution was done in the presence of cross-bridges formed between HMM on the flow cell surface and the actin filaments (Cassell and Tobacman, 1996). The reconstitution procedure used is noted with each in vitro motility experiment.
For both procedures, to maintain well-regulated filaments in MBs, [Tm + Tn] was varied until two conditions were satisfied (Gordon et al., 1997). First, [Tm + Tn] was sufficiently high that at pCa 9.2, no filament gliding motion was detected by eye and <2% of filament paths were characterized by automated analysis as exhibiting uniform motion (see below). Second, [Tm + WT or native Tn] was kept below concentrations that impair motility at pCa 5.
Myosin and HMM
Myosin was prepared from rabbit back muscles and stored in 50% glycerol at −20°C as described (Gordon et al., 1997). For the seven preparations of myosin used in these studies, K-EDTA and Ca2+ ATPase activities (Margossian and Lowey, 1982) were 16.9 ± 0.9 and 3.9 ± 0.2 Pi S1−1 s−1 (mean ± SD), respectively. HMM was made by chymotryptic digestion of myosin according to Kron et al. (1991) and stored at 0–4°C for up to 5 days. At the beginning of each day, ATP-insensitive heads were removed from an aliquot of HMM by ultracentrifugation (Kron et al., 1991) as described previously (Gordon et al., 1997). Competent HMM (supernatant) was then diluted to the final concentration to be used in motility experiments (typically 250 μg ml−1).
In vitro motility assays
Slide preparation and fluorescence microscopy
Motility assays with regulated F-actin were conducted essentially as described previously (Gordon et al., 1997; Regnier et al., 1996). Flow cells were formed on cleaned glass slides from nitrocellulose-coated cover slips and glass spacers. Infused solutions, equilibrated to room temperature (≥2× chamber volume, each) were left for 1 min in the chamber and then flushed with AB before infusing the next solution. HMM was applied first, followed 1 min later by 0.5 mg ml−1 BSA in AB. When the applied concentration of HMM was ≤100 μg ml−1, HMM was diluted with 0.5 mg ml−1 BSA in AB. After the chamber was flushed with AB, unlabeled F-actin (∼100 μg ml−1, sheared by at least 15 rapid passages through a 23-gauge needle) was added, followed 1 min later by a flush with AB solution and then AB with 0.5 mM ATP was added. This dissociated the remaining unlabeled F-actin from competent HMM on the nitrocellulose-coated surface, thus leaving residual dead heads blocked by unlabeled F-actin (Kron et al., 1991; Sellers et al., 1993). After another flush of the chamber with AB, RhPh F-actin or RhPh-labeled regulated F-actin was applied in the absence of ATP. Labeled actin filaments that did not bind to HMM on the surface were flushed from the chamber with a wash buffer (WB) that was either AB for assays with unregulated RhPh F-actin or was AB plus 40–150 nM each Tn and Tm for regulated filaments. When we reconstituted regulated filaments on the flow cell surface, we incubated unregulated RhPh F-actin filaments with AB plus 40–150 nM each Tn and Tm for 3 min. Note that in some experiments, Tn consisted of a mixture containing variable proportions of WT sTn and xxsTnC-sTn.
The final step was to infuse ATP-containing MB into the flow cell. MB for regulated actin filaments was 2 mM MgATP, 1 mM Mg2+, 65 mM Na+ + K+, 10 mM EGTA, 8–29 mM propionate, 28–70 mM 3-[N-morpholino]propanesulfonic acid (MOPS), 0.085 M ionic strength (Γ/2), 0.5–0.75% (w/v) methylcellulose (MC), and pH 7.0 at 30°C, the experimental temperature. Ca(propionate)2 was altered to change the pCa (=−log[Ca2+]) between 9.2 and 4.6 as calculated using the National Institute of Standards and Technology (NIST) Critically Selected Stability Constants of Metal Complexes Database. Motility buffer for assays of regulated F-actin contained 40–150 nM each Tm and Tn. For all motility buffers, 3 mg ml−1 glucose, 100 μg ml−1 glucose oxidase (Sigma, St. Louis, MO), 18 μg ml−1 catalase (Boehringer-Mannheim, Indianapolis, IN), and 40 mM DTT (BioRad, Hercules, CA) were added to minimize photo-oxidation and photobleaching (Kron et al., 1991). To assess variability between days and batches of proteins, at least one assay in each of two conditions was conducted each day using saturating HMM density: RhPh F-actin with AB plus 2 mM ATP and the same antiphotobleaching reagents as the motility buffer, and regulated F-actin (100% WT Tn) at pCa 5.0.
After infusion of MB into the flow cell, the slide was transferred to the stage of a Diastar upright fluorescence microscope (Leica, Deerfield, IL) that is equipped with a 100-W Hg arc lamp (Howard et al., 1993). Flow cell temperature was regulated at 30°C by circulating water through a copper coil wrapped around the 100× objective (Hunt et al., 1994). RhPh-labeled filaments were imaged with a SIT camera (model VE 1000, Dage-MTI, Michigan City, IN) and recorded with an added time-date generator signal (model WJ-810, Panasonic, Secaucus, NJ) on VHS videocassettes (VCR model AG7350, Panasonic) (Gordon et al., 1997). Typically, six fields were recorded for 1 min each in every flow cell.
Data acquisition and analysis
Filament speed distribution statistics were obtained from videocassette recordings by analysis of filament centroids using edge-detection hardware and Expert Vision software from Motion Analysis Systems (Santa Rosa, CA) (Homsher et al., 1996, 1992). Data were sampled at 10 frames per second (fps) and individual filament paths were retained only when the centroid could be unambiguously tracked for at least 2 s. Speed statistics were calculated for each retained path using the Motion Analysis algorithm. The ratio of standard deviation/mean speed was calculated for each path as an indicator of uniformity of motion (Gordon et al., 1997; Homsher et al., 1996, 1992). A filament was accepted as moving uniformly when this ratio was <0.5 for 10 fps sampling (or <0.3 for 2 fps sampling, as described below). For each flow cell (one condition), the fraction of filaments moving uniformly and the unweighted mean speed (±SD) of those uniformly moving filaments were obtained by combining information from all filament paths recorded. To combine data obtained on different days and using different batches of proteins, the mean speed for each flow cell was normalized to that obtained on the same day for regulated F-actin (100% WT Tn) at pCa 5.0 and saturating (250 μg ml−1) HMM density.
Unloaded shortening velocity of permeabilized fibers
Fiber preparation
Chemically permeabilized segments of single fibers from rabbit psoas muscle were isolated and the ends were fixed with glutaraldehyde and wrapped with aluminum foil T-clips for mounting on the mechanical apparatus as described previously (Chase and Kushmerick, 1988; Chase et al., 1993). Images of fiber segments for analysis of sarcomere length, fiber length, and diameter were recorded with an XR-77 CCD (Sony, Japan) interfaced with a DT3155 frame grabber (Data Translation, Marlboro, MA) and analyzed using HLImage++97 software (Western Vision Software, Salt Lake City, UT).
The mechanical apparatus and data acquisition protocols were the same as described previously (Regnier et al., 2002). Fibers were subjected to periodic release/restretch cycles to maintain the structural and functional integrity of fibers during activation (Brenner, 1983). Fibers were held in 200-μl anodized aluminum troughs that were maintained at 15°C (Quest Scientific ATR-4 Adaptable Thermoregulator, North Vancouver, BC, Canada). Compositions of relaxing and activating solutions for fiber mechanics experiments were calculated and solutions were made as described previously (Martyn et al., 1994). The solutions' ionic strengths and pCa values were calculated as described above. Solutions contained 5 mM MgATP, 15 mM phosphocreatine (PCr), 15 mM ethylenebis(oxyethylenenitrilo)tetraacetic acid (EGTA), at least 40 mM MOPS, 1 mM free Mg2+, 117 mM Na+ + K+, 1 mM dithiothreitol, (250 units ml−1 creatine kinase (CK; Sigma, St. Louis, MO), and 4% w/v Dextran T-500 (Pharmacia, Piscataway, NJ). Ca2+ levels of relaxing (pCa 9.2, where pCa = −log[Ca2+]) and maximally activating (pCa 4.5) solutions were established by varying the amount of Ca(propionate)2. Ionic strength (Γ/2) was 0.17 M and pH was 7.0 at 15°C.
Replacement of TnC in permeabilized fibers by purified TnC, xxsTnC, or mixtures of the two was as described (Regnier et al., 2002). TnC extraction reduced force at pCa 4.5 to ≤3% of the initial control value. After TnC extraction, reconstitution time was 5–10 min total.
Velocity of unloaded shortening: slack test
Velocity of unloaded shortening (VUS) was determined at maximal Ca2+ activation using the slack test (Edman, 1979) as described (Martyn et al., 1994). Each slack test consisted of a series of eight length releases (5–12% LF). The slack time—the duration of unloaded shortening for a given length release—was measured for each digitized record. VUS was obtained by linear least squares regression slope of the distance shortened versus slack time relationship.
Statistical analyses
Descriptive statistics, analysis of variance (ANOVA), and linear regression analyses were calculated with Microsoft Excel 97 (Microsoft, Redmond, WA). Averages are given as unweighted mean ± SD. Nonlinear regression analysis was performed using SigmaPlot software (versions 4–5, SPSS, Richmond, CA) to fit [Ca2+] (pCa) dependence of filament sliding speed and fraction of filaments moving uniformly data (y) to the Hill equation:
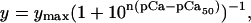 |
(1) |
where regression parameter ymax represents the maximal value obtained at high [Ca2+] (low pCa), pCa50 is equal to the pCa at the midpoint of the relationship (i.e., for y = ymax/2), and n describes the steepness of the relationship around pCa50. For normalized speed data, ymax was constrained to be 1.0.
RESULTS
In vitro motility of unregulated F-actin
The motility of unregulated, RhPh-labeled F-actin in AB plus 2 mM ATP at high HMM density and with no methylcellulose added was determined in at least one flow cell on each experimental day. These control measurements were calculated from >2.6 × 106 individual, frame-to-frame observations comprising 71,591 filament paths that could be traced unambiguously for ≥2 s (see Materials and Methods) in N = 78 flow cells. Fraction of filaments moving uniformly and speed of those filaments meeting the criterion for uniform motion are 0.74 ± 0.14 and 5.22 ± 0.98 μm s−1 and are similar to the values previously reported for comparable conditions (Gordon et al., 1997; Homsher et al., 2000; Sellers et al., 1993).
In vitro motility of regulated F-actin
Two procedures were used to reconstitute regulated, RhPh-labeled F-actin (see Materials and Methods). The first method, in which RhPh F-actin was incubated with Tm plus either purified native sTn or WT sTn and then the regulated filaments were diluted before infusion into the flow cell, gave comparable results to our previous experiments using native sTn (Gordon et al., 1997). With native sTn, the speed at pCa 5 of filaments moving uniformly (6.9 ± 0.7 μm s−1) was 166 ± 34% of the speed of unregulated F-actin measured on the same day. At pCa 9.2, only a small fraction of filaments were moving uniformly (0.02 ± 0.01) and with a speed only 8 ± 12% of that obtained at pCa 5. Similar results were obtained using F-actin-Tm regulated with WT sTn. The speed of uniformly moving filaments at pCa 4.6–5.0 (7.4 ± 1.1 μm s−1) was 138 ± 26% of the speed of unregulated F-actin measured on the same day. At pCa 9.2, the speed of the small fraction (0.02 ± 0.01) of uniformly moving filaments was only 6.8 ± 5.1% of the speed at high [Ca2+]. Similar results were also obtained on eight experimental days when regulated filaments were reconstituted in a rigor flow cell using WT sTn. Speed of filaments moving uniformly at pCa 5 (7.9 ± 0.7 μm s−1) was 136 ± 18% of the speed of unregulated F-actin measured on the same day. At pCa 9.2, the speed of the small fraction of filaments (0.02 ± 0.02) moving uniformly was only 5.9 ± 4.1% of that at pCa 5.
The data presented demonstrate that there was little difference between motility of actin filaments reconstituted with native sTn or with WT sTn or between the two procedures for reconstituting regulated F-actin at this level of analysis; ANOVA showed no significant difference in speeds (absolute or normalized to unregulated F-actin) although the fraction moving data was found to be significantly different (slightly higher with native sTn). The second procedure (reconstitution on the flow cell) is particularly appealing when using mixtures of Tn's because it greatly reduces the amount of protein used when testing a variety of concentrations and proportions. In addition, we experienced fewer problems with filament bundling with this procedure. The data also demonstrate that all three preparations were indeed well regulated (40–150 nM Tn and Tm added to MB) because, at pCa 9.2, <2.2% of filaments met the criteria for uniform motion and the speed of those filaments was substantially slower than at pCa 5, numbers not significantly different from filaments in rigor (no added ATP), an analysis that tests the intrinsic limitations of the system. Finally, these results confirm our previous observation (Gordon et al., 1997) and those of Fraser and Marston (1995) that the maximal speed of regulated F-actin containing Tn and Tm from skeletal muscle is substantially faster than that of unregulated F-actin (also see Fig. 1).
FIGURE 1.

In vitro motility of regulated F-actin with altered density of rabbit skeletal HMM. (A) HMM density dependence of motility of regulated (solid circles; 100% WT sTn) and unregulated (open squares) RhPh-labeled F-actin. Abscissa indicates [HMM] applied to the flow cell (see Materials and Methods). Assay conditions were 0.085 M Γ/2, 0.5% methylcellulose, pCa 5.0, 30°C (see Materials and Methods). Each point is the average ± SD from one individual flow cell (57–739 paths per flow cell), normalized to the average speed obtained for regulated filaments at pCa 5.0 on the same day; error bars are drawn only in one direction for clarity. Lines in panel A were drawn by eye. Ca2+ dependence of in vitro motility speed (B) and fraction of filaments moving uniformly (C) for RhPh-labeled actin filaments regulated with native Tm and sTn from rabbit muscle, studied at 250 μg ml−1 HMM (open triangles) or 75 μg ml−1 HMM (solid inverted triangles) (saturating Ca2+ data for these two HMM densities are circled in panel A). In panels B and C, each point is the unweighted mean ± SD (for clarity, error bars are shown in one direction only) of data from 1 to 11 flow cells (271–6061 paths per flow cell), normalized to the average speed obtained for regulated filaments at pCa 5.0 and 250 μg ml−1 HMM measured on the same day. Lines in panels B and C were drawn according to the nonlinear least squares regression to Eq. 1 (parameter estimates are given in Table 1). Note that Ca2+ dependencies of both speed and fraction moving were shifted to higher [Ca2+] (lower pCa) when HMM density was reduced.
Decreasing cross-bridge number by decreasing the density of motors
In vitro motility
To test the hypothesis that the number of cross-bridges interacting with a regulated actin filament modulates Ca2+-regulation of filament sliding, we reduced HMM density on the motility flow cell surface. Regulated F-actin for these experiments was made by mixing RhPh F-actin with Tn and Tm before infusion into the flow cell (see Materials and Methods). At all HMM densities where both regulated and unregulated filaments (native sTn) were tested, the speed of regulated RhPh F-actin at pCa 5 was greater than that of unregulated filaments. In our assays, application of the higher HMM concentration results in a saturated flow cell surface whereas HMM density is reduced to ∼60% of saturation at 75 μg ml−1 (functional density of HMM estimated by K-EDTA ATPase activity; Gordon et al., 1998). There was only a 20% decrease in the maximal sliding speed of regulated actin filaments (pCa 5.0) when HMM density was decreased from saturating (250 μg ml−1) to 60% saturation (75 μg ml−1; Fig. 1, A and B), but speed dropped off significantly at lower HMM densities (Fig. 1 A). At HMM densities below those shown in Fig. 1 A, regulated filament speed was highly variable with some undergoing stop-and-go motion and others exhibiting diffusional motion.
We therefore chose to compare Ca2+ dependence of regulated F-actin motility at 75 μg ml−1 vs. 250 μg ml−1 HMM (circled data points in Fig. 1 A). The Ca2+ dependencies of both speed and fraction moving were shifted to lower pCa (higher [Ca2+]) when HMM density was reduced to 75 μg ml−1 indicating an apparently lowered sensitivity of motility to Ca2+ (Fig. 1, B and C). Regression analysis of the data in Fig. 1, B and C using the Hill equation (Eq. 1) yielded ΔpCa50 = 0.12–0.20 due to altered HMM density (Table 1). These analyses also showed that the maximal fraction of uniformly moving filaments was 66–70% for both HMM densities (Fig. 1 C) and the maximal speed of uniformly moving filaments at reduced HMM density was 79 ± 3% (±SE) of that at saturating density (Fig. 1, A and B). Data obtained at saturating HMM density (250 μg ml−1) are comparable to our previously published values for similar conditions (Gordon et al., 1997). These results indicate that the number of cross-bridges interacting with a regulated actin filament contributes to the apparent Ca2+ dependence of filament sliding.
TABLE 1.
HMM density dependence of Hill equation (Eq. 1) regression parameter estimates for motility-pCa relationships
Decreasing cross-bridge number by reducing the fraction of Ca2+-activatable regulatory units on thin filaments
In vitro motility
Altered Ca2+ sensitivity of filament sliding with reduced HMM density (Fig. 1, B and C) could indicate that cross-bridges cooperatively activate the thin filament as is clearly demonstrated in measurements of steady-state isometric force (Regnier et al., 2002), but this shift in apparent Ca2+-sensitivity during unloaded sliding has previously been explained in another way (Homsher et al., 1996). When cross-bridge formation is limited, as at low HMM density, filament sliding speed is modulated by the number of cross-bridges even with unregulated F-actin—filament sliding depends on attachment of cross-bridges that is intermittent at low HMM density (Uyeda et al., 1990) and would be even more so when [Ca2+] is also reduced to submaximal levels. To test this we next altered the number of cross-bridges by varying the proportion of available binding sites on maximally Ca2+-activated, regulated actin filaments. To do this, regulated actin filaments were reconstituted with Tm and mixtures of Tn's containing either WT sTnC or the double mutant that is deficient in Ca2+-binding to sites I + II, xxsTnC (see Materials and Methods). Thus we altered in a controlled manner the proportion of thin filament regulatory units that could be activated by Ca2+.
The data in Fig. 2 summarize regulation of filament sliding at saturating [Ca2+] (pCa 5) by varying the proportion of WT sTn-containing regulatory subunits. Filaments containing 100% xxsTnC-sTn and no functional sTn exhibit little or no motility in either the absence (0.05 ± 0.06 of filaments moving) or presence of Ca2+ (0.02 ± 0.01 of filaments moving) and were indistinguishable from filaments reconstituted with 100% WT sTn at pCa 9.2 or from filaments in rigor. This demonstrates that Tm + xxsTnC-sTn bind to thin filaments and are effective inhibitors of motility, consistent with our measurements in permeabilized fibers from rabbit psoas showing that replacement of native sTnC with xxsTnC renders fibers incapable of Ca2+-activated force generation (Regnier et al., 2002) or shortening (see below). Control motility assays validated the necessity for adding regulatory proteins (Tm + WT sTn or xxsTnC-sTn) to MB, suggesting that the initial proportion of WT versus mutant Tn incorporated into the filaments was maintained throughout each measurement.
The data in Fig. 2 A suggest that varying the fraction of activatable regulatory units altered the speed of uniformly moving filaments in a nonlinear manner for both methods of reconstitution (see Materials and Methods) such that the decline in motility is steeper when less than 50% of the regulatory units are functional. The data are less clear on the control of the fraction of moving filaments.
Maximal velocity of unloaded shortening in permeablized fibers from rabbit psoas
To validate the correspondence between the motility assays shown in Fig. 2 with assays of sarcomere shortening in muscle cells (where it is considerably more difficult to reliably vary the number of myosin heads), we measured velocity of unloaded shortening (VU) at pCa 4.5 in permeabilized psoas fibers reconstituted with varying ratios of sTnC and xxsTnC. Fig. 3 shows mechanical (force) slack test records from a single fiber before extraction of endogenous sTnC (A) and after reconstitution of the TnC-depleted fiber with 20% sTnC plus 80% xxsTnC (B; note that, in contrast to motility assays where the whole troponin complex is used for reconstitution, only TnC is replaced in the fiber experiments). In panel B, steady-state isometric force (before length release at t = 0) was reduced to 22% of that in panel A, and slack times were markedly increased for equivalent size length steps (compare solid squares versus open circles and open inverted triangles in inset). In both instances, the slack data were well fit by linear regression (R2 > 0.99). Thus, as is typical for fully activated skinned fibers, we did not have to consider the possibility of multiple, fast, and slow phases of shortening (Martyn et al., 1994; Moss, 1986). The 60% reduction in VU observed with 80% xxsTnC for this example muscle fiber was completely reversed after a second extraction and reconstitution with 100% sTnC (open inverted triangles in inset of Fig. 3 B).
FIGURE 3.

Slack test force records obtained at saturating [Ca2+] (pCa 4.5) in a single, permeabilized fiber from rabbit psoas muscle before extraction of endogenous sTnC (A) and after reconstitution with a mixture of 20% native sTnC and 80% xxsTnC (B). Note the difference in scales for both the ordinate and abscissa in A versus B. (Inset) Summary of data obtained from panels A (open circles) and B (solid squares), and also after a second extraction procedure to remove the 20:80 sTnC:xxsTnC mixture, and reconstitution with 100% native sTnC (open inverted triangles). Velocity of unloaded shortening (VU) was obtained from the slope of these data (3.3 L s−1 before sTnC extraction; 1.3 L s−1 after reconstitution with 20:80 sTnC:xxsTnC mixture; R2 > 0.99). Note that slowing of VU observed in the test condition (open circles) was completely reversed by reconstitution with 100% native sTnC (open inverted triangles).
Fig. 4 summarizes data from 38 fibers reconstituted with 0–100% sTnC (100–0% xxsTnC) and shows that VU decreases with the number of activatable regulatory units in fibers. As with the comparable motility experiments (Fig. 2 A), the relationship between VU and fraction of activatable Tn (Fig. 4 A) was nonlinear. The curves in Figs. 2 A and 4 A were statistically indistinguishable. Thus similar qualitative conclusions can be drawn from both the fiber (Fig. 4) and motility (Fig. 2) assays. Fig. 4 B illustrates that VU was proportional to steady-state isometric force measured during the same activation at pCa 4.5. Force and VU, measured after extraction of endogenous TnC and reconstitution with the appropriate TnC mixture, were normalized to values obtained in the same fiber before TnC extraction. The slope of the linear regression line in Fig. 4 B is slightly greater than unity because fibers reconstituted with 100% native sTnC exhibited, on average, a small decline in isometric force (to 0.93 ± 0.10 of initial control), but no change in VU.
FIGURE 4.

Dependence of VU at maximal Ca2+-activation (pCa 4.5) on the fraction of activatable regulatory units in permeabilized psoas fibers reconstituted with mixtures of native sTnC and xxsTnC (N = 38). VU was determined by the slack test (Fig. 3; see Materials and Methods), and both VU and isometric force were normalized to the values obtained in the same fiber before sTnC extraction (open circles). (A) Nonlinear dependence of VU on the reconstituted fraction of native sTnC. Points are mean ± SE (N = 3–7 fibers). Line was drawn according to the equation y = x + a(1 − x)x where the nonlinear least squares regression parameter estimate for a, obtained from fitting data individual fibers, was 0.58 ± 0.06 (SE). (B) Linear relationship between VU and steady-state isometric force obtained during the same activation. Each point represents data from one fiber. The line is the linear least squares regression constrained to pass through the origin (R2 = 0.928).
Ca2+ dependence of in vitro motility of thin filaments containing 33% Ca2+-activatable troponin
To compare the Ca2+ dependence of motility when the number of cross-bridges is varied either by altering the fraction of myosin-binding sites available on regulated thin filaments or by altering HMM density (Fig. 1), we next recorded motility at high HMM density and various pCa values for regulated F-actin prepared with Tm plus a mixture of 33% WT sTn and 67% xxsTnC-sTn (incubation with 60% WT sTn:40% xxsTnC-sTn mixture to give the 33%:67% proportion in binding since these filaments will be treated with high Ca2+ where Tn containing xxsTnC has a higher affinity for actin-Tm; see Materials and Methods). For motility assays with filaments having a reduced number of activatable regulatory units, the Ca2+ dependence of both speed and fraction moving were shifted to lower pCa (i.e., higher [Ca2+]; Fig. 5, solid squares; Table 2). Regression analysis of the data in Fig. 5 using the Hill equation (Eq. 1) yielded ΔpCa50 = 0.18–0.20 due to the altered number of activatable regulatory units (Table 2). This indicates that there is a similar reduction in apparent sensitivity of motility to Ca2+ when the number of cross-bridges interacting with a filament is reduced by either method tested in these experiments (Figs. 1 and 5; Tables 1 and 2).
FIGURE 5.

In vitro motility assay of Ca2+-dependent speed (A) and fraction of filaments moving uniformly (B) for filaments reconstituted with Tm plus either 100% WT sTn (open triangles) or a mixture of 33% WT sTn plus 67% xxsTnC-sTn (solid squares). Regulated F-actin was reconstituted before infusion into flow cells. Each point represents data obtained from one flow cell (107–2018 paths per flow cell). In panel A, speed (±SD) was normalized to the average speed obtained for regulated filaments containing 100% WT sTn assayed at pCa 5.0 on the same day. Lines were drawn according to the nonlinear least squares regression to Eq. 1 (parameter estimates are given in Table 2). Note that Ca2+ dependencies of both speed and fraction moving were shifted to higher [Ca2+] (lower pCa) when the fraction of activatable regulatory per filament was reduced.
TABLE 2.
Hill equation (Eq. 1) regression parameter estimates for motility-pCa relationships obtained with reconstituted filaments containing either 100% WT sTn or 33% WT sTn (67% xxsTnC-sTn)
| WT sTn (%) | pCa50* | n* | |
|---|---|---|---|
| Fraction of filaments moving uniformly | 100 | 6.03 ± 0.07 | 2.58 ± 0.84 |
| 33 | 5.83 ± 0.06 | 3.25 ± 1.21 | |
| Speed of uniformly moving filaments | 100 | 6.10 ± 0.03 | 2.28 ± 0.39 |
| 33 | 5.92 ± 0.09 | 1.89 ± 0.69 |
Nonlinear least squares regression parameter estimates (±SE) of data in Fig. 5, A (normalized speed) and B (fraction) to the Hill equation (Eq. 1) (see Materials and Methods). Percentage of WT sTn bound to reconstituted filaments was estimated from binding affinity measurements by V. Korman and L. Tobacman (see Materials and Methods).
DISCUSSION
This study led to four major conclusions. The first two confirm and extend observations made previously using the in vitro motility assay to study regulated skeletal muscle F-actin. 1), [Ca2+] modulates both the fraction of regulated actin filaments moving uniformly and the sliding speed of those filaments in the in vitro motility assay. 2), Maximal sliding speed of regulated filaments containing 100% native or WT sTn at maximal [Ca2+] was faster than that of unregulated F-actin. In addition, new results from this study were: 3), At maximal [Ca2+], sliding speed (and fraction moving in motility assays) of regulated filaments was reduced by decreasing cross-bridge numbers. This was accomplished either with lower HMM densities in motility assays (as has also been found with unregulated F-actin) or by reducing the number of activatable regulatory units (with xxsTnC) in both motility assays and permeabilized skeletal muscle fibers. When the number of activatable regulatory units was reduced, the major decrease in sliding speed for both motility assays and fibers occurred when thin filaments contained <50% WT sTnC. In fibers, VU varied in proportion to steady-state isometric force; 4), In motility assays, when the number of cross-bridges was reduced by ∼1/3 by either method, the apparent Ca2+-sensitivity of filament sliding was also reduced.
Taken together, these observations suggest that the primary mode by which Ca2+ controls speed of regulated actin filament sliding is by modulating the number of attached, cycling cross-bridges. However, our results cannot rule out a secondary role for strongly attached, cycling cross-bridges in activating the thin filament during filament sliding.
Modulation of actin filament sliding speed by Ca2+-regulatory proteins
Similar to previous reports (Gordon et al., 1997; Homsher et al., 1996, 2000; Regnier et al., 1996; Sellers et al., 1993), motility of unregulated F-actin in this study was not altered by Ca2+, whereas motility of regulated thin filaments clearly exhibited Ca2+ regulation. This is demonstrated by little or no motion in the absence of Ca2+ (Figs. 1 and 5) and a graded increase in filament motility with increasing [Ca2+] (Figs. 1, B and C and 5). There was little difference between motility of regulated filaments reconstituted with either native sTn or WT sTn recombined from subunits. Additionally, the data confirm previous observations that maximal Ca2+-activated speed of regulated filaments is substantially elevated above the speed of unregulated F-actin (Fig. 1 A) (Fraser and Marston, 1995; Gordon et al., 1998, 1997). Enhancement of both filament speed and force at saturating [Ca2+] has also been observed when actin is reconstituted with bovine cardiac regulatory proteins in motility assays with rabbit skeletal HMM (Homsher et al., 2000). Interestingly, specific point mutations of cTnT, cTnI, and αTm have been reported to result in a further enhancement of regulated filament speed at saturating [Ca2+] (Bing et al., 2000, 1997; Homsher et al., 2000; Köhler et al., 2003; Lin et al., 1996; Sweeney et al., 1998).
On the opposite end of the spectrum, filaments fully reconstituted with Tm and xxsTnC-sTn were immobile in the absence or presence of Ca2+, as expected from previous studies where sTnC with inactivated low-affinity Ca2+-binding sites I and II was incorporated into muscle cells (Butters et al., 1997; Huynh et al., 1996; Putkey et al., 1989; Szczesna et al., 1996).
Implications for mechanism(s) of control of sliding speed by Ca2+-regulatory proteins
What limits in vitro filament sliding speed at saturating [Ca2+], and is this limiting factor the same at subsaturating [Ca2+] and in the intact sarcomere of muscle cells? First, it is clear that Ca2+ does not act directly on actomyosin kinetics, but rather via the regulatory proteins Tn-Tm because Ca2+ does not affect filament sliding speed in the absence of Tn-Tm (Gordon et al., 2000, 1997). Second, viscous drag forces that oppose filament sliding (either extrinsic or intrinsic to the actomyosin system) are not likely to be the primary limiting factor because these forces are too small (and would not vary with Ca2+), though increased viscosity does slow filament sliding of unregulated F-actin (Chase et al., 2000) and sarcomere shortening at saturating [Ca2+] (Chase et al., 1998). This slowing most likely occurs through an effect on a diffusion-controlled process in the actomyosin cycle. Gordon et al. (1997) provided evidence that the drag force from weak cross-bridge binding is not limiting to Ca2+-dependent filament sliding in the motility assay at near-physiological temperature (30°C), although Stehle and Brenner (2000) suggested that weak cross-bridges may limit sarcomere shortening velocity at saturating [Ca2+] and lower temperatures (≤5°C).
It is possible that slower filament sliding at low [Ca2+] results from slower cross-bridge dissociation kinetics (Gordon et al., 2000; LaMadrid et al., 2002), but it is more likely that the major Ca2+-dependent mechanism is the same as for isometric force—control of the probability of attachment of strong cross-bridges (Gordon et al., 2000). These experiments tested the latter possibility, i.e., that subsaturating levels of Ca2+ modulate sliding speed of regulated F-actin by limiting the number of strongly bound cross-bridges that propel filament sliding.
Less data regarding the role of cross-bridge number is available from skinned fibers. One study used p-PDM to inactivate myosin heads and showed a linear relation between VU and isometric force (Chaen et al., 1986), comparable to the results in Fig. 4 B. Our motility assays using xxsTnC-sTn to vary cross-bridge number (Figs. 2 and 5) are directly comparable with the equivalent perturbation in fibers (Fig. 4). Figs. 2 A and 4 A demonstrate that filament sliding speed and sarcomere shortening velocity are both elevated above what is expected from a linear dependence on the fraction of thin filament Ca2+-binding. Furthermore, for filaments containing 60–80% xxsTnC (20–40% sTnC), speed-pCa curves in motility assays (Fig. 5 A) were shifted to the left by 0.27–0.35 pCa units more than force-pCa curves from skinned fibers reconstituted with similar TnC mixtures (Regnier et al., 2002). This supports the idea that fewer cross-bridges are required to attain Ca2+-dependent activation of maximal sliding speed than maximal isometric force (Gordon et al., 1997; Homsher et al., 2000).
Our observation that Ca2+ dependence of regulated thin filament sliding speed is shifted toward higher [Ca2+] (reduced Ca2+-sensitivity) when cross-bridge number is reduced (Figs. 1 B and 5 A; Tables 1 and 2) agrees quantitatively with the results of Homsher et al. (1996), obtained using cardiac troponin. Homsher et al. (1996) hypothesized that when HMM density is reduced more Ca2+ is required to achieve a given sliding speed because more myosin-binding sites must be available to obtain an equal number of strong cross-bridges that propel filament sliding. In this study we tested this hypothesis by reducing cross-bridge number in two ways, reducing HMM density or reducing thin filament myosin-binding access directly. Both methods produced a similar magnitude change of Ca2+ sensitivity of motility (Figs. 1, B and C and 5; Tables 1 and 2).
The band model of Uyeda et al. (1990) predicts there are 15 S1 heads per μm of F-actin at high HMM density and 9 μm−1 (60%) at low density in motility assays (Fig. 1, B and C). This calculation assumes that motors located in a 26-nm wide band can interact with an actin filament (Harris and Warshaw, 1993) and uses surface densities of competent HMM determined in previous ATPase activity measurements (Gordon et al., 1998). In Fig. 5, the fraction of competent regulatory units on thin filaments was decreased to 33% (using mixtures of WT sTn and xxsTnC-sTn). In permeabilized fibers containing mixtures of sTnC and xxsTnC, maximal Ca2+ activated steady-state isometric force was greater than the proportion of sTnC, leading to an estimate of up to 10–12 actin monomers being exposed when each individual regulatory unit is turned on (Regnier et al., 2002). The nonlinear dependence of speed on the fraction of thin filament regulatory units containing sTnC in this study (Figs. 2 A and 4 A) suggests that more than seven actin monomers may also be exposed by Ca2+ binding during unloaded shortening. If so, then 57–66% of myosin-binding sites on F-actin should be available for strong cross-bridge formation in the assays of Fig. 5. These values represent upper limits to the number of strongly bound cross-bridges actually formed. Thus both methods of varying cross-bridge number yielded similar reductions (to ∼60%), which may underlie the quantitatively similar shift in motility-pCa parameters in both cases tested (Tables 1 and 2).
Although both methods of reducing cross-bridge number by ∼40% yielded similar ΔpCa50 values, other changes were observed in motility assays that differed between the two approaches. For example, both the maximal fraction of filaments moving uniformly and the speed of those uniformly moving filaments were significantly reduced for partially inactivated thin filaments (Fig. 5) but not when HMM density was reduced (Fig. 1, B and C). This may have occurred because the effective reduction in cross-bridge number was actually somewhat greater than 40% with partial thin filament inactivation, or it may be related to subtle differences in the two methods at the level of individual regulatory units. Changing HMM density alters the average number of cross-bridges that can form per regulatory unit without affecting the total number of functional regulatory units. On the other hand, partial inactivation of thin filaments does not directly affect the number of cross-bridges that can form within each individual, functional regulatory unit but reduces the number of regulatory units available for cross-bridge binding. The latter procedure also substantially reduced near-neighbor regulatory unit interactions along the thin filament when the fraction of sTnC was <50% (Regnier et al., 2002), similar to the conditions in Fig. 5. Differences in near-neighbor regulatory unit interactions along the thin filament could influence cooperative cross-bridge binding in the motility assay, although this effect seems unlikely to have a significant influence on unloaded sliding because the cross-bridge duty cycle is low and the number of cross-bridges is substantially reduced relative to isometric contraction (Brenner and Chalovich, 1999; Stehle and Brenner, 2000). As seen in Figs. 2 A and 4, filament sliding was substantially inhibited only when the fraction of WT sTn was reduced below 50%. This is similar to results with cardiac troponin containing CBMII on skeletal HMM-coated surfaces, although the latter required a reduction to ∼30% (Homsher et al., 2000). Thus the number of strong cross-bridges appears to be a significant, but perhaps not the sole, factor that limits Ca2+-dependent motility of regulated actin filaments in the motility assay.
Sarcomere shortening velocity at saturating Ca2+ decreased in proportion to the steady-state isometric force when the number of activatable regulatory units was reduced using xxsTnC in fast skeletal muscle fibers (Fig. 4 B). These results agree with observations using aTnC (Martyn et al., 1994) or TnC extraction (Metzger and Moss, 1988; Moss, 1986). If the reduction of steady-state isometric force is proportional to a reduction in the number of cross-bridges formed during filament sliding, then VU may be closely related to cross-bridge number in fibers. The apparent difference in results at saturating Ca2+ shown in Figs. 1 A and 4 B could indicate there are different limiting factors for filament sliding in fibers versus motility assays. It may be that internal loads present in the intact sarcomere are absent in the motility assay. The apparent differences could also result from different numbers of myosin heads available for binding to an actin filament in motility assays than in intact sarcomeres. In the overlap zone there is 1 S1 per 1.8 actin monomer, or 3.9 S1 heads per structural regulatory unit, which is also equivalent to ∼100 S1 μm−1 of actin filament (Gordon et al., 2000), an ∼10-fold greater quantity than in the motility assay (see above). The lower linear density of S1 heads in the motility assay may be partially offset by filaments being approximately an order of magnitude longer than in the sarcomere, which would increase the probability of having at least one cross-bridge per filament at any given time. However, length-dependent differences in sliding speed have only been detected for very short filaments (i.e., shorter than those analyzed in these studies), particularly at low HMM density (LaMadrid et al., 2002; Uyeda et al., 1990).
The results in this study and in previous work (Gordon et al., 1998, 1997; Homsher et al., 1996, 2000; LaMadrid et al., 2002) strongly suggests a common underlying mechanism for thin filament control of regulated F-actin sliding speed for the various conditions studied. In addition to differences in cross-bridge binding distributions along thin filaments, there are different dynamics associated with filaments when 50% activation is achieved by using submaximal levels of Ca2+ with thin filaments containing 100% sTnC versus a mixture of 50% sTnC with 50% xxsTnC at pCa 5. In the latter case, activated regulatory units are distributed randomly along a thin filament and the distribution of activated units is time invariant. For a thin filament containing only sTnC, one would expect a random distribution of activated regulatory units, but that the distribution would change over time due to the stochastic nature of Ca2+ dissociation and rebinding. In fibers, this dynamic aspect of thin filament activation is a major determinant of kinetics of tension redevelopment (kTR) at submaximal [Ca2+] (Brenner and Chalovich, 1999; Chase et al., 1994; Regnier et al., 1996, 1998, 1999). In contrast, control of VU in muscle fibers at submaximal [Ca2+] does not involve dynamic aspects of thin filament regulatory units, but is instead primarily determined by the average level of thin filament activation (Martyn et al., 1994) and thus the number of strongly bound cross-bridges.
Acknowledgments
We thank Drs. E. Homsher, D.A. Martyn, and C. Morris for critical comments; Carol Freitag, Josh Hawkins, Peggy Maker, Martha Mathiason, Robin Mondares, Tony Rivera, Benjamin Shepherd, and Claire Zhang for excellent technical assistance; and Drs. Vicci Korman and Larry Tobacman (University of Iowa College of Medicine) for assays of Tn binding to reconstituted thin filaments.
This work was supported by National Institutes of Health (HL52558 to A.M.G. and HL61683 to M.R.). M. Regnier is an Established Investigator of the American Heart Association.
Bo Liang's present address is Dept. of Kinesiology, Simon Fraser University, Burnaby, BC V5A 1S6, Canada.
Ying Chen's present address is Dept. of Pathology, UMDNJ RWJ Medical School, Piscataway, NJ 08854.
References
- Bing, W., A. Knott, C. Redwood, G. Esposito, I. Purcell, H. Watkins, and S. Marston. 2000. Effect of hypertrophic cardiomyopathy mutations in human cardiac muscle α-tropomyosin (Asp175Asn and Glu180Gly) on the regulatory properties of human cardiac troponin determined by in vitro motility assay. J. Mol. Cell. Cardiol. 32:1489–1498. [DOI] [PubMed] [Google Scholar]
- Bing, W., C. S. Redwood, I. F. Purcell, G. Esposito, H. Watkins, and S. B. Marston. 1997. Effects of two hypertrophic cardiomyopathy mutations in α-tropomyosin, Asp175Asn and Glu180Gly, on Ca2+ regulation of thin filament motility. Biochem. Biophys. Res. Commun. 236:760–764. [DOI] [PubMed] [Google Scholar]
- Brenner, B. 1983. Technique for stabilizing the striation pattern in maximally calcium-activated skinned rabbit psoas fibers. Biophys. J. 41:99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, B., and J. M. Chalovich. 1999. Kinetics of thin filament activation probed by fluorescence of N-((2- (Iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1, 3-diazole- labeled troponin I incorporated into skinned fibers of rabbit psoas muscle: implications for regulation of muscle contraction. Biophys. J. 77:2692–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butters, C. A., J. B. Tobacman, and L. S. Tobacman. 1997. Cooperative effect of calcium binding to adjacent troponin molecules on the thin filament-myosin subfragment 1 MgATPase rate. J. Biol. Chem. 272:13196–13202. [DOI] [PubMed] [Google Scholar]
- Cassell, M., and L. S. Tobacman. 1996. Opposite effects of myosin subfragment 1 on binding of cardiac troponin and tropomyosin to the thin filament. J. Biol. Chem. 271:12867–12872. [DOI] [PubMed] [Google Scholar]
- Chaen, S., M. Shimada, and H. Sugi. 1986. Evidence for cooperative interactions of myosin heads with thin filament in the force generation of vertebrate skeletal muscle fibers. J. Biol. Chem. 261:13632–13636. [PubMed] [Google Scholar]
- Chase, P. B., Y. Chen, K. Kulin, and T. L. Daniel. 2000. Viscosity and solute dependence of F-actin translocation by rabbit skeletal heavy meromyosin. Am. J. Physiol. Cell Physiol. 278:C1088–C1098. [DOI] [PubMed] [Google Scholar]
- Chase, P. B., T. M. Denkinger, and M. J. Kushmerick. 1998. Effect of viscosity on mechanics of single, skinned fibers from rabbit psoas muscle. Biophys. J. 74:1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase, P. B., and M. J. Kushmerick. 1988. Effects of pH on contraction of rabbit fast and slow skeletal muscle fibers. Biophys. J. 53:935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase, P. B., D. A. Martyn, and J. D. Hannon. 1994. Isometric force redevelopment of skinned muscle fibers from rabbit with and without Ca2+. Biophys. J. 67:1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase, P. B., D. A. Martyn, M. J. Kushmerick, and A. M. Gordon. 1993. Effects of inorganic phosphate analogues on stiffness and unloaded shortening of skinned muscle fibres from rabbit. J. Physiol. 460:231–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edman, K. A. P. 1979. The velocity of unloaded shortening and its relation to sarcomere length and isometric force in vertebrate muscle fibres. J. Physiol. 291:143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, I. D. C., and S. B. Marston. 1995. In vitro motility analysis of actin-tropomyosin regulation by troponin and calcium: the thin filament is switched as a single cooperative unit. J. Biol. Chem. 270:7836–7841. [DOI] [PubMed] [Google Scholar]
- Gordon, A. M., Y. Chen, B. Liang, M. LaMadrid, Z. Luo, and P. B. Chase. 1998. Skeletal muscle regulatory proteins enhance F-actin in vitro motility. Adv. Exp. Med. Biol. 453:187–197. [DOI] [PubMed] [Google Scholar]
- Gordon, A. M., E. Homsher, and M. Regnier. 2000. Regulation of contraction in striated muscle. Physiol. Rev. 80:853–924. [DOI] [PubMed] [Google Scholar]
- Gordon, A. M., M. LaMadrid, Y. Chen, Z. Luo, and P. B. Chase. 1997. Calcium regulation of skeletal muscle thin filament motility in vitro. Biophys. J. 72:1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, D. E., and D. M. Warshaw. 1993. Smooth and skeletal muscle myosin both exhibit low duty cycles at zero load in vitro. J. Biol. Chem. 268:14764–14768. [PubMed] [Google Scholar]
- Homsher, E., B. Kim, A. Bobkova, and L. S. Tobacman. 1996. Calcium regulation of thin filament movement in an in vitro motility assay. Biophys. J. 70:1881–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsher, E., D. M. Lee, C. Morris, D. Pavlov, and L. S. Tobacman. 2000. Regulation of force and unloaded sliding speed in single thin filaments: effects of regulatory proteins and calcium. J. Physiol. 524:233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsher, E., F. Wang, and J. R. Sellers. 1992. Factors affecting movement of F-actin filaments propelled by skeletal muscle heavy meromyosin. Am. J. Physiol. 262:C714–C723. [DOI] [PubMed] [Google Scholar]
- Howard, J., A. J. Hunt, and S. Baek. 1993. Assay of microtubule movement driven by single kinesin molecules. Methods Cell Biol. 39:137–147. [DOI] [PubMed] [Google Scholar]
- Hunt, A. J., F. Gittes, and J. Howard. 1994. The force exerted by a single kinesin molecule against a viscous load. Biophys. J. 67:766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh, Q., C. A. Butters, J. M. Leiden, and L. S. Tobacman. 1996. Effects of cardiac thin filament Ca2+: statistical mechanical analysis of a troponin C site II mutant. Biophys. J. 70:1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler, J., Y. Chen, B. Brenner, A. A. M. Gordon, T. Kraft, D. A. Martyn, M. Regnier, A. J. Rivera, C.-K. Wang, and P. B. Chase. 2003. Familial hypertrophic cardiomyopathy mutations in troponin I (K183Δ, G203S, K206Q) enhance filament sliding. Physiological Genomics. 14:117–128. [DOI] [PubMed] [Google Scholar]
- Kron, S. J., Y. Y. Toyoshima, T. Q. P. Uyeda, and J. A. Spudich. 1991. Assays for actin sliding movement over myosin-coated surfaces. Methods Enzymol. 196:399–416. [DOI] [PubMed] [Google Scholar]
- LaMadrid, M. A., P. B. Chase, and A. M. Gordon. 2002. Motility assays of calcium regulation of actin filaments. Results Probl. Cell Differ. 36:133–148. [DOI] [PubMed] [Google Scholar]
- Liang, B., Y. Chen, C.-K. Wang, P. B. Chase, and A. M. Gordon. 2000. Skeletal muscle regulated actin filament sliding: control by calcium and crossbridge number. Biophys. J. 78:399A. [Google Scholar]
- Liang, B., Y. Chen, C.-K. Wang, M. Regnier, P. B. Chase, and A. M. Gordon. 1999. Calcium control of skeletal muscle regulated actin filament sliding: role of myosin crossbridge number. Biophys. J. 76:A155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, D., A. Bobkova, E. Homsher, and L. S. Tobacman. 1996. Altered cardiac troponin T in vitro function in the presence of a mutation implicated in familial hypertrophic cardiomyopathy. J. Clin. Invest. 97:2842–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margossian, S. S., and S. Lowey. 1982. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 85:55–71. [DOI] [PubMed] [Google Scholar]
- Martyn, D. A., P. B. Chase, J. D. Hannon, L. L. Huntsman, M. J. Kushmerick, and A. M. Gordon. 1994. Unloaded shortening of skinned muscle fibers from rabbit activated with and without Ca2+. Biophys. J. 67:1984–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger, J. M., and R. L. Moss. 1988. Thin filament regulation of shortening velocity in rat skinned skeletal muscle: effects of osmotic compression. J. Physiol. 398:165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, C. A., L. S. Tobacman, and E. Homsher. 2001. Modulation of contractile activation in skeletal muscle by a calcium-insensitive troponin C mutant. J. Biol. Chem. 276:20245–20251. [DOI] [PubMed] [Google Scholar]
- Moss, R. L. 1986. Effects on shortening velocity of rabbit skeletal muscle due to variations in the level of thin-filament activation. J. Physiol. 377:487–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss, R. L., J. D. Allen, and M. L. Greaser. 1986. Effects of partial extraction of troponin complex upon the tension-pCa relation in rabbit skeletal muscle. J. Gen. Physiol. 87:761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee, J. D., and J. A. Spudich. 1982. Purification of muscle actin. Methods Enzymol. 85:164–181. [DOI] [PubMed] [Google Scholar]
- Potter, J. D. 1982. Preparation of troponin and its subunits. Methods Enzymol. 85:241–263. [DOI] [PubMed] [Google Scholar]
- Putkey, J. A., H. L. Sweeney, and S. T. Campbell. 1989. Site-directed mutation of the trigger calcium-binding sites in cardiac troponin C. J. Biol. Chem. 264:12370–12378. [PubMed] [Google Scholar]
- Regnier, M., D. A. Martyn, and P. B. Chase. 1996. Calmidazolium alters Ca2+ regulation of tension redevelopment rate in skinned skeletal muscle. Biophys. J. 71:2786–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier, M., D. A. Martyn, and P. B. Chase. 1998. Calcium regulation of tension redevelopment kinetics with 2-deoxy-ATP or low [ATP] in rabbit skeletal muscle. Biophys. J. 74:2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier, M., A. J. Rivera, M. A. Bates, C.-K. Wang, P. B. Chase, and A. M. Gordon. 2002. Thin filament near-neighbor regulatory unit interactions affect rabbit skeletal muscle steady state force-Ca2+ relations. J. Physiol. 540:485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier, M., A. J. Rivera, P. B. Chase, L. B. Smillie, and M. M. Sorenson. 1999. Regulation of skeletal muscle tension redevelopment by troponin C constructs with different Ca2+ affinities. Biophys. J. 76:2664–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers, J. R., G. Cuda, F. Wang, and E. Homsher. 1993. Myosin-specific adaptations of the motility assay. Methods Cell Biol. 39:23–49. [DOI] [PubMed] [Google Scholar]
- Smillie, L. B. 1982. Preparation and identification of α- and β-tropomyosins. Methods Enzymol. 85:234–241. [DOI] [PubMed] [Google Scholar]
- Stehle, R., and B. Brenner. 2000. Cross-bridge attachment during high-speed active shortening of skinned fibers of the rabbit psoas muscle: implications for cross-bridge action during maximum velocity of filament sliding. Biophys. J. 78:1458–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney, H. L., H. S. Feng, Z. Yang, and H. Watkins. 1998. Functional analyses of troponin T mutations that cause hypertrophic cardiomyopathy: insights into disease pathogenesis and troponin function. Proc. Natl. Acad. Sci. USA. 95:14406–14410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesna, D., G. Guzman, T. Miller, J. Zhao, K. Farokhi, H. Ellemberger, and J. D. Potter. 1996. The role of the four Ca2+ binding sites of troponin C in the regulation of skeletal muscle contraction. J. Biol. Chem. 271:8381–8386. [DOI] [PubMed] [Google Scholar]
- Uyeda, T. Q. P., S. J. Kron, and J. A. Spudich. 1990. Myosin step size estimation from slow sliding movement of actin over low densities of heavy meromyosin. J. Mol. Biol. 214:699–710. [DOI] [PubMed] [Google Scholar]
- Vandenboom, R., D. R. Claflin, and F. J. Julian. 1998. Effects of rapid shortening on rate of force regeneration and myoplasmic [Ca2+] in intact frog skeletal muscle fibres. J. Physiol. 511:171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]