

Abstract
Sugars are known to stabilize proteins. This study addresses questions of the nature of sugar and proteins incorporated in solid sugar films. Infrared (IR) and Raman spectroscopy was used to examine trehalose and sucrose films and glycerol/water solvent. Proteins and indole-containing compounds that are imbedded in the sugar films were studied by IR and optical (absorption, fluorescence, and phosphorescence) spectroscopy. Water is able to move in the sugar films in the temperature range of 20–300 K as suggested by IR absorption bands of HOH bending and OH stretching modes that shift continuously with temperature. In glycerol/water these bands reflect the glass transition at ∼160 K. The fluorescence of N-acetyl-L-tryptophanamide and tryptophan of melittin, Ca-free parvalbumin, and staphylococcal nuclease in dry trehalose/sucrose films remains broad and red-shifted over a temperature excursion of 20–300 K. In contrast, the fluorescence of these compounds in glycerol/water solvent shift to the blue as temperature decreases. The fluorescence of the buried tryptophan in Ca-bound parvalbumin in either sugar film or glycerol/water remains blue-shifted and has vibronic resolution over the entire temperature range. The red shift for fluorescence of indole groups exposed to solvent in the sugars is consistent with the motion of water molecules around the excited-state molecule that occurs even at low temperature, although the possibility of static complex formation between the excited-state molecule and water or other factors is discussed. The phosphorescence yield for protein and model indole compounds is sensitive to the matrix glass transition. Phosphorescence emission spectra are resolved and shift little in different solvents or temperature, as predicted by the small dipole moment of the excited triplet state molecule. The conclusion is that the sugar film maintains the environment present at the glass formation temperature for surface Trp and amide groups over a wide temperature excursion. In glycerol/water these groups reflect local changes in the environment as temperature changes.
INTRODUCTION
Many extracellular proteins are glycosylated. Although sugar groups of these proteins are often involved in processes such as recognition in cell-cell communication and in the immune system, they also serve a role in protein stability (Helenius and Aebi, 2001). Stabilization of proteins by sugar is seen in another situation in biology. A wide variety of organisms use either the disaccharide α,α-trehalose or sucrose to survive extreme temperatures and dehydration. These species can be restored to activity after rehydration (Crowe et al., 1992).
The mechanism by which sugars interact with proteins is, therefore, of importance for the overall question of protein stability. Trehalose has been shown to replace water molecules that form hydrogen bonds to the surface of the protein (Carpenter and Crowe, 1989; Prestrelski et al., 1993). Sugar glasses can also be used as an experimental tool because they provide a means to immobilize the protein, allowing the internal dynamics of proteins to be studied (Cordone et al., 1998; Gottfried et al., 1996; Khajehpour et al., 2003; Prabhu et al., 2002).
We have recently shown that films made from a mixture of trehalose and sucrose are stable and transparent, and that proteins retain their structure when incorporated in them (Wright et al., 2002). The glycerol/water solvent system has been widely used as a cryosolvent for proteins (Douzou, 1977), and in the work here it serves as a comparison to amorphous sugar films. In our study we wish to see how the solvent matrix interacts with the protein. The first measurement directly examines the sugar film and the protein by IR spectroscopy. The basis of this measurement is that molecules and groups rearrange to lower energy states, since lowering temperature gives increasing H-bonding strengths. Consequently, the IR absorption bands of groups involved in H-bonding shift with temperature (Jeffrey, 1997) and the temperature dependence of these bands permits us to infer what motions are allowed in the sugar substrate. The second measurement examines how the environment influences the emission properties of the indole derivative N-acetyl-l-tryptophanamide (NATA) and Trp in single Trp-containing proteins. It is widely understood that indole has a larger dipole moment in the excited state than in the ground state (Hahn and Callis, 1997). Time-resolved studies indicate that as water molecules relax around the excited-state molecule there is a shift to the red in the emission spectra (Brand and Toptygin, 2000; Hahn and Callis, 1997; Lakowicz and Cherek, 1980; Montoro et al., 1988). The red shift, therefore, could indicate the movement of water molecules. Phosphorescence of Trp is another emission parameter that is influenced by dynamics. At high temperatures in fluid solutions, phosphorescence is quenched by atmospheric O2 (Papp and Vanderkooi, 1989). Phosphorescence yield is also influenced by the rigidity of the solution: the more rigid the environment, the higher the efficiency of phosphorescence (Gonnelli and Strambini, 1993). The excited triplet state has lower dipole moment compared with the excited singlet state, and consequently phosphorescence spectra are less sensitive than fluorescence to a dipolar environment, but spectral shifts of phosphorescence can also indicate solvent effects.
MATERIALS AND METHODS
Materials
Sigma Chemical Co. (St. Louis, MO) supplied α−D-glucopyranosyl-α-D-glucopyranoside (trehalose), α-D-glucopyranosyl-β-D-fructofuranoside (sucrose), N-acetylglycine (AG), NATA, honeybee melittin, and staphylococcal nuclease (micrococcal endonuclease) from Staphylococcus aureus. Parvalbumin was prepared from frozen codfish (Sudhakar et al., 1995). D2O and n-pentane (spectroscopic grade) were obtained from Aldrich Chemical Co. (Milwaukee, WI).
Methods
IR spectra were obtained with a Bruker IFS 66 Fourier transform IR instrument (Brookline, MA) as previously described (Kaposi et al., 1999). All spectra were taken in the transmission mode, except for the crystals (Fig. 3) where the ATR mode was used. Raman spectra were taken with a Bruker Raman-IR IFS 66V Fourier transform instrument. A Hitachi Perkin-Elmer U-3000 (Newtown, PA) spectrophotometer was used to take visible absorption spectra. Steady-state emission spectra were obtained with a Fluorolog-3-21 Jobin-Yvon Spex instrument equipped with a R2658P Hamamatsu photomultiplier and using front-face geometry (Edison, NJ).
FIGURE 3.

(a) Trehalose amorphous film at 44% relative humidity; (b) trehalose crystal. HOH bending absorption indicated. Temperature: 20°C.
Widths and maximal positions of spectral bands were determined using PeakFit (Jandel Scientific Software, San Rafael, CA).
The sample temperature for IR and UV absorption and for fluorescence was maintained using an APD closed cycle Helitran cryostat (Advanced Research Systems, Allentown, PA). A holder for these windows was constructed to minimize strain on the windows due to contraction at low temperature (Research Instrumentation Shop, University of Pennsylvania School of Medicine, Philadelphia, PA). For IR measurements, the outer cryostat windows were made of CaF2. The inner cryostat windows, which experience the temperature gradient, were 2 mm thick and were made of ZnSe (Janos Technology, Townshend, VT). For UV absorption or fluorescence measurements, the outer windows were made of quartz and the inner windows were made of sapphire. The temperature was measured with a silicon diode near the sample, and the temperature was controlled using a Model 9650 temperature controller (Scientific Instruments, Palm Beach, FL). Cryogenic temperature profiles were carried out from high to low temperature, with the temperature being measured every 10 degrees.
Sugar film formation
Crystals can form during the course of an experiment in films made of pure trehalose (Librizzi et al., 1999). A mixture of sugars alleviates this problem. Sugar film was prepared as follows. For mixed sugar films (TS), trehalose (300 mg) and sucrose (300 mg) were dissolved in 500 ml of distilled water to form the stock sugar solution. For pure sugar films, 600 mg of the desired sugar was dissolved in the water. The sugar solution was heated to ∼65°C to ensure complete dissolving of the sugar. The solution was cooled, and ∼1 mg of dry protein or 5 μl of 4 mM NATA was added to 100 μl of the sugar and 400 μl of 10 mM potassium phosphate, pH 7.0. A transparent film was formed by evaporation of water from the sugar solutions. In one film preparation that is suitable for UV/vis absorption measurements, 100 μl of the stock sugar solution was added to 400 ml phosphate buffer (pH 7.0), and the solution was pipetted to cover a 25-mm round quartz plate. Quartz plates were obtained from Esco Products Inc. (Oak Ridge, NJ), and they were of 1 mm thickness. For IR measurements, 10 μl of the sugar solution was diluted with water to 500 μl and the solution was plated on a CaF2 plate (Janos Technology). This resulted in a thickness of the sugar film of ∼6–20 μ. The samples were allowed to dry at 65°C. During drying, the sample temperature was maintained using a VWR Scientific Products Heat Block.
The sugar films were hard to the touch and optically clear. Examination of the films under crossed polarizers showed no indication of crystal formation. The mole ratio of water to trehalose in the film was determined by the extinction coefficients of water and trehalose. The extinction coefficient of water was determined to be 21.4 M−1 cm−1 at 1641 cm−1 at 20°C. This value was obtained by measuring the absorption of water using a spacer that was calibrated to be 6 μ thick and is the absorptivity found by other workers (Venyaminov and Prendergast, 1997). The spacers used were Teflon, and since the spacing is influenced by the tightness of the screws holding the plates, the actual spacing was determined by interference patterns as described in textbooks (Stuart, 1996). The CH stretching frequency was used to determine the trehalose and sucrose content. In this case a solution that was 0.9 molar in trehalose and sucrose was examined for a sample at known spacing as described for water. The extinction for the sugar at 1932 cm−1 was determined to be 24.7 M−1 cm−1. Beer's Law was assumed to apply when a mixture of sugar and water was used. The method used may have systematic errors, but since the ratio of the two absorption peaks were used to determine the water/sugar content, the errors will tend to cancel. Fig. 1 gives IR spectra of the TS film at two levels of hydration. Trehalose and sucrose show similar bands in the C-H stretch region (∼2930 cm−1). The off-scale band at 3400 cm−1 represents absorption from the OH stretch mode that arises from the sugar and water. The peaks marked νCH and σHOH are the peaks used to determine the water/sugar ratio. A background correction was applied to obtain the absorbance of the peaks.
FIGURE 1.

IR spectra of TS amorphous film in two humidity conditions. Trehalose/sucrose glass at 1/1 molar ratio. Spacer: 6 μ. Percent relative humidity: (a) 0%; (b) 65%. Temperature: 20°C.
After the film was formed, the hydration of the sugar was varied by exposing the sample to an atmosphere of known humidity for 2 h for thin samples and 24 h for thick samples. Since the films were typically 6–20 μ thick, this time was sufficient for hydration equilibration. The relative humidity of air at 20°C over saturated solutions of salts is as follows: potassium sulfate, 97%; ammonium nitrate, 65%; potassium carbonate, 44%; potassium acetate, 22%; lithium chloride, 12%. The original sources for these values are given in the thesis of Gruner (1977). For the fluorescence measurements at room temperature, where the time of measurement was rather long, a solution of the salts was placed in the cell compartment, so that the film maintained hydration. For temperature-dependent measurements, the sample was placed between two plates, so that it was not exposed to the surrounding gases during the measurement. The variation of water content in the film with exposure to air at given relative humidity is shown in Fig. 2. The molar ratio scatter plot of water to sugar suggests that under usual atmospheric humidity conditions, the amount of water in the trehalose film is ∼2 mol of water per mol of sugar. This amount of water also corresponds to the two molecules of water of crystallization per molecule of trehalose (Akao et al., 2001).
FIGURE 2.

Water:sugar ratio in TS glass at different relative humidity. Temperature: 20°C.
To exchange the water of hydration of the film with D2O, the sugar was initially dissolved in D2O, rather than in H2O. The D2O was evaporated at 70°C and the film hydrated over D2O.
To obtain IR absorption from trehalose crystals, the trehalose solution was placed on an ATR ZnSe IR plate and allowed to sit in the atmosphere (40–60% relative humidity) for two days. The sample became noticeably crystalline, which was confirmed by its absorption under crossed polarizers. The absorption was measured in the ATR mode.
RESULTS
Characterization of sugar films
In Fig. 3 we contrast IR spectra of trehalose in amorphous and crystalline states. The absorption of crystalline sugar has more fine structure than seen for the glassy sugar. The region between 1000 and 1200 cm−1 is especially diagnostic. What appears as one peak at 1050 cm−1 in the glass resolves into two peaks in the crystal. The absorption of water in the sample is indicated. The water content was slightly higher in the crystal than in the sugar film, but this is a function of the sample preparation and exposure to water vapor (see Fig. 2). It is noteworthy that the absorption peak of νHOH is slightly different in the two conditions. The peak of HOH bending is at 1647 cm−1 in the glass and 1639 cm−1 in the crystal.
IR absorption spectra of trehalose and sucrose glassy films are shown in Fig. 4. The spectra of the two sugars differ in the region between ∼1000–1200 cm−1. Trehalose/sucrose (TS) film resembles the summed average of trehalose and sucrose films taken separately. This fact is interpreted to show that there are no specific interactions between molecules, an observation consistent with the glassy (i.e., amorphous) nature of the sample.
FIGURE 4.

IR spectra of sugar films. Trehalose (T), sucrose (S), and mixed trehalose/sucrose (TS) glasses. Glasses made at 65°C as described in Materials and Methods. T + S is the weighted sum of the individual trehalose and sucrose spectra. Temperature: 20°C.
Vibrational spectra of trehalose in water solution and in film were next compared. Both IR and Raman spectra were obtained. In the two types of spectroscopy the peaks occur at the same position, but in the Raman spectrum, peaks of vibrational modes with a strongly dipolar nature, such as the OH stretch at 3400 cm−1, are relatively greatly reduced (Fig. 5, a and b). The Raman spectrum also allows for examination at frequencies below 1000 cm−1. The Raman peaks of the sugar in the solid films and in water solution are very similar (Fig. 5, b and c), a result that is also consistent with the amorphous nature of the sample.
FIGURE 5.

IR and Raman spectra of trehalose and sugar in glass and in solution. (a) IR absorption of TS glass at 22% relative humidity. (b) Raman spectrum of TS film at 22% relative humidity on CaF2. (c) Raman spectrum of 0.9 M trehalose and 0.9 M sucrose in H2O. All spectra taken at 20°C. The resolution of the Raman measurement was 1 cm−1 and a 3.5-mm aperture was used. 2000 scans were averaged.
Temperature dependence of IR bands in TS films and glycerol/water
The sugar film sample is a solid, but it is well recognized that groups within solids have a variety of motions. Fig. 6 summarizes the temperature dependence of the HOH bend and OH stretch absorption for the TS film and the glycerol/water solvent. The OH stretch absorption peaks are off-scale so the midpoint value was used for a plot. (The OH region has contributions from sugar or glycerol, as well as water, and water, in turn, exhibits absorption from both symmetric and antisymmetric modes). As temperature decreases, the OH stretch midpoint absorption frequency goes lower and the bend absorption band goes to higher frequency. The OH stretch in trehalose film that is nearly devoid of water shifted ∼20 cm−1 over the temperature range from 300 to 20 K. Remarkably, even down to 40 K, there are changes with temperature, indicating that the water in the film still has motion at this low temperature. For glycerol/water, the shift in the HOH bending mode has a break at ∼160 K, the temperature of the glass transition for this solvent. The shift of the OH stretch midpoint of glycerol/water solvent was ∼80 cm−1.
FIGURE 6.

Temperature dependence of OH stretch and HOH bend frequencies. Squares, TS film; Circles, glycerol/water.
IR absorption of protein in TS film
We now examine the protein in TS film. IR spectra of parvalbumin in TS film at 22% humidity are shown in Fig. 7 at 20 and 300 K. The absorbance in the region from 1000 to 1200 cm−1 is dominated by the sugars. In the first and second derivative spectra, it can be seen that there is very little change in the IR absorption over the temperature range of almost 300 degrees. This region is sensitive to the condition of the sugar (see Fig. 3), and therefore it can be concluded that the film is stable over this temperature range.
FIGURE 7.
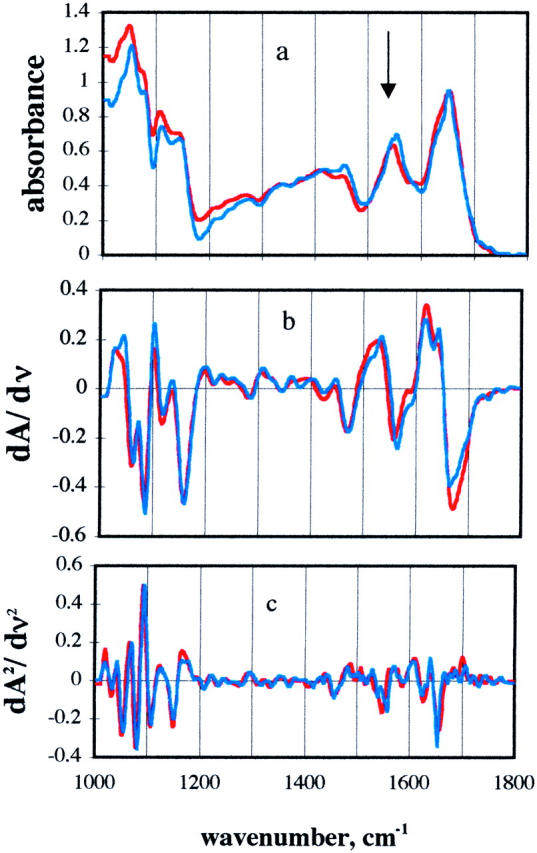
(a) Infrared spectra of Ca-parvalbumin. Ca-parvalbumin in TS glass at 22% relative humidity at 20 K (blue) or 300 K (red); (b) first derivative; (c) second derivative.
The small peak at around 1585 cm−1 is at the position of the antisymmetric stretch of −COO−(Nara et al., 1994). The peak at 1650 cm−1 is identified as the amide I. The position of the amide I band is indicative of folded protein. Previous work by Carpenter and Crowe showed by IR that protein in freeze-dried sugar had native-like conformation (Carpenter and Crowe, 1989). The amide I band position has little temperature dependence from 20 to 300 K. The constancy of this region for parvalbumin in TS film shows that the protein retains its overall folding over this temperature range. In the derivative spectra for the protein in TS there can be seen some changes in the IR spectrum in the amide I region, but the overall positions of the major bands are maintained, and the band is indicative of a folded protein. The measurements also were repeated with a dry sample, and the same conclusion was made.
The amide II peak occurs at 1550 cm−1. Close examination of Fig. 7 reveals that this peak changes with temperature, as was previously reported (Wright et al., 2002). The temperature dependence of the peak position in TS film is shown in Fig. 8, upper panel. The figure also shows that the amide II peak of AG shifts in the same manner as in the protein. To compare the amide peak in glycerol/water we used a deuterated sample. The amide II′ of AG shifts 15 cm−1 to higher frequency as temperature decreases (Fig. 8, lower panel). The amide II peak is predominately due to an NH bending mode and has similar temperature dependence as the HOH bending mode (see Fig. 6).
FIGURE 8.

Frequency of IR absorption bands. (Upper) Square, Amide II of bending mode for AG in TS film at 22% humidity; triangle, amide II absorption band of Ca-parvalbumin in TS glass at 22% humidity. (Lower) Amide II′ of AG in perdeuterated glycerol/D2O (60/40).
Characterization of NATA and proteins in sugars and glycerol
Absorption and fluorescence of indole and its derivatives in solvents
We now examine how the glassy matrix influences the absorption and emission of Trp in proteins and indole model compounds. In Fig. 9 the absorption of NATA in TS film and in glycerol/water solvent is given for room temperature and 20 K. The absorption of 3-methyl indole in pentane is also given for comparison. The absorption band at 290 nm of NATA narrows with lower temperature in TS, but the narrowing was more pronounced in glycerol/water. The absorbance band is narrowest for 3-methyl indole in pentane. The highest energy fluorescence band coincides with the absorption band, as has been reported before (Meech et al., 1983). For NATA in glycerol/water, an arrow indicates a shoulder on fluorescence peak, which would represent the highest energy emission band. In this case, there is no coincidence of absorption and emission, consistent with the emission being from the 1La state.
FIGURE 9.

Spectra of indole compounds. (Upper) NATA in TS film; (middle) NATA in glycerol/water; (bottom) 3-methyl indole in n-pentane. Solid lines, 290 K; dotted lines, 20 K. Spectra to the left are absorption spectra; spectra to the right are fluorescence spectra. Emission band-pass, 2 nm. Arrow indicates the position of a shoulder.
The emission spectra of NATA, N-methyl indole, 3-methyl indole, Ca-parvalbumin and Ca-free parvalbumin in different solvents at room temperature are compared in Fig. 10. The emission maximum of NATA, N-methyl indole, and 3-methyl indole in water is at 358, 355, and 370 nm, respectively, and the emission spectra are broad. The values of the maximum in the TS film are somewhat blue-shifted relative to those found for the compounds in glycerol/water. When TS film is hydrated, the emission maximum is intermediate between the aqueous glycerol solution and totally dry film. In pentane, the fluorescence spectra of N-methyl indole and 3-methyl indole are blue-shifted with indication of vibronic structure (Fig. 10, B and C). The shift to the blue for indole in hydrophobic solvent is well-known (Konev, 1967).
FIGURE 10.
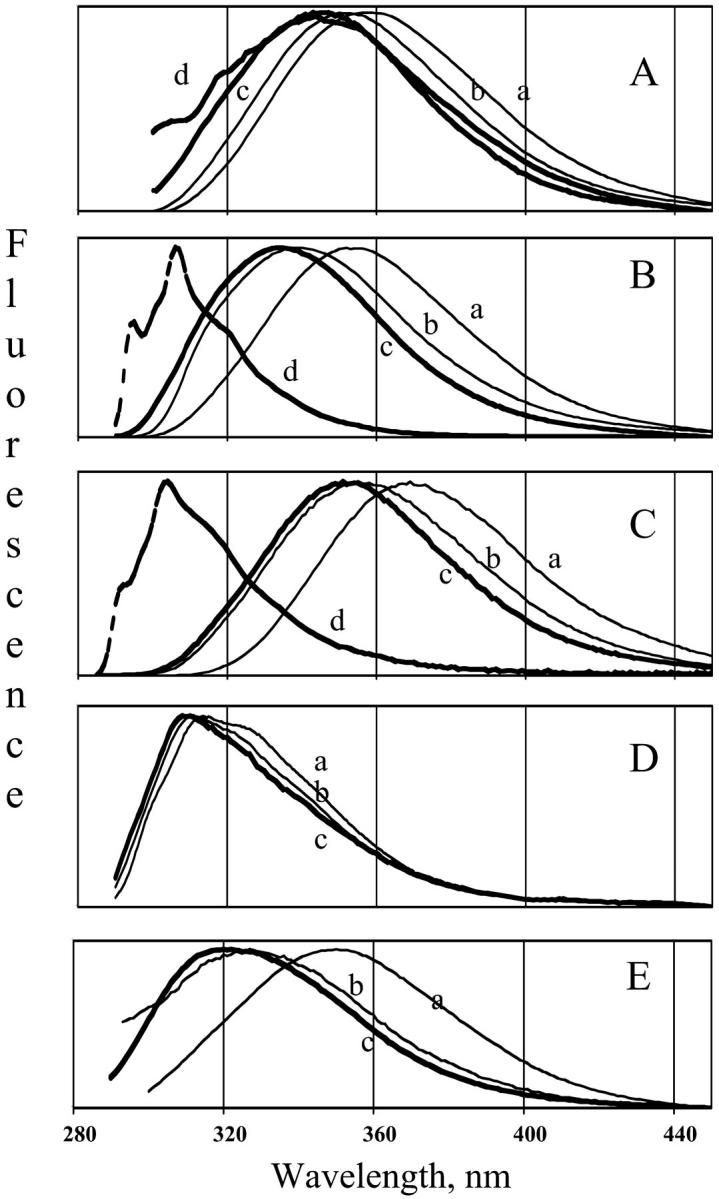
Fluorescence spectra of indole compounds in TS glass and in solution at 20°C. (A) NATA; (B) N-methyl indole; (C) 3-methyl indole; (D) Ca-parvalbumin; (E) Ca-free parvalbumin. The concentration of the compounds was ∼30 μM. The solvents are as follows: (a) water, (b) TS film at 97% humidity, (c) dry TS film, (d) n-pentane.
Panels D and E show emission from Trp in parvalbumin. The removal of Ca from parvalbumin induces large changes in the protein conformation. The Trp fluorescence maximum shifts from ∼325 to ∼350 nm when Ca is removed (Eftink and Wasylewski, 1989; Ferreira, 1989; Permyakov et al., 1980; Sudhakar et al., 1993, 1995). In the glass, the emission of Ca-parvalbumin shows a blue-shifted emission spectrum (Fig. 10 D), resembling 3-methyl indole in pentane. In contrast, the emission of the Ca-free protein is broad in all solvents. The emission shifts to the blue with dehydration in the TS film (Fig. 10 E) but never becomes as resolved as the emission of the buried Trp in Ca-parvalbumin.
Absorption, fluorescence, and phosphorescence spectra of NATA as a function of temperature
Fig. 11 shows the emission of NATA in glycerol/water at temperatures ranging from room temperature to 20 K. The fluorescence maximum of NATA shifts to lower wavelength as the temperature decreases, and the emission band begins to be structured. The difference in wavenumber between the fluorescence maximum in fluid solution (359 nm) and at 20 K (312 nm) is 4200 cm−1. At low temperature the fluorescence of NATA in glycerol/water manifests some vibrational resolution although the resolution is still low. As temperature decreases, the phosphorescence of the indole group is apparent as vibronically-resolved emission bands from 385 to 500 nm build in, with 190 K being the highest temperature where phosphorescence was seen in the steady-state emission spectrum.
FIGURE 11.

Emission of NATA in glycerol/water. Temperature was 290 to 12 K in 20 K increments. Representative temperatures are indicated. Excitation, 280 nm; band-pass, 2 nm. Emission band-pass, 2 nm.
The emission spectrum of NATA in TS film was examined at different hydration of the film. For hydrated film the emission spectra are shown in Fig. 12. For dry film, the emission is shown in Fig. 13. The fluorescence emission remains red-shifted as temperature decreases for the dry film with a small shift for the hydrated film. The red shift is in contrast to the emission of NATA in glycerol/water (see Fig. 11). At higher temperature, the fluorescence maximum of NATA is 359, 338, and 337 nm for glycerol/water, hydrated TS, and dehydrated TS film, respectively. Phosphorescence of NATA is apparent at room temperature in the steady-state spectrum for the dry film (Fig. 13).
FIGURE 12.

Emission from NATA in TS glass. Hydration, 97%. Temperature was 290 to 12 K in 20 K increments. Excitation, 280 nm; band-pass, 2 nm. Emission band-pass, 2 nm.
FIGURE 13.

Emission from NATA in TS glass. Hydration, dry. Temperature was 295 K and then 290 K to 12 K in 20 K increments. Excitation, 280 nm; band-pass, 2 nm. Emission band-pass, 2 nm.
The shift of fluorescence is an important feature for us. We considered several means to describe this. It would be most reliable to plot the shift of the S0,0 transition. This transition is unambiguously seen only in the hydrophobic environment (see Fig. 9), and therefore it cannot be used. Where the emission is purely a single electronic transition, it would be correct to use the barycenter, but the “exciplex”-like broad emission is shifted in aqueous solution so that the phosphorescence and fluorescence overlap in some spectra (see Fig. 11). Therefore, we used the fluorescence maximum, a value that is easily reproduced by other workers. The fluorescence maxima for NATA are plotted for glycerol/water and TS in Fig. 14. The shift in fluorescence of NATA with change in temperature is more pronounced for the compound in the glycerol/water solvent as compared to dry TS film. The trend is that when the emission is red-shifted at room temperature, it becomes blue-shifted at low temperature.
FIGURE 14.

Fluorescence peak positions for NATA in TS (squares) or in 60:40 v/v glycerol/water (circles).
One interpretation of a red shift of the emission spectrum is that water relaxes around the excited state of tryptophan. We reasoned that if so, the water must be mobile on the ns time scale in the film at very low temperatures. For this to occur, the mechanism must be a tunneling reaction, well known for protons. A tunneling reaction should be much slower for deuterium than for hydrogen. With this reasoning, we examined the temperature dependence of NATA emission in TS film that had D2O exchanged for H2O. We did not see that the spectrum of NATA becomes blue-shifted, as for glycerol/water. As seen in Fig. 15, the spectrum resembles that for film hydrated with H2O.
FIGURE 15.

Fluorescence of NATA in deuterated TS film. Deuteration was achieved as described in Materials and Methods.
Fluorescence spectra of Trp in proteins
Trp of Ca-parvalbumin in TS film shows fluorescence with structured features (Fig. 16). There is some increase in the spectral resolution as temperature is lowered below ∼160 K, but no large shift in the band positions occurs over the temperature range of 10 to 300 K. The spectrum of Ca-parvalbumin in glycerol/water is very similar to that for the protein in TS film (Fig. 17). In Fig.16 the spectra were not normalized, to emphasize the fact that as temperature decreases there is an increase in fluorescence intensity. (The increase in fluorescence cannot be accounted for by an increased concentration of sample due to shrinkage by lowering the temperature because the change in absorption was less than or equal to ∼10%). In Fig. 17 the spectra were normalized in fluorescence to emphasize that as temperature decreases, the ratio of phosphorescence to fluorescence increases.
FIGURE 16.

Emission of Ca-parvalbumin in TS film taken at temperatures from 290 to 11 K in 20 K increments. Excitation, 280 nm; band-pass, 2 nm. Emission band-pass, 2 nm.
FIGURE 17.

Emission of Ca-parvalbumin in 60:40 v/v glycerol/water taken at temperatures from 290 to 11 K in 20 K increments. Excitation, 280 nm; band-pass, 2 nm. Emission band-pass, 2 nm.
Emission spectra of melittin in glycerol/water are shown as a function of temperature in Fig. 18. The emission features for the melittin show broad emission at high temperature, and as temperature decreases, the bands change in resolution such that at low temperature the emission resembles that for the buried tryptophan of parvalbumin. Melittin at low concentrations is a monomer in a random coil to helical equilibrium, whereas in higher concentrations it is a tetramer in α-helix (Quay and Condie, 1983; Tatham et al., 1983). We verified that the protein is helical by the position of the amide I IR absorption band in independent studies. Our experiments were done at high protein concentrations where the protein is helical; the similarity of the spectra with NATA suggests that local events, not the global rearrangement of the peptide, are causing the shifts. Fig. 19 shows the spectrum of melittin in the TS sheets. In this case, like NATA, the emission remained red-shifted over the temperature range.
FIGURE 18.

Emission of melittin taken at temperatures from 290 to 11 K in 20 K increments. 50 mg protein/ml of 60:40 v/v glycerol/water. Excitation at 275 nm, with 5-nm bandpass. Emission band pass, 1 nm.
FIGURE 19.

Emission of melittin in TS glass taken at temperatures from 290 to 11 K in 20 K increments. Excitation at 275 nm, with 5-nm bandpass. Emission band pass, 1 nm.
The emission of staphylococcal nuclease in glycerol/water is shown in Fig. 20. The same pattern is seen for melittin in glycerol/water (Fig. 18). The fluorescence of nuclease in TS remained red-shifted at low temperature (not shown). However, it was noted that exposure to the UV lamp produced deterioration of the Trp. The Trp of nuclease has positive charges nearby that red shift its emission spectrum (Vivian and Callis, 2001). Whether this environment also influences its photoreactivity is not known. The fact that the spectrum blue shifts at low temperature in the glycerol/water solvent may indicate that there is unfolding of the protein in this solvent. Ca-free parvalbumin showed the general pattern of nuclease and melittin: broad, red-shifted emission at high temperature in glycerol/water and blue shift at low temperature (data not shown). The loss of some structural features—cold denaturation—is a well known phenomenon for many proteins, including nuclease (Griko et al., 1988). In examining the IR spectra of various proteins and peptides in glycerol/water, we came to the conclusion that the α-helix was retained at low temperature, although the H-bonding to the solvent for exposed amide groups increased as temperature lowered (Manas et al., 2000; Walsh et al., 2003). Nevertheless, it is still a possibility that the features that we see may be due to global changes in the protein, in addition to local changes at the Trp site.
FIGURE 20.

Emission of staphylococcal nuclease taken at temperatures from 290 to 11 K in 20 K increments. 28 mg protein/ml of 60:40 v/v glycerol/water. Excitation at 275 nm, with 6-nm bandpass. Emission band pass, 1 nm.
The fluorescence maxima of NATA and single Trp-containing proteins observed for different conditions are given in Table 1.
TABLE 1.
Fluorescence maxima, nm, of NATA and single-Trp proteins
| Compound | Solvent/matrix | 290 K | 20 K |
|---|---|---|---|
| NATA | TS glass, dry | 337 | 325 |
| TS glass, 97% | 337 | 321 | |
| Glycerol/water | 359 | 312 | |
| Ca-parvalbumin | TS glass, dry | 310 | 308 |
| TS glass, 97% | 310 | 308 | |
| Glycerol/water | 312 | 312 | |
| Ca-free parvalbumin | TS glass, dry | 328 | 328 |
| Glycerol/water | 335 | 313 | |
| Melittin | TS glass, 97% | 331 | 332 |
| Glycerol/water | 344 | 314 | |
| Staphylococcal nuclease | TS glass, 97% | 316 | 316 |
| Glycerol/water | 337 | 313 |
97%: the sample was incubated at this relative humidity during preparation.
Phosphorescence of NATA and proteins
The appearance of phosphorescence as temperature is lowered is another informative parameter since the absolute intensities are a function of the quantum yield of luminescence and intersystem crossing. The emission maximal positions and widths are given in Table 2.
TABLE 2.
Phosphorescence parameters: position and width (half maximum)
| Compound | Solvent/matrix | Temperature, K | Position, nm | Width, nm |
|---|---|---|---|---|
| NATA | TS glass | 270 | 412 | 7.0 |
| 12 | 410 | 8.3 | ||
| Glycerol/water | 190 | 413 | 10 | |
| 11 | 406 | 6.6 | ||
| Ca-parvalbumin | TS glass | 280 | 410 | 6.2 |
| 10 | 409 | 5.6 | ||
| Glycerol/water | 210 | 411 | 7.6 | |
| 10 | 409 | 7.4 | ||
| Melittin | TS glass | 270 | 412 | 7.4 |
| 12 | 409 | 4.9 | ||
| Glycerol/water | 200 | 412 | 7.5 | |
| 20 | 410 | 6.0 | ||
| Staphylococcal nuclease | TS glass | 295 | 408 | 4.3 |
| 11 | 406 | 3.5 | ||
| Glycerol/water | 200 | 407 | 4.5 | |
| 13 | 406 | 4 |
In the range of temperature scans given it can be seen that as temperature decreases there is an increase in fluorescence intensity (Examples: Figs. 16, 18, and 19). Intersystem crossing from S1 to T1 may be relatively temperature independent, and so the phosphorescence/fluorescence (P/F) ratio can give information on how the sugar film affects the indole chromophore transition T1 to S0. The phosphorescence intensity is much smaller than the fluorescence at room temperature, so the phosphorescence is not apparent in the steady-state spectra. The phosphorescence lifetime can be measured, and NATA in glass has a phosphorescence lifetime of ∼30 ms in TS (Wright et al., 2002). The phosphorescence lifetimes of the indole compounds were all 5–6 s at low temperature. Fig. 21 gives the P/F ratios for NATA and Ca-parvalbumin in three conditions. The phosphorescence increases at the glass transition of glycerol/water for both NATA and parvalbumin (Fig. 21 C). In case of the TS, the film is stable over the temperature range, but the phosphorescence increases as temperature decreases. Focusing on the line at 200 K, one can see the differences in the samples. The hydrated sample shows an increase in the NATA P/F ratio below this temperature, whereas NATA in the dry TS film exhibits phosphorescence at temperatures above this.
FIGURE 21.

Phosphorescence/ fluorescence (P/F) ratio for the indole-containing compounds. NATA (squares) and Ca-parvalbumin (circles). Upper: dry TS glass. Middle: TS at ∼97% relative humidity. Bottom: glycerol and water, 60:40 v/v. For parvalbumin in dry and 97% TS film, the phosphorescence intensity at 410 nm was used to calculate the P/F ratio. For other samples, the phosphorescence intensity maximum (436–440 nm) was used for phosphorescence. The fluorescence maximum was used for the fluorescence intensity.
DISCUSSION
The sugar films are hard to the touch, do not flow, and are transparent. The sugar films are suitable for incorporation of proteins, and therefore the condition of proteins in this form of sugar is a subject of general interest. We show here that the sugar film is stable and remains amorphous over a wide temperature range. Although the glass appears physically solid, motion occurs. In these films of amorphous sugar, the OH stretch frequency shifts with lowering temperature even to very low temperature. In glycerol/water, the OH stretch frequency shifts with lowering temperature until the glass transition; the shift is much larger in liquid glycerol/water than for the sugar and residual water of the TS films (Fig. 6). The bending mode of water is also temperature dependent, and again the temperature dependence is stronger in the liquid glycerol/water than in the TS glass. The results are interpreted as follows. As thermal motion decreases, the solvent rearranges to give favorable H-bonding angles and distances. An increase in H-bonding strength with lowering temperature would shift the peaks in the observed way. It follows from the fact that the spectral bands shift with temperature that there is motion within the glass to allow for the more favorable H-bonding interactions at lower temperatures.
Protein interactions with the sugar
Because the glass water content is low, the amide I and II bands of incorporated proteins can be examined without significant water interference. The amide I band arises primarily from the C=O stretch of the amide group (Krimm and Bandekar, 1986). The amide I band positions of the protein parvalbumin in TS is nearly constant over the temperature range (Fig. 7), although the peaks become sharper at lower temperature. The frequency of the amide I band depends upon its H-bonding, and in glycerol/water there are large changes in the amide I region as a function of temperature (Kaposi et al., 1999; Manas et al., 2000). As thermal motion of water decreases with lowering of temperature the H-bonding between water and the carbonyl increases, resulting in a shift to lower frequency for the amide I band (Walsh et al., 2003). H-bonding is highly dependent upon angle, and so a rotation of the water group to maximize the interaction as the temperature decreases will be reflected in the amide I band shift to lower frequency.
The amide II band of protein incorporated into glass and the HOH bending mode band go to higher frequency as temperature decreases, and these bands have very similar temperature dependence (Fig. 8). The amide II band consists of NH in-plane bending combined with CH stretching (Krimm and Bandekar, 1986). The band at 1650 is the νHOH bending mode. The implication is that there is still motion of water and the protein within the film over a large temperature range.
Fluorescence of indole
The fluorescence maximum of tryptophan in proteins ranges from ∼310 to 360 nm depending upon exposure of the group to water. Fluorescence lifetimes and quantum yields are also very sensitive to the environment. The large fluorescence spectral shift of Trp in different proteins and indole in different solvents is related to the large difference in dipole in the ground and excited state of the molecule. With the availability of a sizeable dataset of protein structures, it is possible to sort out the protein environmental effects producing the Stokes shift. The charges in the protein and the field of water both contribute to an internal electric field (Pierce and Boxer, 1995; Vivian and Callis, 2001). The large dipole moment of the excited-state molecule makes a dipolar solvent more interactive with the excited-state than the ground-state molecule. This interaction with solvent results in very large shifts in the emission seen in proteins. We should note that Trp has two emitting states, the La and Lb states, which can complicate analysis. Many experiments and calculations led to the conclusion that for all or nearly all Trp in proteins the fluorescence emission is from the 1La state (Callis, 1991; Callis et al., 1995; Eftink et al., 1990; Valeur and Weber, 1977; Vivian and Callis, 2001). A red shift in spectra maximum in fluid solution arises in dipolar solvents when solvent molecules can rearrange around the excited state. The fluorescence of Trp is a measure of how the solvent responds to a change in the charge distribution of indole upon excitation. Observed Stokes shifts are therefore functions of both the field and the rearrangement of charged groups around the molecule in response to the altered electron density upon its excitation.
To summarize the results of Trp fluorescence:
For the buried Trp in Ca-parvalbumin, the emission spectrum remains blue-shifted and shows vibronic resolution both in glycerol/water and in the solid TS film. The spectrum changes very little with temperature. The Trp in Ca-parvalbumin is buried, and therefore not directly exposed to the solvent. It can be concluded that both solvents leave the Trp buried in a hydrophobic environment.
In glycerol/water, the fluorescence spectra of NATA and proteins that have partially exposed Trp (Ca-free parvalbumin, mellitin, and staphylococcal nuclease) are blue-shifted at low temperature and red-shifted at high temperature.
In contrast, for all of these samples in the TS sheet the emission is broad and red-shifted. For very dry samples, the peak position is constant with temperature; with samples containing more water, there is a shift with temperature, but less than seen in the glycerol/water sample.
The conclusion seems to be that the TS glass maintains the environment of the exposed indole chromophore over a wide temperature range. How can this be, when the glycerol/water system shows large shifts with temperature? There are various ways to explain the results. We discuss them below.
First, we consider that the response of the solvent is due to a dynamic rearrangement around the excited-state molecule. As noted above, there is a large change in dipole moment upon excitation for indole, and this causes the solvent molecules to rearrange around the excited-state molecule and produces a red shift. This explains the red shift at higher temperatures for exposed Trp in glycerol/water, as seen in NATA, Ca-parvalbumin, Ca-free parvalbumin, and melittin. Longworth (1971) showed a similar fluorescence spectrum for adrenocorticotropin. The Trp in nuclease is shown from x-ray to be partly buried. Demchenko et al. (1993) and Longworth (1971) have shown that the solvent is able to relax around the Trp in staphylococcal nuclease.
But how can this explain why in the TS glass the fluorescence of Trp is broad and red-shifted even at low temperature? There is always water in the sample, and there may be water located around the indole chromophore. The sugars are larger and less flexible than glycerol. As TS glass is formed, the smaller water molecules would remain at the protein surface or around the surface of the model compound NATA. Consequently, there may be a layer of fluid water around the chromophore that rearranges upon excitation. Relevant to the view that there is motion of water in the glass, single molecules of a fluorescent cytochrome c derivative in trehalose films were found to undergo large angular motions on the timescale of seconds, also supporting the idea of motion of proteins incorporated into glass (Mei et al., 2003).
For this model to work out, this water must have the character of being mobile on the timescale of the excited singlet state (ns) and the mobility must be temperature independent down to the lowest temperatures studied (∼20 K). It has been known for a long time that turning of the protons around the oxygen atom in water is a low-energy step (Bjerrum, 1952). The protons do not have to move very far to change the orientation of the dipole. Since the fluorescence spectrum of NATA in TS glass remains red-shifted and temperature independent, a mechanism for motion at low temperature may be tunneling (Hammes-Schiffer, 2001). Tunneling effects on Trp in solution have been suggested by quantum calculations (Simonson et al., 1997). In ice systems, the dielectric relaxation is very sensitive to the isotope (Bruni et al., 1993), as is the case for reactions involving tunneling of hydrogen. In our case, substituting with D2O did not lead to resolution (Fig. 15). This does not necessarily rule out the solvent tunneling mechanism to explain the spectrum, however, since we would need to monitor carefully the rates of relaxation. The rotation of water in the sugar films remains an intriguing possibility.
Second, we take another view and consider that the solvent in the TS glass is static. In this model the OH residues from the solvent are close enough to interact with the long lived (nsec) excited-state molecule. In this model, we consider that there are interactions with the electrons of neighboring groups, without the requirement of diffusive atomic displacements. Relevant to this model, we note that absorption and emission are not reverse processes. The absorption of light is a stimulated process. The observed fluorescence arises from a spontaneous emission process. There is a loss of coherence in the process, and the finite lifetime of the excited-state molecule means that there is a different electron distribution from the ground-state molecule, and the molecule can interact with neighboring groups. The interaction of the excited-state molecules with electrons of the surrounding groups has been proposed as a mechanism for broadening of the emission of indole compounds (Lassser et al., 1977), and this electric field interaction would extend over distance.
A model without the rearrangement of the atoms around the chromophore explains the TS glass results, since the fluorescence spectrum of NATA in dry TS glass changes very little with temperature.
But does this explain the fluorescence blue shift of NATA in glycerol/water as temperature decreases? The concentration of OH is larger in glycerol/water than in TS solvent, and if the solvent molecules are randomly arranged, then there should also be the possibility for OH in the glycerol/water to interact with the indole ring. For the static model to work, we consider the possibility that the solvent around the ground-state molecule changes. As temperature decreases, the liquid solvent is expected to rearrange around the chromophore to form the lowest, least energetic arrangement. Models of glycerol indicate that the lowest energy form is such that one side is hydrophobic (Challi et al., 1999). We speculate that this side would tend to be exposed to the aromatic indole ring as the lowest energy structure is obtained. A contributing factor to this ordering effect around the chromophore would be that as temperature decreases, the H-bonding between the OH's of water and glycerol would tend to increase, thereby excluding the interaction between the glycerol OH and the aromatic ring. This would increase the likelihood that the environment around the indole becomes more hydrophobic. For the solid, sugar film, the sugar is stable over the entire range (Fig. 7), and so the likelihood of “complexes” existing at room temperature is retained at low temperature.
Third, we consider “other” effects. Excess energy of excitation must be accommodated by the surroundings. The major peak of excitation is at 289 nm, and the apparent S0,0 peak of emission is at 305 nm for NATA in glycerol and water (Fig. 9). The energy difference is 1800 cm−1. Because there are so many degrees of freedom in the solvent, this vibrational energy is rapidly dissipated into the solvent bath, as shown by examination of probe molecules at short times (Castner et al., 1987). However, the energy dissipation may be different in the two solvents. This was suggested by spectral diffusion dynamics of cytochrome c in trehalose and glycerol/water at low temperature (Ponkratov et al., 2002). The H-bonding is different in the two matrices, as indicated by HOH bend and stretch frequencies (Fig. 6). The local electric field at the indole ring may be different, which could affect the excited-state geometry. This effect was invoked to rationalize the spectrum of heme in cytochrome c (Rasnik et al., 2001), although the smaller size of Trp relative to heme may make this less significant.
The above possibilities are not mutually exclusive, and there may be more than one factor causing the difference between the two solvents.
The main observation of the fluorescence data remains: NATA or Trp of proteins in TS at low temperature acts as if it is held in an environment similar to that at room temperature.
Phosphorescence used to characterize the protein interactions with sugar and glycerol: comparison with fluorescence and IR spectroscopy
The phosphorescence of all the indole-group molecules in all the solvents studied shows resolution, and, unlike the fluorescence spectrum, lacks a large spectral shift under different conditions. It is well known that the lesser sensitivity of phosphorescence to solvent is due to the smaller dipole for indole triplet state. The experimental dipole moment for the indole in the 1So state is 2.13 D and in the 1La state it is 5.4 D. The lowest triplet state of indole, 3La, has a calculated dipole moment of ∼1.5 D (Hahn and Callis, 1997). The low dipole moment means that the triplet state does not interact strongly with the dipolar solvent.
In the absence of O2, the lifetime of excited-state molecules is related to flexibility. This is also true for Trp phosphorescence. The phosphorescence lifetime of Trp is seconds long in a rigid environment but becomes only tens of microseconds in a fluid environment (Bent and Hayon, 1975). The O2 molecule is a potent diffusional quencher of phosphorescence from the indole group (Papp and Vanderkooi, 1989). At low temperature in solid samples, O2 can no longer diffuse and so its presence does not cause quenching. Using O2-sensitive dyes it has been demonstrated that O2 can not diffuse through the dry glass at room temperature (Khajehpour et al., 2003). Phosphorescence from Trp can be seen in sugar glasses without deoxygenation (Fig. 13). The ratio of phosphorescence/fluorescence intensity of NATA in glycerol water tracks the glass transition. As seen in Fig. 21, bottom, the glass transition as seen by NATA and parvalbumin for glycerol/water is at ∼160 K, and very little phosphorescence intensity can be seen above 200 K. At 200 K in the wet glass (Fig. 21, middle) the phosphorescence is about half of what it is at low temperature. In dry glass (Fig. 21, top) at 200 K, the phosphorescence is about equivalent to what it is at low temperature. Motion of the chromophore is still implied by the decrease of the phosphorescence at high temperature.
It is concluded, then, that the phosphorescence yield indicates motion in the glass at temperatures ranging from 200 to 300 K. Considering that the flexibility of the indole ring appears related to its phosphorescence lifetime, it is reasonable to consider that large-amplitude low-frequency modes of the solvent influence phosphorescence lifetime.
The many weak bonds that determine protein structure each have temperature dependencies that have overall consequences on protein stability and function. Many studies indicate that proteins behave like glasses in terms of varying flexibility of groups and complexity of reaction kinetics. There are suggestions that proteins may undergo a glass transition, but also that dynamical features of the protein are determined by the glass (Paciaroni et al., 2002; Prestrelski et al., 1993; Vitkup et al., 2000). In the case of comparison between TS and glycerol/water, the protein IR modes, for the most part, resemble the matrix, but the phosphorescence data also indicate motion within the protein that is independent of the solvent (Fig. 21). Prahbu et al. (2000) examined the influence of the glass on the absorbance of buried heme groups in cytochrome c. When the temperature of glass formation was high, the optical absorption band showed an increase width, showing that the glass trapped the large-scale fluctuations that occur at high temperature, but the major contribution to the line-width was due to internal solvent-independent motions. The retention of motion in heme proteins in sugar glasses is also shown by the recombination of CO after photodissociation (Doster et al., 1986). These data would also support the idea that motion in the interior of the protein is allowed in the interior of proteins, but this motion is still influenced by the solvent matrix.
SUMMARY
Water in hydrated TS film shows temperature-dependent features in its HOH bending and OH stretching modes. The conclusion is that water in hydrated amorphous sugar has flexibility. The fluorescence spectra of indole derivatives are sensitive to the glass transition of glycerol/water and the fluorescence shifts to the red at high temperature, consistent with known relaxation of solvent molecules around the excited-state molecule. The fluorescence of NATA in TS glass remains relatively constant with change in temperature, indicating that the glass maintains the same environment over a wide range of temperatures.
Acknowledgments
The authors thank Drs. Bogumil Zelent, Kent Blaisie, Sergio Dalosto, Paul Angiolillo, and Mazdak Khajehpour for helpful discussions. The authors also thank Drs. K.S. Reddy and Chris Moser for help with the Raman spectra.
This work was supported by National Institutes of Health grant PO1 GM 48130.
Abbreviations used: NATA, N-acetyl-l-tryptophanamide; TS, trehalose/sucrose; AG, N-acetylglycine.
References
- Akao, K., Y. Okubo, N. Askawa, Y. Inoue, and M. Sukurai. 2001. Infrared spectroscopic study on the properties of the anhydrous form II of trehalose. Implications for the functional mechanism of trehalose as a biostabilizer. Carbohydr. Res. 334:233–241. [DOI] [PubMed] [Google Scholar]
- Bent, D. V., and E. Hayon. 1975. Excited state chemistry of aromatic amino acids and related peptides. III. Tryptophan. J. Am. Chem. Soc. 97:2612–2619. [DOI] [PubMed] [Google Scholar]
- Bjerrum, N. 1952. Structure and properties of ice. Science. 115:385–390. [DOI] [PubMed] [Google Scholar]
- Brand, L., and D. Toptygin. 2000. Spectrally and time-resolved fluorescence emission of indole during solvent relaxation. A quantitative model. Chem. Phys. Lett. 322:492–502. [Google Scholar]
- Bruni, F., G. Consolini, and G. Careri. 1993. Temperature dependence of dielectric relaxation in H2O and D2O ice. A dissipative quantum tunneling approach. J. Chem. Phys. 99:538–547. [Google Scholar]
- Callis, P. R. 1991. Molecular orbital theory of the 1Lb and 1La states of indole. J. Chem. Phys. 95:4230–4240. [Google Scholar]
- Callis, P. R., J. T. Vivian, and L. S. Slater. 1995. Ab initio calculation of vibronic spectra for indole. Chem. Phys. Lett. 244:53–58. [Google Scholar]
- Carpenter, J. F., and J. H. Crowe. 1989. An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry. 28:3916–3922. [DOI] [PubMed] [Google Scholar]
- Castner, E. W., Jr., J. Maroncelli, and G. R. Fleming. 1987. Subpicosecond resolution studies of solvation dynamics in polar aprotic and alcohol solvents. J. Chem. Phys. 86:1090–1097. [Google Scholar]
- Challi, R., P. Procacci, G. Cardini, and S. Califano. 1999. Glycerol condensed phases. Part II. A molecular dynamics study of the conformation structure and hydrogen bonding. Phys. Chem. Chem. Phys. 1:879–885. [Google Scholar]
- Cordone, L., P. Galajda, E. Vitrano, A. Gassmann, A. Ostermann, and F. Parak. 1998. A reduction of protein specific motions in co-ligated myoglobin embedded in a trehalose glass. Eur. Biophys. J. 27:173–176. [DOI] [PubMed] [Google Scholar]
- Crowe, J. H., F. A. Hoekstra, and L. M. Crowe. 1992. Anhydrobiosis. Annu. Rev. Physiol. 54:579–599. [DOI] [PubMed] [Google Scholar]
- Demchenko, A. P., K. Gryczynski, Z. Gryczynski, W. Wiczk, H. Malak, and M. Fishman. 1993. Intramolecular dynamics in the environment of the single tryptophan residue in staphylococcal nuclease. Biophys. Chem. 48:39–48. [DOI] [PubMed] [Google Scholar]
- Doster, W., A. Bachleitner, R. Dunau, M. Hiebl, and E. Luscher. 1986. Thermal properties of water in myoglobin crystals and solutions at subzero temperatures. Biophys. J. 50:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douzou, P. 1977. Cryobiochemistry. An Introduction. Academic Press, London.
- Eftink, M. R., L. A. Selvidge, P. R. Callis, and A. A. Rehms. 1990. Photophysics of indole derivatives: experimental resolution of La and Lb transitions and comparison with theory. J. Phys. Chem. 94:3469–3479. [Google Scholar]
- Eftink, M. R., and Z. Wasylewski. 1989. Fluorescence lifetime and solute quenching studies with the single tryptophan containing protein parvalbumin from codfish. Biochemistry. 28:382–391. [DOI] [PubMed] [Google Scholar]
- Ferreira, S. T. 1989. Fluorescence studies of the conformational dynamics of parvalbumin in solution: lifetime and rotational motions of the single tryptophan residue. Biochemistry. 28:10066–10072. [DOI] [PubMed] [Google Scholar]
- Gonnelli, M., and G. B. Strambini. 1993. Glycerol effects on protein flexibility: a tryptophan phosphorescence study. Biophys. J. 65:131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried, D. S., E. S. Peterson, A. G. Sheikh, J. Wang, M. Yang, and J. M. Friedman. 1996. Evidence for damped hemoglobin dynamics in a room temperature trehalose glass. J. Phys. Chem. B. 100:12034–12042. [Google Scholar]
- Griko, Y. V., P. L. Privalov, J. M. Sturtevant, and S. Y. Venyaminov. 1988. Cold denaturation of staphylococcal nuclease. Proc. Natl. Acad. Sci. USA. 85:3343–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruner, S. M. 1977. The Application of an Efficient X-Ray Detector to Diffraction from Retinal Rod Outer Segment Membrane. Princeton University, Princeton, NJ.
- Hahn, D. K., and P. R. Callis. 1997. Lowest triplet state of indole: an ab initio study. J. Phys. Chem. A. 101:2686–2691. [Google Scholar]
- Hammes-Schiffer, S. 2001. Theoretical perspectives on proton coupled electron transfer reactions. Acc. Chem. Res. 34:273–281. [DOI] [PubMed] [Google Scholar]
- Helenius, A., and M. Aebi. 2001. Intracellular functions of N-linked glycans. Science. 291:2364–2369. [DOI] [PubMed] [Google Scholar]
- Jeffrey, G. A. 1997. An Introduction to Hydrogen Bonding. Oxford University Press, New York.
- Kaposi, A. D., J. Fidy, E. S. Manas, J. M. Vanderkooi, and W. W. Wright. 1999. Horseradish peroxidase monitored by infrared spectroscopy: effect of temperature, substrate and calcium. Biochim. Biophys. Acta. 1435:41–50. [DOI] [PubMed] [Google Scholar]
- Khajehpour, M., T. Troxler, and J. M. Vanderkooi. 2003. The effect of protein dynamics upon reactions that occur in the heme-pocket of horseradish peroxidase. Biochemistry. 42:2672–2679. [DOI] [PubMed] [Google Scholar]
- Konev, S. V. 1967. Fluorescence and Phosphorescence of Proteins and Nucleic Acids. Plenum Press, New York. 9–11.
- Krimm, S., and J. Bandekar. 1986. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 38:181–364. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. R., and H. Cherek. 1980. Dipolar relaxation in proteins on the naosecond timescale observed by wavelength-resolved phase fluorometry of tryptophan fluorescence. J. Biol. Chem. 255:831–834. [PubMed] [Google Scholar]
- Lasser, N., J. Feitelson, and R. Lumry. 1977. Exciplex formation between indole derivatives and polar solutes. Isr. J. Chem. 16:330–334. [Google Scholar]
- Librizzi, F., E. Vitrano, and L. Cordone. 1999. Dehydration and crystallization of trehalose and sucrose glasses containing carbonmonoxy-myoglobin. Biophys. J. 76:2727–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth, J. W. 1971. Luminescence of polypeptides and proteins. In Excited States of Proteins and Nucleic Acids. F. F. Steiner and I Weinryb, editors. Plenum Press, New York. 319–484.
- Manas, E. S., Z. Getahun, W. W. Wright, W. F. DeGrado, and J. M. Vanderkooi. 2000. Infrared spectra of amide groups in α-helical proteins: evidence for hydrogen bonding between helices and water. J. Am. Chem. Soc. 122:9883–9890. [Google Scholar]
- Meech, S. R., D. Phillips, and A. G. Lee. 1983. On the nature of the fluorescent state of methylated indole derivatives. Chem. Phys. 80:317–328. [Google Scholar]
- Mei, E., J. Tang, J. M. Vanderkooi, and R. M. Hochstrasser. 2003. Motions of single molecules and proteins in trehalose glass. J. Am. Chem. Soc. 125:2730–2735. [DOI] [PubMed] [Google Scholar]
- Montoro, T., M. Chabbert, J. Tryzyk, and H. Lami. 1988. Subnanosecond-time-resolved emission spectroscopy of 1-methylindole and 2,3-dimethylindole in n-butanol. J. Chem. Phys. 89:2712–2719. [Google Scholar]
- Nara, M., M. Tasumi, M. Tanokura, T. Kiraoki, M. Yazawa, and A. Tsutsumi. 1994. Infrared studies of interaction between metal ions and Ca2+-binding proteins. Marker bands for identifying the types of coordination of the side-chain COO− groups to metal ions in pike parvalbumin (pI = 4.10). FEBS Lett. 349:84–88. [DOI] [PubMed] [Google Scholar]
- Paciaroni, A., S. Cinelli, and G. Onori. 2002. Effect of the environment on the protein dynamical transition: a neutron scattering study. Biophys. J. 83:1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp, S., and J. M. Vanderkooi. 1989. Tryptophan phosphorescence at room temperature as a tool to study protein structure and dynamics. Photochem. Photobiol. 49:775–784. [DOI] [PubMed] [Google Scholar]
- Permyakov, E. A., V. V. Yarmolenko, V. I. Emelyanenko, E. A. Burstein, J. Closset, and C. Gerday. 1980. Fluorescence studies of the calcium binding to whiting (Gadus merlangus) parvalbumin. Eur. J. Biochem. 109:307–315. [DOI] [PubMed] [Google Scholar]
- Pierce, D. W., and S. G. Boxer. 1995. Stark effect spectroscopy of tryptophan. Biophys. J. 68:1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponkratov, V. V., J. Friedrich, and J. M. Vanderkooi. 2002. Solvent effects on conformational dynamics of proteins: cytochrome c in a dried trehalose film. J. Chem. Phys. 117:4594–4601. [Google Scholar]
- Prabhu, N. V., S. D. Dalosto, K. A. Sharp, W. W. Wright, and J. M. Vanderkooi. 2002. Optical spectra of Fe(II) cytochrome c interpreted using molecular dynamics simulations and quantum mechanical calculations. J. Phys. Chem. B. 106:5561–5571. [Google Scholar]
- Prestrelski, S. J., N. Tedeschi, T. Arakawa, and J. F. Carpenter. 1993. Dehydration-induced conformational transitions in proteins and their inhibition by stabilizers. Biophys. J. 655:661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quay, S. C., and C. C. Condie. 1983. Conformational studies of aqueous melittin: thermodynamic parameters of the monomer-tetramer self-association reaction. Biochemistry. 22:695–700. [DOI] [PubMed] [Google Scholar]
- Rasnik, I., K. A. Sharp, J. A. Fee, and J. M. Vanderkooi. 2001. Spectral analysis of cytochrome c: effect of heme conformation, axial ligand, peripheral substituents, and local electric fields. J. Phys. Chem. B. 105:282–286. [Google Scholar]
- Simonson, T., C. F. Wong, and A. T. Brunger. 1997. Classical and quantum simulations of tryptophan in solution. J. Phys. Chem. A. 101:1935–1945. [Google Scholar]
- Stuart, B. 1996. Modern Infrared Spectroscopy. John Wiley & Sons, Chichester, UK.
- Sudhakar, K., C. M. Phillips, C. S. Owen, and J. M. Vanderkooi. 1995. Dynamics of parvalbumin studied by fluorescence emission and triplet absorption spectroscopy of tryptophan. Biochemistry. 34:1355–1363. [DOI] [PubMed] [Google Scholar]
- Sudhakar, K., C. M. Phillips, S. A. Williams, and J. M. Vanderkooi. 1993. Excited states of tryptophan in cold parvalbumin: identification of a short-lived emitting triplet state at room temperature. Biophys. J. 64:1503–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatham, A. S., R. C. Hider, and A. F. Drake. 1983. The effect of counterions on melittin aggregation. Biochem. J. 211:683–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeur, B., and G. Weber. 1977. Resolution of the fluorescence excitation spectrum of indole into the 1La and 1Lb excitation bands. Photochem. Photobiol. 25:441–444. [DOI] [PubMed] [Google Scholar]
- Venyaminov, S. Y., and F. G. Prendergast. 1997. Water (H2O and D2O) molar absorptivity in the 1000–4000 cm−1 range and quantitative infrared spectroscopy of aqueous solutions. Anal. Biochem. 248:234–245. [DOI] [PubMed] [Google Scholar]
- Vitkup, D., D. Ringe, G. A. Petsko, and M. Karplus. 2000. Solvent mobility and the protein ‘glass’ transition. Nat. Struct. Biol. 7:34–38. [DOI] [PubMed] [Google Scholar]
- Vivian, J. T., and P. R. Callis. 2001. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys. J. 80:2093–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, S. T. R., R. P. Cheng, V. Daggett, J. M. Vanderkooi, and W. F. DeGrado. 2003. The hydration of amides in helices: a comprehensive picture from molecular dynamics, IR, and NMR. Protein Sci. 12:520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, W. W., C. J. Baez, and J. M. Vanderkooi. 2002. Mixed trehalose/sucrose glasses used for protein incorporation as studied by infrared and optical spectroscopy. Anal. Biochem. 307:167–172. [DOI] [PubMed] [Google Scholar]